
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enantioselective synthesis of cyanohydrins catalysed by hydroxynitrile lyases – a review
Paula
Bracco†
a,
Hanna
Busch†
a,
Jan
von Langermann
b and
Ulf
Hanefeld
*a
aGebouw voor Scheikunde, Biokatalyse, Afdeling Biotechnologie, Technische Universiteit Delft, Julianalaan 136, 2628BL Delft, The Netherlands. E-mail: u.hanefeld@tudelft.nl
bInstitute of Chemistry, University of Rostock, Albert-Einstein-Str. 3a, 18059 Rostock, Germany
First published on 1st June 2016
Abstract
The first enantioselective synthesis was the selective addition of cyanide to benzaldehyde catalysed by a hydroxynitrile lyase (HNL). Since then these enzymes have been developed into a reliable tool in organic synthesis. HNLs to prepare either the (R)- or the (S)-enantiomer of the desired cyanohydrin are available and a wide variety of reaction conditions can be applied. As a result of this, numerous applications of these enzymes in organic synthesis have been described. Here the examples of the last decade are summarised, the enzyme catalysed step is discussed and the follow-up chemistry is shown. This proves HNLs to be part of main stream organic synthesis. Additionally the newest approaches via immobilisation and reaction engineering are introduced.
 Paula Bracco | Paula Bracco obtained her M.Sc. Pharm. Chemistry degree from the Universidad de la República, Montevideo (Uruguay). During her period as research assistant at the organic chemistry department, Prof. Seoane introduced her to a new area of interest: biocatalysis. In this field she obtained her first scholarship from the OPCW which was held at Surrey University (UK) during 2007. In 2010, she moved to Aachen (Germany) where she received her Ph.D. title from RWTH Aachen, under the supervision of Prof. Schallmey. After her first postdoctoral position at Universiteit van Amsterdam during 2014, she joined Prof. Hanefeld's group where she is involved in biocatalysis and protein engineering related projects. |
 Hanna Busch | Hanna Busch received her M.Sc. degree in chemistry from the University of Rostock in 2015 where Dr Jan von Langermann introduced her to the field of hydroxynitrile lyases. During her postgraduate studies she visited the Manchester Institute of Biotechnology (2014–2015) working in the laboratories of Professor Sabine Flitsch and Professor Nicholas Turner. In 2015, she started her Ph.D. studies at the Delft University of Technology under the guidance of Professor Ulf Hanefeld studying the stereoselective, enzyme-catalysed water addition to double bonds. |
 Jan von Langermann | Jan von Langermann obtained his PhD (chemical engineering) under the supervision of Prof. Udo Kragl in 2008 from the University of Rostock. From 2008 to 2011 he joined the research group of Prof. Andreas Seidel-Morgenstern as a Postdoctoral Fellow at the Max-Planck-Institute in Magdeburg, Germany. Afterwards he moved to the University of Minnesota, USA for further postdoctoral studies at the Biotechnology Institute in the research group of Prof. Romas Kazlauskas. Since 2013 he has been a Junior Research Group Leader at the Institute of Chemistry at the University of Rostock. His research is focused on the development of compartmented biocatalytic processes, especially for cascade reactions, and crystallization techniques within integrated reaction systems. |
 Ulf Hanefeld | Ulf Hanefeld was born in 1966 in Köln, Germany, and grew up in (West) Berlin and London. In 1993 he received his PhD from the Georg-August-Universität zu Göttingen, having performed the research both with Prof. H. Laatsch (Göttingen) and Prof. H. G. Floss (Seattle). After postdoctoral years with Prof. C. W. Rees (Imperial College London), Prof. J. Staunton (Cambridge) and Prof. J. J. Heijnen and Dr A. J. J. Straathof (TU Delft), he received a fellowship from the Royal Netherlands Academy of Arts and Sciences (KNAW). He rose through the ranks at the Technische Universiteit Delft and his research in Delft focuses on enzymes, in particular HNLs, their immobilisation and application in organic synthesis. |
1. Introduction
Enantiopure cyanohydrins are valuable and versatile building blocks in organic chemistry. With their two functional groups they are readily converted to yield α-hydroxy aldehydes or ketones, β-amino alcohols or α-fluorocyanides and many other compounds. They are therefore of central importance both for the synthesis of fine and bulk chemicals.1 Consequently they are a mainstay in academic and industrial research.The synthesis of enantiopure cyanohydrins is performed by the addition of nucleophilic cyanide to a prochiral carbonyl compound catalysed by a chiral catalyst that induces the enantioselective addition.2 The cyanide can either be obtained from inorganic sources (i.e. KCN) or be released in situ from organic compounds, for instance acetone cyanohydrin. A broad range of chemical and enzymatic catalytic systems has been developed and extensively reviewed over the years.2–7 Out of all the different systems the (R)- and (S)-selective hydroxynitrile lyases (HNLs, in the older literature also known as oxynitrilases) are the catalysts of choice. This is exemplified by their well-established industrial application.7–9 Hydroxynitrile lyases (EC 4.1.2.X, X = 10, 11, 46, 47) belong to the group of C–C lyases and are typically found in plants, but recent investigations have also revealed the presence of HNLs in bacteria and arthropods.10–15
Already in 1837 the founding fathers of organic chemistry Wöhler and Liebig described an HNL catalysed reaction. They reported the emulsin (an extract of bitter almonds) catalysed generation of HCN – by decomposition of a cyanohydrin.16 In 1908 Rosenthaler performed the reverse reaction. In the first enantioselective synthesis ever he prepared optically pure mandelonitrile. This emulsin-catalysed enantioselective addition of HCN to benzaldehyde is a milestone in both organic chemistry and catalysis.17
Today HNLs are part of organic text book chemistry and the disconnection approach.18,19 Compared to chemical approaches, they are straightforward to use in aqueous media or water saturated organic solvents without an inert gas atmosphere. They offer high reaction rates, excellent enantioselectivities for either (R)- or (S)-stereoisomers while using environmentally benign reaction conditions. A stern warning on the hazards of HCN has to be added at this place. HCN is volatile, can be adsorbed through the skin and is toxic. All procedures involving hydrogen cyanide should therefore be performed in a well-ventilated fume hood equipped with a HCN detector. HCN-containing waste should be neutralised with commercial bleach and stored independently over a large excess of bleach for disposal. Therefore in situ release, as described above, should be applied and immediate treatment of the reaction waste is essential.4,51 Industrially the handling of such toxic compounds and in particular of HCN is no problem and consequently HNLs are the catalyst of choice in industry.7–9,15,20–22 Various reviews concentrate on all aspects of catalyst preparation, modification and mechanisms, including the enantioselectivity and its modification in HNLs;4,6 similar reviews exist for chemical catalysts.2,23,24 While HNL immobilisation for reaction engineering has recently been reviewed,7 we herein introduce the asymmetric synthesis catalysed by HNLs and the essential background information for their successful application.
The nucleophilic attack of the cyanide on the carbonyl is a base-catalysed equilibrium reaction whereby the equilibrium strongly depends on the substrate structure. This reaction is not stereoselective.25 It needs to be suppressed in order to ensure that only the enantioselective, HNL-catalysed reaction takes place. By lowering the pH below 5 this can typically be achieved but also lower temperatures can help.26 When using aldehydes as substrates, the equilibrium in general lies on the product side while ketones often show a more unfavourable balance due to their higher steric demand.7,9 A change in the thermodynamic reaction conditions, i.e. an excess of HCN, in situ product removal or lower temperature can influence the yield positively.5,7,26–33 In this context the improvement in yield by switching from a mono-phasic to a bi-phasic reaction system should be mentioned. The mono-phasic reaction medium in HNL-catalysed reactions is an aqueous buffer solution which allows two parameters to be optimised, pH and temperature.34 Adding water-miscible organic solvents to increase substrate concentrations tends to destabilise the enzyme.35 A powerful tool in the optimisation process is the use of water-immiscible solvents in an organic–aqueous bi-phasic system. Here, the enzyme catalysed reaction takes place in the aqueous phase while the substrate and product are in the water-immiscible organic solvent where no chemical background reaction occurs. As a result, product separation as well as recycling of the enzyme in the aqueous phase is simplified. Subsequently, low substrate and product concentrations in the aqueous phase reduce the probability of inhibitory effects. To take this to the extreme the HNLs can often be used in wet organic solvents, however, then enzyme recycling can be more difficult.7,26 This will be discussed in detail in section 3 of this review.
Overall, for the asymmetric synthesis of cyanohydrins, (S)- and (R)-selective hydroxynitrile lyases are readily available, straightforward to use and standardised in textbooks as well as in industry. The cyanohydrins synthesised with their help offer a broad range of possible follow-up reactions to give a wide spectrum of products. We herein review recent examples of syntheses which are based on the formation of chiral cyanohydrins as the key step (Scheme 1).
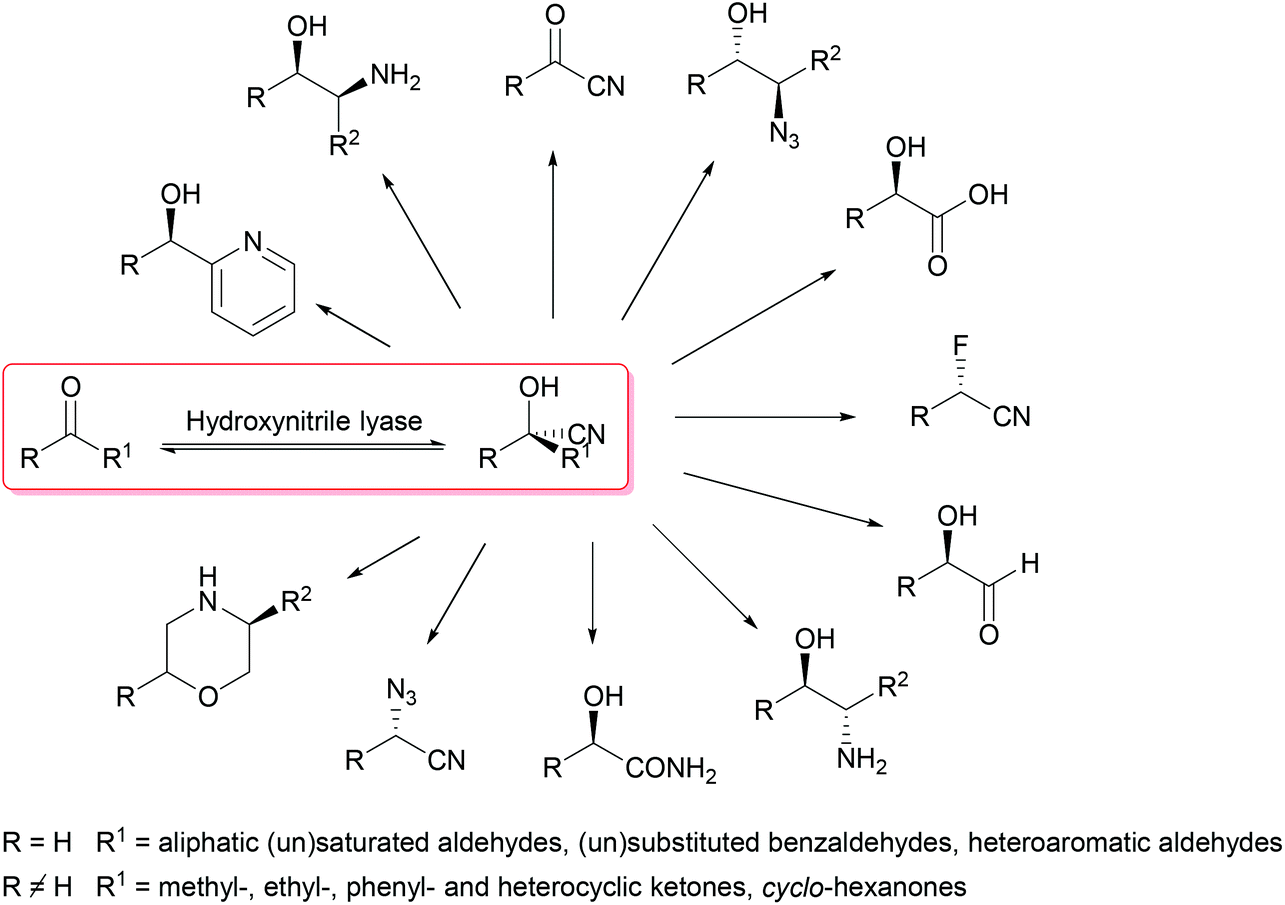 | ||
| Scheme 1 Enzymatic synthesis and follow-up chemistry of cyanohydrins. | ||
2. Applications
2.1 (Imino)sugars
By utilising a straightforward HNL in a buffer saturated solvent (low pH buffer) system, the synthesis of eight 1-deoxynojirimycin isomers was developed.36 (R)-Selective Prunus amygdalus HNL (PaHNL, almond), the enzyme that Wöhler and Liebig had already used, catalysed the cyanide addition with ee = 99%. Protection, followed by a one-pot DIBAL-H/transamination/NaBH4 reduction sequence using (S)-benzyloxyvinylglycinol in the transamination step and immediate N′-Boc protection gave the starting material for ring-closing metathesis with the trans-substituted ring as product 4 (Scheme 2). Switching to (R)-benzyloxyvinylglycinol and following the same protocol resulted in the cis-substituted heterocycles. Based on the selective 3,4-cis-Upjohn-dihydroxylation of precursor 4 followed by multi-step synthesis yielded deoxynojirimycin. The trans-isomer led to four isomers D-altro- (5), D-galacto- (6), D-allo- and D-talo-1-deoxynojirimycin while the cis-substituted intermediate led to the respective L-series.37,38 | ||
| Scheme 2 Synthesis of 1-deoxynojirimycin isomers.37,38 | ||
The de novo synthesis of D- and L-pentoses was achieved with (S)-selective Hevea brasiliensis hydroxynitrile lyase (HbHNL, Brazilian rubber tree) and (Z)-2-buten-1,4-diol (7) as starting materials.39 Here bi-phasic reaction conditions at 0 °C with a low pH in the aqueous layer were chosen. To ensure high rates and optimal phase transfer the reaction was performed in an emulsin of the two layers. By varying the protecting groups (PG, 8, Scheme 3) the ee could be enhanced up to 92%. Once the cyanohydrin was formed, the bulky TBDPS-protection group was strategically used to enhance the directing effect of the chiral centre in the asymmetric dihydroxylation. In addition to lower cytotoxicity and higher stability, these non-natural compounds 11 are of high importance since they can exhibit even higher antiviral activity than their D-counterparts 12 when incorporated into nucleoside analogues.
 | ||
| Scheme 3 Synthesis of pentoses.39 | ||
In the de novo synthesis of non-natural nucleosides, deoxysugars were prepared based on an oxidation–hydrocyanation protocol leading to enantiopure, protected cyanohydrins.40 The mild oxidation of primary alcohols 13 with TEMPO and PhI(OAc)2 gave β,γ-unsaturated aldehydes without isomerisation of the double bond (Scheme 4A). Subsequent hydrocyanation with either (S)-selective HbHNL or (R)-selective PaHNL yielded γ,δ-unsaturated cyanohydrins 14 which were afterwards protected. Again a bi-phasic strategy with an emulsin and low pH was applied. 3′-deoxyribolactones are known to be suitable precursors for 3′-deoxyribose, an important building block in the synthesis of pharmaceutically interesting nucleosides. Treating (R)-14 with Oxone® led to the diastereomeric mixture of epoxide 15. Subsequent separation of diastereomers via column chromatography and hydrolysis-induced lactonisation resulted in the formation of 5′-chloro-methyl lactones (16 and 17) instead of desired 3′-deoxyribolactones due to an attack of the chloride to the primary carbon center (C1). This induced the retention of the C2 stereocenter. To prove the applicability of the follow-up chemistry it was also applied to racemic cyanohydrin 19, generated from dec-3-ynal (18) and TBSCN (Scheme 4B). Subsequent hydrogenation of the triple bond with the Lindlar catalyst to yield the respective (Z)-alkene followed by epoxidation with Oxone® resulted in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 diastereomeric mixture. Final hydrolysis-induced lactonisation gave a 1:5:6:1 mixture of desired 3′-deoxyribolactone stereoisomers 20 in moderate yields.41
:1 diastereomeric mixture. Final hydrolysis-induced lactonisation gave a 1:5:6:1 mixture of desired 3′-deoxyribolactone stereoisomers 20 in moderate yields.41
 | ||
| Scheme 4 Synthesis of (A) 5′-chloro-3′-deoxyribolactones and (B) 3′-deoxy-5′-hexyl lactones.40,41 | ||
Acetone cyanohydrin was used as a cyanide source for (R)-PaHNL in the chemo-enzymatic synthesis of D- and L-(2-cyano-2-hydroxy/acetoxy)mannosides (23, 24). This synthesis involves also lipase PS-D as the second enzyme for diastereomeric kinetic resolution by acylation.42PaHNL releases HCN from acetone cyanohydrin and later catalyses its addition via an enantioselective transcyanation with D- and L-mannosylacetaldehydes (21, Scheme 5) in a bi-phasic system. Unfortunately, only racemic cyanohydrins 22 were obtained. Experiments with and without HNL at different water contents and low pHs revealed the enzyme to be the main reason for the unselective cyanide addition. The unselective cyanide generation and addition catalysed by PaHNL was now performed in buffer saturated toluene. This allowed the combination with an enantioselective lipase for an interesting synthetic two-step method for the production of four mannosides from their respective D- and L-aldehydes. Extending this research, the modification of monosaccharides at position C-6 was studied.43 HNLs are overall very versatile catalysts, but as this example shows, other catalysts are occasionally a better choice to induce chirality.
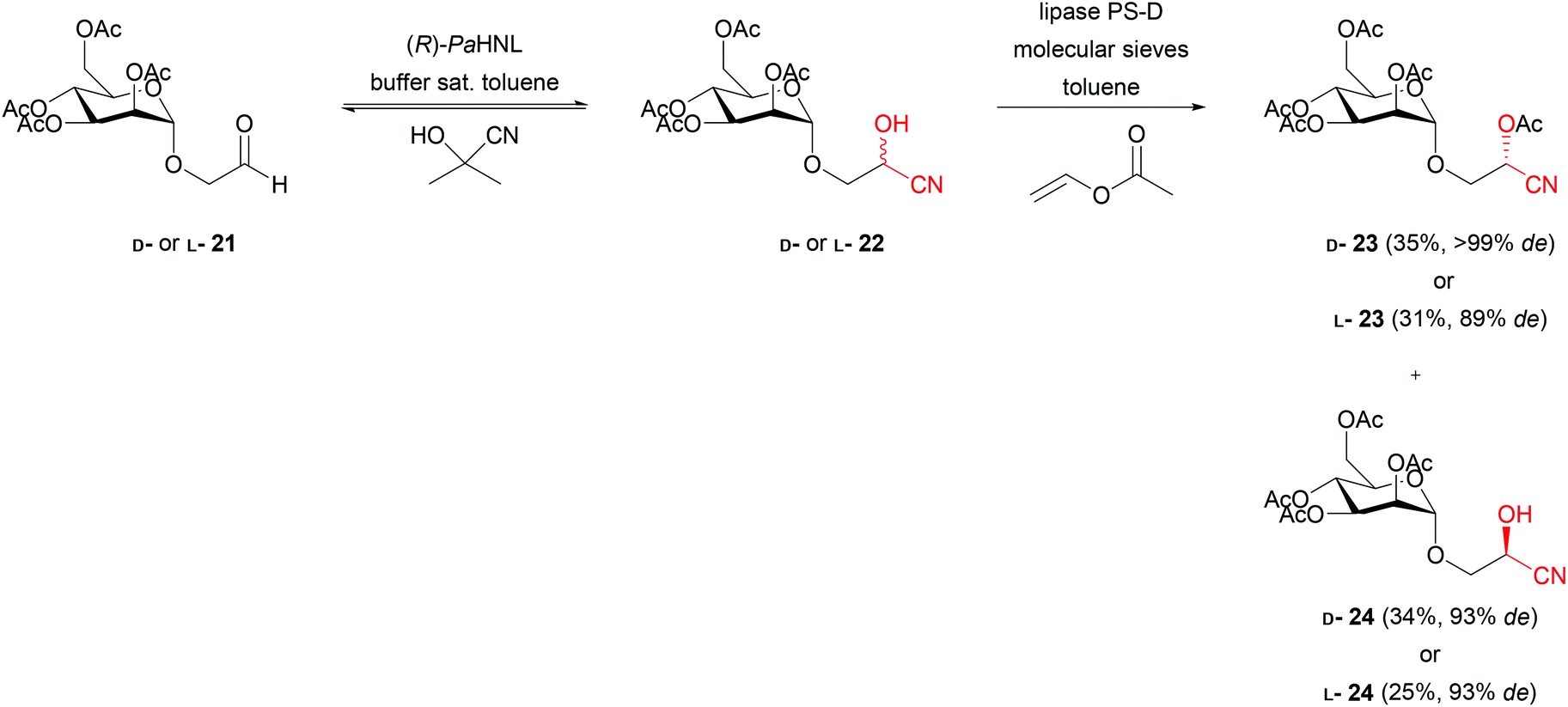 | ||
| Scheme 5 Chemo-enzymatic synthesis of D- and L-(2-cyano-2-hydroxy/acetoxy)mannosides.42,43 | ||
To take this even further, when starting from highly functionalised and protected sugars 25 the stereocentres present can readily suffice to induce the stereochemistry of the cyanohydrin 26. Straightforward base-catalysis allowed the preparation of modified nucleosides 27 (Scheme 6).44
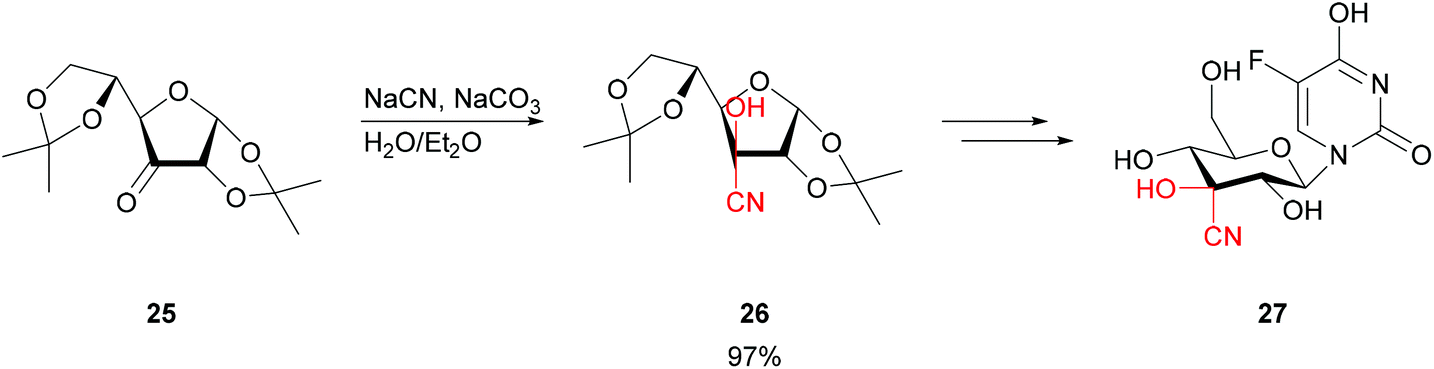 | ||
| Scheme 6 Synthesis of 3′-C-cyano nucleosides.44 | ||
2.2 N′-containing heterocycles
Various synthesis routes to obtain N′-containing heterocycles like trans-2,3-disubstituted aziridines 32, cis- and trans-2,5-disubstituted morpholines 35 and functionalised 4-hydroxy-2-pyrrolines 38 have been described. All of them are based on the stereoselective cyanohydrin 29 formation catalysed by (R)- or (S)-selective HNLs as a key step. In all cases a bi-phasic procedure was used, with the HNL in the aqueous layer at a low pH to avoid the racemic, chemical background reaction and an organic layer to enable high substrate loading. Immediate protection of the cyanohydrins prevented degradation to the respective aldehydes.45–47Aziridines are known to be highly reactive and they occur in a number of natural products which show antibiotic and antitumor activities due to their ability to cross-link DNA. trans-2,3-Disubstituted aziridines 32 (Scheme 7) were obtained in a five-step procedure via the formation of anti-amino alcohols 30 using a one-pot Grignard addition–reduction sequence. Subsequent Cu(II)-catalysed diazotransfer led to the formation of anti-azido alcohols 31 which were converted to trans-aziridines 32 by a triphenylphosphine-mediated reductive cyclisation in good yields and excellent enantioselectivities.45
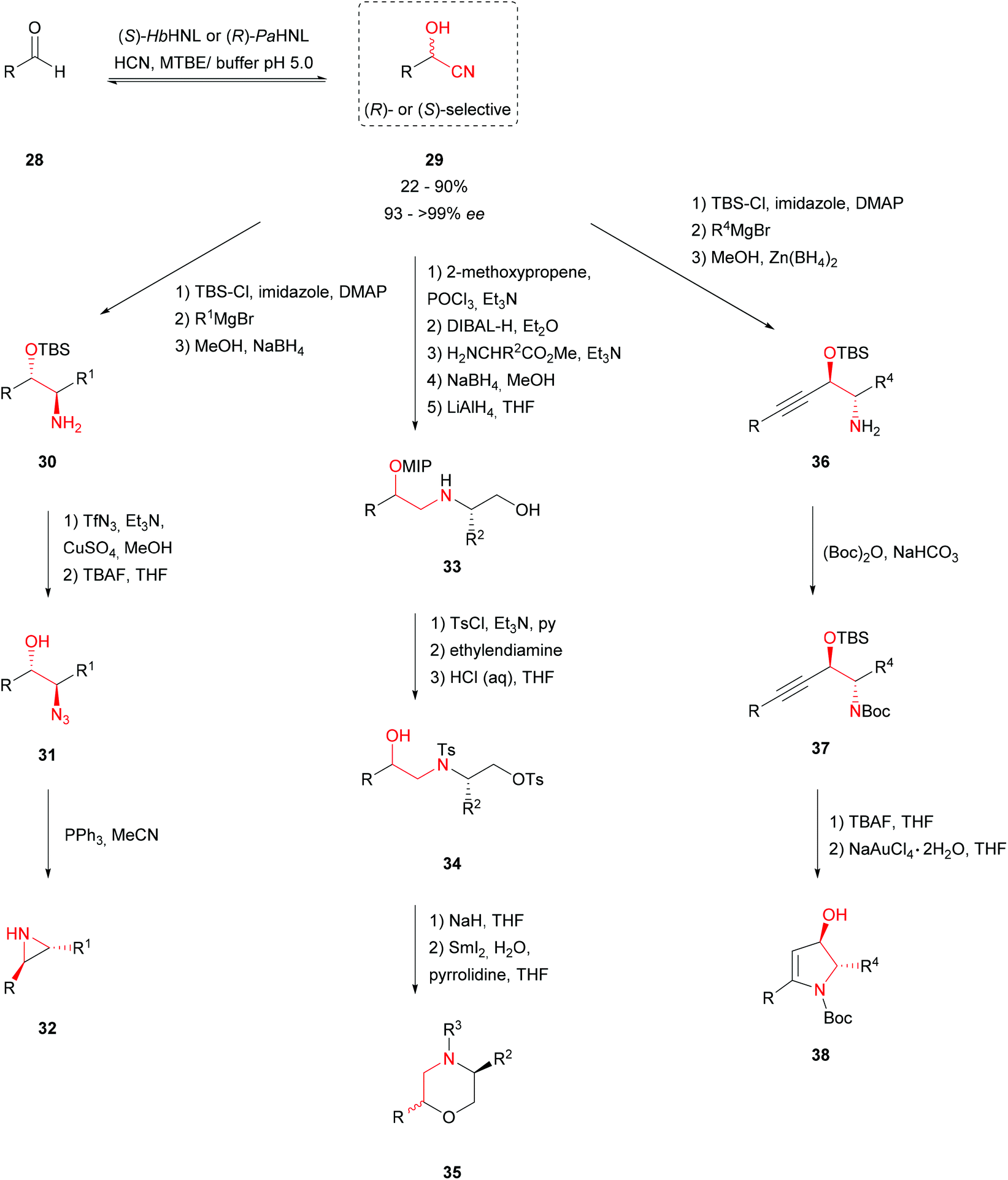 | ||
| Scheme 7 Cyanohydrins as key intermediates in the synthesis of N‘-containing heterocycles.45–47 | ||
Substituted morpholines have been shown to be valuable in the production of both therapeutical compounds and agrochemical fungicides and bactericides. A high yielding synthesis route to cis- and trans-2,5-disubstituted morpholines 35 with excellent diastereoselectivities was therefore developed. In a one-pot reduction–transimination–reduction sequence the cyanohydrin was treated with amino esters to obtain substituted amino esters. Reduction of the ester resulted in the formation of amino alcohols 33. Tosylation of the primary alcohol and secondary amine followed by selective deprotection of the secondary alcohol led to cyclisation precursor 34. Subsequent treatment with NaH induced the cyclisation. Depending on the protection and activation strategy a wide range of enantiopure morpholines 35 was generated.46
Encouraged by these results, an elegant procedure combining HNL and gold (NaAuCl4·2H2O) catalysis to synthesise functionalised 4-hydroxy-2-pyrrolines 38 was developed. These compounds are known to occur in biologically active and pharmaceutically interesting compounds. Formylating terminal alkynes with DMF gave α,β-acetylenic aldehydes being used as substrates in the enzymatic hydrocyanation. Both PaHNL and HbHNL were employed at low temperature; thus (R)- and (S)-enantiomers could be obtained. Subsequent protection and the one-pot Grignard addition–Zn(BH4)2 reduction sequence resulted in acetylene-containing amino alcohols 36. A 5-endo-dig gold-catalysed cyclisation gave aryl-substituted 4-hydroxy-2-pyrrolines 38 in reasonable yields and good diastereomeric ratios.47
Also enantiomerically pure tetrahydroisoquinolines 43, often occurring natural motifs, can be prepared by combining HNL and gold-catalysed reactions (Scheme 8). Based on earlier investigations that demonstrated that furylic substrates can be converted by HbHNL and PaHNL, i.e. into the (R)- and the (S)-enantiomer, a furfural derivative 39 was utilised.48 It is worth mentioning that due to the CIP nomenclature system the (R)-HNL gives the (S)-product and vice versa as the furan oxygen has a higher priority than the nitrile nitrogen atom. The reaction was again performed in a bi-phasic mixture with PaHNL. Protection of the hydroxyl function 40, selective reduction with DIBAL-H to amines 41, tosylation and subsequent propargylation resulted in enantiomerically pure alkynes 42. The [(Ph3PAu)2Cl]BF4-catalysed cycloisomerisation completed the route to give hydroxytetrahydroisoquinolines 43 in good to excellent yields and enantioselectivities.49
 | ||
| Scheme 8 Synthesis of tetrahydroisoquinolines.49 | ||
2.3 Lactones
The HNL-catalysed synthesis of (R)-pantolactone (46), a precursor in the vitamin B5 production, has the potential to be applied industrially (Scheme 9).32 Conversion of hydroxypivalaldehyde (44) followed by a hydrolytic ring closure gave (R)-pantolactone (46) in quantitative yield with excellent enantioselectivity. A problem in this reaction was that even at a low pH value of 4 the racemic cyanide addition takes place. By applying site-saturation mutagenesis a highly stable and active mutant (V317A) of (R)-selective PaHNL5 was developed. This allowed performing the reaction at pH 2.5, completely suppressing the “base”-catalysed side reaction. In an aqueous mono-phasic system an excellent yield and ee = 99% were obtained. This system has so far been rated as the most efficient biocatalyst in the vitamin B5 synthesis route.32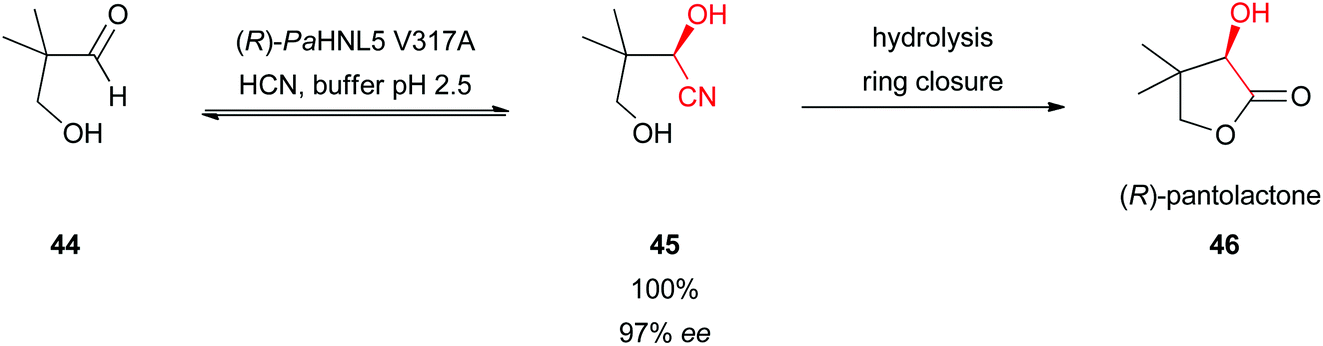 | ||
| Scheme 9 Total synthesis of (R)-pantolactone.32 | ||
α-Methylene-γ-butyrolactones 50 are widely spread in biologically active natural products and they additionally serve as versatile building blocks in organic chemistry. An organocatalysed one-pot allylic-alkylation–cyclisation reaction to obtain them with various substituents at the β- and γ-positions was recently described.50 Adding THP-protected cyanohydrin 48 to Morita–Baylis–Hillman (MBH) adduct 49 with catalytic amounts of 1,8-diazabicyclo[5.4.0]undec-7-ene in DCM followed by the addition of TFA resulted in the formation of α-methylene-γ-butyrolactones in good yields (Scheme 10). The synthetic potential of 50 was proven by follow-up reactions leading to i.e. the respective amides 51 or α,β-unsaturated-γ-butyrolactones 52. This synthesis was performed with racemic THP protected cyanohydrins. Very efficient HNL catalysed preparations of both (R)- and (S)-cyanohydrins, protected under either acidic or basic conditions with several different protection groups, have already been described. This included THP ethers. In all the cases the HNL was employed in wet organic solvents that could be replaced by dry solvents of choice without work-up before the protection step.51,52 This strategy allows for placing the above described synthesis on a chiral footing. An example is the total chemo-enzymatic synthesis of (−)-ephedrine, using benzaldehyde and immobilised PaHNL, yielding the desired product with moderate yield and excellent enantioselectivity.52
 | ||
| Scheme 10 Synthesis of α-methylene-γ-butyrolactones.50,52 | ||
2.4 Suzuki coupling
The complete stereoinversion of a protected, chiral cyanohydrin was recently presented.53 The stereospecific Suzuki cross-coupling of alkyl α-cyanohydrin triflates 53 with a range of aryl and alkenyl boronic acids 54 was achieved, under mild reaction conditions with moderate to excellent yields and excellent enantioselectivities. Triflate 53 has been synthesised chemically with a Lewis base–Lewis acid based approach using TMSCN followed by basic protection with TfO2 (Scheme 11), but is equally accessible via HNL-catalysis.51,54 As both the transmetalation and reductive elimination proceed with retention, the coupling takes place with complete inversion of the configuration induced by the oxidative addition of the triflate protected cyanohydrin 53 to the Pd.53 | ||
| Scheme 11 Pd-catalysed Suzuki cross-coupling of alkyl α-cyanohydrin triflates.53,54 | ||
2.5 Total syntheses
The (S)-HbHNL enabled the total synthesis of (+)-febrifugine (61) and its analogues.55 The desired compound, known for its powerful antimalarial activity, was obtained with an overall yield of 2.5% over 15 chemoenzymatic steps, including the formation of a lactam intermediate 58 from the enantiopure cyanohydrin 57 applying N-acyliminium ion chemistry (Scheme 12). The HNL catalysed step was performed in a bi-phasic system and was scaled successfully to 25 g. In addition, a second purely chemical synthetic strategy with only 10 steps and improved overall yield (32%) was developed. However limited access to the starting material and fewer possibilities of the latter for the synthesis of analogues make the chemoenzymatic route significantly more attractive. The first strategy to form N-acyliminium ions based on functionalised cyclic N,N-acetals was presented as a key transformation in the total synthesis of (+)-epiquinamide (62), involving the same key chemoenzymatic steps (Scheme 12).56 By applying the (R)-selective PaHNL the opposite enantiomer could be synthesised in excellent yield and enantioselectivity (95%, 95% ee), however when scaling up to 25 g the enantiomeric excess was significantly reduced. | ||
| Scheme 12 Total synthesis of (+)-febrifugine and (+)-epiquinamide.55,56 | ||
Another (R)-selective HNL (Prunus communis L. var. dulcis Borkh) from kernels of Badamu (almond from Xinjiang) was used in the total chemoenzymatic synthesis of broad spectrum antibiotics from the chloramphenicol family, thiamphenicol (65) and florphenicol (66) (Scheme 13).57 The latter possesses considerably lower side effects and 10 times higher bacterial potency than 65. The key step, the formation of enantiopure 4-methylsulfanyl-mandelonitrile intermediate 64, was performed in a wet organic solvent. This allowed a high substrate loading and straightforward work-up; the insoluble enzyme was removed by filtration. Overall 65 and 66 were synthesised with excellent enantiomeric excess and yields of 26% and 18%, respectively.
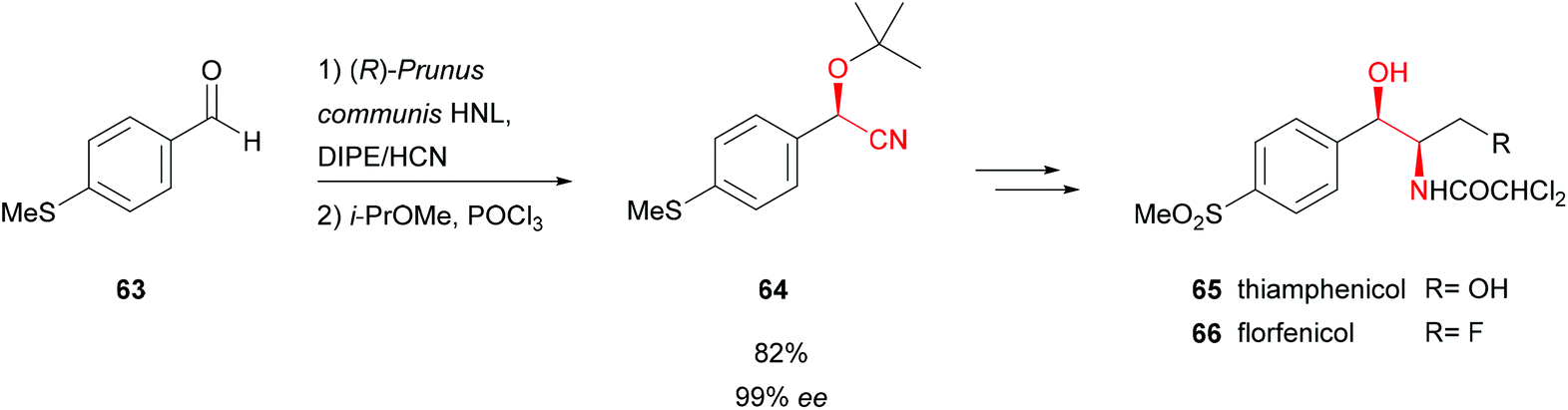 | ||
| Scheme 13 Total synthesis of thiamphenicol and florfenicol.57 | ||
Several chemoenzymatic approaches for the asymmetric total synthesis of nonenolides (also known as decanolides), naturally occurring compounds in fungi, were catalysed by (R)-selective Prunus armeniaca HNL (ParsHNL), isolated from an apricot species. All reactions performed with this HNL were bi-phasic in an emulsin to ensure that mass transfer could not limit the rate of reaction.58–61Scheme 14A shows the use of aliphatic short chain aldehydes as starting materials to obtain the respective enantiopure (R)-cyanohydrins 68a–b.61 Subsequent reduction of the nitrile with DIBAL-H yields the respective aldehyde intermediates 69a–b, accomplishing after 12 and 15 steps the total synthesis of stagonolide-B (70), (Z)-cytospolide E (71) and D (72) respectively, including ring-closing metathesis (Scheme 14A).58,59
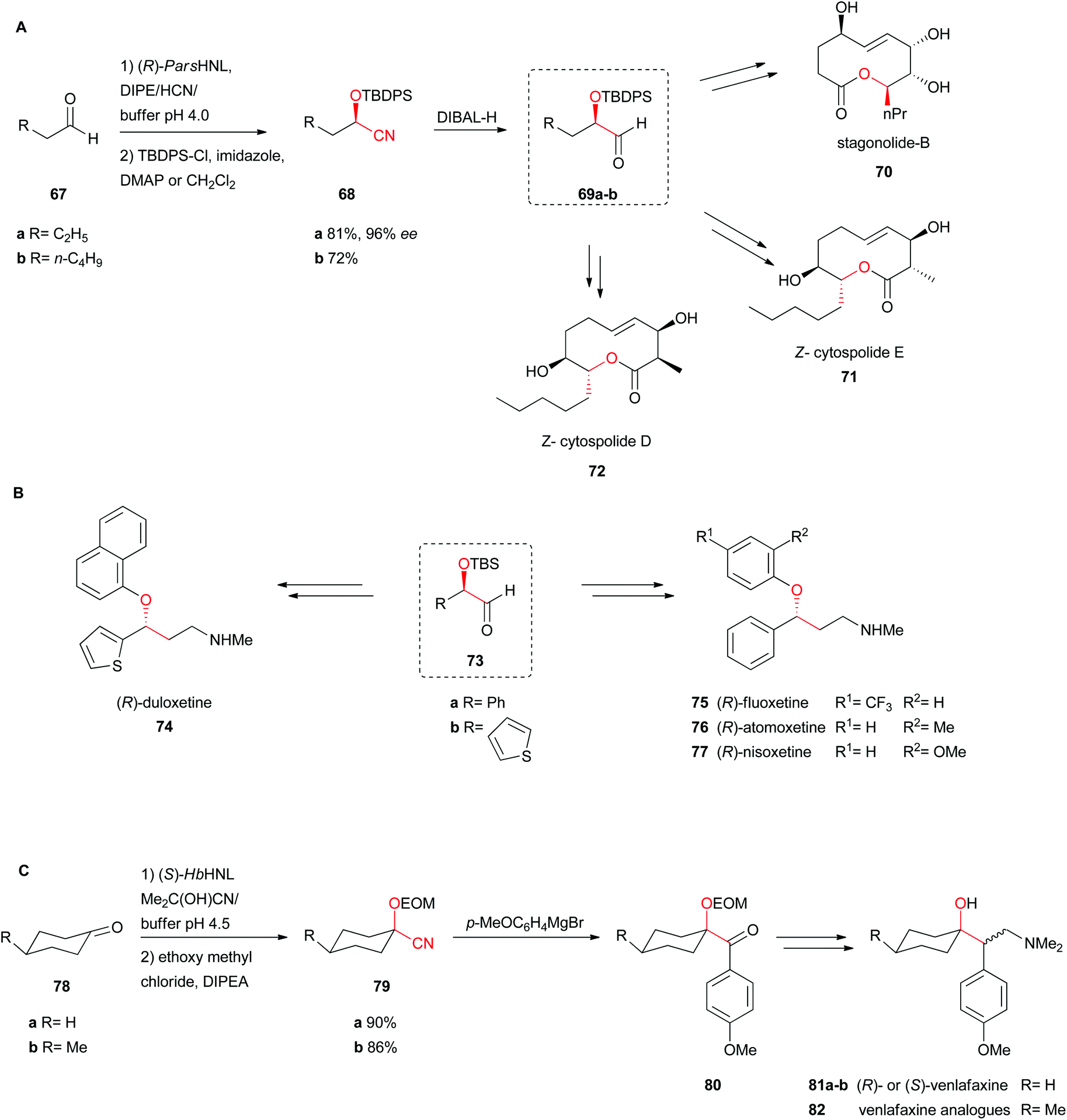 | ||
| Scheme 14 Total synthesis of (A) (Z)-cytospolide D and E, stagonolide-B, (B) (R)-fluoxetine, (R)-atomoxetine, (R)-nisoxetine and (R)-duloxetine, and (C) (R)- and (S)-venlafaxine.58–60,62 | ||
Utilising the same chemoenzymatic approach with ParsHNL described above, several well-known antidepressants from the serotonin-specific reuptake inhibitor (SSRI) and serotonin-norepinephrine reuptake inhibitor (SNRI) classes, including the block buster fluoxetine (US$350 million a year), atomoxetine, nisoxetine and duloxetine have also been synthesised (Scheme 14B).60 After DIBAL reduction of the enantiopure cyanohydrin, Wittig olefination, and hydroboration, standard transformation including the final Mitsunobu reaction with the relevant phenol yielded 75, 76 and 77 with 23, 25 and 24% overall yields, respectively. For the synthesis of (R)-duloxetine a similar reaction scheme was applied, starting from (S)-2-hydroxy-2-(thiophen-2-yl)acetonitrile (73b).
Venlafaxine is an antidepressant where both enantiomers have a roll in its activity. The (+)-enantiomer belongs to the SSRIs and the (−)-enantiomer to the SNRIs. In the chemoenzymatic synthesis of venlafaxine and several derivatives, cyclohexanone (78a), a rather hindered ketone, was used as the substrate. (S)-HbHNL was employed as a catalyst in a bi-phasic approach, again an emulsin. Only one diastereomer was formed by the syn-addition of the CN group (Scheme 14C).62 Protection of the hydroxyl group 79 with ethoxymethyl chloride (EOM-Cl), followed by Grignard, Wittig reaction and hydroboration yielded a racemic alcohol. In another biocatalytic step, the lipase-catalysed kinetic resolution finally yielded both diastereomers 81a–b in pure form (Scheme 14C).
In a new strategy for the synthesis of polyketides based on the ortho-diphenylphosphino benzoate-directed (o-DPPB) allylic substitution chiral alcohols were needed.63 The key enantiomerically pure allylic o-DPPB ester 85 or 86 was obtained in five steps (Scheme 15). HNL catalysed addition of cyanide to crotonaldehyde (83) and the subsequent Pinner reaction of the nitrile group, then reduction of the newly generated ester 84 and protection of the resulting primary alcohol allowed the introduction of the o-DPPB group on the secondary alcohol. (R)-selective PaHNL induced the desired stereochemistry; buffer saturated organic solvents were used to achieve high substrate loading. The o-DPPB ester served as a building block for iterative propionate insertion occurring with complete control of chemo-, regio-, and stereochemistry. The mentioned approach was applied in the syntheses of several polyketide precursors 87 and 88 as well as in the total syntheses of natural 89 and unnatural 90 isomers of tribolure, known as aggregation pheromones of the red flour beetle (Scheme 15).63 Using the same strategy, later the total synthesis of another aggregation pheromone used for plant protection, (+)-vittatalactone (91, Scheme 15) was reported in 11 steps with total yield of 18%.64
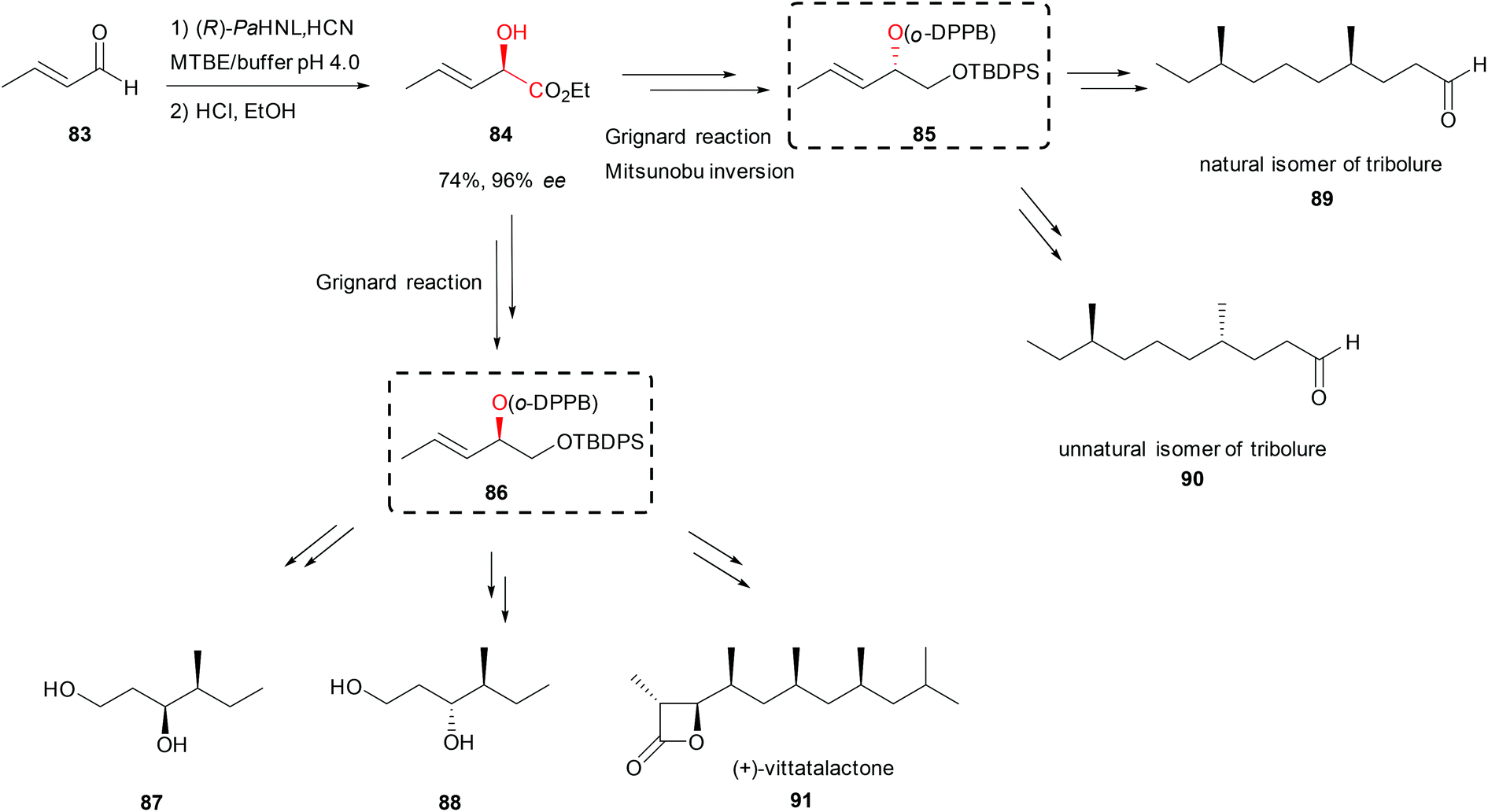 | ||
| Scheme 15 Total synthesis of (+)-vittatalactone and tribolure natural and unnatural isomers.63,64 | ||
3. Enzyme immobilisation
In addition to the traditional use of soluble HNLs in bi-phasic reaction media, immobilisation of HNLs is a frequently used powerful technique for the efficient synthesis of enantiopure cyanohydrins. In general the technique is categorised into non-covalent immobilisation on a carrier, covalent immobilisation on solid surfaces and encapsulation within an inorganic or polymer matrix (Fig. 1).7,9 | ||
| Fig. 1 Concepts of hydroxynitrile lyase immobilisation. | ||
Typically an efficient use of immobilised HNLs is mainly obtained by the use of buffer/water-saturated solvents such as methyl tert-butyl ether, diisopropylether or hydrocarbons as the reaction medium with a dispersed immobilised biocatalyst.9 These organic solvent systems allow very high substrate solubility within the liquid phase and also efficiently suppress the undesired non-selective addition of cyanide to the carbonyl compound.26 For this purpose numerous systems of covalent and non-covalent immobilisation concepts of HNLs were investigated in the past, which were also recently reviewed in detail.7
A special case hereof, the immobilisation of hydroxynitrile lyases as crossed-linked enzyme aggregates (CLEAs), without the need for any solid carrier, was introduced. Here physical aggregates of precipitated proteins are formed, which are later cross-linked by using bi-functional reagents such as glutaraldehyde.65 Within the resulting cross-linked aggregates the enzymes maintain their catalytic structure, but are basically rendered insoluble in various solvents due to the huge increase in size. Further modifications thereof have also been described.51,66–70
As an example, HbHNL-CLEA facilitates the synthesis of (S)-mandelonitrile with >95% conversion and >97% enantiomeric excess at very high substrate loadings in buffer-saturated solvents, as mentioned above.51 Within the CLEAs structurally relevant water molecules still remain, which explains the excellent activity in such solvent systems.71 Furthermore, HNLs can also be co-immobilised with other enzymes as a combi-CLEA-preparation, which can be used in single step bi-enzymatic cascade reactions for the synthesis of enantiopure α-hydroxycarboxylic acids and corresponding amides (using a nitrile hydratase).72 Even catalytically active triple-CLEA-preparations were reported involving a HNL, a nitrilase, and an amidase in a one-pot cascade reaction from benzaldehyde (92) to (S)-mandelic acid (95) at excellent conversion and enantiomeric excess (Scheme 16).73
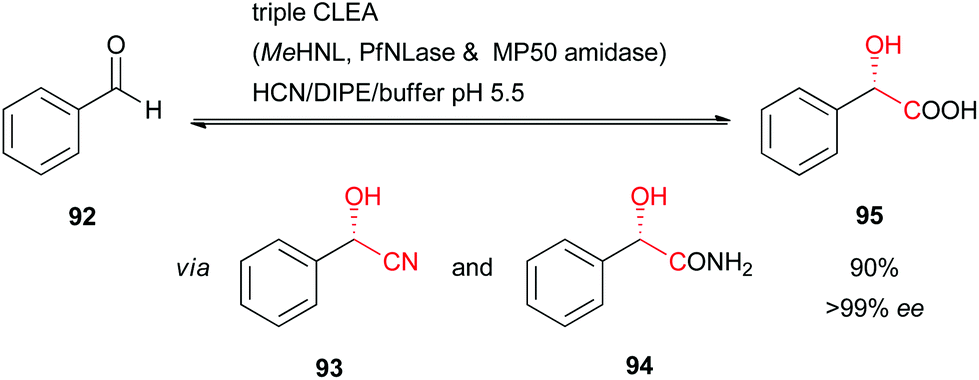 | ||
| Scheme 16 Triple-CLEA-catalysed synthesis of (S)-mandelic acid.73 | ||
Recently a related but rather counter-intuitive approach to immobilise AtHNL without the need for a solid support was reported. By fusion of a coiled-coil domain to AtHNL catalytically active inclusion bodies were formed. This insoluble enzyme preparation shows an improved stability at low pH, which is a weak point of the native form of Arabidopsis thaliana hydroxynitrile lyase (AtHNL). This strategy facilitated the synthesis of (R)-mandelonitrile with excellent enantiomeric excess in a buffer saturated solvent system. Furthermore, the fused protein was recycled multiple times without any noticeable loss of enzymatic activity and enantioselectivity.74
A powerful alternative to covalent immobilisation or the direct use of insoluble protein-approaches is the encapsulation of HNLs into inorganic or organic materials, such as sol gels or silicone. Here the enzyme is physically entrapped in the dense network and a potential harmful chemical bond formation to the solid matrix is not required. HbHNL was encapsulated in a sol–gel matrix by gelation of a precursor (sol) with subsequent aging of the sol–gel, and eventually ground into a fine powder.75 The catalytic material obtained was successfully used for the synthesis of aliphatic and aromatic cyanohydrins with high conversion and enantiomeric excesses of up to 99%. Recycling of the sol gel was successfully performed up to 4 cycles and was also applied, albeit less successfully, to AtHNL.76
The encapsulation of MeHNL into silicone-based spheres, which contained aqueous enzyme solution inclusions in a polymer matrix, proved more successful.77 Here, the biocatalyst remains in solution, while the silicone precursor polymerises and thus cures around the aqueous droplets. The resulting polymer preparation was subsequently dispersed in diisopropylether with the dissolved substrates and was afterwards easily removed by filtration. Again the undesired spontaneous formation of racemic cyanohydrin was efficiently suppressed due to the absence of a large continuous aqueous phase. This efficient immobilisation by inclusion was also scaled-up for the synthesis of various mandelonitrile derivatives at a multi-gram scale. Subsequent saponification gave the corresponding mandelic acid derivatives with high chemical purity and enantiomeric excess.
Recently a very interesting flow chemistry concept was successfully introduced with immobilised AtHNL. This facilitated the continuous synthesis of protected (R)-cyanohydrins (Scheme 17).78 Immobilised CalB (Candida antarctica lipase B as Novozym 435) starts the orthogonal biocatalytic reaction sequence by cleaving ethyl cyanoformate (96) and thus releasing hydrogen cyanide, greatly improving laboratory safety. Subsequently benzaldehyde (92) was introduced, and the mixture flows to the second biotransformation step. Immobilised AtHNL catalyses the formation of the desired enantiopure cyanohydrin (97). This relatively unstable compound was afterwards chemically protected by a classical acylation with acetic anhydride and pyridine, forming the desired, stable O-acetylcyanohydrin (98). The reaction concept was also successfully used for a series of other aromatic substrates with high conversions and enantiomeric excesses. Similar approaches were reported combining classical bi-phasic reaction systems with the flow chemistry concept.79,80 However, these reaction systems require an additional separator, which is not required with an immobilised HNL.
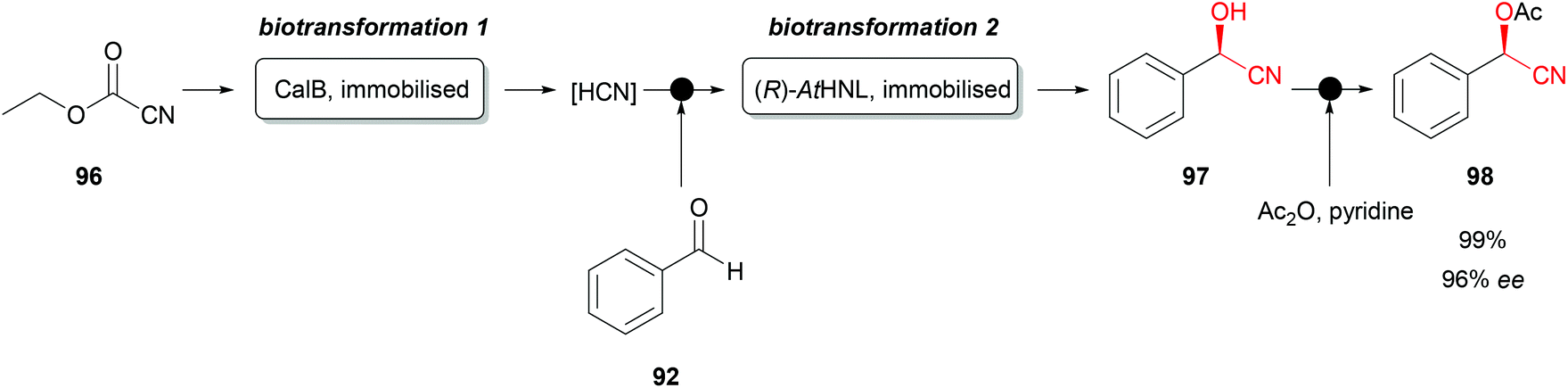 | ||
| Scheme 17 Orthogonal biocatalytic synthesis of chiral O-acetylcyanohydrins with immobilised CalB and AtHNL.78 | ||
4. Conclusions & outlook
The HNL catalysed enantioselective synthesis of cyanohydrins is a well-established technique that is a key step in many syntheses. Purely aqueous, bi-phasic and wet organic solvent systems are all used with great success, giving the chemist ample room to find suitable reaction conditions for the envisaged synthesis, as exemplified in this review.The challenges for the future lie in the efficient engineering of the reaction to enable even more straightforward applications of these versatile enzymes in organic chemistry. Here immobilisation in combination with smart flow-through systems open up entirely new routes for synthesis. As demonstrated in Scheme 17 a plug and play approach for organic chemistry, including enantioselective HNLs, is the next stage of development.
Acknowledgements
Generous funding to P. B. (BE-BASIC; grant number FES-0905) and H. B. (STW; grant number 14170) is acknowledged. J. v. L. kindly thanks the German Federal Ministry of Education and Research (BMBF – Bundesministerium für Bildung und Forschung) (project number 031A123).References
- M. Gruber-Khadjawi, M. H. Fechter and H. Griengl, in Enzyme Catalysis in Organic Synthesis, ed. K. Drauz, H. Gröger and O. May, Wiley-VCH Verlag GmbH & Co. KGaA, 2012, pp. 947–990 Search PubMed.
- M. North, D. L. Usanov and C. Young, Chem. Rev., 2008, 108, 5146–5226 CrossRef CAS PubMed.
- M. Brovetto, D. Gamenara, P. Saenz Méndez and G. A. Seoane, Chem. Rev., 2011, 111, 4346–4403 CrossRef CAS PubMed.
- K. Steiner, A. Glieder and M. Gruber-Khadjawi, in Science of Synthesis: Biocatalysis in Organic Synthesis, Georg Thieme Verlag, 2015, vol. 2, pp. 1–30 Search PubMed.
- J. Holt and U. Hanefeld, Curr. Org. Synth., 2009, 6, 15–37 CrossRef CAS.
- M. Dadashipour and Y. Asano, ACS Catal., 2011, 1, 1121–1149 CrossRef CAS.
- U. Hanefeld, Chem. Soc. Rev., 2013, 42, 6308–6568 RSC.
- D. J. Ager and O. May, Spec. Chem. Mag., 2008, 28, 30–32 CAS.
- E. Lanfranchi, K. Steiner, A. Glieder, I. Hajnal, R. A. Sheldon, S. van Pelt and M. Winkler, Recent Pat. Biotechnol., 2013, 7, 197–206 CrossRef CAS PubMed.
- M. A. Kassim and K. Rumbold, Biotechnol. Lett., 2014, 36, 223–228 CrossRef CAS PubMed.
- M. Dadashipour, M. Yamazaki, K. Momonoi, K. Tamura, K. Fuhshuku, Y. Kanase, E. Uchimura, G. Kaiyun and Y. Asano, J. Biotechnol., 2011, 153, 100–110 CrossRef CAS PubMed.
- M. Asif and T. C. Bhalla, Catal. Lett., 2016, 146, 1118–1127 CrossRef CAS.
- I. Hajnal, A. Łyskowski, U. Hanefeld, K. Gruber, H. Schwab and K. Steiner, FEBS J., 2013, 280, 5815–5828 CrossRef CAS PubMed.
- M. Zagrobelny, S. Bak and B. L. Møller, Phytochemistry, 2008, 69, 1457–1468 CrossRef CAS PubMed.
- M. Dadashipour, Y. Ishida, K. Yamamoto and Y. Asano, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 10605–10610 CrossRef CAS PubMed.
- F. Wöhler and J. Liebig, Ann. Pharm., 1837, 22, 1–24 CrossRef.
- L. Rosenthaler, Biochem. Z., 1908, 14, 238–253 CAS.
- P. Wyatt and S. Warren, in Organic Synthesis Strategy and Control, John Wiley & Sons, Ltd., 2007, pp. 652–679 Search PubMed.
- J. Clayden, N. Greeves and S. Warren, in Organic Chemistry, Oxford University Press Inc., New York, 2012, pp. 1102–1133 Search PubMed.
- R. K. Henderson, C. Jimenez-Gonzalez, C. Preston, D. J. C. Constable and J. M. Woodley, Ind. Biotechnol., 2008, 4, 180–192 CrossRef CAS.
- R. Hatti-Kaul, U. Törnvall, L. Gustafsson and P. Börjesson, Trends Biotechnol., 2007, 25, 119–124 CrossRef CAS PubMed.
- D. J. Pollard and J. M. Woodley, Trends Biotechnol., 2007, 25, 66–73 CrossRef CAS PubMed.
- W. Wang, X. Liu, L. Lin and X. Feng, Eur. J. Org. Chem., 2010, 4751–4769 CrossRef CAS.
- N. H. Khan, R. I. Kureshy, S. H. R. Abdi, S. Agrawal and R. V. Jasra, Coord. Chem. Rev., 2008, 252, 593–623 CrossRef CAS.
- D. T. Mowry, Chem. Rev., 1948, 42, 189–283 CrossRef CAS PubMed.
- J. N. Andexer, J. V. Langermann, U. Kragl and M. Pohl, Trends Biotechnol., 2009, 27, 599–607 CrossRef CAS PubMed.
- Z. Liu, B. Pscheidt, M. Avi, R. Gaisberger, F. S. Hartner, C. Schuster, W. Skranc, K. Gruber and A. Glieder, ChemBioChem, 2008, 9, 58–61 CrossRef CAS PubMed.
- M. Avi, R. M. Wiedner, H. Griengl and H. Schwab, Chem. – Eur. J., 2008, 14, 11415–11422 CrossRef CAS PubMed.
- R. Wiedner, B. Kothbauer, T. Pavkov-Keller, M. Gruber-Khadjawi, K. Gruber, H. Schwab and K. Steiner, ChemCatChem, 2015, 7, 325–332 CrossRef CAS.
- Y. Asano and N. Kawahara, Ind. Biotechnol., 2016, 12, 91–97 CrossRef CAS.
- J. von Langermann, J.-K. Guterl, M. Pohl, H. Wajant and U. Kragl, Bioprocess Biosyst. Eng., 2008, 31, 155–161 CrossRef CAS PubMed.
- B. Pscheidt, Z. Liu, R. Gaisberger, M. Avi, W. Skranc, K. Gruber, H. Griengl and A. Glieder, Adv. Synth. Catal., 2008, 350, 1943–1948 CrossRef CAS.
- W. F. Willeman, U. Hanefeld, A. J. J. Straathof and J. J. Heijnen, Enzyme Microb. Technol., 2000, 27, 423–433 CrossRef CAS PubMed.
- J.-K. Guterl, J. N. Andexer, T. Sehl, J. von Langermann, I. Frindi-Wosch, T. Rosenkranz, J. Fitter, K. Gruber, U. Kragl, T. Eggert and M. Pohl, J. Biotechnol., 2009, 141, 166–173 CrossRef CAS PubMed.
- J. von Langermann, A. Mell, E. Paetzold, T. Daußmann and U. Kragl, Adv. Synth. Catal., 2007, 349, 1418–1424 CrossRef CAS.
- P. Zandbergen, J. van der Linden, J. Brussee and A. van der Gen, Synth. Commun., 1991, 21, 1387–1391 CrossRef CAS.
- A. M. C. H. van den Nieuwendijk, M. Ruben, S. E. Engelsma, M. D. P. Risseeuw, R. J. B. H. N. van den Berg, R. G. Boot, J. M. Aerts, J. Brussee, G. A. van der Marel and H. S. Overkleeft, Org. Lett., 2010, 12, 3957–3959 CrossRef CAS PubMed.
- A. M. C. H. van den Nieuwendijk, R. J. B. H. N. van den Berg, M. Ruben, M. D. Witte, J. Brussee, R. G. Boot, G. A. van der Marel, J. M. F. G. Aerts and H. S. Overkleeft, Eur. J. Org. Chem., 2012, 3437–3446 CrossRef CAS.
- M. Avi, R. Gaisberger, S. Feichtenhofer and H. Griengl, Tetrahedron, 2009, 65, 5418–5426 CrossRef CAS.
- D. J. Vugts, L. Veum, K. al-Mafraji, R. Lemmens, R. F. Schmitz, F. J. J. de Kanter, M. B. Groen, U. Hanefeld and R. V. A. Orru, Eur. J. Org. Chem., 2006, 1672–1677 CrossRef CAS.
- D. J. Vugts, H. Aktas, K. Al-Mafraji, F. J. J. de Kanter, E. Ruijter, M. B. Groen and R. V. A. Orru, Eur. J. Org. Chem., 2008, 1336–1339 CrossRef CAS.
- A. Hietanen, F. S. Ekholm, R. Leino and L. T. Kanerva, Eur. J. Org. Chem., 2010, 6974–6980 CrossRef CAS.
- A. Hietanen and L. T. Kanerva, Eur. J. Org. Chem., 2012, 2729–2737 CrossRef CAS.
- C. Kiritsis, S. Manta, V. Parmenopoulou, A. Dimopoulou, N. Kollatos, I. Papasotiriou, J. Balzarini and D. Komiotis, Carbohydr. Res., 2012, 364, 8–14 CrossRef CAS PubMed.
- B. Ritzen, M. C. M. van Oers, F. L. van Delft and F. P. J. T. Rutjes, J. Org. Chem., 2009, 74, 7548–7551 CrossRef CAS PubMed.
- B. Ritzen, S. Hoekman, E. D. Verdasco, F. L. van Delft and F. P. J. T. Rutjes, J. Org. Chem., 2010, 75, 3461–3464 CrossRef CAS PubMed.
- B. Ritzen, G. J. J. Richelle, L. Brocken, F. L. van Delft and F. P. J. T. Rutjes, Synlett, 2014, 270–274 CAS.
- F. Effenberger, Angew. Chem., Int. Ed., 1994, 33, 1555–1564 CrossRef.
- A. S. K. Hashmi, F. Ata, P. Haufe and F. Rominger, Tetrahedron, 2009, 65, 1919–1927 CrossRef CAS.
- Y. Yan, D. En, Z. Zhuang, Y. Guo and W.-W. Liao, Tetrahedron Lett., 2014, 55, 479–482 CrossRef CAS.
- F. L. Cabirol, A. E. C. Lim, U. Hanefeld and R. A. Sheldon, Org. Process Res. Dev., 2010, 14, 114–118 CrossRef CAS.
- F. L. Cabirol, Doctoral Thesis, Technische Universiteit, Delft, ISBN 978-0-615-38055-1, 2010. link: http://repository.tudelft.nl/view/ir/uuid%3Ad0f437b4-7df4-454f-9dad-88e279650784/.
- A. He and J. R. Falck, J. Am. Chem. Soc., 2010, 132, 2524–2525 CrossRef CAS PubMed.
- Y. Hamashima, D. Sawada, M. Kanai and M. Shibasaki, J. Am. Chem. Soc., 1999, 121, 2641–2642 CrossRef CAS.
- M. A. Wijdeven, R. J. F. van den Berg, R. Wijtmans, P. N. M. Botman, R. H. Blaauw, H. E. Schoemaker, F. L. van Delft and F. P. J. T. Rutjes, Org. Biomol. Chem., 2009, 7, 2976–2980 CAS.
- M. A. Wijdeven, R. Wijtmans, R. J. F. van den Berg, W. Noorduin, H. E. Schoemaker, T. Sonke, F. L. van Delft, R. H. Blaauw, R. W. Fitch, T. F. Spande, J. W. Daly and F. P. J. T. Rutjes, Org. Lett., 2008, 10, 4001–4003 CrossRef CAS PubMed.
- W. Lu, P. Chen and G. Lin, Tetrahedron, 2008, 64, 7822–7827 CrossRef CAS.
- T. Das, T. Mahapatra and S. Nanda, Tetrahedron Lett., 2012, 53, 1186–1189 CrossRef CAS.
- R. K. Rej and S. Nanda, Eur. J. Org. Chem., 2014, 860–871 CrossRef CAS.
- R. K. Rej, T. Das, S. Hazra and S. Nanda, Tetrahedron: Asymmetry, 2013, 24, 913–918 CrossRef CAS.
- R. Bhunya, T. Mahapatra and S. Nanda, Tetrahedron: Asymmetry, 2009, 20, 1526–1530 CrossRef CAS.
- R. Bhunya and S. Nanda, Tetrahedron Lett., 2012, 53, 1990–1992 CrossRef.
- T. Reiss and B. Breit, Chem. – Eur. J., 2009, 15, 6345–6348 CrossRef CAS PubMed.
- Y. Schmidt, K. Lehr, U. Breuninger, G. Brand, T. Reiss and B. Breit, J. Org. Chem., 2010, 75, 4424–4433 CrossRef CAS PubMed.
- C. Mateo, J. M. Palomo, L. M. van Langen, F. van Rantwijk and R. A. Sheldon, Biotechnol. Bioeng., 2004, 86, 273–276 CrossRef CAS PubMed.
- D. Costes, E. Wehtje and P. Adlercreutz, J. Mol. Catal. B: Enzym., 2001, 11, 607–612 CrossRef CAS.
- R. A. Sheldon, Org. Process Res. Dev., 2011, 15, 213–223 CrossRef CAS.
- R. A. Sheldon, Appl. Microbiol. Biotechnol., 2011, 92, 467–477 CrossRef CAS PubMed.
- G. Torrelo, N. van Midden, R. Stloukal and U. Hanefeld, ChemCatChem, 2014, 6, 1096–1102 CrossRef CAS.
- D. Yildirim, S. S. Tükel and D. Alagöz, Biotechnol. Prog., 2014, 30, 818–827 CrossRef CAS PubMed.
- M. Paravidino, M. J. Sorgedrager, R. V. A. Orru and U. Hanefeld, Chem. – Eur. J., 2010, 16, 7596–7604 CrossRef CAS PubMed.
- F. van Rantwijk and A. Stolz, J. Mol. Catal. B: Enzym., 2015, 114, 25–30 CrossRef CAS.
- A. Chmura, S. Rustler, M. Paravidino, F. van Rantwijk, A. Stolz and R. A. Sheldon, Tetrahedron: Asymmetry, 2013, 24, 1225–1232 CrossRef CAS.
- M. Diener, B. Kopka, M. Pohl, K.-E. Jaeger and U. Krauss, ChemCatChem, 2016, 8, 142–152 CrossRef CAS.
- L. Veum, U. Hanefeld and A. Pierre, Tetrahedron, 2004, 60, 10419–10425 CrossRef CAS.
- D. Okrob, M. Paravidino, R. V. A. Orru, W. Wiechert, U. Hanefeld and M. Pohl, Adv. Synth. Catal., 2011, 353, 2399–2408 CrossRef CAS.
- J. von Langermann and S. Wapenhesch, Adv. Synth. Catal., 2014, 356, 2989–2997 CrossRef CAS.
- A. Brahma, B. Musio, U. Ismayilova, N. Nikbin, S. Kamptmann, P. Siegert, G. E. Jeromin, S. V. Ley and M. Pohl, Synlett, 2016, 27, 262–266 CAS.
- M. M. E. Delville, K. Koch, J. C. M. van Hest and F. P. J. T. Rutjes, Org. Biomol. Chem., 2015, 13, 1634–1638 CAS.
- K. Koch, R. J. F. van den Berg, P. J. Nieuwland, R. Wijtmans, H. E. Schoemaker, J. C. M. van Hest and F. P. J. T. Rutjes, Biotechnol. Bioeng., 2008, 99, 1028–1033 CrossRef CAS PubMed.
Footnote |
| † Both authors have contributed equally. |
| This journal is © The Royal Society of Chemistry 2016 |