
A mild and chemoselective CALB biocatalysed synthesis of sulfoxides exploiting the dual role of AcOEt as solvent and reagent†
Silvia
Anselmi
 a,
Siyu
Liu
a,
Seong-Heun
Kim
a,
Sarah M.
Barry
b,
Thomas S.
Moody
*cd and
Daniele
Castagnolo
*a
a,
Siyu
Liu
a,
Seong-Heun
Kim
a,
Sarah M.
Barry
b,
Thomas S.
Moody
*cd and
Daniele
Castagnolo
*a
aSchool of Cancer and Pharmaceutical Sciences, King's College London, 150 Stamford Street, SE1 9NH, London, UK. E-mail: daniele.castagnolo@kcl.ac.uk
bDepartment of Chemistry, Faculty of Natural & Mathematical Sciences, King's College London, Britannia House, 7 Trinity Street, London, SE1 1DB, UK
cAlmac Sciences, Department of Biocatalysis & Isotope Chemistry, 20 Seagoe Industrial Estate, BT63 5QD, Craigavon, UK. E-mail: tom.moody@almacgroup.com
dArran Chemical Company Limited, Unit 1 Monksland Industrial Estate, Athlone, Co. Roscommon N37 DN24, Ireland
First published on 6th November 2020
Abstract
A mild, chemoselective and sustainable biocatalysed synthesis of sulfoxides has been developed exploiting CALB and using AcOEt with a dual role of more environmentally friendly reaction solvent and enzyme substrate. A series of sulfoxides, including the drug omeprazole, have been synthesised in high yields and with excellent E-factors.
Sulfoxides are a ubiquitous class of organic compounds that play pivotal roles in organic synthesis as chiral auxiliaries,1 synthons for C–C bond forming reactions,2 directing groups in C–H bond functionalisation3 and can partake in numerous other functionalisation reactions.4 The sulfoxide moiety is also widely found in many pharmaceutical agents, including the blockbuster antacid agent omeprazole 1 and the dopamine reuptake inhibitor modafinil 2,5 as well as in nature, for example in the garlic components alliin 3 and ajoene 4, which is crucial for their antimicrobial and antifungal activity (Fig. 1).6 Sulfoxides can be easily obtained through oxidation of the corresponding sulfides using nitric acid,7 hypohalites such as NaIO4 and NaOCl,8 peroxides such as tert-butyl hydroperoxide (TBHP),9meta-chloroperbenzoic acid (mCPBA)10 and oxone.9c,11 However these methods have limited industrial use as they require potentially shock sensitive or explosive reagents or expensive metal-based catalysts making them unsuitable for large scale production. Recently, several protocols exploiting borax,12 2,2,2-trifluoroacetophenone13 and enzymes like cytochromes P450, monooxygenases, chloroperoxidases, laccase, and reductive enzymes14 have been proposed to provide more sustainable, cheaper and ultimately safer methodologies to access sulfoxide compounds.15,14b However, despite the excellent conversions, these approaches still have poor industrial applicability because of the low recyclability, high costs and stability of the enzymes. In addition, some enzymatic oxidations also require toxic and flammable additives and effective aeration of the system, all of which represent drawbacks in industry.16
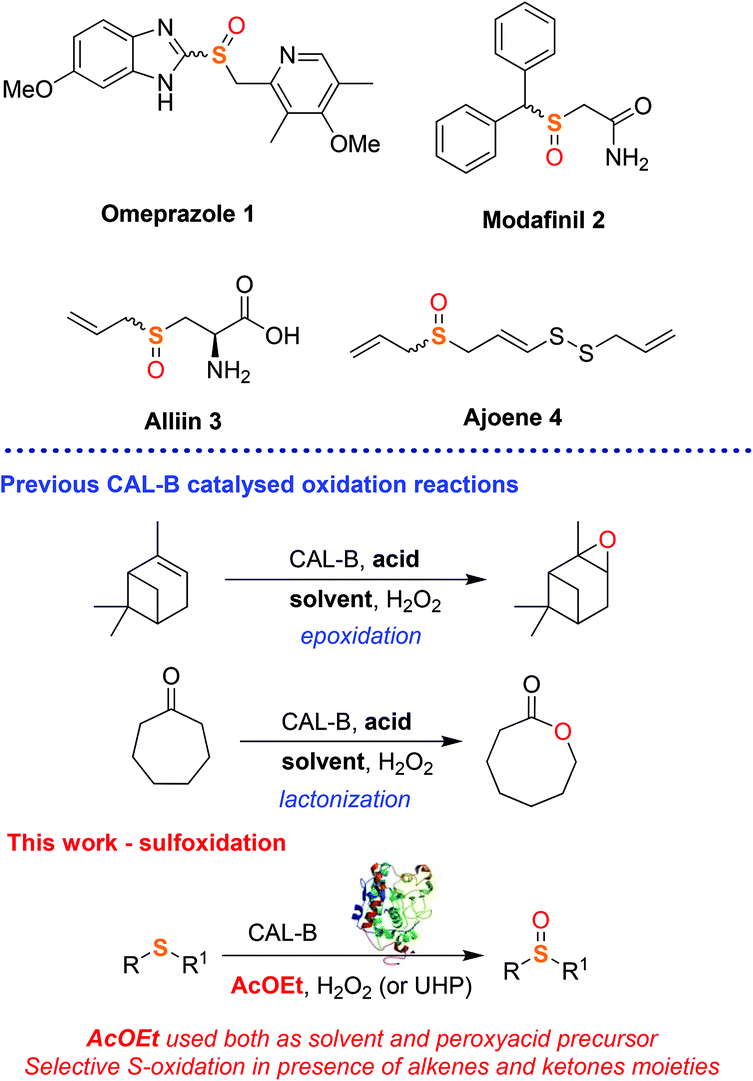 | ||
| Fig. 1 Sulfoxide containing drugs and natural products and an overview of the work. | ||
Immobilised Candida antarctica lipase B (CALB) is a robust and versatile enzyme which retains its activity in aqueous and organic solvents and it is already widely used in industry for both hydrolytic and acylation reactions of esters, alcohols and amines.17 Currently, CALB is one of the few enzymes that finds application and offers a real sustainable alternative to chemocatalysis in industry.17b In addition to its natural hydrolytic activity, CALB has been recently used as a biocatalyst in oxidative reactions, including epoxidations,18 Baeyer–Villiger lactonizations/esterifications19 and amine oxidation.20 These reactions exploit the ability of CALB to catalyse the in situ generation of peroxyacid oxidants from carboxylic acids under mild reaction conditions (Fig. 1). Surprisingly, to the best of our knowledge, the use of CALB as a biocatalyst in the oxidation of sulfide substrates into sulfoxides has never been investigated. Following our interest in the development of new and industrially applicable green methodologies for the synthesis of drugs and drug-like synthons, herein we report a facile, chemoselective and scalable biocatalytic protocol for the synthesis of sulfoxides using CALB. The method proves to be cost effective, robust and selective with few side-reactions (epoxidation and esterification). Furthermore, we exploit AcOEt in the dual role of solvent and CALB substrate, thus avoiding the use of extra acid additives. The choice of AcOEt as solvent/reagent improves the industrial sustainability of the method. In fact, when considering all factors in choosing a solvent for a chemical process such as the health, environment and safety scores,21 AcOEt is considered a safer and more economical alternative to other widely used solvents, such as halogenated or high boiling point solvents or even ionic liquids. Thus, AcOEt is ideal for the development of this sulfoxidation methodology, where it can serve as a solvent and CALB substrate, in turn contributing to the atom economy of the process.
The commercially available methyl phenyl sulfide 5a was selected as substrate to develop the CALB biocatalysed sulfoxidation methodology. Sulfide 5a was initially treated with CALB (20% w/w) and 1.1 equivalents of H2O2 in EtOAc (400 mM) leading to an 83![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :17 mixture of the desired sulfoxide 6a and the over-oxidation sulfone by-product 7a within 24 h (entry 1, Table 1). Replacement of H2O2 with urea hydrogen peroxide (UHP),22 which is often used as a more stable alternative to H2O2, led to the full oxidation of 5a in only 2 h and to the formation of the sulfoxide 6a as the major product in improved 93:7 ratio against 7a (entry 2). Reducing the concentration of 5a to 200 mM (entry 3) led to a small improvement in the sulfoxide/sulphone ratio (94:6), while a lower ratio (92:8) was observed in more concentrated conditions (entry 4). Thus, the optimal reaction concentration of 5a was kept at 400 mM. All the reactions were carried out under open air conditions. In order to confirm that the oxidation of sulfide 5a was biocatalysed by CALB rather than being promoted by UHP only or by air, a series of control experiments (entries 5–7) were performed. Upon the removal of the CALB and in the presence of UHP only, both in stoichiometric amount and in excess (5.0 eq.), negligible formation of 6a was observed, clearly accounting for the key role of CALB for the in situ generation of the peroxyacid oxidant intermediate 8a (Scheme 1).19a,23 Similarly, when UHP was omitted from the reaction, only a small amount of 6a was obtained and 96% of 5a was recovered. Finally, no sulfoxidation was observed when DCM or toluene (entries 8 and 9) were used as solvents, further corroborating the key dual role of AcOEt as solvent and CALB substrate and precursor of peroxyacid 8a. Remarkably, compared to traditional methods based on the use of a peroxyacid such as the industrially unappealing mCPBA,10b very little over-oxidation to the undesired sulfone was observed by proton NMR, clearly showing that the peroxyacid formed in situ rapidly oxidises the more reactive sulfide to sulfoxide and is converted back to the corresponding acid for a new oxidation cycle.
:17 mixture of the desired sulfoxide 6a and the over-oxidation sulfone by-product 7a within 24 h (entry 1, Table 1). Replacement of H2O2 with urea hydrogen peroxide (UHP),22 which is often used as a more stable alternative to H2O2, led to the full oxidation of 5a in only 2 h and to the formation of the sulfoxide 6a as the major product in improved 93:7 ratio against 7a (entry 2). Reducing the concentration of 5a to 200 mM (entry 3) led to a small improvement in the sulfoxide/sulphone ratio (94:6), while a lower ratio (92:8) was observed in more concentrated conditions (entry 4). Thus, the optimal reaction concentration of 5a was kept at 400 mM. All the reactions were carried out under open air conditions. In order to confirm that the oxidation of sulfide 5a was biocatalysed by CALB rather than being promoted by UHP only or by air, a series of control experiments (entries 5–7) were performed. Upon the removal of the CALB and in the presence of UHP only, both in stoichiometric amount and in excess (5.0 eq.), negligible formation of 6a was observed, clearly accounting for the key role of CALB for the in situ generation of the peroxyacid oxidant intermediate 8a (Scheme 1).19a,23 Similarly, when UHP was omitted from the reaction, only a small amount of 6a was obtained and 96% of 5a was recovered. Finally, no sulfoxidation was observed when DCM or toluene (entries 8 and 9) were used as solvents, further corroborating the key dual role of AcOEt as solvent and CALB substrate and precursor of peroxyacid 8a. Remarkably, compared to traditional methods based on the use of a peroxyacid such as the industrially unappealing mCPBA,10b very little over-oxidation to the undesired sulfone was observed by proton NMR, clearly showing that the peroxyacid formed in situ rapidly oxidises the more reactive sulfide to sulfoxide and is converted back to the corresponding acid for a new oxidation cycle.
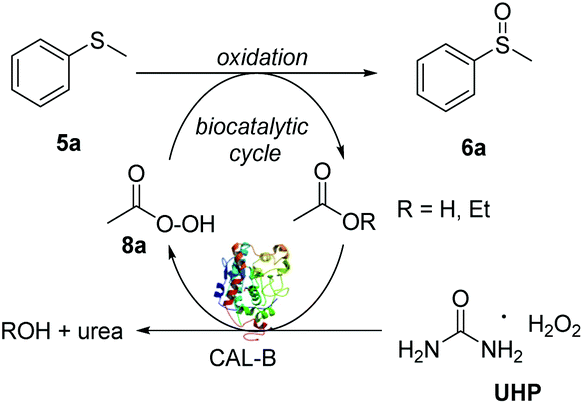 | ||
| Scheme 1 Proposed mechanism for the CALB biocatalysed sulfoxidation. | ||
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | 5a (mM) | CALB (% w/w) | Peroxidea | Solvent | Acid additivea | Time (h) | Conv.b (%) | Ratio 6a/7ac |
| a 1.1 eq. of H2O2 or UHP were used unless indicated differently. b Determined by analysis of the 1H NMR crude mixture and referred to the conversion of 5a into 6a–7a together. c Determined by 1H NMR. d Compound 6a was obtained with 89% isolated yield. | ||||||||
| 1 | 400 | 20 | H2O2 | AcOEt | — | 24 | 99 | 83:17 |
| 2 | 400 | 20 | UHP | AcOEt | — | 2 | >99 |
93![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :7 :7
|
| 3 | 200 | 20 | UHP | AcOEt | — | 2 | >99 | 94:6 |
| 4 | 600 | 20 | UHP | AcOEt | — | 2 | >99 | 92:8 |
| 5 | 400 | — | UHP | AcOEt | — | 2 | 8 | ND |
| 6 | 100 | — | UHP (5.0 eq.) | AcOEt | — | 24 | 30 | ND |
| 7 | 400 | 20 | — | AcOEt | — | 2 | 4 | ND |
| 8 | 400 | 20 | UHP | DCM | — | 2 | 1 | ND |
| 9 | 400 | 20 | UHP | Toluene | — | 2 | 4 | ND |
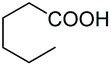
| 10 | 400 | 20 | H2O2 | DCM |
|
20 | 44 | ND |
| 11 | 400 | 20 | UHP | Toluene |
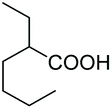
|
2 | 17 | ND |
| 12 | 160 | 20 | UHP | Toluene |
|
20 | 81 | 66:34 |
| 13 | 160 | 20 | UHP | Toluene |
|
2 | 16 | ND |
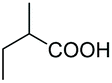
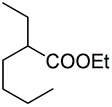
As a further confirmation of the key dual functionality of AcOEt over the use of inert solvents such as DCM and toluene, a series of experiments using acid additives as precursors of the peroxyacid oxidant was carried out. The treatment of 5a with CALB in DCM or toluene in presence of stoichiometric hexanoic acid and 2-ethylhexanoic acid (entries 10 and 11) led to 6a with poor conversion after 2 h. Interestingly, in the presence of 2-methylbutyric acid, 5a was converted at 81%, but with poor 66:34 sulfoxide/sulphone ratio (entry 12), while in the presence of the ester additive ethyl (2-ethyl)-hexanoate in toluene, 6a was obtained in low amount (entry 13).
Following identification of the best reaction conditions, the scope of the CALB biocatalysed sulfoxidation was investigated. A series of alkyl-aryl(benzyl) sulfides 5a–o was synthesised from the appropriate thiophenol or benzylthiol precursors 9a–h through the reaction of the appropriate alkyl halide in water under microwave irradiation. All substrates 5 were converted into the corresponding sulfoxides 6 with high yields as shown in Table 2. In most cases, only a low amount of the sulphone by-product was formed and high isolated yields were obtained regardless the size of the alkyl substituent (Me, Et, Pr) on the sulfoxide moiety. Remarkably, derivatives 6l–n bearing a double bond were also obtained selectively with high yields (entries 11–13). It is widely reported that CALB can catalyse the epoxidation of double bonds when in presence of peroxides and acid substrates.18 However, no traces of epoxide (by)-products were detected from the reaction of 5l–n, highlighting the chemoselectivity of this reaction and the preferred oxidation of sulfur over the alkenes. Finally, excellent conversions and high yields were obtained also for the benzyl sulfoxides 6o–p (entries 14 and 15), the chiral nitrile 6q (entry 16) and the dialkyl derivative 6r (entry 17).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Compound | Conv.a (%) | Yieldb,c,d (%) | Ratio SO/SO2 | |
|
a Determined by analysis of the 1H NMR crude mixture and referred to the conversion of 5 in 6–7 together.
b All the reactions were carried out for 24 h, unless completed before as revealed by TLC.
c Isolated yields are reported; isolated yields refer to the pure sulfoxides.
d Isolated yields refer to the biocatalytic step only.
e The reaction was completed in 2 h.
f Obtained as a 3:2 mixture of diastereoisomers.
|
|||||
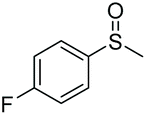
| 1 | 6b |
|
>99e | 85 | 88.12 |
| 2 | 6c |
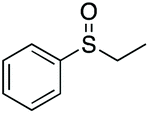
|
99 | 63 | 66:34 |
| 3 | 6d |
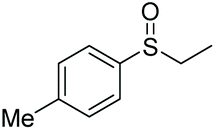
|
90 | 71 | 80:20 |
| 4 | 6e |
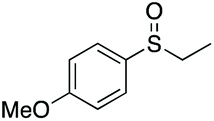
|
97e | 60 | 70:30 |
| 5 | 6f |
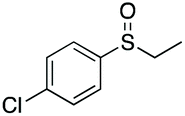
|
90 | 67 | 80:20 |
| 6 | 6g |
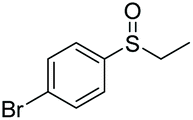
|
97 | 67 | 76:24 |
| 7 | 6h |
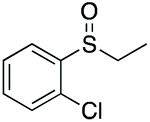
|
99 | 81 | 83:17 |
| 8 | 6i |
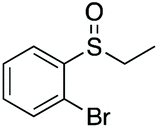
|
99 | 80 | 90:10 |
| 9 | 6j |
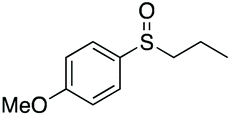
|
98 | 79 | 80:20 |
| 10 | 6k |
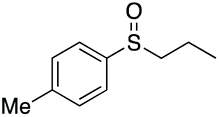
|
88 | 68 | 80:20 |
| 11 | 6l |
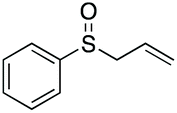
|
90 | 68 | 100:0 |
| 12 | 6m |
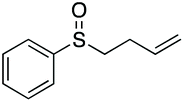
|
67 | 58 | 100:0 |
| 13 | 6n |
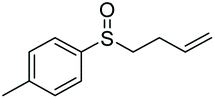
|
85 | 73 | 100:0 |
| 14 | 6o |
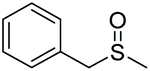
|
90 | 82 | 83:17 |
| 15 | 6p |
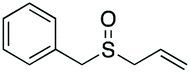
|
96 | 86 | 92:8 |
| 16 | 6q |
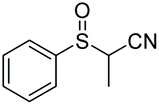
|
94 | 78f | 95:5 |
| 17 | 6r |
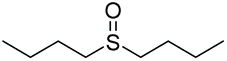
|
>99e | 89 | 94:6 |
It is documented that CALB can also catalyse the Baeyer–Villiger oxidation of ketone substrates when in the presence of peroxides.19 Thus, with the aim to further investigate the chemoselectivity of our transformation, namely the S-oxidation versus the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O oxidation to corresponding ester, a series of sulfide substrates 12a–f bearing a carbonyl moiety was synthesised as described in Table 3. All the carbonyl containing substrates 12a–f were selectively oxidised at the sulphur atom, as determined by NMR, affording the corresponding sulfoxides 13a–f with excellent conversions (up to 99%) and high yields after 24 h. The only exception was represented by the aldehyde substrate 12c (entry 3) which degraded during the reaction and no sulfoxide or other oxidation by-products were obtained from the reaction mixture. In all cases, the oxidation was highly selective towards the formation of the sulfoxide over the sulphone. Remarkably, no Baeyer–Villiger oxidation side products were observed in any reaction, further proving the high chemoselectivity of the methodology.
O oxidation to corresponding ester, a series of sulfide substrates 12a–f bearing a carbonyl moiety was synthesised as described in Table 3. All the carbonyl containing substrates 12a–f were selectively oxidised at the sulphur atom, as determined by NMR, affording the corresponding sulfoxides 13a–f with excellent conversions (up to 99%) and high yields after 24 h. The only exception was represented by the aldehyde substrate 12c (entry 3) which degraded during the reaction and no sulfoxide or other oxidation by-products were obtained from the reaction mixture. In all cases, the oxidation was highly selective towards the formation of the sulfoxide over the sulphone. Remarkably, no Baeyer–Villiger oxidation side products were observed in any reaction, further proving the high chemoselectivity of the methodology.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Cmpd | R | R1 | n | Conv.a (%) | Yieldb,c (%) | Ratio SO/SO2 |
| a Determined by analysis of the 1H NMR crude mixture and referred to the conversion of 12 in SO/SO2 products together. b Isolated yields are reported; isolated yields refer to the pure sulfoxides. c Isolated yields refer to the biocatalytic step only. d 30% w/v H2O2 used as peroxide. | |||||||
| 1 | 13a | Me | H | 1 | 89 | 71 | 100:0 |
| 2 | 13b | Me | Me | 1 | 68d | 42 | 100:0 |
| 3 | 13c | H | Me | 1 | 0 | 0 | 100:0 |
| 4 | 13d | Me | H | 0 | 99 | 67 | 80:20 |
| 5 | 13e | Et | H | 0 | 96 | 81 | 85:15 |
| 6 | 13f | iPr | H | 0 | 93 | 68 | 89:11 |
| 7 | 15a | Me | H | 1 | 99 | 67 | 76:24 |
| 8 | 15b | H | Me | 1 | 99 | 74 | 76:24 |
Finally, the alcohol derivatives 14a–b, in turn obtained from 12a and 12c, were oxidised leading to products 15a–b with excellent conversions and yields (entries 7 and 8).24 Interestingly, in the case of 15b, CALB also catalysed the acetylation of the primary hydroxyl group in addition to the S-oxidation.
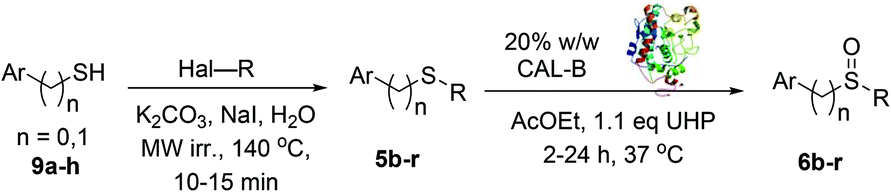
In order to prove the applicability of the methodology to pharmaceutical ingredients, the synthesis of omeprazole 1 was carried out (Scheme 2a). The substrate 16 was treated with CALB and UHP under standard conditions25 and the selective oxidation of the sulphur to sulfoxide was accomplished within 2 hours, leading to omeprazole 1 with 73% isolated yield. No trace of the sulphone by-product were observed. Gratifyingly, the E-factor of the transformation was found to be 35, confirming the high industrial applicability of the CALB oxidation method. Interestingly, most of the current approaches reported in literature for the synthesis of omeprazole 1 are carried out under harsher reaction conditions and longer reaction times,12,13,15,26 highlighting the potential impact of this method on the synthesis of sulfoxide containing pharmaceutical ingredients at industrial level.
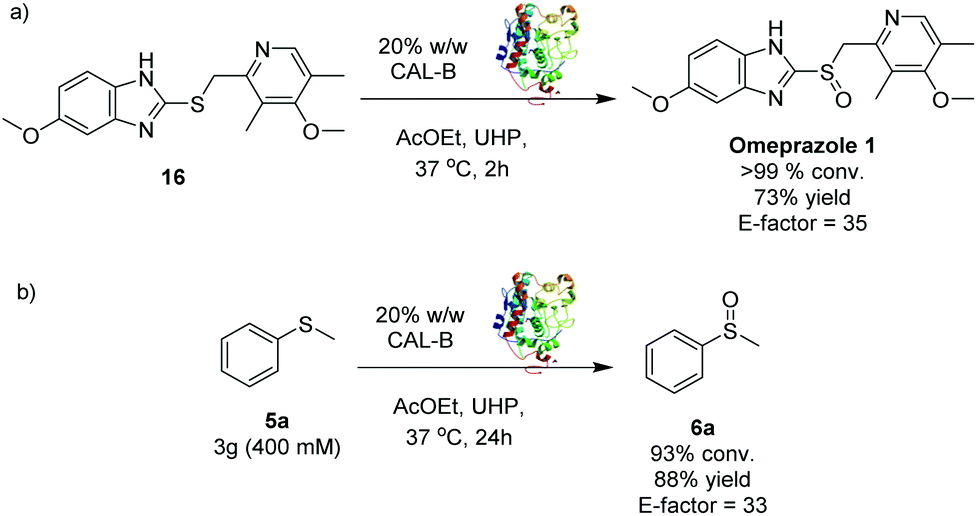 | ||
| Scheme 2 (a) Synthesis of omeprazole 1via CALB biocatalysed oxidation; (b) Gram-scale synthesis of sulfoxide 6a. | ||
One of the main drawbacks of the synthetic methodologies developed within an academic environment is that they often fail to perform at an industrial scale. The scalability of the CALB S-oxidation was thus investigated through the oxidation at multi-gram scale of 5a27 (Scheme 2b). The sulfoxide 6a was obtained with 93% conversion and 88% isolated yield with an excellent E-factor of 33.
Finally, a series of enzyme recycling experiments were performed to further confirm the industrial potentiality of the methodology. Sulfide 5a was dissolved in AcOEt (400 mM) with 20% w/w CALB and 1.1 equivalents of UHP and stirred for 24 h. At the end of the reaction, CALB was filtered off and washed with a 9:1 mixture of CH3CN/water (9:1) to remove leftover urea from UHP. CALB, obtained with a recovery rate of 75–96%, was then re-used in a subsequent sulfoxidation reaction of 5a. The catalytic activity of CALB was maintained through four reaction cycles without significant loss in oxidation activity (99–94% conversions of 5a, Fig. 2). A drop to 39% of activity was observed in the fifth cycle.28
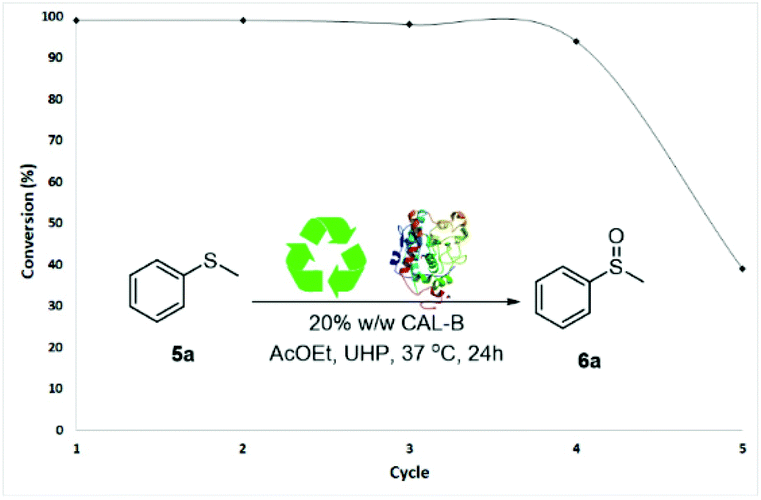 | ||
| Fig. 2 Recycling experiments of CALB. | ||
Conclusions
In conclusion, a novel, mild and selective methodology for the synthesis of sulfoxide compounds in high yields was developed using CALB biocatalyst and UHP. The oxidation of sulfide substrates occurs exploiting AcOEt in dual roles of solvent and CALB co-substrate in the generation of the peroxyacid reactive intermediate 8a. Sulfide substrates bearing different functional groups such as alkenes and carbonyls were also selectively oxidised at the sulfur atom, proving the chemoselectivity of the methodology over side reactions like epoxidations and Baeyer–Villiger oxidations. The methodology was applied to the synthesis of the drug omeprazole and investigated at gram scale on the substrate 5a, showing excellent yields and E-factors. These data, in addition to the robustness and the recyclability of CALB biocatalyst, demonstrate the high translation potential of this methodology for industrial applications.Conflicts of interest
There are no conflicts to declare.Notes and references
- X. Salom-Roig and C. Bauder, Synthesis, 2020, 52, 964–978 CrossRef CAS.
- F. Colobert, A. Tito, N. Khiar, D. Denni, M. A. Medina, M. Martin-Lomas, J. L. G. Ruano and G. Solladié, J. Org. Chem., 1998, 63, 8918–8921 CrossRef CAS.
- K. X. Tang, C. M. Wang, T. H. Gao, L. Chen, L. Fan and L. P. Sun, Adv. Synth. Catal., 2019, 361, 26–38 CrossRef CAS.
- (a) M. C. Carreno, Chem. Rev., 1995, 95, 1717–1760 CrossRef CAS; (b) H. Pellissier, Tetrahedron, 2006, 62, 5559–5601 CrossRef CAS; (c) I. Fernández and N. Khiar, Chem. Rev., 2003, 103, 3651–3706 CrossRef; (d) K. Peng and Z.-B. Dong, Eur. J. Org. Chem., 2020, 34, 5488–5495 Search PubMed.
- (a) K. A. Scott and J. T. Njardarson, Top. Curr. Chem., 2018, 376, 5 CrossRef; (b) M. J. Kendall, Aliment. Pharmacol. Ther., 2003, 17, 1–4 CrossRef CAS; (c) F. E. Held, K. A. Stingl and S. B. Tsogoeva, Symmetry, 2017, 9, 88 CrossRef.
- (a) K. H. Kyung, Curr. Opin. Biotechnol., 2012, 23, 142–147 CrossRef CAS; (b) S. G. Santhosha, P. Jamuna and S. N. Prabhavathi, Food Biosci., 2013, 3, 59–74 CrossRef CAS.
- (a) A. K. Misra and G. Agnihotri, Synth. Commun., 2006, 34, 1079–1085 CrossRef; (b) A. R. Suárez, L. I. Rossi and S. E. Martín, Tetrahedron Lett., 1995, 36, 1201–1204 CrossRef; (c) A. R. Hajipour, B. Kooshki and A. E. Ruoho, Tetrahedron Lett., 2005, 46, 5503–5506 CrossRef CAS; (d) F. Gasparrini, M. Giovannoli, D. Misiti, G. Natile and G. Palmieri, J. Org. Chem., 1990, 55, 1323–1328 CrossRef CAS.
- (a) V. Pace, L. Castoldi and W. Holzer, Tetrahedron Lett., 2012, 53, 967 CrossRef CAS; (b) P. Kowalski, K. Mitka, K. Ossowska and Z. Kolarska, Tetrahedron, 2005, 61, 1933–1953 CrossRef CAS; (c) N. J. Leonard and C. R. Johnson, J. Org. Chem., 1962, 27, 282–284 CrossRef CAS; (d) M. Kirihara, T. Okada, Y. Sugiyama, M. Akiyoshi, T. Matsunaga and Y. Kimura, Org. Process Res. Dev., 2017, 21, 1925–1937 CrossRef CAS; (e) A. Sudalai, A. Khenkin and R. Neumann, Org. Biomol. Chem., 2015, 13, 4374–4394 RSC.
- (a) F. Di Furia, G. Modena and R. Curci, Tetrahedron Lett., 1976, 17, 4637–4638 CrossRef; (b) F. Di Furia, G. Modena and R. Seraglia, Synthesis, 1984, 325–326 CrossRef CAS; (c) P. J. Kropp, G. W. Breton, J. D. Fields, J. C. Tung and B. R. Loomis, J. Am. Chem. Soc., 2000, 122, 4280–4285 CrossRef CAS; (d) K. Kaczorowska, Z. Kolarska, K. Mitka and P. Kowalski, Tetrahedron, 2005, 61, 8315–8327 CrossRef CAS; (e) A. Russo and A. Lattanzi, Adv. Synth. Catal., 2009, 351, 521–524 CrossRef CAS; (f) M. Mokhtary, M. Qandalee and M. R. Niaki, J. Chem., 2012, 9, 863–868 CAS; (g) S. Matavos-Aramyan, S. Soukhakian and M. H. Jazebizadeh, Phosphorus, Sulfur Silicon Relat. Elem., 2020, 195, 181–193 CrossRef CAS.
- (a) A. Bottoni, M. Calvaresi, A. Ciogli, B. Cosimelli, G. Mazzeo, L. Pisani, E. Severi, D. Spinelli and S. Superchi, Adv. Synth. Catal., 2013, 355, 191–202 CrossRef CAS; (b) H. Hussain, A. Al-Harrasi, I. R. Green, I. Ahmed, G. Abbas and N. U. Rehman, RSC Adv., 2014, 4, 12882–12917 RSC.
- (a) B. Yu, A. H. Liu, L. N. He, B. Li, Z. F. Diao and Y. N. Li, Green Chem., 2012, 14, 957–962 RSC; (b) R. V. Kupwade, S. S. Khot, U. P. Lad, U. V. Desai and P. P. Wadgaonkar, Res. Chem. Intermed., 2017, 43, 6875–6888 CrossRef CAS.
- S. Hussain, S. K. Bharadwaj, R. Pandey and M. K. Chaudhuri, Eur. J. Org. Chem., 2009, 3319–3322 CrossRef CAS.
- E. Voutyritsa, I. Triandafillidi and C. G. Kokotos, Synthesis, 2017, 49, 917–924 CAS.
- (a) J. Han, V. A. Soloshonok, K. D. Klika, J. Drabowicz and A. Wzorek, Chem. Soc. Rev., 2018, 47, 1307–1350 RSC; (b) W. Maczka, K. Wińska and M. Grabarczyk, Catalysts, 2018, 8, 624 CrossRef; (c) F. Garzón-Posse, L. Becerra-Figueroa, J. Hernández-Arias and D. Gamba-Sánchez, Molecules, 2018, 23, 1265 CrossRef; (d) S. Anselmi, N. Aggarwal, T. S. Moody and D. Castagnolo, ChemBioChem, 2020 Search PubMed, in press; (e) A. Rostami, B. Mohammadi, Z. Shokri and S. Saadati, Catal. Commun., 2018, 111, 59–63 CrossRef CAS; (f) E. O'Reilly, M. Corbett, S. Hussain, P. P. Kelly, D. Richardson, S. L. Flitsch and N. J. Turner, Catal. Sci. Technol., 2013, 3, 1490–1492 RSC.
- J. P. Colomer, M. Traverssi and G. Oksdath-Mansilla, J. Flow Chem., 2020, 10, 123–138 CAS.
- (a) R. A. Sheldon and D. Brady, Chem. Commun., 2018, 54, 6088–6104 RSC; (b) M. D. Truppo, ACS Med. Chem. Lett., 2017, 8, 476–480 CrossRef CAS; (c) G. Hughes and J. C. Lewis, Chem. Rev., 2018, 118, 1–3 CrossRef CAS; (d) P. N. Devine, R. M. Howard, R. Kumar, M. P. Thompson, M. D. Truppo and N. J. Turner, Nat. Rev. Chem., 2018, 2, 409–421 CrossRef.
- (a) V. Gotor-Fernandez, E. Busto and V. Gotor, Adv. Synth. Catal., 2006, 348, 797–812 CrossRef CAS; (b) M. T. Reetz, Curr. Opin. Chem. Biol., 2002, 6, 145–150 CrossRef CAS; (c) J. Dulęba, K. Czirson, T. Siódmiak and M. P. Marszałł, Med. Res. J., 2019, 4, 174–177 CrossRef; (d) A. Basso and S. Serban, Mol. Catal., 2019, 479, 110607 CrossRef CAS; (e) C. Ortiz, M. L. Ferreira, O. Barbosa, J. C. S. dos Santos, R. C. Rodrigues, Á. Berenguer-Murcia, L. E. Briand and R. Fernandez-Lafuente, Catal. Sci. Technol., 2019, 9, 2380–2420 RSC; (f) P. Domínguez de María, G. de Gonzalo and A. R. Alcántara, Catalysts, 2019, 9, 802 CrossRef.
- (a) P. Zhou, X. Wang, B. Yang, F. Hollmann and Y. Wang, RSC Adv., 2017, 7, 12518–12523 RSC; (b) M. De Torres, G. Jiménez-Osés, J. A. Mayoral, E. Pires, R. M. Blanco and O. Fernández, Catal. Today, 2012, 195, 76–82 CrossRef CAS; (c) V. Skouridou, H. Stamatis and F. N. Kolisis, J. Mol. Catal. B: Enzym., 2003, 21, 67–69 CrossRef CAS; (d) M. Rusch gen Klaas and S. Warwel, J. Am. Oil Chem. Soc., 1996, 73, 1453–1457 CrossRef; (e) M. Steinhagen, A. Gräbner, J. Meyer, A. E. W. Horst, A. Drews, D. Holtmann and M. B. Ansorge-Schumacher, J. Mol. Catal. B: Enzym., 2016, 133, S179–S187 CrossRef.
- (a) G. Chávez, R. Hatti-Kaul, R. A. Sheldon and G. Mamo, J. Mol. Catal. B: Enzym., 2013, 89, 67–72 CrossRef; (b) D. González-Martínez, M. Rodríguez-Mata, D. Méndez-Sánchez, V. Gotor and V. Gotor-Fernández, J. Mol. Catal. B: Enzym., 2015, 114, 31–36 CrossRef; (c) A. Drożdż, K. Erfurt, R. Bielas and A. Chrobok, New J. Chem., 2015, 39, 1315–1321 RSC; (d) A. Drożdża and A. Chrobok, Chem. Commun., 2016, 52, 1230–1233 RSC.
- D. Méndez-Sánchez, I. Lavandera, V. Gotor and V. Gotor-Fernández, Org. Biomol. Chem., 2017, 15, 3196–3201 RSC.
- (a) P. G. Jessop, Green Chem., 2011, 13, 1391–1398 RSC; (b) C. J. Clarke, W.-C. Tu, O. Levers, A. Bröhl and J. P. Hallett, Chem. Rev., 2018, 118, 747–800 CrossRef CAS; (c) F. P. Byrne, S. Jin, G. Paggiola, T. H. M. Petchey, J. H. Clark, T. J. Farmer, A. J. Hunt, C. R. McElroy and J. Sherwood, Sustainable Chem. Processes, 2016, 4, 7 CrossRef.
- R. Matyáš, J. Selesovsky, V. Pelikán, M. Szala, S. Cudziło, W. A. Trzciński and M. Gozin, Propellants, Explos., Pyrotech., 2017, 42, 198–203 CrossRef.
- The formation of the peroxyacid intermediate 8a has been confirmed by titration according to ref. 19a.
- Compounds 15a–b were obtained as 1:1 mixture of diastereoisomers as confirmed by HPLC analysis using a Chiralpak® IG column.
- Due to the poor solubility of 16, 1% DCM was added to fully solubilise the substrate in the reaction mixture. The addition of DCM was taken into account in the calculation of the E-factor.
- (a) T. Neveselý, E. Svobodová, J. Chudoba, M. Sikorski and R. Cibulka, Adv. Synth. Catal., 2016, 358, 1654–1663 CrossRef; (b) C. Yang, Q. Jin, H. Zhang, J. Liao, J. Zhu, B. Yu and J. Deng, Green Chem., 2009, 11, 1401–1405 RSC; (c) S. Hou, N. Chen, P. Zhang and S. Dai, Green Chem., 2019, 21, 1455–1460 RSC; (d) D. A. Alonso, C. Nájera and M. Varea, Tetrahedron Lett., 2002, 43, 3459–3461 CrossRef CAS; (e) P. Tomanová, J. Šturala, M. Buděšínský and R. Cibulka, Molecules, 2015, 20, 19837–19848 CrossRef; (f) F. Shi, M. K. Tse, H. M. Kaiser and M. Beller, Adv. Synth. Catal., 2007, 349, 2425–2430 CrossRef CAS.
- In order to avoid the over-oxidation of 5a due to high amount of UHP used, as well as for safety reasons, the peroxide was added to the reaction mixture in two portions every 30 min.
- No appreciable increase in the over-oxidation of 6a to sulfone 7a was observed during the recycling experiments.
Footnote |
| † Electronic supplementary information (ESI) available: Full characterization of sulfoxide compounds and copies of 1H NMR and 13C NMR spectra. See DOI: 10.1039/d0ob01966f |
| This journal is © The Royal Society of Chemistry 2021 |