
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe effect of the donor moiety of DPP based polymers on the performance of organic electrochemical transistors†
Yazhou
Wang‡
a,
Amer
Hamidi-Sakr‡
b,
Jokubas
Surgailis‡
b,
Yecheng
Zhou
 a,
Hailiang
Liao
a,
Junxin
Chen
a,
Genming
Zhu
a,
Zhengke
Li
a,
Sahika
Inal
*b and
Wan
Yue
*a
a,
Hailiang
Liao
a,
Junxin
Chen
a,
Genming
Zhu
a,
Zhengke
Li
a,
Sahika
Inal
*b and
Wan
Yue
*a
aState Key Laboratory of Optoelectronic Materials and Technologies, Key Laboratory for Polymeric Composite and Functional Materials of Ministry of Education, Guangzhou Key Laboratory of Flexible Electronic Materials and Wearable Devices, School of Materials and Engineering, Sun Yat-Sen University, Guangzhou 510275, China. E-mail: yuew5@mail.sysu.edu.cn
bKing Abdullah University of Science and Technology (KAUST), Biological and Environmental Science and Engineering Division, Organic Bioelectronics Laboratory, Thuwal 23955-6900, Saudi Arabia. E-mail: sahika.inal@kaust.edu.sa
First published on 2nd September 2021
Abstract
Organic mixed (ionic and electronic) charge conductors are the building blocks of state-of-the-art bioelectronic devices, including the organic electrochemical transistors (OECTs). Despite the interest in OECTs, the library of polymers that show efficient mixed charge transport is still narrow. In this work, we developed two donor–acceptor (D–A) type polymers based on the glycolated thiophene diketopyrrolopyrrole (TDPP) as the acceptor unit. We combined the acceptor with two donor units distinguished with different electron-donating strengths, i.e., a glycolated thienylenevinylene (gTVT) and a glycolated thiophene–benzothiadiazole–thiophene (gTBTT), leading to the polymers TDPP-gTVT and TDPP-gTBTT, respectively. Using spectral, gravimetric, structural, and electrical characterization techniques combined with density functional theory calculations, we sought to understand the effect of the donor unit on the mixed conduction performance of these D–A type polymers. We found that the stronger electron-donating nature of gTVT vs. gTBTT endowed TDPP-gTVT with a lower backbone curvature, leading to films with a tighter lamellar packing, and thereof, a higher degree of crystallinity compared to TDPP-gTBTT. The combination of a high product of electronic mobility and volumetric capacitance (μC* = 205 F cm−1 V−1 s−1) and a low threshold voltage (−0.36 V) rendered TDPP-gTVT based OECTs superior to TDPP-gTBTT devices. Our work introduces the selection of donor units with higher electron-donating power as a means to tune the film's microstructure, providing an effective approach to optimize mixed conduction properties of D–A type OECT materials.
Introduction
Organic mixed ionic-electronic conductors (OMIECs) transport ionic charges coupled with electronic ones. Mixed conductivity rendered them ideal for developing devices that have an electrolyte interface, such as biosensors, bioactuators, neuromorphic circuits, energy storage, or conversion devices.1,2 A state-of-the-art bioelectronic device that utilizes an OMIEC at the interface with a biological medium is the organic electrochemical transistor (OECT). Compared with the organic field-effect transistor (OFET), which relies on the interfacial accumulation of charges at the channel/dielectric interface, the charge generation in the channel of the OECT occurs in the whole volume of the OMIEC. The volumetric ion-electron coupling in the OECT channel leads to high device transconductance at low gate voltages, typically below 1 V.3 Leveraging the volumetric ion transport in the channel, the OECT acts as a powerful transducer of biochemical events and electrophysiological signals.4–6 Conjugated polymers are typical OECT channel materials due to their relatively intact morphology against ion motion and the possibility of tuning their transport properties by chemical synthesis. Operation in aqueous media however requires the conjugated polymers to have oxidation/reduction potentials within a certain electrochemical window to reduce the possibility of side reactions in water. Despite their high transconductance, OECTs typically suffer from slow switching speeds, on the contrary to OFETs, limiting their use in detecting fast-occurring events. However, with proper optimization of mixed conductivity, conjugated polymers are promising for high performance and stable OECTs, sought after for real-world applications.7The steady-state performance of OECTs is evaluated in terms of their transconductance (gm) at saturation conditions, expressed as follows:3,8
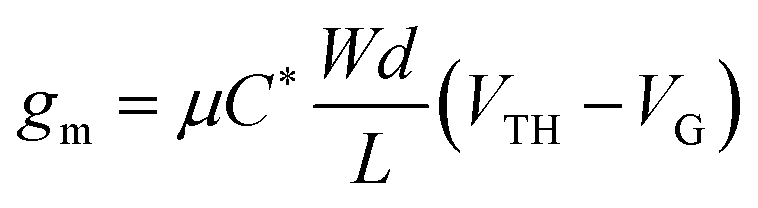 | (1) |
So far, most of the OECTs have relied on the conducting polymer poly(3,4-ethylenedioxythiophene) doped with poly(styrene sulfonate) (PEDOT:PSS) due to its high [μC*], stability, and commercial availability.8–10 These transistors operate in the depletion mode as PEDOT:PSS is electrically conducting in its pristine state. The application of VG turns the OECT OFF. The enhancement mode OECTs, on the other hand, operate in the reverse regime as they use materials that are insulating in their pristine state. In the enhancement mode, the device is turned ON upon application of a VG, which drives dopant ions inside the film to compensate with electronic charges injected from the contacts. The high ON/OFF current ratios achieved with the enhancement-mode devices reduce the background noise, and the zero-level current in the OFF state demands low power.10,11 The abrupt increase in channel current with very low gate voltages is ideal for biosensor applications. Consequently, research efforts in the past years have focused on developing conjugated polymers with maximized [μC*] product for high-performance enhancement-mode OECTs. One common design theme involved rigid and planar electron-rich heterocycles in the backbone.12–15 Such an architecture has resulted in high electronic charge carrier mobility because of the enhanced molecular interactions and order in the film.14,16 To facilitate ion transport, polar side-chains, such as those comprising various lengths of ethylene glycol (EG) units, were grafted on the backbone, which increased the film's hydrophilicity.17,18 As it is the case for OFETs, the side-chain engineering and conjugated backbone chemistry are critical for determining the OECT performance as they govern the solubility, crystallinity, and charge transport properties of the films.16,19–22 However, unlike the OFETs, where the relationships between the chemical structure, morphology of the film and device performance are mostly well-established23,24 such concepts are yet to be extended to OECTs.25,26
An electron-deficient building block with an easily modifiable skeleton, widely used in OFET and organic photovoltaic devices, is the diketopyrrolopyrrole (DPP).9,27–31 DPPs, especially thiophene flanked DPPs (TDPPs), owe their popularity to the high OFET mobilities (up to 10 cm2 V−1 s−1) of their films stemming from their densely π–π stacked units and planar backbones.32,33 The backbone planarity of the DPP-based films could be further tuned by attaching various flanked, electron-donating aromatic units.28 Despite the promise of DPPs in OECT applications, only a few examples exist for the DPPs developed as OMIECs. Du et al. reported a DPP polymer functionalized with lysine side chains.34 These side chains improved the redox properties of the polymer films and enhanced neural cell adhesion. Reynolds et al. reported OECTs comprised of a DPP based polymer with cleavable substituents.35 The polymer was printed from green solvents, and the resulting devices yielded a μC* of 22 F cm−1 V−1 s−1. Giovannitti et al. designed pyridine-flanked DPP polymers with a large ionization potential.36 Although these polymers avoided side reactions with oxygen during the device operation, they showed a low electronic charge carrier mobility. McCulloch et al. have recently reported three donor–acceptor (D–A) type glycolated TDPPs and highlighted the relationship between the polaron delocalization and the OECT mobility. One of these DPP polymers had a high μC* of about 300 F cm−1 V−1 s−1 but a high threshold voltage of 0.52 V.37–41
Thienylenevinylene (TVT) and thiophene–benzothiadiazole–thiophene (TBTT) are two popular electron-donating units used to form D–A type polymers with high charge-carrier mobilities in OFETs.42–44 Incorporating the highly π-extended TVT and TBTT units in the main chain with DPP promotes intermolecular π–π stacking.42,43 These two donors have different electron-donating strengths, which are known to influence their interactions with the acceptor, and consequently, the backbone conformation, intramolecular interactions, and the packing behavior of the chains.42–45 Motivated by these distinct properties that the donor unit can introduce to the D–A type polymer films,44 we developed two new electron-donating moieties, i.e., the glycolated thiophene-vinyl-thiophene (gTVT) and thiophene–benzothiadiazole–thiophene (gTBTT), to construct high-performance OMIECs. We combined these units with a glycolated TDPP and generated two novel D–A type semiconducting polymers. We grafted a suitable length of EG chains on each monomer unit, which improved the polymers' solubility in common organic solvents and endowed the film with redox activity in aqueous electrolytes. By comparing the properties of the two polymers using a combination of solid-state and electrochemical techniques, we systemically elucidated the impact of backbone engineering on the polymer chain conformation, film microstructure, water uptake, surface morphology, and OECT performance. We found that we can tune the backbone curvature via the choice of the door unit, which enabled us to develop an OMIEC with a low threshold voltage (−0.36 V) and high OECT performance (μC* of 205 F cm−1 V−1 s−1) competing with that of the PEDOT:PSS-based devices.
Results and discussion
Synthesis
Two novel monomers of bis(trimethylstannyl)-TVT and bis(trimethylstannyl)-TBTT were synthesized, and their chemical structures and intermediate products (1–6) are shown in Scheme 1. The bis(trimethylstannyl)-TVT 4 begins with nucleophilic substitution of 3-bromothiophene to obtain the glycolated-thiophene 1, followed by the bromination with NBS at low temperature, giving glycolated 2-bromo thiophene 2.46 A Stille coupling between 2 and bis(tributylstannyl)ethene afforded the glycolated-TVT unit (3) and then stannization reaction produced the key monomer 4. Bis(trimethylstannyl)-TBTT 6 was synthesized by methods similar to 4,46,47 except that we used the reagent 4,7-bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2yl)2,1,3-benzothiadiazole. We chose to have two EG units on the side chains to enhance the polymer's solubility in common organic solvents and aid in the uptake of hydrated ions of the electrolyte without causing dissolution or delamination of the films in water.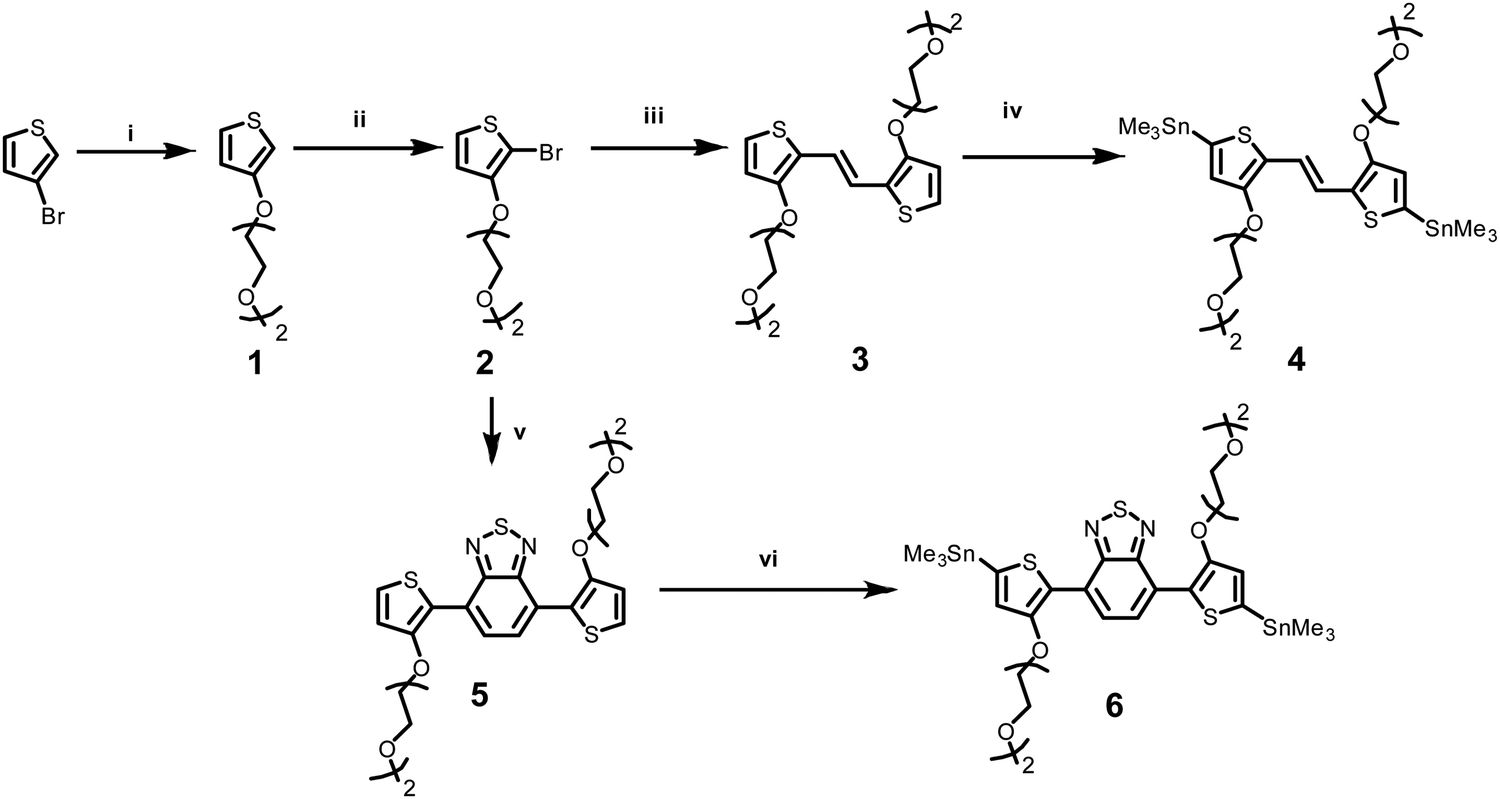 | ||
| Scheme 1 The synthetic route of bis(trimethylstannyl) glycolated TVT, bis(trimethylstannyl) glycolated TBTT monomers. Reaction conditions: (i) t-BuOK, 2-(2-methyloxyethoxy)ethanol, RT, then 100 °C; (ii) NBS, THF/Hexane, 0 °C; (iii) 2-bis(tributylstannyl)ethene, Pd2(dba)3, P(o-tol)3, 100 °C; (iv) n-BuLi, THF, −78 °C, SnClMe3, then RT; (v) 4,7-Bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2yl)2,1,3-benzothiadiazole, Pd2(dba)3, P(o-tol)3, tricaprylylmethylammonium chloride, aq.K3PO4, toluene; (vi) n-BuLi, THF, −78 °C, SnClMe3, then RT. | ||
Besides functionalizing the electron-donating monomers with EG units, we incorporated linear EG chains (n = 4) to the DPP with the anticipation to enhance the films' capacitance (Scheme 2). Tosylation and subsequent glycolation with DPP afforded the non-halogenated thiophene DPP, which was finally brominated to give our acceptor unit. Stille polymerization of the glycolated thiophene dibromoDPP and 2,5-bis(trimethylstannyl)-TVT or 2,5-bis(trimethylstannyl)-TBTT was then used to generate the two polymers, namely, TDPP-gTVT and TDPP-gTBTT, in high yields (Scheme 2). The linear oligo EG side-chains of TVT and TBTT had the same lengths. Like the EG functionalized polymers reported before,47 the polymers' high polarity did not allow the molecular weight analysis by gel permeation chromatography (GPC) in solvents such as chlorobenzene, chloroform, or dimethylformamide. We, therefore, used the matrix-assisted laser desorption/ionization time-of-flight spectrometry (MALDI-TOF) and estimated comparable molecular weights ranging from 4.0 kDa to 8.0 kDa (Fig. S1, ESI†). The resulting polymers showed excellent solubility in common solvents, such as dichloromethane and chloroform, (≈10 mg mL−1). The materials also exhibited good thermal stability with the degradation temperature above 340 °C (Fig. S2, ESI†).
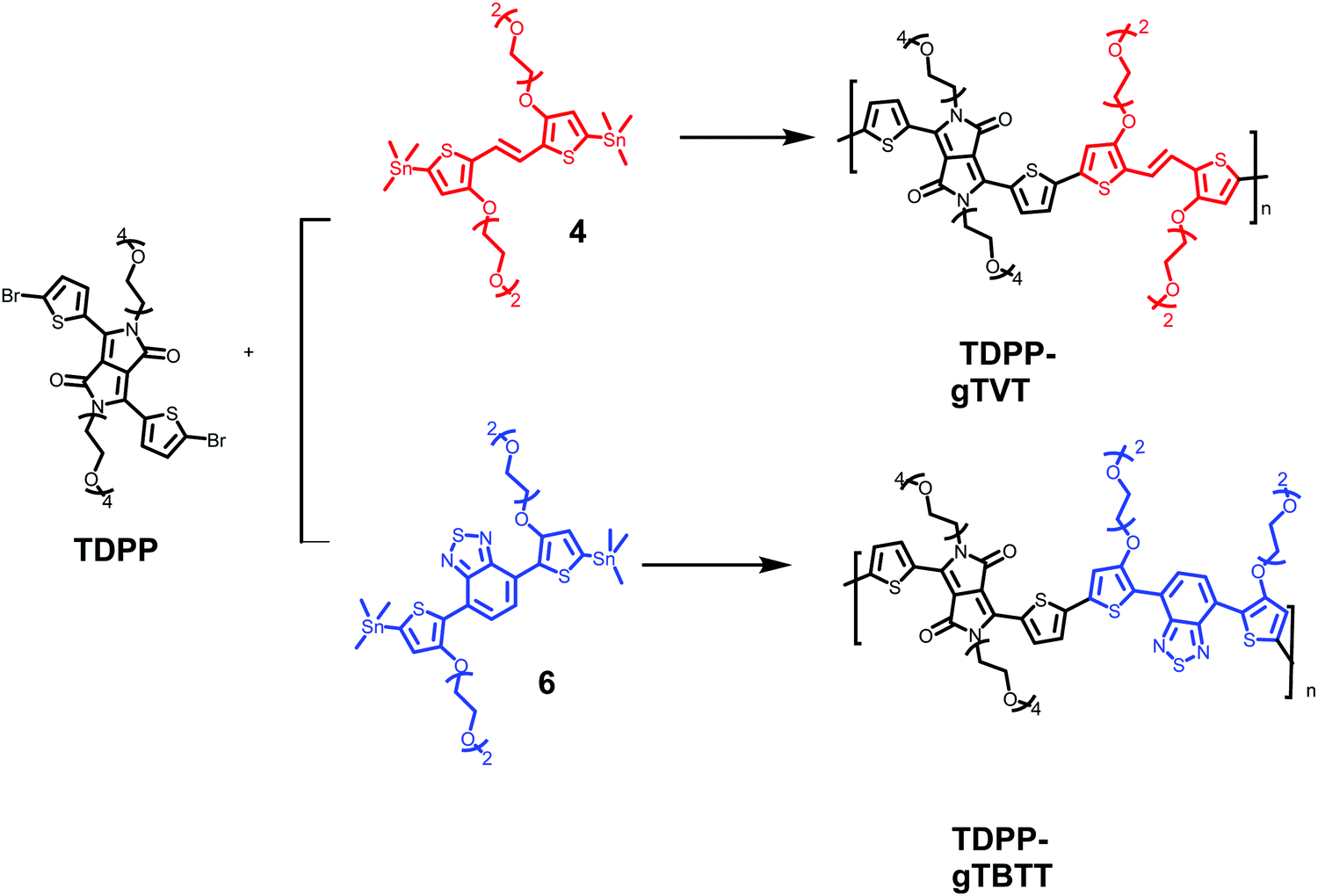 | ||
| Scheme 2 The synthetic route of the glycolated DPP polymers: TDPP-gTVT and TDPP-gTBTT. | ||
Optoelectrical properties
To study the effect of backbone chemistry on the polymers' aggregation properties, we recorded the UV-Vis-NIR absorption spectra of conjugated polymers in solution and as thin films. All the absorption spectra show a similar profile with dual-band absorption (see Fig. 1a for the thin film spectra and Fig. S3 (ESI†) for the solution spectra). We observe a high energy band spanning from 350 nm to 500 nm in all spectra, attributed to the π–π* transition. The low-energy bands originate from an intramolecular charge transfer (ICT) between the DPP and the donors. In solution, we record an absorption maximum (λmax) at 853 nm for TDPP-gTVT, and 785 nm for TDPP-gTBTT (Table 1). When going from the solution to the thin film, the spectra of both films display a red-shift, i.e., the λmax of TDPP-gTVT and TDPP-gTBTT shifts to 878 nm and 813 nm, respectively, indicating that some degree of aggregation or y π–π stacking occurs in the solid-state.48 Notably, while the change in λmax when going from the solution-to-thin film is larger for TDPP-gTBTT, the absorption maximum is at higher wavelengths for TDPP-gTVT, both in solid and solution state. The red-shifted absorption of TDPP-gTVT stems from the stronger donating strength due to the more electron-rich nature of gTVT compared to gTBTT. This trend is consistent with their all-alkylated polymer analogues.42,45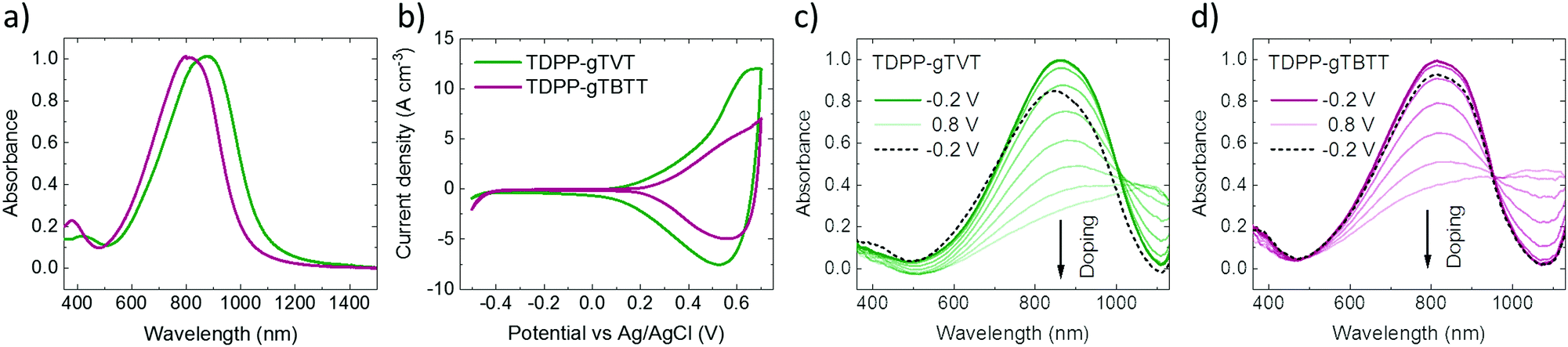 | ||
| Fig. 1 (a) Normalized UV-Vis-NIR absorption spectra and (b) cyclic voltammograms of the polymer films spin-cast from chloroform on ITO substrates. The CV curves were recorded with a scanning speed of 80 mV in−1 aqueous NaCl solution (0.1 M). The 10th cycle is shown. (c and d) UV-Vis absorption spectra of the films during the forward doping sweep (−0.2 V to 0.8 V) direction and as they were de-doped (black dashed spectrum, at −0.2 V). All measurements were performed in aqueous NaCl solution (0.1 M) and voltages were applied vs. Ag/AgCl. The film thickness was in the range of 80–125 nm. | ||
| Polymer | λ solu [nm] | λ film [nm] | IPc [eV] | EAd [eV] | E ox [V] | E opt [eV] |
|---|---|---|---|---|---|---|
| a In chloroform. b As thin films. c Ionization potentials (IPs) were estimated from the onset of the first oxidation peak (Eoxvs. Fc/Fc+) in n-Bu4NPF6 using the equation IP (eV) = 4.8 − Eox. d Electron affinities (EAs) were estimated according to EA (eV) = IP − Eopt. e Estimated from the onset of the first oxidation peak in 0.1 M aqueous NaCl. f Determined from the absorption onset in thin-film UV-Vis-NIR spectra. | ||||||
| TDPP-gTVT | 853 | 878 | 4.44 | 3.30 | 0.21 | 1.14 |
| TDPP-gTBTT | 785 | 812 | 4.60 | 3.37 | 0.29 | 1.23 |
To elucidate the effect of the donor unit on the electron density distribution and energetics, we carried out density functional theory (DFT) calculations on methyl-substituted DPP and methoxy-substituted gTVT and gTBTT of TDPP-gTVT and TDPP-gTBT by Gaussian 09 with the level of B3LYP/6-31G. The geometry optimized structures reveal that each trimer approaches planarity with very small inter-ring torsion (Fig. S4, ESI†). However, the backbone curvature based on the through-space distance is distinctive across the two polymers (Fig. S5, ESI†). Our calculations show that the degree of curvature of the conjugated backbone differs depending on the donor unit, with the dihedral angle between the comonomers in the following order: TDPP-gTVT (157°) > TDPP-gTBTT (134°). We expect the TDPP-gTVT with lower backbone curvature to exhibit stronger interchain interactions, leading to a higher degree of crystallinity.33
We next extracted the energy levels of our polymer films using cyclic voltammetry (CV) measurements performed in 0.1 M tetrabutylammonium hexafluorophosphate (n-Bu4NPF6) in acetonitrile (Fig. S6, ESI†). The highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) energy levels are −4.44 eV and −3.30 eV for TDPP-gTVT, and −4.60 eV and −3.37 eV for TDPP-gTBTT (Table 1). The simulated HOMO/LUMO levels are −4.28 eV and −2.88 eV for TDPP-gTVT, and −4.61 eV and −3.16 eV for TDPP-gTBTT (Fig. S5, ESI†). The trend of the calculated HOMO and LUMO is consistent with experimental results, i.e, both simulated levels of TDPP-gTVT are higher than those of TDPP-gTBTT. As expected from the electron-withdrawing property of BT, TDPP-gTBTT shows lower HOMO and LUMO values than the TDPP-TVT that bears the vinylene motif. These results agree well with the DFT calculations summarized in Fig. S5 (ESI†). Our calculations suggest that HOMO of both polymers is delocalized over the backbone while LUMO localizations differ. The LUMO of TDPP-gTVT is mainly located on the TDPP and the neighboring thiophene of the TVT. As for TDPP-gTBTT, LUMOs are located on the TDPP, the adjacent thiophene, as well as the BT moiety of the TBTT. Therefore, while the HOMO levels of the two polymers are affected by both donor and acceptor units, the LUMO level of TDPP-gTBTT is affected more by the acceptor unit, which can be ascribed to the electron-accepting nature of BT. Finally, the high HOMO levels of these polymers suggest that they are promising to build p-type enhancement-mode OECTs.
Electrochemical analysis and OECT characterization
We recorded the CV curves of the films in 0.1 M NaCl solution to characterize the extent of electrochemical switching between the neutral and oxidized states (Fig. 1b). Both polymers show an increase in the current in the p-type region with a low onset of oxidation below 0.3 V and redox peaks appearing at ca. 0.5 V versus the reference electrode (Ag/AgCl). The polymers show electrochemical stability, deduced from their relatively constant current profiles over ten CV cycles (Fig. S7, ESI†). The gTVT analog exhibits a lower onset (0.21 V vs. 0.29 V) and higher oxidation currents than gTBTT. Fig. 1c and d show the UV-VIS absorption spectrum changes as the polymer films are electrochemically doped in 0.1 M NaCl. We see that, for both films, when they are biased at positive voltages versus Ag/AgCl, the broad absorption band extending from 600 nm to 1000 nm is bleached, while a new band appears above 1000 nm. We attribute the latter to the formation of polarons as anions are injected into the films. Neither of the spectra, however, seem to fully recover upon de-doping at a bias of opposite polarity (i.e., −0.2 V), while the electrochromic property of TDPP-gTBTT appears to be slightly more reversible than TDPP-TVT.As the films showed promising HOMO levels and electrochemical doping performance in the aqueous electrolyte, we next fabricated micron-scale OECTs to assess the effect of the donor units on device performance. The films were patterned on OECT channels that had identical geometry (W = 100 μm, L = 10 μm). The OECT output, transfer and transconductance curves are shown in Fig. 2. While both films switched ON upon application of a negative VG, TDPP-gTVT OECTs showed much better device characteristics. For instance, TDPP-TVT OECTs generated about 4-fold higher ON-currents than the TDPP-gTBTT devices and exhibited a geometry normalized transconductance, gm/(Wd/L), of 47.8 ± 2.8 S cm−1, at least one order of magnitude higher than the TDPP-gTBTT devices (3.65 ± 0.13 S cm−1). TDPP-gTVT has also a lower threshold voltage (VTH = −0.36 V vs. VTH = −0.42 V). The ON/OFF ratios measured are in the order of 104–105, which is at the same range with the recently reported D–A type OECT polymers.34–37 Using eqn (1) above and the steady-state characteristics recorded, we found that TDPP-gTVT has a higher [μC*] of 205.2 ± 12 F cm−1 V−1 s−1 than TDPP-gTBTT ([μC*] of 21.5 ± 0.7 F cm−1 V−1 s−1). The steady-state performance of TDPP-gTVT OECTs is comparable to the performance of other recently published high-performance D–A polymers or all donor polymers.34–37 Importantly, the top OECT performer in this work showed a lower threshold voltage (VTH = −0.36 V) than those of the previously reported D–A polymers, such as p(gDPP-T2) (VTH = −0.52 V) and PDPP-DT (VTH = −0.59 V) (Table S1, ESI†).37,49 When we recorded the transient behaviour of these devices, we found a response time of around 7.3 ms and 8.7 ms for TDPP-gTVT and TDPP-gTBTT, respectively (Fig. S8a and b, ESI†), indicating that the TVT variant has a faster response than TBTT. The devices could however not withstand to the application of square shaped voltage pulses at the gate electrode, i.e., the ID decreased over time (Fig. S8c and d, ESI†). TDPP-gTBTT devices were more stable than TDPP-gTVT, which can also be linked to higher electrochromic reversibility of the TBTT variant (Fig. 1c and d). Note, however, that CV curves of both polymers showed only a negligible change upon cycling (Fig. S6 and S7, ESI†). This discrepancy can be explained by the fact that in OECTs, the electronic conductivity in the channel is the first to suffer from the side effects of swelling, as interconnectivity between electronically conductive regions may become disrupted due to a morphology change upon ion/water uptake. The CV profile may be less affected by such morphological changes as the current response is not limited to horizontal charge transport and governed fully by the interconnectivity of chains. It is also the case that films with higher degree of aggregation tend to be more susceptible to electronic disruption due to electrolyte uptake,50 which can explain why the TDPP-gTVT OECT appears to break down faster than that of TDPP-gTBTT. To prevent this degradation and obtain OECTs robust against pulsing, one may consider increasing the conjugated polymer length, while retaining the film's characteristic electrochemical properties.
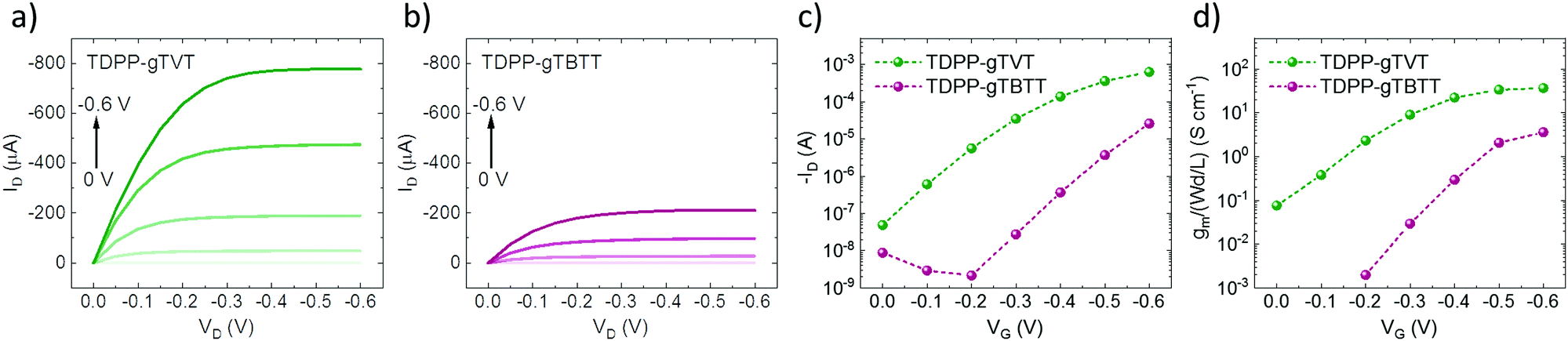 | ||
| Fig. 2 OECT output (a and b), transfer (c) and transconductance curves (d) curves. VG changed from 0 V to −0.6 V with a step of −0.1 V. Transconductance (gm) values were calculated at a drain voltage of VD = −0.6 V and normalized with channel dimensions. The gate electrode was Ag/AgCl and NaCl (0.1 M, aqueous) was used as the electrolyte. The channels had a width of 100 μm and a length of 10 μm. The channel thicknesses were 73 nm and 45 nm, for TDPP-gTVT and TDPP-gTBTT, respectively. | ||
To understand which polymer property governs the OECT behavior and how it is affected by the donor unit in the structure, we investigated our materials' ion uptake and transport abilities. We, first, recorded the electrochemical impedance spectra of these films (Fig. S9 and S10, ESI†). As the films were electrochemically doped at 0.6 V vs. Ag/AgCl, the impedance magnitude decreased, and the phase gained a capacitive character for two polymers. The data in the doped state could be fit with Rs(Rp‖C) equivalent circuit, allowing us to extract the capacitance values of each film (Fig. S11, ESI†). The C* values at low frequencies exhibit a perfect plateau (Fig. S11, ESI†), and C* at f = 0.1 Hz agree with those calculated from the equivalent circuit fit. TDPP-gTVT has a C* of ca. 173.5 F cm−3, higher than that of TDPP-gTBTT (122.5 F cm−3). Calculating the C* allows to estimate the electronic mobility of each polymer in the OECT channel (μOECT). The μOECT of TDPP-gTVT is about an order of magnitude higher than TDPP-gTBTT (ca. 1.1 cm−1 V−1 s−1vs. 0.18 cm−1 V−1 s−1). Since the C* values are relatively comparable, these results show that the high performance of TDPP-gTVT in OECTs stems mainly from the film's high charge carrier mobility. We summarize the OECT characteristics and the materials figures of merit in Table 2.
| Polymer |
I
D![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ON:OFF ON:OFF
|
V TH (V) | μ OECT (cm2 V−1 s−1) |
C*b (F cm−3) |
μC*c (F cm−1 V−1 s−1) |
g m (Wd L−1)d (S cm−1) | I G leakage currente (nA) |
|---|---|---|---|---|---|---|---|
| a OECT hole mobility of the films calculated using eqn (1) by using the C* values. b Calculated from the electrochemical impedance spectra using Rs(RpC) fits. c Estimated using the eqn (1), at maximum gm. d The highest gm values normalized with the channel thickness. Error bars for the capacitance measurements arise from thickness variations. For OECTs measurements, at least 3 channels were measured. e Gate current recorded at VG = VD = −0.6 V. | |||||||
| TDPP-gTVT | 105 | −0.36 | 1.1 ± 0.07 | 173.5 ± 5 | 205.2 ± 12 | 47.8 ± 2.80 | −89.7 |
| TDPP-gTBTT | 105 | −0.42 | 0.18 ± 0.006 | 122.5 ± 15 | 21.5 ± 0.7 | 3.65 ± 0.13 | −28.8 |
It is interesting to note that the CV curves, the polymer energetics or the spectroelectrochemistry data do not predict such a large difference in the OECT performance of these two polymers. To elucidate the origin of the high performance of TVT unit versus TBTT, we performed a combination of morphological and structural studies. First, we used electrochemical quartz crystal microbalance with dissipation monitoring (EQCM-D) technique to evaluate whether these differences are related to the electrolyte uptake behavior of the polymer films. EQCM-D measured the mass accumulating inside the polymer films as they were subject to doping potentials. We observe an initial mass increase for both films upon exposure to the electrolyte due to passive film swelling. TDPP-gTVT, however, exhibits passive swelling of nearly double that of TDPP-gTBTT (22.5% vs. 13.2%, respectively (Fig. S12, ESI†). As we start applying doping potentials, both films start gaining mass. Fig. 3a shows that TDPP-gTVT exhibits an earlier onset of electrolyte uptake into the film (0.3 V) compared to TDPP-gTBTT (0.5 V) and takes up more than 3 times the amount of electrolyte at a doping potential of 0.7 V. The enhanced electrolyte uptake of the TDPP-gTVT film is accompanied with an earlier and greater charge generation (Fig. 3b). The charging behavior in TDPP-gTVT is thus in line with its high C*. As the film swells more readily, ion penetration upon biasing is easier, promoting a greater extent of ionic-electronic charge coupling.
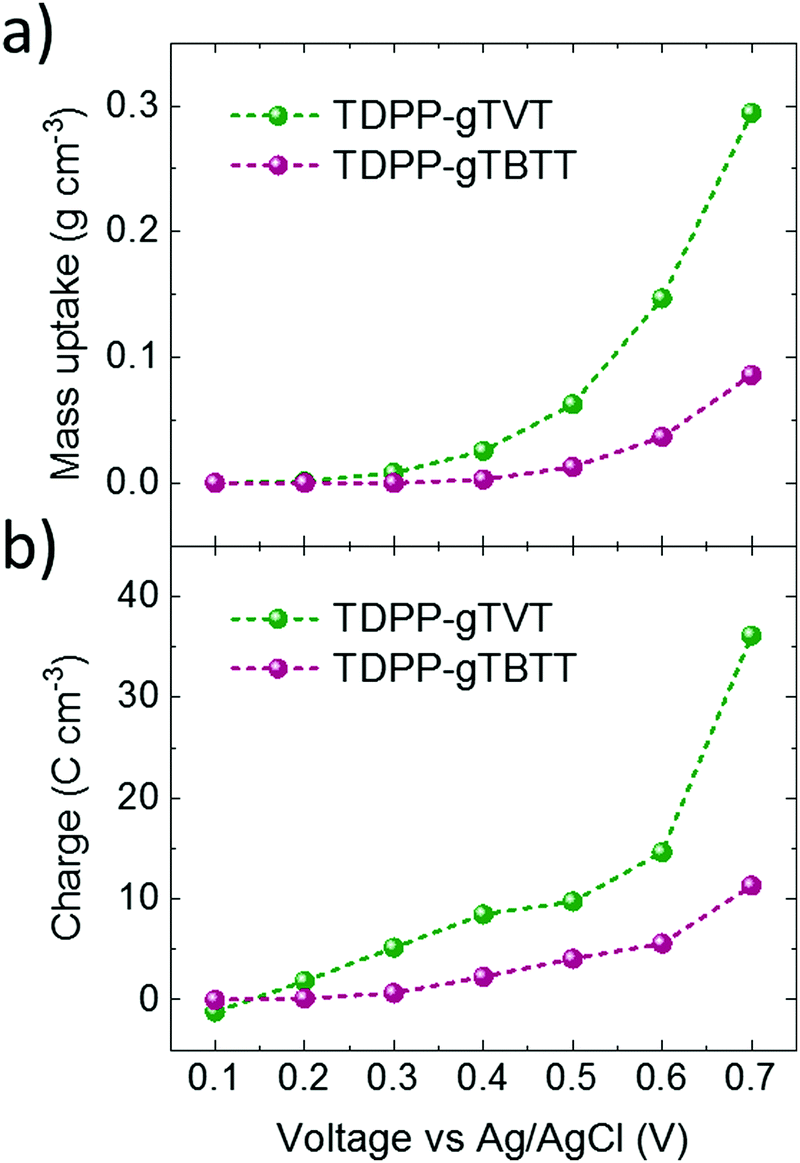 | ||
| Fig. 3 Electrochemical quartz crystal microbalance with dissipation monitoring (EQCM-D) results of TDPP-gTVT and TDPP-gTBTT. The differences in (a) the electrolyte uptake and (b) charge generation per unit volume during the application of doping potentials from 0.1 V to 0.7 V vs. Ag/AgCl with a step of 0.1 V. | ||
To understand what kind of film morphology leads to the improved electrolyte uptake, C* and μOECT, we next performed tapping-mode atomic force microscopy (AFM). AFM scanned the overall surface morphology of the films in their as-cast, hydrated in 0.1 NaCl solution, and after two oxidative CV cycles (Fig. 4). AFM images show that both films have nanofibrillar intercalated structures in their pristine state. The root-mean-square roughness (RMS) of the films is also similar: TDPP-gTVT shows an RMS of 1.06 nm and TDPP-gTBTT has an RMS of 0.92 nm. However, the surface morphology of TDPP-gTVT changes significantly in the hydrated state with a reduction in the RMS (0.90 nm), in contrast to the TDPP-gTBTT film, which shows negligible changes when going from a dry-to-swollen state. The smoothened surface is indicative of a film swollen by ions and water. After doping–dedoping cycles were completed, TDPP-gTVT exhibited fibrillar features characterized by a much rougher height profile (RMS = 1.22 nm). TDPP-gTBTT, on the other hand, became smoother, evidenced by a low RMS of 0.60 nm. We argue that the surface becomes rougher because of ion and water penetration and ejection cycles upon doping and de-doping.51 Formation of distinct regions with different textures is a sign of ion movement in and out of the film, in agreement with the EQCM-D findings.
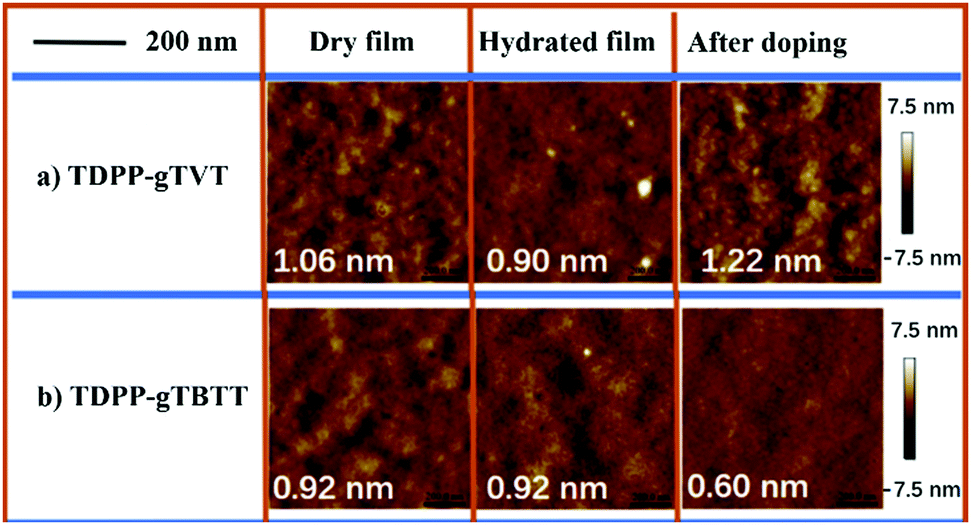 | ||
| Fig. 4 Atomic force microscopy (AFM) height images of (a) TDPP-gTVT and (b) TDPP-gTBTT, along with the RMS values in dry (as-cast), electrolyte swollen and after being subject to two oxidative CV cycles. | ||
Ex situ cycled and swollen state AFM images evidence the changes TDPP-gTVT undergoes due to effective electrochemical doping. However, since the film morphologies of the two films are very similar in their pristine state, AFM does not provide any insight on why one polymer outperforms the other in OECTs. To investigate the packing behavior of chains, we performed two-dimensional grazing incident wide-angle X-ray scattering (GIWAXS) studies (Fig. 5, see the packing parameters summarized in Table S2, ESI†). Scattering profiles show that TDPP-gTVT and TDPP-gTBTT exhibit a lamellar diffraction peak (100) along the in-plane direction at qxy = 0.332 and 0.329 Å−1, which corresponds to a lamellar d-spacing of 18.92 and 19.09 Å, respectively. The shorter lamellar stacking distance of TDPP-gTVT suggests a tighter packing in the lamellar direction. TDPP-gTVT and TDPP-gTBTT also exhibit a significant out-of-plane (010) π–π scattering peak at qz = 1.75 and 1.73 Å−1, corresponding to a π spacing of 3.59 and 3.63 Å, respectively. Together with the lamellar diffraction peak (100) peak in the in-plane direction, this peak suggests a predominantly face-on orientation where π–π stacks are perpendicular to the substrate. Using (010) diffractions, we also calculated the crystal coherence lengths (CCLs) according to the Scherrer equation. We summarized these values in Table S2 (ESI†). The CCLOOP,010 of TDPP-gTVT in the out-of-plane direction is estimated to be 23.5 Å, larger than TDPP-gTBTT (20.2 Å). The larger CCL of TDPP-gTVT suggests higher crystallinity.
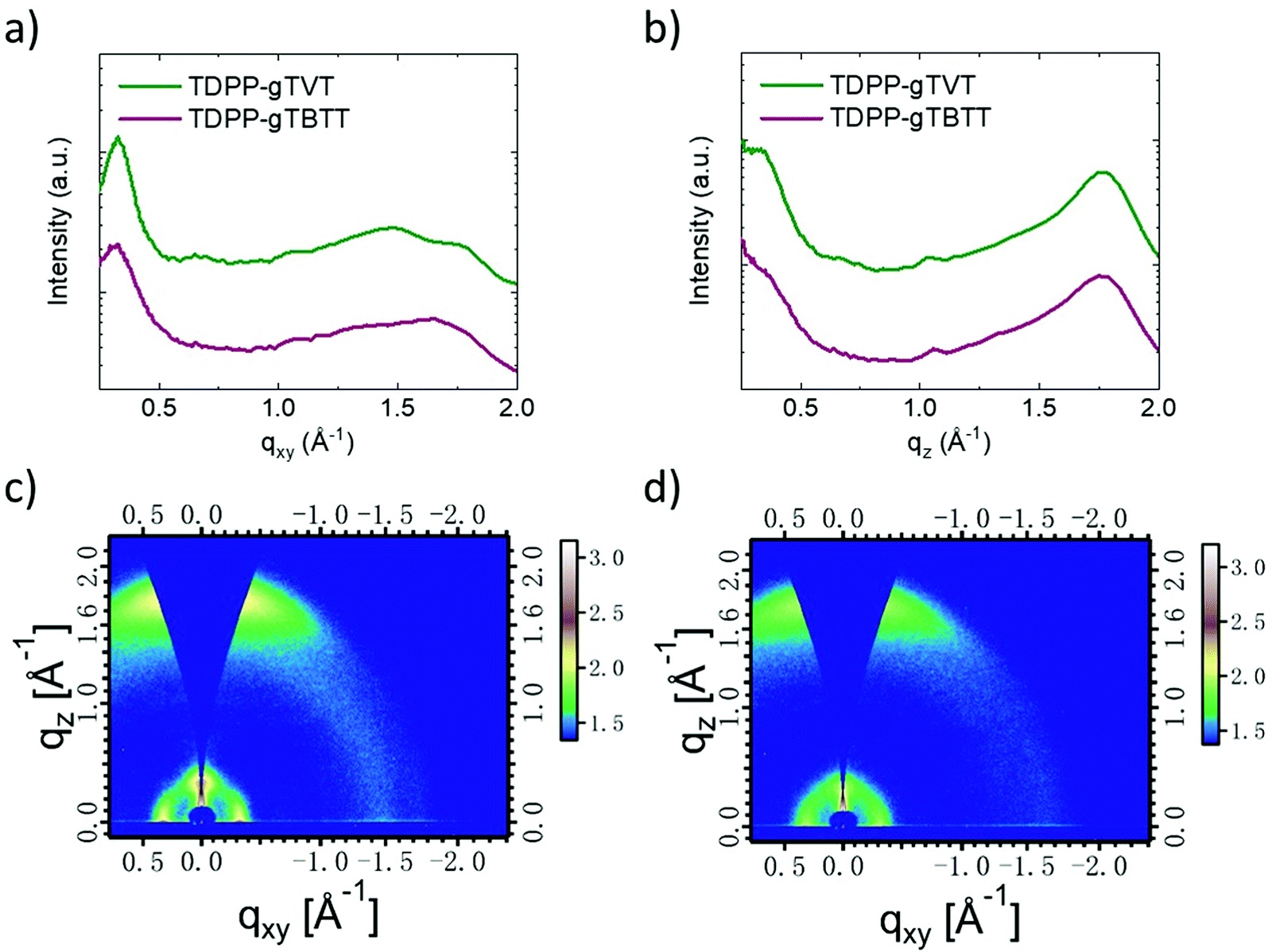 | ||
| Fig. 5 Line-cut profiles of TDPP-gTVT and TDPP-gTBTT films obtained by integration along the (a) in-plane (qxy) and (b) out-of-plane (qz) direction. 2D grazing-incidence wide-angle X-ray scattering (GIWAXS) images of (c) TDPP-gTVT and (d) TDPP-gTBTT films; qz and qxy are the perpendicular and parallel wave vector transfers with respect to the substrate surface. | ||
Although TDPP-gTVT and TDPP-gTBTT form films with similar chain orientation with respect to the substrate, TDPP-gTVT exhibits a higher order in the lamellar stacking direction, a denser π-stacking, and longer CCL, resulting in a higher charge carrier mobility. Recalling the red-shifted spectrum of TDPP-gTVT (Fig. 1a), we suggest that this film has a microstructure with better connected or larger aggregates. Moreover, the lower backbone curvature of TDPP-gTVT, revealed by the DFT calculations, indicates stronger intermolecular interactions, which agrees well with the scattering results. Taken together, these results suggest that by carefully choosing the donor unit, we can fine-tune the packing behavior of the D–A polymer chains through the backbone curvature, which affects their charge-transport ability. Surprisingly, the TVT unit also enables larger swelling upon exposure to electrolyte and electrochemical doping, which leads to higher C*. For this polymer, mobility, promoted by crystallinity, does not come at the expense of ion/water uptake and high capacitance. We postulate that improving both of the parameters that lead to high-performance OECTs was possible by including EG units on both the donor and the acceptor (which promotes ion uptake) while maintaining a rather rigid backbone (which allows for electronic charge transport). Note also that our polymers have relatively short chains, which help form better-stacked aggregates compared with other OMIECs.
Conclusion
In this study, we developed two novel glycolated DPP based copolymers by introducing gTVT and gTBTT units on the backbone. We examined how the donating strength of the donor unit affects the polymer film optical, morphological, electrochemical, and water swelling properties as well as the steady-state performance in microfabricated OECTs. TDPP-gTVT film took up more electrolyte when immersed in it, changed its morphology upon electrochemical doping–dedoping cycles and accumulated more mass upon doping compared to TDPP-gTBTT. TDPP-gTVT exhibited a red-shifted absorption and denser π-stacking. In OECTs, TDPP-gTVT exhibited a relatively high C* of 173.5 F cm−3, μC* of 205 F cm−1 V−1 s−1 and a low VTH of −0.36 V. The superior performance of TDPP-gTVT over TDPP-gTBTT is ascribed to (1) the enhanced affinity to the electrolyte and larger amount of charge generated, (2) the low backbone curvature, which leads to a high degree of crystallinity, promoting high hole carrier mobility. Both polymers however show low device stability, revealed from the decrease in the drain current upon application of voltage pulses at the gate electrode. The instability can be addressed by increasing the molecular weight of the polymers produced. Our results suggest that it is possible to balance capacitance and mobility, which often requires contracting structural features (soft-amorphous content and high crystallinity, respectively) by tuning the donating strength of polymers with minimized backbone curvature and sufficient hydrophilicity.Author contributions
The manuscript was written through the contributions of all authors. Y. Wang, A. Hamidi-Sakr, and Jokubas Surgailis have contributed equally to this work. Wan Yue and Sahika Inal contributed equally to this work.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the National Research Foundation of China (Grant No. 21875291 and 21702240) and China Postdoctoral Foundation (Grant No. 2021M693580). S. I., J. S., and A. H.-S. acknowledge the support from KAUST.References
- B. D. Paulsen, K. Tybrandt, E. Stavrinidou and J. Rivnay, Nat. Mater., 2020, 19, 13–26 CrossRef CAS PubMed.
- D. Ohayon and S. Inal, Adv. Mater., 2020, 32, 2001439 CrossRef CAS PubMed.
- J. Rivnay, S. Inal, A. Salleo, R. M. Owens, M. Berggren and G. G. Malliaras, Nat. Rev. Mater, 2018, 3, 17086 CrossRef CAS.
- S. Wustoni, C. Combe, D. Ohayon, M. H. Akhtar, I. McCulloch and S. Inal, Adv. Funct. Mater., 2019, 29, 1904403 CrossRef CAS.
- E. Macchia, P. Romele, K. Manoli, M. Ghittorelli, M. Magliulo, Z. M. Kovács-Vajna, F. Torricelli and L. Torsi, Anal. Bioanal. Chem., 2020, 412, 5005–5014 CrossRef CAS PubMed.
- D. Khodagholy, T. Doublet, P. Quilichini, M. Gurfinkel, P. Leleux, A. Ghestem, E. Ismailova, T. Hervé, S. Sanaur, C. Bernard and G. G. Malliaras, Nat. Commun., 2013, 4, 1575 CrossRef PubMed.
- S. Inal, J. Rivnay, A. Suiu, G. G. Malliaras and I. McCulloch, Acc. Chem. Res., 2018, 51, 1368–1376 CrossRef CAS PubMed.
- S. Inal, G. G. Malliaras and J. Rivnay, Nat. Commun., 2017, 8, 1767 CrossRef PubMed.
- Y. Fu, N. Wang, A. Yang, H. W. Law, L. Li and F. Yan, Adv. Mater., 2017, 29, 1703787 CrossRef PubMed.
- W. Lee, D. Kim, N. Matsuhisa, M. Nagase, M. Sekino, G. G. Malliaras, T. Yokota and T. Someya, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 10554–10559 CrossRef CAS PubMed.
- A. M. Pappa, D. Ohayon, A. Giovannitti, I. P. Maria, A. Savva, I. Uguz, J. Rivnay, I. McCulloch, R. M. Owens and S. Inal, Sci. Adv., 2018, 4, eaat0911 CrossRef PubMed.
- E. Zeglio and O. Inganäs, Adv. Mater., 2018, 30, 1800941 CrossRef PubMed.
- C. B. Nielsen, A. Giovannitti, D. T. Sbircea, E. Bandiello, M. R. Niazi, D. A. Hanif, M. Sessolo, A. Amassian, G. G. Malliaras, J. Rivnay and I. McCulloch, J. Am. Chem. Soc., 2016, 138, 10252–10259 CrossRef CAS PubMed.
- A. Giovannitti, D. T. Sbircea, S. Inal, C. B. Nielsen, E. Bandiello, D. A. Hanif, M. Sessolo, G. G. Malliaras, I. McCulloch and J. Rivnay, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 12017–12022 CrossRef CAS PubMed.
- E. Zeglio, J. Eriksson, R. Gabrielsson, N. Solin and O. Inganäs, Adv. Mater., 2017, 29, 1605787 CrossRef PubMed.
- M. Moser, J. F. Ponder Jr, A. Wadsworth, A. Giovannitti and I. McCulloch, Adv. Funct. Mater., 2019, 29, 1807033 CrossRef.
- Y. Wang, E. Zeglio, H. Liao, J. Xu, F. Liu, Z. Li, I. P. Maria, D. Mawad, A. Herland, I. McCulloch and W. Yue, Chem. Mater., 2019, 31, 9797–9806 CrossRef CAS.
- Y. Wang, Y. Yu, H. Liao, Y. Zhou, I. McCulloch and W. Yue, Acc. Chem. Res., 2020, 53, 2855–2868 CrossRef CAS PubMed.
- C. B. Nielsen, M. Turbiez and I. McCulloch, Adv. Mater., 2013, 25, 1859–1880 CrossRef CAS PubMed.
- J. Mei and Z. Bao, Chem. Mater., 2014, 26, 604–615 CrossRef CAS.
- M. Moser, T. C. Hidalgo, J. Surgailis, J. Gladisch, S. Ghosh, R. Sheelamanthula, Q. Thiburce, A. Giovannitti, A. Salleo, N. Gasparini, A. Wadsworth, I. Zozoulenko, M. Berggren, E. Stavrinidou, S. Inal and I. McCulloch, Adv. Mater., 2020, 2002748 CrossRef CAS PubMed.
- P. Schmode, A. Savva, R. Kahl, D. Ohayon, F. Meichsner, O. Dolynchuk, T. Thurn-Albrecht, S. Inal and M. Thelakkat, ACS Appl. Mater. Interfaces, 2020, 12, 13029–13039 CrossRef PubMed.
- E. J. W. Crossland, K. Tremel, F. Fischer, K. Rahimi, G. Reiter, U. Steiner and S. Ludwigs, Adv. Mater., 2012, 24, 839–844 CrossRef CAS PubMed.
- A. Hamidi-Sakr, L. Biniek, S. Fall and M. Brinkmann, Adv. Funct. Mater., 2016, 26, 408–420 CrossRef CAS.
- J. W. Onorato and C. B. Luscombe, Mol. Syst. Des. Eng., 2019, 4, 310–324 RSC.
- J. H. Kim, S. Kim, G. Kim and M. Yoon, Macromol. Biosci., 2020, 2000211 CrossRef CAS PubMed.
- L. R. Savagian, A. M. Österholm, J. F. Ponder Jr., K. J. Barth, J. Rivnay and J. R. Reynolds, Adv. Mater., 2018, 1804647 CrossRef PubMed.
- X. Guo, A. Facchetti and T. J. Marks, Chem. Rev., 2014, 114, 8943–9021 CrossRef CAS PubMed.
- R. S. Ashraf, I. Meager, M. Nikolka, M. Kirkus, M. Planells, B. C. Schroeder, S. Holliday, M. Hurhangee, C. B. Nielsen, H. Sirringhaus and I. McCulloch, J. Am. Chem. Soc., 2015, 137, 1314–1321 CrossRef CAS PubMed.
- I. Meager, R. S. Ashraf, S. Mollinger, B. C. Schroeder, H. Bronstein, D. Beatrup, M. S. Vezie, T. Kirchartz, A. Salleo, J. Nelson and I. McCulloch, J. Am. Chem. Soc., 2013, 135, 11537–11540 CrossRef CAS PubMed.
- Z. Liu, G. Zhang and D. Zhang, Acc. Chem. Res., 2018, 51, 1422–1432 CrossRef CAS PubMed.
- I. Kang, H.-J. Yun, D. S. Chung, S.-K. Kwon and Y.-H. Kim, J. Am. Chem. Soc., 2013, 135, 14896–14899 CrossRef CAS PubMed.
- J. Y. Back, H. Yu, I. Song, I. Kang, H. Ahn, T. J. Shin, S.-K. Kwon, J. K. Oh and Y.-H. Kim, Chem. Mater., 2015, 27, 1732–1739 CrossRef CAS.
- W. Du, D. Ohayon, C. Combe, L. Mottier, I. P. Maria, R. S. Ashraf, H. Fiumelli, S. Inal and I. McCulloch, Chem. Mater., 2018, 30, 6164–6172 CrossRef CAS.
- B. Schmatz, A. W. Lang and J. R. Reynolds, Adv. Funct. Mater., 2019, 29, 1905266 CrossRef CAS.
- A. Giovannitti, R. B. Rashid, Q. Thiburce, P. D. Paulsen, C. Cendra, K. Thorley, D. Moia, J. T. Mefford, D. Hanif, D. Weiyuan, M. Moser, A. Salleo, J. Nelson, I. McCulloch and J. Rivnay, Adv. Mater., 2020, 32, 1908047 CrossRef CAS PubMed.
- M. Moser, A. Savva, K. Thorley, B. D. Paulsen, T. C. Hidalgo, D. Ohayon, H. Chen, A. Giovannitti, A. Marks, N. Gasparini, A. Wadsworth, J. Rivnay, S. Inal and I. McCulloch, Angew. Chem., Int. Ed., 2021, 60, 7777–7785 CrossRef CAS PubMed.
- G. Krauss, F. Meichsner, A. Hochgesang, J. Mohanraj, S. Salehi, P. Schmode and M. Thelakkat, Adv. Funct. Mater., 2021, 2010048, DOI:10.1002/adfm.202010048.
- H. Jia, Z. Huang, P. Li, S. Zhang, Y. Wang, J. Wang, X. Gu and T. Lei, J. Mater. Chem. C, 2021, 9, 4927–4934 RSC.
- N. Wang, L. Xie, H. Ling, V. Piradi, L. Li, X. Wang, X. Zhu and F. Yan, J. Mater. Chem. C, 2021, 9, 4260–4266 RSC.
- J. J. Samuel, A. Garudapalli, A. A. Mohapatra, C. Gangadharappa, S. Patil and N. P. B. Aetukuri, Adv. Funct. Mater., 2021, 2102903 CrossRef.
- H. Chen, Y. Guo, G. Yu, Y. Zhao, J. Zhang, D. Gao, H. Liu and Y. Liu, Adv. Mater., 2012, 24, 4618–4622 CrossRef CAS PubMed.
- K. Shi, W. Zhang, D. Gao, S. Zhang, Z. Lin, Y. Zou, L. Wang and G. Yu, Adv. Mater., 2018, 30, 1705286–1705293 CrossRef PubMed.
- H. Huang, Z. Chen, R. P. Ortiz, C. Newman, H. Usta, S. Lou, J. Youn, Y.-Y. Noh, K.-J. Baeg, L. X. Chen, A. Facchetti and T. Marks, J. Am. Chem. Soc., 2012, 134(26), 10966–10973 CrossRef CAS PubMed.
- J. D. Yuen, J. Fan, J. Seifter, B. Lim, R. Hufschmid, A. J. Heeger and F. Wudl, J. Am. Chem. Soc., 2011, 133, 20799–20807 CrossRef CAS PubMed.
- J. Miao, J. Wang, B. Meng, J. Liu and L. Wang, Mater. Chem. Front., 2018, 2, 2333–2339 RSC.
- A. Giovannitti, C. B. Nielsen, D. Sbircea, S. Inal, M. Donahue, M. R. Niazi, D. A. Hanifi, A. Amassian, G. G. Malliaras, J. Rivnay and I. McCulloch, Nat. Commun., 2016, 7, 1–10 Search PubMed.
- E. Wang, Z. Ma, Z. Zhang, K. Vandewal, P. Henriksson, O. Inganas, F. Zhang and M. R. Andersson, J. Am. Chem. Soc., 2011, 133, 14244–14247 CrossRef CAS PubMed.
- X. Wu, Q. Liu, A. Surendran, S. E. Bottle, P. Sonar and W. L. Leong, Adv. Electron. Mater, 2020, 2000701 Search PubMed.
- L. Q. Flagg, C. G. Bischak, J. W. Onorato, R. B. Rashid, C. K. Luscombe and D. S. Ginger, J. Am. Chem. Soc., 2019, 141(10), 4345–4354 CrossRef CAS PubMed.
- A. Savva, R. Hallani, C. Cendra, J. Surgailis, T. C. Hidalgo, S. Wustoni, R. Sheelamanthula, X. Chen, M. Kirkus, A. Giovannitti, A. Salleo, I. McCulloch and S. Inal, Adv. Funct. Mater., 2020, 1907657 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis and all experimental procedures, characterization of all monomers and polymers (TGA, NMR, MALDI-TOF), DFT calculations, UV-Vis-NIR absorption spectra, electrochemical impedance spectra, cyclic voltammograms, GIWAX data, EQCM-D, OECT performance comparison, response time and stability data, and solid-state packing parameters. See DOI: 10.1039/d1tc02994k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |