
Infinite building blocks for directed self-assembly of a supramolecular polyoxometalate–cyclodextrin framework for multifunctional oxidative catalysis†
Xiaohui
Liu‡
a,
Jinlin
Zhang‡
a,
Yuxin
Lan
a,
Qi
Zheng
 *b and
Weimin
Xuan
*a
*b and
Weimin
Xuan
*a
aCollege of Chemistry and Chemical Engineering & State Key Laboratory for Modification of Chemical Fibers and Polymer Materials, Donghua University, Shanghai 201620, P. R. China. E-mail: weiminxuan@dhu.edu.cn
bState Key Laboratory for Modification of Chemical Fibers and Polymer Materials & College of Materials Science and Engineering, Donghua University, Shanghai 201620, P. R. China. E-mail: qi.zheng@dhu.edu.cn
First published on 2nd November 2022
Abstract
The utility of infinite building blocks (IBBs) has encouraged the facile construction of porous organic–inorganic hybrid materials such as metal–organic frameworks. However, this strategy remains less explored for building functional supramolecular frameworks that are assembled by weak supramolecular interactions. Here, we report the self-assembly of a supramolecular polyoxometalate–cyclodextrin (POM–CD) framework directed by IBBs of ({MnV12Ce6}{PW10V2}2)∞ for multifunctional oxidative catalysis. The IBBs exhibit an infinite 1D rod shape embedded by {MnV12} and {PW10V2} POM clusters which specifically interact with the outer walls of α-CD because of a weak chaotropic effect and size-mismatch. This has allowed the ordered packing of α-CD between adjacent IBBs via hydrogen bonding and van der Waals contacts, leading to a porous framework containing interconnected channels along two dimensions, with the largest opening up to 12 × 17 Å. The incorporation of catalytically active POM clusters on the porous channels enables the high performance of the POM–CD framework towards selective oxidation of sulfides to sulfoxides. Moreover, cross-linking the POM–CD framework with hexamethylene diisocyanate afforded a more stable porous framework that can be used as an authentically heterogeneous catalyst for not only sulfoxidation but also oxidative cleavage of styrenes under harsher conditions. The robust cross-linked framework can preserve its structural integrity after cycling 5 times with little loss of catalytic performance. This work may pave the way for the rational design of porous organic–inorganic hybrid materials with customized functionalities based on the IBB strategy.
Introduction
The biological system showcases how an ordered bottom-up self-assembly of simple functional building blocks can result in intricate structures with targeted functions to support the very complicated living system.1,2 Learning form nature, the controlled construction of periodic organic–inorganic hybrids bearing pre-designed functionalities is the central topic of chemistry and materials science.3–6 With the aid of strong interactions such as covalent and coordination, porous frameworks including MOFs (metal–organic frameworks) and COFs (covalent organic frameworks) can be rationally fabricated based on the reticular chemistry-guided assembly of molecular building blocks (MBBs),7,8 enabling a myriad of applications in catalysis, energy storage and conversion, gas storage and separation as well as sensors.3,9 In contrast, less attention has been paid to weak supramolecular interactions, although these trivial forces collectively are critical to elucidating the origin of self-assembly and have been used as versatile tools to build supramolecular functional materials based on the directionality of supramolecular interactions such as π–π and host–guest interactions.10,11Polyoxometalates (POMs) represent a class of discrete metal-oxo clusters that exhibit unmatched structural diversity.12–16 Owing to the inherent strong acidity, reversible redox and multiple electron storage and transfer properties, they are extensively employed as functional MMBs to afford crystalline porous materials such as POMOFs (polyoxometalate-based metal–organic frameworks) and cluster-derived COFs in which the assembly is facilitated by strong interactions.17–19 On the other hand, although the relatively weak ion-dipole and van der Waals interactions have already been identified as the primary driving forces to construct supramolecular porous hybrid solids from POM MMBs and macrocyclic calixarene/cucurbituril for more than a decade,20,21 it is not until recently that the discovery of the superchaotropic effect of POMs in solution has unveiled the key role of such solvent-mediated weak interactions in guiding the architecture design of POM-CD (cyclodextrin) complexes.22–25 Since the pioneering work of developing archetypal Keggin and Dawson-CD assemblies,26,27 the scope of POM clusters has evolved from smaller Strandberg, Lindqvist and Anderson to bigger Preyssler, and even to gigantic Mo blue.25,28–31 In spite of these prominent advancements, the deliberate fabrication of porous ordered POM–CD systems is still challenging because of the dominant interacting modes between the POM clusters and native CDs mainly involving host–guest inclusion occurring at the primary or secondary face of CDs, which leads to easy blocking of the cavity of CDs and thus resulting in the formation of non-porous networks.
To address the above-mentioned issues, two approaches have recently been developed by Yang and Cadot based on either the size-mismatch between POMs and CDs or using POMs with an intrinsic weak chaotropic nature (Scheme 1).25,28,32 The former makes use of POM MBBs with bigger size that cannot fit into the cavity of parent CDs,32 while the stable inclusion complex is precluded from forming due to the insufficient chaotropic driving force in the latter case (Scheme 1).25,28 Overall, both approaches rely on the specific interaction of POM MMBs with the outer wall of CDs via weak supramolecular interactions. Nevertheless, only the cavity of CDs is available for the size-mismatch approach, while the presence of free counterions such as alkaline metal ions in the POM–CD frameworks constructed by the second method will in general partially reduce the accessible pore volume. Following the same idea of the two MMB approaches and adopting the concept of “infinite building blocks (IBBs)” in MOF chemistry,7,33 it is reasonable to hypothesize that the transformation of zero-periodic POM-based MMBs into 1D/2D rigid IBBs may facilitate the generation of open POM-CD frameworks owing to their much bigger sizes compared with that of CDs and the judicious selection of weakly chaotropic POM MMBs (Scheme 1). Indeed, as demonstrated by the ready generation of porous all-inorganic POM-OFs via the connection of POM-metal ions IBBs,34,35 the concurrent introduction of oxophilic metal centers into the POM–CD system holds great potential to produce IBBs in situ. Since the metal ions are a part of the IBBs and behave as both the connectors and counterions, they do not occupy the cavities/channels so that the as-obtained pore structures can be accessed to the maximum extent.
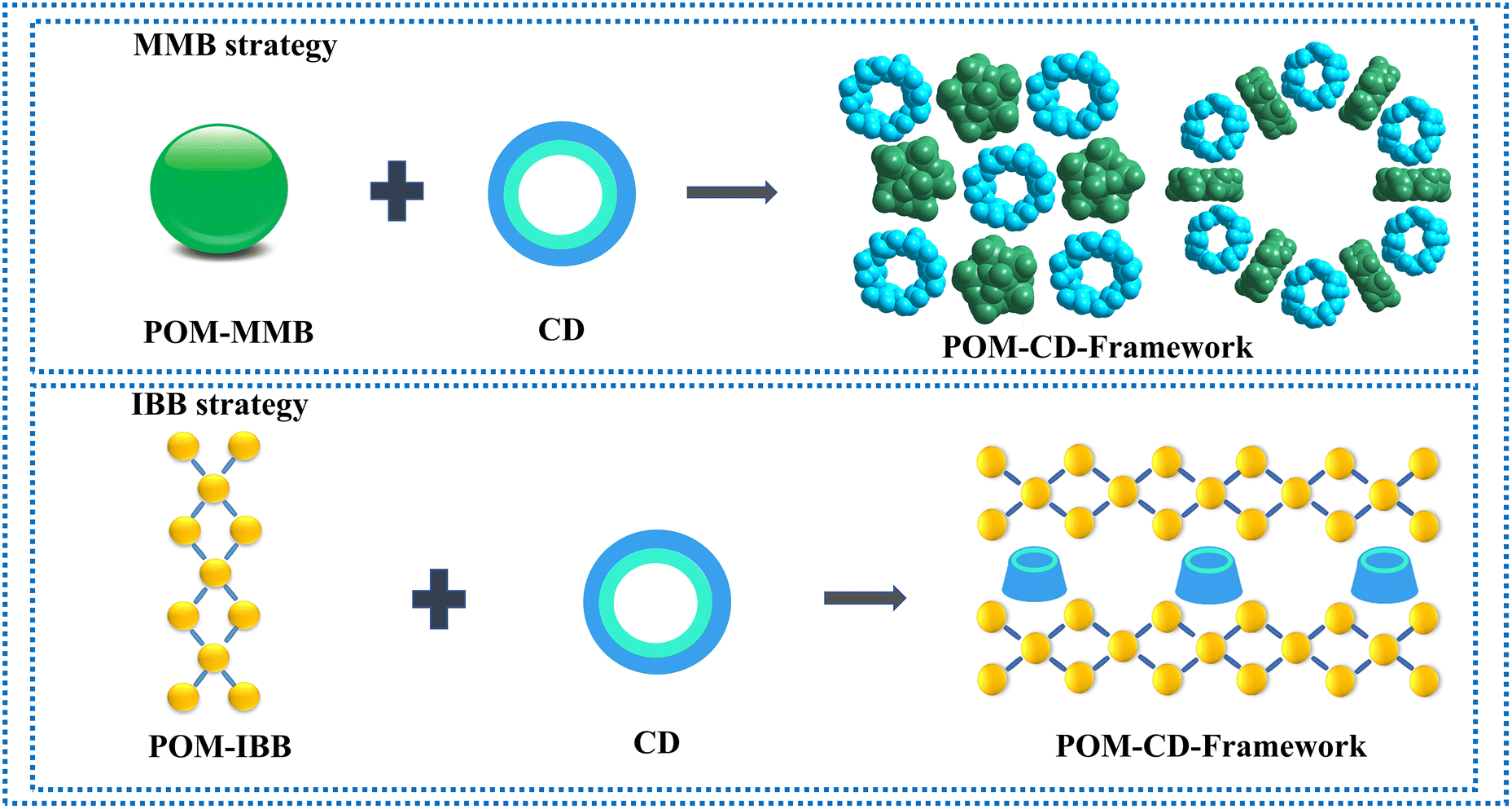 | ||
| Scheme 1 Schematic representation of adopting the MBB strategy and IBB strategy based on size mismatch and chaotropic effect to build POM-CD frameworks. | ||
The intelligent integration of catalytically active POMs and toroidal CDs into supramolecular assemblies has made them excellent catalysts towards organic transformations.36–39 However, one drawback arises from the high solubility of POMs in water, which causes the deconstruction of the complexes in aqueous media or when using aqueous agents such as H2O2.36 To solidify the POM–CD complexes, cross-linking of CDs has been proved to be highly efficient in the construction of POM–CD nanowires, functional 2D membranes and porous frameworks for sieving molecules/nano-objects and sensing pollutants.40–42 Regarding the porous POM-CD supramolecular frameworks, cross-linking of adjacent CDs will further reinforce the frameworks to prevent leakage of POMs while keeping the pore structure accessible even under harsh conditions. This simple method provides numerous options for developing porous POM–CD supramolecular frameworks as heterogenous catalysis platforms that can potentially compete with well-established POMOFs.
Herein, we report the synthesis of one supramolecular POM-CD framework H2Ce3(H2O)13(MnV12O38)0.5(PW10V2O40)(C36H60O 30)·20H2O (1) based on the in situ formed IBBs of ({MnV12Ce6}{PW10V2}2)∞. Due to the 1D rod-like shape and weak chaotropic effect of {MnV12} and {PW10V2}, the IBBs specifically interact with the external surfaces of α-CD and thus give rise to a stable framework showing open channels along both the a- and c-axes, with the largest opening up to 12 × 17 Å. The permanent porosity of 1 was confirmed by N2 and CO2 absorption as well as dye uptake in solution. Coupled with evenly distributed {MnV12} and {PW10V2} on the interior surfaces, 1 showed high performance for selective oxidation of sulfides to sulfoxides at room temperature with high selectivity (up to >99%). Moreover, hexamethylene diisocyanate (HDI) was applied to cross-link α-CDs and 1-CL0.5 (0.5 denotes the molar ratio of HDI to the 18 hydroxyl groups on α-cyclodextrin) was obtained, which showed not only accessible porosity inherited from 1 but also comparable catalytic activity towards sulfoxidation. Notably, the enhanced stability of the cross-linked 1-CL0.5 enabled facile oxidative transformation of styrene and derivatives to the corresponding aldehydes even at 80 °C. The heterogeneous nature of 1-CL0.5 was verified by cycling ICP detection of the reaction mixture and hot-filtration experiments, and the structural integrity was supported by IR and solid-state NMR studies.
Experimental
Synthesis of 1 and 1-CL0.5
Results and discussion
Synthesis of 1
Compound 1 was synthesized from a one-pot reaction of Na7[MnV13O38]·18H2O, Na9[α-PW9O34]·7H2O, Ce(NO3)3·6H2O and α-CD. Heating at 40 °C is critical to drive the construction of 1. An unidentified precipitate will appear soon when heated at 60 °C, while the reaction mixture was turbid at r.t. and resulted in an amorphous solid. Besides, the adoption of α-CD is indispensable for the formation of 1, as using β-CD or γ-CD under the same conditions only resulted in crystalline solids. Moreover, using K5PW10V2O40 instead of Na9[α-PW9O34]·7H2O gave rise to a new POM-CD compound consisting of {MnV13} and α-CD. This indicates that lacunary {α-PW9O34} is essential for the self-assembly of 1.Crystal structure of 1
Compound 1 crystallizes in the orthorhombic P21212 space group. Single-crystal X-ray diffraction structural analysis indicates that the asymmetric unit contains half {MnV12Ce6}, one {PW10V2} and α-cyclodextrin (Fig. 1a). {MnV12} is coordinated with six Ce3+ ions to form an octahedral {MnV12Ce6} building block (Fig. 1b). All the Ce3+ ions adopt a mono-capped square antiprism configuration (Fig. S1†). The coordination sphere of four Ce3+ in the equatorial plane is fulfilled by four terminal oxygen atoms from {MnV12}, one terminal oxygen atom from {PW10V2} and four water molecules, while the two apical Ce3+ are surrounded by four terminal oxygen atoms from {MnV12} and five coordination water molecules. In this manner, {MnV12Ce6} can be regarded as a 4-conneted node which links four adjacent {PW10V2} to form 1D rod-like ({MnV12Ce6}{PW10V2}2)∞ IBBs (Fig. 1c). The IBBs pack in a staggered arrangement along the c-direction with α-CDs intercalated between {PW10V2} units. In fact, {MnV12Ln6} (Ln = La–Pr) have been well established as versatile building blocks: both a 2D inorganic framework and 1D organic–inorganic hybrid were reported based on the 4-connectivity of this rigid building block.43,44 Multiple hydrogen bonds are formed between the outer wall of α-CD and {PW10V2} with O⋯H–C in the range of 2.872–3.028 Å (Fig. S2†). In addition, the coordination water molecules on Ce ions are also involved in strong hydrogen bonding with α-CD (2.875–2.933 Å). As such, 1D open channels are generated along the a- and c-axes, with openings of ∼6 Å × 8 Å and 12 Å × 17 Å, respectively (Fig. 2). Moreover, XPS analysis indicates that all the Mn, V, W and Ce centers adopt +4, +5, +6 and +3 oxidation states, respectively (Fig. S3†).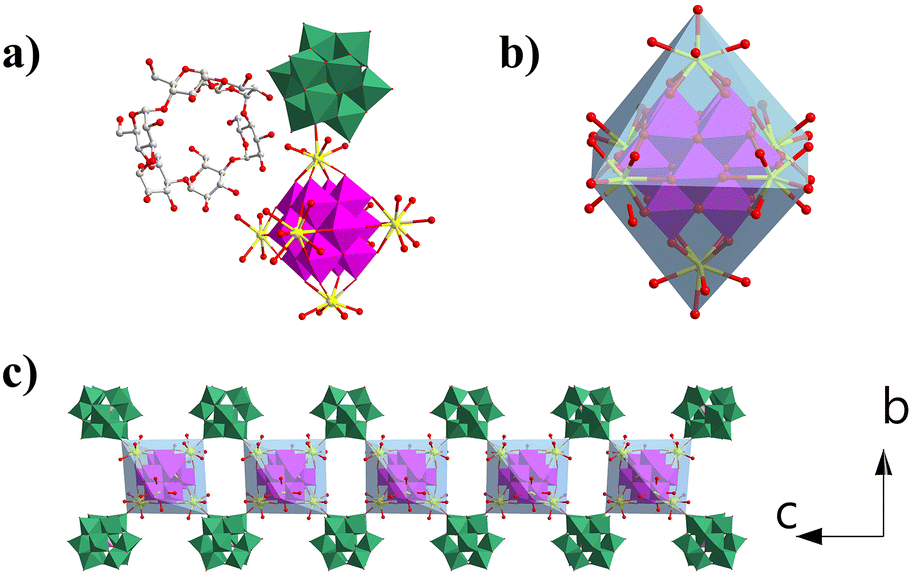 | ||
| Fig. 1 (a) The basic building units of 1 with H atoms removed for clarity, (b) octahedral structure of {MnV12Ce6}, (c) 1D IBBs formed by {MnV12Ce6} and {PW10V2}. Colour codes: Mn blue polyhedron, V purple polyhedron, P pink polyhedron, W/V green polyhedron, Ce yellow ball, O red ball, C grey ball. | ||
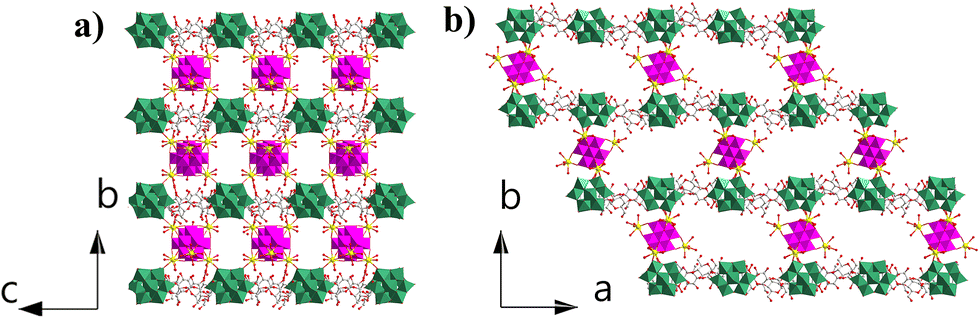 | ||
| Fig. 2 (a) View of the open channels constructed from IBBs and α-CD along a (a) and c (b) directions. The colour codes are the same as in Fig. 1. | ||
Compared with the previously reported porous POM-CD supramolecular frameworks using POMs as MBBs,22–321 represents the first example featuring interconnected channels along two dimensions constructed from ({MnV12Ce6}{PW10V2}2)∞ IBBs. First, the infinite nature of such IBBs makes it hard for them to approach the primary/secondary faces of α-CD due to the size mismatch; secondly, the embedded {PW10V2} bears a negative charge of −5, which makes it to preferentially interact with the external surface of α-CD according to the reported Hofmeister series of Keggin-type POMs where [BW12]5− is located outside γ-CD due to the weak Hofmeister efffect.22 As such, the porous framework of 1 is successfully constructed by IBBs and α-CD. Moreover, protons are proposed as the counterions since negligible sodium ions (0.02%) were detected by ICP (see the ESI† for details). In view of the rather small volume of H+ or H3O+, the pore structure can be accessed to the maximum extent. Taken together, the formation of IBBs enables the ordered fabrication of porous architectures, and this approach may have unique advantages in building porous POM–CD supramolecular frameworks.
Structural characterization of 1 and 1-CL0.5
Due to the poor solubility of 1 in D2O, 0.2 M DCl was used to digest the framework of 1 for 1H NMR and 31P NMR study. As shown in Fig. S4,† the signals corresponding to the protons of H1–H6 on α-CD can be clearly identified. Interestingly, three peaks appear in the 31P NMR spectrum of 1 (Fig. S5†), and their chemical shifts fit well with those of {PW9V3}(δ 13.78 ppm), {PW10V2}(δ 14.45 ppm) and {PW11V}(δ 14.56 ppm) according to the reported literature, respectively.45,46 Because the synthesis of 1 started from the trilacunary {PW9}, the decomposition of {MnV13} on the one hand promoted the structural transformation to {MnV12}, and on the other hand provided a V source to assemble with {PW9} to afford a mixture of {PW9V3}, {PW10V2} and {PW11V} with an average composition of {PW10V2}. Accordingly, three peaks are observed. This is further supported by the ICP analysis of 1 which shows a W/V ratio quite close to 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8. Also, it is consistent with the crystal data where all the metal sites in {PW10V2} show statistical disorder of W and V in a ratio of 10:2. In addition to solution NMR, solid-state 13C NMR and 31P NMR were also performed to elucidate the structure of 1. The characteristic signals of methylene and methine carbon derived from α-CD (60.0–120.0 ppm) can be clearly identified in the 13C NMR spectrum, and the broad peak observed at −18 ppm in the 31P NMR spectrum can be assigned to the {PW10V2} (Fig. S6 and S7†). On the whole, the NMR study has unambiguously demonstrated the structural composition of 1.
:8. Also, it is consistent with the crystal data where all the metal sites in {PW10V2} show statistical disorder of W and V in a ratio of 10:2. In addition to solution NMR, solid-state 13C NMR and 31P NMR were also performed to elucidate the structure of 1. The characteristic signals of methylene and methine carbon derived from α-CD (60.0–120.0 ppm) can be clearly identified in the 13C NMR spectrum, and the broad peak observed at −18 ppm in the 31P NMR spectrum can be assigned to the {PW10V2} (Fig. S6 and S7†). On the whole, the NMR study has unambiguously demonstrated the structural composition of 1.
As shown in the IR spectrum, the peaks at 2923 cm−1 and 1150 cm−1–1000 cm−1 represent the vibration peaks of –CH2–, C–O–C and C–O bonds, respectively (Fig. S8†). The characteristic peaks of W![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (952 cm−1), W–O–W (792 cm−1), VO (916 cm−1) and V–O–V (616 cm−1) as well as V–O–Mn (423 cm−1) can also be clearly identified from the IR spectrum,47,48 further confirming that the structure of compound 1 contains α-CD, {MnV12} and {PW10V2} (Fig. S8†). The N2 sorption isotherm indicated that the BET surface of 1 is 45 m2 g−1 (Fig. S9†). CO2 adsorption and desorption isotherms measured at 25 °C showed a moderate absorption capacity of 9.95 mL g−1 (Fig. S10†), further confirming the permanent porosity of 1. Moreover, 1 could readily adsorb 0.20 rhodamine B (1.39 nm × 0.98 nm × 0.62 nm) and 0.33 methylene blue (1.39 nm × 0.59 nm × 0.15 nm) per formula unit in solution (Fig. S11†). This indicates that the open channels of 1 are fully accessible in solution, which is critical for heterogenous catalysis. The phase purity of 1 could be proved by the good agreement between experimental and simulated PXRD pattern (Fig. S12†).
O (952 cm−1), W–O–W (792 cm−1), VO (916 cm−1) and V–O–V (616 cm−1) as well as V–O–Mn (423 cm−1) can also be clearly identified from the IR spectrum,47,48 further confirming that the structure of compound 1 contains α-CD, {MnV12} and {PW10V2} (Fig. S8†). The N2 sorption isotherm indicated that the BET surface of 1 is 45 m2 g−1 (Fig. S9†). CO2 adsorption and desorption isotherms measured at 25 °C showed a moderate absorption capacity of 9.95 mL g−1 (Fig. S10†), further confirming the permanent porosity of 1. Moreover, 1 could readily adsorb 0.20 rhodamine B (1.39 nm × 0.98 nm × 0.62 nm) and 0.33 methylene blue (1.39 nm × 0.59 nm × 0.15 nm) per formula unit in solution (Fig. S11†). This indicates that the open channels of 1 are fully accessible in solution, which is critical for heterogenous catalysis. The phase purity of 1 could be proved by the good agreement between experimental and simulated PXRD pattern (Fig. S12†).
In order to improve the structural stability and extend the application scope of 1, 1-CL0.5 was prepared by the reaction of hydroxyl groups on α-CD with hexamethylene diisocyanate (HDI).49 As depicted in Fig. 3, HDI can span {PW10V2} and {MnV12Ce6} to crosslink adjacent α-CDs into an organic framework that encapsulates the inorganic IBBs. The successful crosslinking is confirmed by the insolubility of 1-CL0.5 in water/common organic solvents and a variety of spectroscopic studies in a similar way to that of 1.
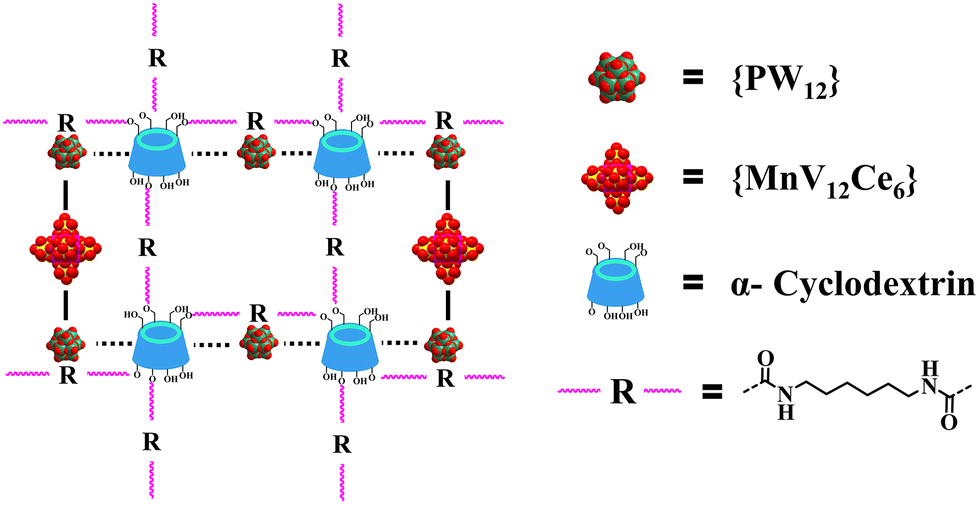 | ||
| Fig. 3 Schematic representation of the structure of 1-CL0.5. | ||
The solid state 13C NMR spectrum of 1-CL0.5 showed the characteristic peaks corresponding to α-CD (60–120 ppm), while the signals between 20 and 50 ppm are attributed to the methylene carbon on HDI and the peak at 160.07 ppm is ascribed to the carbon of amino ether group (–NH–CO–O–), both of which are consistent with the reported values for HDI and amino ether groups in cross-linked CD (Fig. S6†).49 Similar to 1, a broad peak was observed at −18 ppm for {PW10V2} in the solid-state 31P NMR spectrum, indicating the structural integrity of POM clusters in 1-CL0.5 (Fig. S7†). Compared with 1, the sharp and enhanced vibration peaks between 2975–2845 cm−1 are indicative of the presence of considerable methylene groups. Meanwhile, the appearance of new and characteristic signals of CO (1569 cm−1) verify the formation of amino ether groups after cross-linking (Fig. S14†).49 The presence of hexamethylene diamino ether is also reflected by TGA analysis where 1-CL0.5 showed a weight loss ∼8% higher than that of 1 between 50 and 500 °C (Fig. S13†). After confirming the structure, the porosity of 1-CL0.5 was probed by N2 and CO2 adsorption. 1-CL0.5 exhibits a BET surface of 5 m2 g−1 and CO2 absorption capacity of 8.91 mL g−1 (Fig. S15 and S16†). This is caused by the introduction of hydrophobic hexamethylene groups on the inner surfaces, which partially reduce the accessible surfaces. Therefore, the cross-linking afforded a stable framework of 1-CL0.5 with apparent porosity. The enhanced stability is proved by the excellent resistance towards aqueous solutions with a wide pH range (3–10), as negligible amounts of Ce/Mn/V/W were detected by the ICP analysis. The improvement of stability is however at the expense of porosity reduction. As indicated by dye absorption, 1-CL0.5 could hardly adsorb rhodamine B but it could adsorb 0.14 methylene blue per formula unit in solution (Fig. S11†). This value is much lower than in 1, implying that the pore sizes of 1-CL0.5 diminish so that only substrates with a size similar to or smaller than that of methylene blue can diffuse into the open channels. The PXRD analysis of 1-CL0.5 demonstrated that the cross-linked framework still partially preserved the crystallinity of the original 1, as several prominent peaks before 10° can be clearly observed (Fig. S12†).
Catalytic oxidation of sulfides by 1
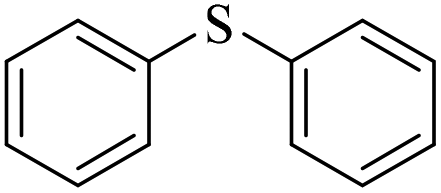
Sulfoxides, as key intermediates in the synthesis of many natural products, have a broad application prospect in medicine, chemistry, industry and other fields.50–52 In fact, POMs have been well established as green and highly efficient catalysts for the synthesis of sulfoxides.53,54 Due to the presence of {MnV12} and {PW10V2} clusters and open channels in 1, we decided to explore the sulfoxidation using 1 as a catalyst. Initially, the catalysis was performed with thioanisole (2a) as the model substrate. First, the solvents, the amount of catalyst and H2O2 were screened (Tables S3–S5†). Methyl phenyl sulfoxide can be produced in a high yield (95%) and selectivity (95%) using 0.4 mol% compound 1 in methanol at room temperature for 6 h with H2O2 (1.05 eq.) as the oxidant. In contrast, the precursors of 1 or stoichiometric mixtures of the precursors showed lower yield of the targeted 3a (Table S6†), demonstrating the synergistic effect derived from {MnV12Ce6}, {PW10V2} and α-CD in a porous supramolecular framework.With the optimal reaction conditions in hand, the substrate scope was then examined. Thioanisoles bearing both electron-donating and electron-withdrawing substituents could be converted to the corresponding sulfoxides with a high yield and selectivity (Table 1, entries 2–5), although longer time is required for 4-nitrothioanisole (3e). Switching from the methyl to ethyl group, overoxidation happened even when the reaction time was reduced to 4 h, with a yield of 82% for 3f (Table 1, entry 6). Similar to thionanisoles, alkyl sulfides can also be smoothly transformed to sulfoxides in a high yield (91%, Table 1 entry 7). In contrast, a heterocyclic substrate was obtained in relatively lower yield even after reacting for 48 h (Table 1, entry 8). Moreover, sulfoxidation of a bulky substrate, diphenyl sulfide, can also give 3i in a good yield of 80% (Table 1, entry 9). Overall, 1 manifested general applicability towards a variety of sulfides. After the catalysis, the recovered catalyst 1 was characterized to confirm the structural integrity. PXRD showed that the diffraction peaks of recovered 1 become very broad, indicative of the partial loss of crystallinity (Fig. S18†). The IR spectrum and elemental analysis result of recovered 1 are almost the same as pristine 1 (Fig. S19 and Table S7†), implying that the basic structure of 1 was intact after catalysis. However, the ICP detection of the reaction mixture after removing 1 revealed a slight leashing of metal ions from {MnV12Ce6} and {PW10V2}, accounting for a weight loss of ∼2.3%. This means that the catalysis did not completely proceed in a heterogenous manner.
|
|
|||
|---|---|---|---|
| Entry | Substrate | Yieldb (%) | |
| a Reaction conditions: 2a (0.2 mmol), 1 (0.4 mol%), 30% H2O2 (1.05 eq.), r.t., methanol (2 mL), 6 h. b Yields are determined by GC-MS using mesitylene as an internal standard. c 8 h. d 12 h. e 4 h. f 24 h. g 8 h. The “n.d.” means “not detected”. | |||
| 1 |
|
R = H | 3a/4a = 95/5 |
| 2 | R = CH3 | 3b/4b = 96/4 | |
| 3c | R = OCH3 | 3c/4c = 93/4 | |
| 4 | R = Cl | 3d/4d = 93/trace | |
| 5d | R = NO2 | 3e/4e = 93/n.d. | |
| 6e |
|
3f/4f = 82/18 | |
| 7 |
|
3g/4g = 91/9 | |
| 8f |
|
3h/4h = 85/n.d. | |
| 9g |
|
3i/4i = 80/15 | |
Catalytic oxidation of sulfides and oxidative cleavage of styrenes by 1-CL0.5
In view of the fact that 1 can be slightly dissolved in the catalytic system, this prompted us to explore the utilization of more stable 1-CL0.5 as a heterogeneous catalyst under the same conditions. In most cases, lower conversion of sulfides was observed even after extending the reaction time to 19 h (Table S8†). Therefore, we sought to promote the conversion via tuning the reaction temperature (Table S9†). Upon a slight increase of temperature to 40 °C, most sulfide substrates can be facilely converted to the related sulfoxides with comparable catalytic efficiency and selectivity to 1 under otherwise identical conditions (Table 2). The lower yield of 3e and 3h may be caused by the introduction of hexamethylene groups to the inner surfaces of 1-CL0.5 (Table 2, entries 5 and 8), which can exert a detrimental impact on the sulfides containing either a strong electron-withdrawing group or a heterocycle skeleton. Also, the amino ether groups generated by cross-linking may be involved in hydrogen bonding with 4-nitrothioanisole and 2-sulfidomethyl pyridine, which might forbid the interaction of these substrates with catalytically active sites. Interestingly, the bulky substrate diphenyl sulphide can be converted into sulfoxide in better selectivity as compared with 1 (Table 2, entry 9). This may originate from the enhanced confinement effect from cross-linking, which has been long known to facilitate the selectivity in porous materials such as zeolites and MOFs.|
|
|||
|---|---|---|---|
| Entry | Substrate | Yieldb (%) | |
| a Reaction conditions: 2a (0.2 mmol), 1-CL0.5 (0.4 mol%), 30% H2O2 (1.10 eq.), 40 °C, methanol (2 mL), 6 h. b Yields are determined by GC-MS using mesitylene as an internal standard. c 8 h. d 12 h. e 4 h. f 24 h. g 8 h. The “n.d.” means “not detected”. | |||
| 1 |
|
R = H | 3a/4a = 95/5 |
| 2 | R = CH3 | 3b/4b = 97/3 | |
| 3c | R = OCH3 | 3c/4c = 89/7 | |
| 4 | R = Cl | 3d/4d = 95/5 | |
| 5d | R = NO2 | 3e/4e = 63/5 | |
| 6e |
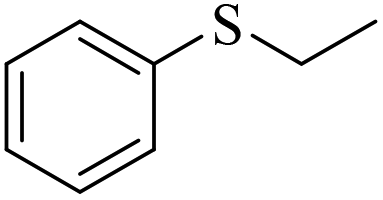
|
3f/4f = 83/10 | |
| 7 |
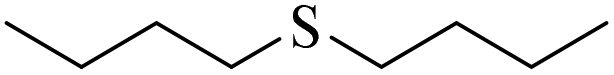
|
3g/4g = 90/10 | |
| 8f |
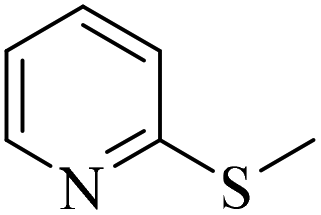
|
3h/4h = 74/n.d. | |
| 9g |
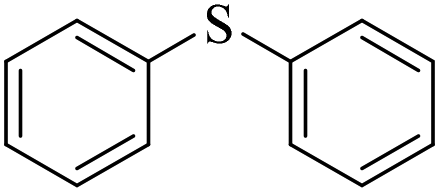
|
3i/4i = 79/5 | |
In a similar way, IR spectrum and elemental analysis also confirmed that the structure of 1-CL0.5 was intact after sulfoxidation (Fig. S20 and Table S11†). In contrast to 1, the ICP analysis of the filtered solution indicated negligible amounts of metal ions (<0.01%). Moreover, the heterogeneity of the 1-CL0.5 catalyst was verified by the hot-filtration experiment, and no additional oxidation product was observed after the filtration of 1-CL0.5 (Fig. S21†). All these results suggested that 1-CL0.5 was robust enough to heterogeneously catalyse the oxidation without structure decomposition.
Inspired by the successful application of 1-CL0.5 for heterogenous sulfoxidation, we then further examined its potential for other important catalytic transformations under harsher conditions. The selective oxidative cleavage of CC was chosen due to its wide application in the synthesis of carbonyl compounds such as aldehydes and ketones.55–57 In addition, this reaction in general requires a higher temperature to cleave the double bond.58,59 Initially, a model reaction is performed with 4-chlorostyrene (5a) using 1-CL0.5 as the catalyst. After systematically screening the solvents, temperature, reaction time, the amount of catalyst and H2O2 (Tables S12–S16†), 4-chlorobenzaldehyde can be obtained in a yield of 88% with the selectivity of 88/5/7 for aldehyde/ketone/benzoic acid when 0.05 mol% 1-CL0.5 was used to perform oxidation in acetonitrile at 80 °C with H2O2 (2.5 eq.) as oxidant for 4 h. When the starting materials or individual components of 1-CL0.5 were used as the catalysts for styrene oxidation, much lower yields of 6a were obtained, with 51%, 24%, 28% and 15% for Na7[MnV13O38]·18H2O, K5PW10V2O40, Ce(NO3)3·6H2O and α-CD (Table S17†). Next, we used the mixture of starting materials or individual components for the catalysis, and found that the yields of 6e generally improved but were still inferior to that with 1-CL0.5 (Table S17†). This clearly demonstrated the advantage of integrating multiple catalytic sites into an open framework, which provides 1-CL0.5 with a synergetic effect.
Once the optimal reaction conditions were established, the general applicability was extended for various styrene substrates. Styrenes containing electron-withdrawing groups gave comparable yields (76% to 88%) and selectivities (Table 3, entries 1–4). The reaction activity of styrene was inferior to substituted styrene, affording benzaldehyde in a yield of 68% with high selectivity (Table 3, entry 5). For styrenes containing electron-donating groups p-CH3 and p-OCH3, the corresponding aldehydes can be obtained in much improved yields of 86% and 84%, respectively. However, the substrates were more likely to be overoxidized to benzoic acids as compared with 5a–5d, resulting in lower selectivity (Table 3, entries 6 and 7). Bulky p-Ph substituted styrene can also be smoothly converted to the related aldehyde in a high yield (Table 3, entry 8), indicating that the pore structure is still accessible to bigger substrates. As expected, the introduction of the methyl group at the α-position completely excluded the generation of both aldehyde and benzoic acid, giving rise to acetophenone in a yield of 90% (Table 3, entry 9).
|
|
|||
|---|---|---|---|
| Entry | Substrate | Yieldsb (%) | |
| a Reaction conditions: 5a (0.6 mmol), 1-CL0.5 (0.05 mol%), 30% H2O2 (2.5 eq.), 80 °C, acetonitrile (2 mL), 3.5 h. b Yields are determined by GC-MS using mesitylene as an internal standard. c 3 h. d 4 h. e 3 h. The “n.d.” means “not detected”. | |||
| 1c | R = Cl | 6a/7a/8a = 88/5/7 | |
| 2 |
|
R = Br | 6b/7b/8b = 81/12/4 |
| 3d | R = F | 6c/7c/8c = 76/7/5 | |
| 4 | R = NO2 | 6d/7d/8d = 87/7/n.d. | |
| 5c | R = H | 6e/7e/8e = 68/n.d./n.d. | |
| 6 | R = CH3 | 6f/7f/8f = 86/3/11 | |
| 7d | R = OCH3 | 6g/7g/8g = 84/3/10 | |
| 8e | R = ph | 6h/7h/8h = 78/3/15 | |
| 9 |
|
6i/7i/8i = n.d./90/n.d. | |
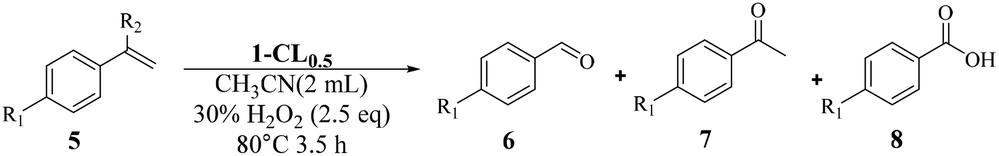
On the whole, 1-CL0.5 demonstrated general applicability towards a variety of styrenes.
IR and elemental analysis showed that 1-CL0.5 had little change before and after reaction, indicating that 1-CL0.5 maintained its structural integrity (Fig. S22 and Table S18†). Also, the ICP analysis of the filtered solution indicated negligible amounts of metal ions (<0.01%) (Table S18†). The presence of all the characteristic peaks in both solid-state 13C NMR and 31P NMR spectra after catalysis further confirms the structural robustness of 1-CL0.5 (Fig. S23 and S24†). Although the hot-filtration experiment showed that ∼16% 4-chlorostyrene could still undergo oxidative cleavage in the absence of the catalyst (Fig. S25†), it was supposed that this was caused by the direct oxidation of H2O2 as a yield of 25% was observed in the blank reaction (Table S17,† entry 2). In addition, the retrieved catalyst could be reused at least five times without an appreciable loss of its high catalytic performance (Fig. S26†). As such, 1-CL0.5 represents the first crosslinked POM–CD hybrid porous material that can be applied as a heterogeneous catalyst towards a series of selective oxidative catalysis.
Conclusions
In summary, a supramolecular POM–CD framework 1 is rationally assembled by using 1D inorganic IBBs. Compared with the general MBB strategy, the evolution of POM MBBs to IBBs enables the facile formation of open channels along the a- and c-axes, with catalytically active {MnV12Ce6} and {PW12} clusters lined evenly on the inner surfaces. The porous structure can be readily accessible both by gases and substrates in solution, thus rendering 1 as a good platform for catalytic transformation. Accordingly, 1 could efficiently promote the sulfoxidation of a variety of sulfides at room temperature with high selectivity (up to 99%). Upon cross-linking with hexamethylene diisocyanate, the as-synthesized 1-CL0.5 not only preserved the high catalytic activity for selective oxidation of sulfides but also facilitated the selective oxidation cleavage of styrene and its derivatives to the corresponding aldehydes with high efficiency. Due to the enhanced stability, the reaction proceeded in a heterogeneous manner, and 1-CL0.5 can be easily recovered and reused for at least 5 cycles with little loss of catalytic performance. This work provides a feasible IBB strategy to build porous organic–inorganic hybrid materials with customized functionalities.Author contributions
W. X. designed research. Q. Z gave suggestions and instructions to perform the experiments. X. L., J. Z. and Y. L. contributed to the synthesis, characterization and property study of the samples. X. L., Q. Z and W. X. wrote the manuscript with contribution from all the authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 92161111, 21901037, 21901038, and 21871042), Shanghai Pujiang Program (No. 19PJ1400200), and the Program for Professor of Special Appointment (Eastern Scholar) at Shanghai Institutions of Higher Learning and International Cooperation Fund of Science and Technology Commission of Shanghai Municipality (No. 21130750100). We also thank the staff from BL17B beamline of National Facility for Protein Science in Shanghai (NFPS) at Shanghai Synchrotron Radiation Facility, for assistance during data collection.References
- K. Das, L. Gabrielli and L. J. Prins, Chemically fueled self-assembly in biology and chemistry, Angew. Chem., Int. Ed., 2021, 60, 20120–20143 CrossRef CAS PubMed.
- A. C. Mendes, E. T. Baran, R. L. Reis and H. S. Azevedo, Self-assembly in nature: using the principles of nature to create complex nanobiomaterials, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2013, 5, 582–612 Search PubMed.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, The chemistry and applications of metal-organic frameworks, Science, 2013, 341, 1230444 Search PubMed.
- A. G. Slater and A. I. Cooper, Function-led design of new porous materials, Science, 2015, 348, aaa8075 Search PubMed.
- T. R. Cook and P. J. Stang, Recent developments in the preparation and chemistry of metallacycles and metallacages via coordination, Chem. Rev., 2015, 115, 7001–7045 Search PubMed.
- M. M. Smulders, I. A. Riddell, C. Browne and J. R. Nitschke, Building on architectural principles for three-dimensional metallosupramolecular construction, Chem. Soc. Rev., 2013, 42, 1728–1754 RSC.
- H. Jiang, D. Alezi and M. Eddaoudi, A reticular chemistry guide for the design of periodic solids, Nat. Rev. Mater., 2021, 6, 466–487 CrossRef CAS.
- K. Geng, T. He, R. Liu, S. Dalapati, K. T. Tan, Z. Li, S. Tao, Y. Gong, Q. Jiang and D. Jiang, Covalent organic frameworks: design, synthesis, and functions, Chem. Rev., 2020, 120, 8814–8933 Search PubMed.
- M. X. Wu and Y. W. Yang, Applications of covalent organic frameworks (COFs): from gas storage and separation to drug delivery, Chin. Chem. Lett., 2017, 28, 1135–1143 CrossRef CAS.
- Z. Liu, S. K. M. Nalluri and J. F. Stoddart, Surveying macrocyclic chemistry: from flexible crown ethers to rigid cyclophanes, Chem. Soc. Rev., 2017, 46, 2459–2478 RSC.
- Z. Li and Y. W. Yang, Functional materials with pillarene struts, Acc. Mater. Res., 2021, 2, 292–305 CrossRef CAS.
- A. M. Cronin, Themed issue: polyoxometalate cluster science, Chem. Soc. Rev., 2012, 41, 7325–7648 RSC.
- M. R. Horn, A. Singh, S. Alomari, S. Goberna-Ferrón, R. Benages-Vilau, N. Chodankar, N. Motta, K. Ostrikov, J. MacLeod, P. Sonar, P. Gomez-Romero and D. Dubal, Polyoxometalates (POMs): from electroactive clusters to energy materials, Energy Environ. Sci., 2021, 14, 1652–1700 RSC.
- S. S. Wang and G. Y. Yang, Recent advances in polyoxometalate-catalyzed reactions, Chem. Rev., 2015, 115, 4893–4962 CrossRef CAS PubMed.
- D. L. Long, R. Tsunashima and L. Cronin, Polyoxometalates: building blocks for functional nanoscale systems, Angew. Chem., Int. Ed., 2010, 49, 1736–1758 CrossRef CAS PubMed.
- M. D. Symes, B. Rausch, G. Chisholm and L. Cronin, Decoupled catalytic hydrogen evolution from a molecular metal oxide redox mediator in water splitting, Science, 2014, 345, 1326–1330 CrossRef PubMed.
- D. Y. Du, J. S. Qin, S. L. Li, Z. M. Su and Y. Q. Lan, Recent advances in porous polyoxometalate-based metal-organic framework materials, Chem. Soc. Rev., 2014, 43, 4615–4632 RSC.
- W. Xu, X. Pei, C. S. Diercks, H. Lyu, Z. Ji and O. M. Yaghi, A metal-organic framework of organic vertices and polyoxometalate linkers as a solid-state electrolyte, J. Am. Chem. Soc., 2019, 141, 17522–17526 Search PubMed.
- M. Lu, M. Zhang, J. Liu, T. Y. Yu, J. N. Chang, L. J. Shang, S. L. Li and Y. Q. Lan, Confining and highly dispersing single polyoxometalate clusters in covalent organic frameworks by covalent linkages for CO2 photoreduction, J. Am. Chem. Soc., 2022, 144, 1861–1871 Search PubMed.
- S. Bhattacharya, A. Barba-Bon, T. A. Zewdie, A. B. Muller, T. Nisar, A. Chmielnicka, I. A. Rutkowska, C. J. Schurmann, V. Wagner, N. Kuhnert, P. J. Kulesza, W. M. Nau and U. Kortz, Discrete, cationic palladium(II)-oxo clusters via f-metal ion incorporation and their macrocyclic host-guest interactions with sulfonatocalixarenes, Angew. Chem., Int. Ed., 2022, 61, e202203114 Search PubMed.
- X. Fang, P. Kögerler, L. Isaacs, S. Uchida and N. Mizuno, et al., Cucurbit[n]uril-polyoxoanion hybrids, J. Am. Chem. Soc., 2009, 131, 432–433 CrossRef CAS PubMed.
- S. Yao, C. Falaise, A. A. Ivanov, N. Leclerc, M. Hohenschutz, M. Haouas, D. Landy, M. A. Shestopalov, P. Bauduin and E. Cadot, Hofmeister effect in the Keggin-type polyoxotungstate series, Inorg. Chem. Front., 2021, 8, 12–25 RSC.
- S. Yao, C. Falaise, S. Khlifi, N. Leclerc, M. Haouas, D. Landy and E. Cadot, Redox-responsive host-guest association between gamma-cyclodextrin and mixed-metal Keggin-type polyoxometalates, Inorg. Chem., 2021, 60, 7433–7441 CrossRef CAS PubMed.
- C. Falaise, S. Khlifi, P. Bauduin, P. Schmid, W. Shepard, A. A. Ivanov, M. N. Sokolov, M. A. Shestopalov, P. A. Abramov, S. Cordier, J. Marrot, M. Haouas and E. Cadot, “Host in host” supramolecular core-shell type systems based on giant ring-shaped polyoxometalates, Angew. Chem., Int. Ed., 2021, 60, 14146–14153 CrossRef CAS PubMed.
- S. Khlifi, J. Marrot, M. Haouas, W. E. Shepard, C. Falaise and E. Cadot, Chaotropic effect as an assembly motif to construct supramolecular cyclodextrin-polyoxometalate-based frameworks, J. Am. Chem. Soc., 2022, 144, 4469–4477 Search PubMed.
- Y. Wu, R. Shi, Y. L. Wu, J. M. Holcroft, Z. Liu, M. Frasconi, M. R. Wasielewski, H. Li and J. F. Stoddart, Complexation of polyoxometalates with cyclodextrins, J. Am. Chem. Soc., 2015, 137, 4111–4118 CrossRef CAS PubMed.
- G. Izzet, M. Menand, B. Matt, S. Renaudineau, L. M. Chamoreau, M. Sollogoub and A. Proust, Cyclodextrin-induced auto-healing of hybrid polyoxometalates, Angew. Chem., Int. Ed., 2012, 51, 487–490 Search PubMed.
- P. Yang, B. Alshankiti and N. M. Khashab, Intrinsically porous molecular building blocks for metal organic frameworks tailored by the bridging effect of counter cations, CrystEngComm, 2020, 22, 2889–2894 RSC.
- C. Falaise, M. A. Moussawi, S. Floquet, P. A. Abramov, M. N. Sokolov, M. Haouas and E. Cadot, Probing dynamic library of metal-oxo building blocks with gamma-cyclodextrin, J. Am. Chem. Soc., 2018, 140, 11198–11201 CrossRef CAS PubMed.
- N. Leclerc, M. Haouas, C. Falaise, S. Al Bacha, L. Assaud and E. Cadot, Supramolecular association between gamma-cyclodextrin and preyssler-type polyoxotungstate, Molecules, 2021, 26, 5126 Search PubMed.
- M. A. Moussawi, M. Haouas, S. Floquet, W. E. Shepard, P. A. Abramov, M. N. Sokolov, V. P. Fedin, S. Cordier, A. Ponchel, E. Monflier, J. Marrot and E. Cadot, Nonconventional three-component hierarchical host-guest assembly based on Mo-blue ring-shaped giant anion, gamma-cyclodextrin, and Dawson-type polyoxometalate, J. Am. Chem. Soc., 2017, 139, 14376–14379 Search PubMed.
- P. Yang, W. Zhao, A. Shkurenko, Y. Belmabkhout, M. Eddaoudi, X. Dong, H. N. Alshareef and N. M. Khashab, Polyoxometalate-cyclodextrin metal-organic frameworks: from tunable structure to customized storage functionality, J. Am. Chem. Soc., 2019, 141, 1847–1851 CrossRef CAS PubMed.
- A. Schoedel, M. Li, D. Li, M. O'Keeffe and O. M. Yaghi, Structures of metal-organic frameworks with rod secondary building units, Chem. Rev., 2016, 116, 12466–12535 CrossRef CAS PubMed.
- H. N. Miras, L. Vila-Nadal and L. Cronin, Polyoxometalate based open-frameworks (POM-OFs), Chem. Soc. Rev., 2014, 43, 5679–5699 Search PubMed.
- S. G. Mitchell, C. Streb, H. N. Miras, T. Boyd, D. L. Long and L. Cronin, Face-directed self-assembly of an electronically active Archimedean polyoxometalate architecture, Nat. Chem., 2010, 2, 308–312 Search PubMed.
- L. Ni, H. Li, H. Xu, C. Shen, R. Liu, J. Xie, F. Zhang, C. Chen, H. Zhao, T. Zuo and G. Diao, Self-assembled supramolecular polyoxometalate hybrid architecture as a multifunctional oxidation catalyst, ACS Appl. Mater. Interfaces, 2019, 11, 38708–38718 Search PubMed.
- C. Zou, P. Zhao, J. Ge, Y. Qin and P. Luo, Oxidation/adsorption desulfurization of natural gas by bridged cyclodextrins dimer encapsulating polyoxometalate, Fuel, 2013, 104, 635–640 CrossRef CAS.
- H. Ge, Y. Leng, C. Zhou and J. Wang, Direct hydroxylation of benzene to phenol with molecular oxygen over phase transfer catalysts: cyclodextrins complexes with vanadium-substituted heteropoly acids, Catal. Lett., 2008, 124, 324–329 Search PubMed.
- J. Wang, H. Yu, Z. Wei, Q. Li, W. Xuan and Y. Wei, Additive-mediated selective oxidation of alcohols to esters via synergistic effect using single cation cobalt catalyst stabilized with inorganic ligand, Research, 2020, 2020, 3875920 Search PubMed.
- W. Guan, G. Wang, J. Ding, B. Li and L. Wu, A supramolecular approach of modified polyoxometalate polymerization and visualization of a single polymer chain, Chem. Commun., 2019, 55, 10788–10791 Search PubMed.
- L. Yue, S. Wang, D. Zhou, H. Zhang, B. Li and L. Wu, Flexible single-layer ionic organic-inorganic frameworks towards precise nano-size separation, Nat. Commun., 2016, 7, 10742 Search PubMed.
- Q. Li, D. Wang, X. Fang, X. Wang, S. Mao and K. Ostrikov, Function–targeted lanthanide–anchored polyoxometalate–cyclodextrin assembly: discriminative sensing of inorganic phosphate and organophosphate, Adv. Funct. Mater., 2021, 31, 2104572 CrossRef CAS.
- S. X. Liu, D. H. Li, L. H. Xie, H. Y. Cheng, X. Y. Zhao and Z. M. Su, Two-dimensional lanthanide heteropolyvanadates of manganese(IV) and nickel(IV) containing two types of heteropoly anions with 1:13 and 1:12 stoichiometry, Inorg. Chem., 2006, 45, 8036–8040 CrossRef CAS PubMed.
- H. Y. An, J. Zhang, S. Z. Chang, Y. J. Hou and Q. S. Zhu, 2D hybrid architectures constructed from two kinds of polyoxovanadates as efficient heterogeneous catalysts for cyanosilylation and knoevenagel condensation, Inorg. Chem., 2020, 59, 10578–10590 Search PubMed.
- P. J. Domaille, 1- and 2-dimensional tungsten-183 and vanadium-51 NMR characterization of isopolymetalates and heteropolymetalates, J. Am. Chem. Soc., 1984, 106, 7677–7687 CrossRef CAS.
- R. I. Maksimovskaya, L. I. Kuznetsova and O. A. Subocheva, Synthesis of phosphorovanadotungsten heteropolycomplexes PVnW12−nO 40(3−n)−(n = 1–4) monitored by 31P and 51V NMR, Russ. Chem. Bull., 1987, 36, 427–431 CrossRef.
- R. Thouvenot, M. Fournier, R. Franck and C. Rocchiccioli-Deltcheff, Vibrational investigations of polyoxometalates. 3. Isomerism in molybdenum(VI) and tungsten(VI) compounds related to the Keggin structure, Inorg. Chem., 1984, 23, 598–605 CrossRef CAS.
- W. Guo, J. Bacsa, J. van Leusen, K. P. Sullivan, H. Lv, P. Kogerler and C. L. Hill, A layered manganese(IV)-containing heteropolyvanadate with a 1:14 stoichiometry, Inorg. Chem., 2015, 54, 10604–10609 Search PubMed.
- M. H. Mohamed, L. D. Wilson and J. V. Headley, Design and characterization of novel beta-cyclodextrin based copolymer materials, Carbohydr. Res., 2011, 346, 219–229 Search PubMed.
- N. Wang, P. Saidhareddy and X. Jiang, Construction of sulfur-containing moieties in the total synthesis of natural products, Nat. Prod. Rep., 2020, 37, 246–275 Search PubMed.
- P. Devendar and G. F. Yang, Sulfur-containing agrochemicals, Top. Curr. Chem., 2017, 375, 82 Search PubMed.
- M. Feng, B. Tang, S. H. Liang and X. Jiang, Sulfur containing scaffolds in drugs: synthesis and application in medicinal chemistry, Curr. Top. Med. Chem., 2016, 16, 1200–1216 CrossRef CAS PubMed.
- L. Huang, S. S. Wang, J. W. Zhao, L. Cheng and G. Y. Yang, Synergistic combination of multi-Zr(IV) cations and lacunary Keggin germanotungstates leading to a gigantic Zr24-cluster-substituted polyoxometalate, J. Am. Chem. Soc., 2014, 136, 7637–7642 CrossRef CAS PubMed.
- Y. Chen, H. An, S. Chang, Y. Li, Q. Zhu, H. Luo and Y. Huang, A POM-based porous supramolecular framework for efficient sulfide–sulfoxide transformations with a low molar O/S ratio, Inorg. Chem. Front., 2022, 9, 3282–3294 RSC.
- S. Velusamy, T. Punniyamurthy and J. Iqbal, Recent advances in transition metal catalyzed oxidation of organic substrates with molecular oxygen, Chem. Rev., 2005, 105, 2329–2363 Search PubMed.
- A. Rubinstein, P. Jimenez-Lozanao, J. J. Carbo, J. M. Poblet and R. Neumann, Aerobic carbon-carbon bond cleavage of alkenes to aldehydes catalyzed by first-row transition-metal-substituted polyoxometalates in the presence of nitrogen dioxide, J. Am. Chem. Soc., 2014, 136, 10941–10948 Search PubMed.
- B. Liu, P. Wang, A. Lopes, L. Jin, W. Zhong, Y. Pei, S. L. Suib and J. He, Au–carbon electronic Iinteraction mediated selective oxidation of styrene, ACS Catal., 2017, 7, 3483–3488 Search PubMed.
- Y. Ma, H. Peng, J. Liu, Y. Wang, X. Hao, X. Feng, S. U. Khan, H. Tan and Y. Li, Polyoxometalate-based metal-organic frameworks for selective oxidation of aryl alkenes to aldehydes, Inorg. Chem., 2018, 57, 4109–4116 Search PubMed.
- Q. X. Gu, X. L. Zhao, M. Meng, Z. Y. Shao, Q. Zheng and W. M. Xuan, Crystalline porous ionic salts assembled from polyoxometalates and cationic capsule for the selective photocatalytic aerobic oxidation of aromatic alcohols to aldehydes, Chin. Chem. Lett., 2022 DOI:10.1016/j.cclet.2022.04.042.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2209404. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2qi02085h |
| ‡ These authors contributed equally. |
| This journal is © the Partner Organisations 2022 |