
Aggregation and interactions of chemical mechanical planarization nanoparticles with model biological membranes: role of phosphate adsorption†
Xitong
Liu
and
Kai Loon
Chen
*
Department of Geography and Environmental Engineering, Johns Hopkins University, Baltimore, Maryland 21218-2686, USA. E-mail: kailoon.chen@jhu.edu; Tel: +(410) 516 7095
First published on 2nd November 2015
Abstract
In the semiconductor industry, chemical mechanical planarization (CMP) slurries are used in large quantities for the planarization of wafers and the release of CMP nanoparticles (NPs) into the natural environment may pose a threat to human health. In this study, the aggregation behavior of four model CMP NPs, namely, colloidal SiO2, fumed SiO2, CeO2, and Al2O3 NPs, was investigated through dynamic light scattering in the presence and absence of phosphate at pH 7.4. The colloidal and fumed SiO2 NPs were observed to have remarkable stability under neutral pH conditions both in the presence and absence of phosphate. Phosphate adsorption enhanced the negative surface charge of CeO2 NPs and reversed the charge of Al2O3 NPs, resulting in an increase in the stability of both NPs. In order to evaluate the propensity for the CMP NPs to attach to and disrupt cell membranes, the interactions of these NPs with supported lipid bilayers (SLBs) and lipid vesicles composed of zwitterionic 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) were examined using a quartz crystal microbalance. The attachment efficiencies of both SiO2 NPs on SLBs increased to 1 when the NaCl concentration was raised to 100 mM in the presence of phosphate. In contrast, favorable attachment of CeO2 and Al2O3 NPs to SLBs was not achieved in the presence of phosphate even at elevated NaCl concentrations, likely due to the enhancement of nanoparticle surface charge resulting from phosphate adsorption. When the supported vesicles were exposed to the SiO2 NPs, no disruption of vesicles was detected despite favorable conditions for nanoparticle–membrane association.
Nano impactWith the extensive use of CMP slurries in the semiconductor industry, there is a growing concern on their effect on human health. In this study, the surface charge and colloidal stability of CMP NPs, as well as their interactions with model membranes, were investigated, with an emphasis on the role of phosphate adsorption. Our results show that the presence of phosphate altered the surface charge of CeO2 and Al2O3 NPs, which in turn enhanced their stability and reduced their attachment to lipid bilayers. Since phosphate is a key constituent in biological fluids, these findings will allow for a better understanding of the aggregation behavior of CMP NPs and their association with cell membranes when they enter the human body. |
Introduction
Chemical mechanical planarization (CMP) is a key process in the semiconductor industry for the production of integrated circuit devices.1 In CMP processes, slurries of abrasive particles, including SiO2, CeO2, and Al2O3, are employed to planarize wafers through a combination of mechanical abrasion and chemical etching.2 The planarized wafers are subsequently rinsed with ultra-pure water, resulting in the generation of CMP wastewater.3,4 Although CMP wastewaters are generally subjected to on-site wastewater treatment (e.g., electrocoagulation5 and microfiltration6) and/or municipal biological wastewater treatment, the wastewater effluent from municipal wastewater treatment plants may still contain some unremoved CMP NPs which can be discharged, together with the treated effluent, into surface waters.7 In their study on the removal efficiency of CeO2 NPs by a model wastewater treatment plant, Limbach et al.8 reported that about 6 wt% of the added CeO2 NPs was found in the outflow of the model plant. The release of CMP NPs into the natural environment potentially poses a threat to the ecosystem and human health. In addition to the risks associated with the environmental release of CMP NPs, the exposure of workers to CMP NPs at the workplace may give rise to health risk concerns.9,10There have been extensive studies concerning the toxicity of SiO2, CeO2, and Al2O3 NPs. The SiO2 NPs used in CMP processes mainly include colloidal silica (c-SiO2) and fumed silica (f-SiO2).7 Colloidal SiO2 is synthesized through the hydrolysis and condensation of a Si precursor (e.g., silicon alkoxide),11 while fumed SiO2 is produced by pyrolysis of silicon tetrachloride (SiCl4) at high temperatures (1800 °C).12,13 It has been reported that f-SiO2 has a greater ability than c-SiO2 to generate hydroxyl radicals and cause red blood cells (RBCs) to undergo lysis due to their strained three-membered rings and chainlike aggregate structure.14 The CeO2 NPs used in CMP processes are made from high temperature calcination (>300 °C).7 CeO2 NPs have been shown to induce oxidative stress in human cancer cells and subsequently result in the damage of cell membranes.15 Finally, Al2O3 NPs can be produced through dehydration of γ-AlOOH (boehmite) at high temperatures (500–700 °C).16 Previous studies have reported that Al2O3 NPs can show inhibitory effect on human bronchial epithelial cells and cause hemolysis of RBCs.17,18
While the mechanisms for the cytotoxicity of different classes of nanomaterials are still under investigation, studies have shown that the direct association of NPs with cell membranes is often a critical step that precedes the subsequence biological responses.19,20 The attachment of NPs on cell membranes can also result in damage of the membranes.20 Therefore, a detailed assessment of the interactions of CMP NPs with cell membranes is expected to improve our understanding on the biological effects of these nanomaterials. Due to the inherent heterogeneity and diversity of cell membranes, the use of model (or artificial) cell membranes that are composed of lipid bilayers is an attractive approach to gain fundamental insights into the interactions between NPs and cell membranes.20–22 A common approach to investigate the attachment of NPs to cell membranes involves the employment of supported lipid bilayers (SLBs) to evaluate the NP's propensity to attach on lipid bilayers.23,24 Since lipid bilayer is a key component of cell membranes, the use of SLBs will enable the elucidation of the nonspecific interactions between NPs and cell membranes at the nano–bio interface.19,20,24–28 A recent study utilizing model membranes showed that the presence of proteins can decrease the attachment of NPs to membranes and hence reduce the uptake of NPs by human epithelial cells.29 Additionally, lipid bilayer vesicles have been used as model cell membranes to probe the propensity of NPs to disrupt bilayers.20,30–32
The objective of this work is to investigate the interactions of model CMP NPs with model biological membranes which include SLBs and supported vesicular layers (SVLs). Since phosphate is a key constituent in biological fluids, such as human blood serum (0.81–1.45 mM) and saliva (5–14 mM),33 the role of phosphate in these interactions will be studied. In addition, we will examine the aggregation behavior of CMP NPs in model biological media since the cellular uptake of NPs, as well as the subsequent biological responses, has been shown to be dependent on the aggregation state of the NPs.34,35 The results of this study will provide new insights into the nonspecific interactions of CMP NPs with cell membranes when they enter biological systems.
Experimental methods
CMP NP suspensions
Four model CMP NPs, namely, c-SiO2, f-SiO2, CeO2, and Al2O3, were provided by Cabot Microelectronics Corporation (Aurora, Illinois, USA) as slurries. These NP slurries are shared within a research consortium and have been extensively characterized.7 Their physicochemical properties (including shape, size, crystalline structure, and surface charge) are presented in a recently published paper by Speed et al.7 These slurries were maintained colloidally stable by pH adjustment and did not contain organic stabilizers.7 Here, we highlight some of the properties of the CMP NPs reported by Speed et al.7 Through transmission electron microscopy (TEM) imaging, the c-SiO2 NPs were observed to be mostly spherical, while the f-SiO2 NPs appeared irregular-shaped and fused together. X-ray diffraction (XRD) analysis showed that both c-SiO2 and f-SiO2 NPs had amorphous SiO2 structures. The CeO2 NPs were rectangular and the XRD patterns of the CeO2 slurry revealed the cubic fluorite structure of the NPs.36 Finally, the Al2O3 NPs contained a broad range of particles sizes and XRD results indicated that the Al2O3 NPs were γ-Al2O3.37 The mean diameters of the c-SiO2, CeO2, and Al2O3 NPs determined through TEM imaging were 36, 39, and 38 nm, respectively. The size of the f-SiO2 NPs could not be determined through TEM due to their coalesced state. By employing dynamic light scattering (DLS), the hydrodynamic diameter of the f-SiO2 NPs was determined to be ca. 148 nm. All slurries were used directly after being diluted in deionized (DI) water (18 MΩ cm, Millipore, MA) without further purification.Solution chemistry
ACS-grade NaCl, NaH2PO4, Na2HPO4, and NaHCO3 were used for the preparation of stock solutions. All stock solutions were filtered using 0.1 μm polyvinylidene fluoride syringe filters (Millipore, MA). The pH of the diluted CMP dispersions was maintained at 7.4 with either a phosphate (1.13 mM NaH2PO4 and 3.87 mM Na2HPO4) or bicarbonate (0.4 mM NaHCO3) buffer. Three buffers containing N-(2-hydroxyethyl)-piperazine-N′-(2-ethanesulfonic acid) (HEPES, Sigma-Aldrich) were also used in the experiments using a quartz crystal microbalance with dissipation monitoring (QCM-D). They are denoted as Buffer A (10 mM HEPES, 150 mM NaCl, pH adjusted to 7.4), Buffer B (10 mM HEPES, 100 mM NaCl, with unadjusted pH ca. 5.5), and Buffer C (20 mM HEPES, 150 mM NaCl, pH adjusted to 7.4).Electrophoretic mobility measurements
The electrophoretic mobilities (EPMs) of the CMP NPs were measured (ZetaPALS, Brookhaven Instruments Corp., Holtsville, NY) over a range of NaCl concentrations in either phosphate or bicarbonate buffer at pH 7.4. The concentrations of the CMP NPs used in the EPM measurements are presented in Table S1 of the ESI.† Three samples were measured at each electrolyte concentration. For most of the experimental conditions, ten measurements were conducted for each sample. At high NaCl concentrations, only five measurements were conducted in order to minimize the effect of NP aggregation.Phosphate adsorption by CMP NPs
The adsorption of phosphate on the CMP NPs was determined after an exposure time period of 10 min. The concentration of phosphate was measured using the colorimetric technique of Murphy and Riley.38 The details for the adsorption experiments are provided in the ESI.†Dynamic light scattering
Time-resolved DLS measurements were conducted with a light scattering unit to investigate the aggregation kinetics of four CMP NPs in phosphate and bicarbonate buffers. This unit employs an argon laser with a wavelength of 488 nm (Lexel 95, Cambridge Lasers Laboratories Ltd., CA).39 The details for the time-resolved DLS measurements are provided in our previous publications39,40 and also in the ESI.† The early-stage aggregation kinetics of CMP NPs can be derived from time-resolved DLS measurements using eqn (1):41,42 | (1) |
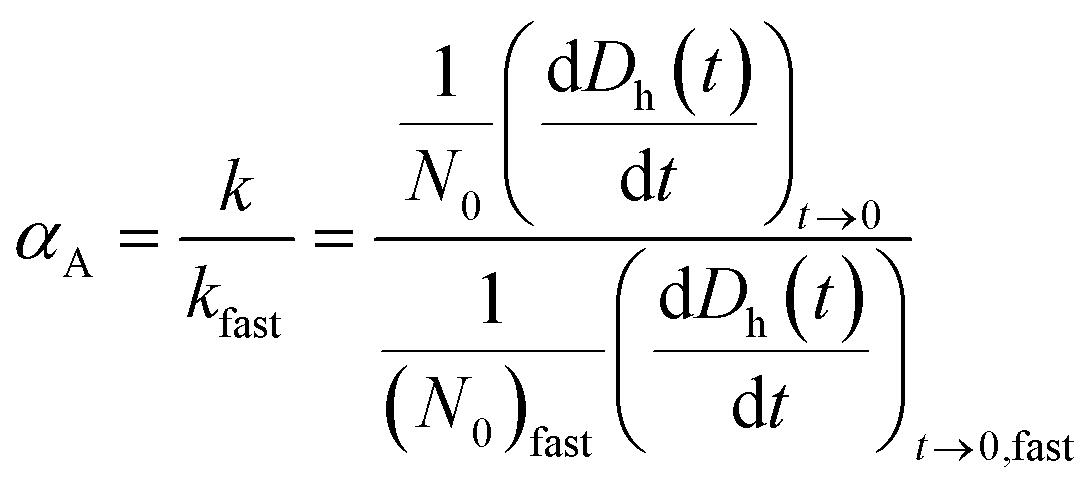 | (2) |
Preparation of vesicle stock suspensions
Two types of vesicles were used in the QCM-D experiments: 1) zwitterionic 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) vesicles for the formation of SLBs to investigate the propensity for CMP NPs to attach to lipid bilayers, and 2) DOPC vesicles encapsulated with carboxyfluorescein (CF) dye (to be referred to as CF-DOPC vesicles) to assess the disruption of lipid membranes by the NPs. For the preparation of DOPC vesicles, 0.2 mL of 25 g L−1 DOPC chloroform solution (Avanti Polar Lipids, Inc., Alabaster, AL) was dried in an Erlenmeyer flask using a gentle stream of ultrapure nitrogen gas. A DOPC thin film was formed at the bottom of the flask and the film was subjected to vacuum desiccation for at least 4 h. The DOPC lipids were then re-suspended in 5 mL Buffer A and the solution was magnetically stirred for ca. 30 min. To obtain unilamellar vesicles, the DOPC solution was extruded using a mini-extruder (Avanti Polar Lipids) through a 50 nm polycarbonate membrane (Whatman) for at least 15 times.47 For the preparation of CF-DOPC vesicles, the aforementioned DOPC thin film was hydrated with a 100 mM CF solution prepared in Buffer C and was subsequently subjected to the same extrusion treatment.32 All vesicle suspensions were stored in glass vials at 4 °C and used within four days after preparation.Quartz crystal microbalance with dissipation monitoring
The interactions of CMP NPs with DOPC SLBs and vesicles were studied using a QCM-D E4 setup (Biolin Scientific, Västra Frölunda, Sweden) that is equipped with four measurement chambers. Each chamber housed a 5 MHz quartz crystal sensor with either a silica-coated (QSX 303) or gold-coated (QSX 301) surface. The sensors were cleaned prior to the QCM-D experiments according to a protocol that was described in our previous paper.48 The temperature inside the QCM-D chambers was maintained at 25 °C. The rate of frequency shift at the 3rd overtone, dΔf(3)/dt, was used to quantify the deposition rates of GO on SLBs.48,49 Duplicate experiments were carried out for each condition.For the deposition of CMP NPs on SLBs, a SLB was first formed on the surface of a silica-coated sensor using the method of Keller and Kasemo.50 The sensor was rinsed with Buffer A until a stable baseline was achieved (i.e., frequency signal drifts of less than 0.2 Hz per 10 min). Following that, a 0.07 g L−1 DOPC vesicle suspension was flowed through the QCM-D chamber for ca. 5 min. The vesicles deposited on the silica surface and subsequently ruptured on the surface to form a SLB.50 The formation of a DOPC SLB resulted in a characteristic frequency shift of −25 ± 0.5 Hz and a simultaneous dissipation shift of 0.1 × 10−6.50 After the SLB was formed, the chamber was rinsed with Buffer A for ca. 10 min to remove the unadsorbed vesicles. A NaCl solution with phosphate or bicarbonate buffer was then flowed through the measurement chamber to obtain a stable baseline, followed by the introduction of CMP NPs in the same solution chemistry to initiate the deposition process. The NP concentrations used in the QCM-D experiments are presented in Table S3 of the ESI.† The flow rate was maintained at 0.10 ± 0.002 mL min−1 using a peristaltic pump (ISM935C, Ismatec SA, Zürich, Switzerland). This flow rate resulted in a laminar flow in the QCM-D chamber.49
The deposition kinetics of CMP NPs on SLBs were quantified using the deposition attachment efficiency, αD, which can be calculated by normalizing the rate of frequency shift on SLBs to the rate under favorable condition obtained at the same solution chemistry:40,51
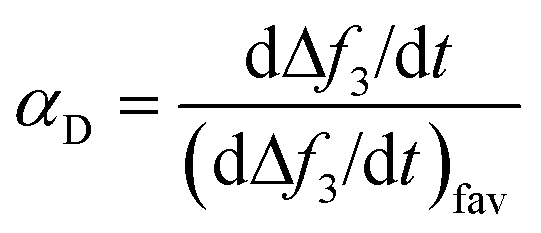 | (3) |
Detection of dye leakage from supported vesicles
A supported CF-DOPC vesicular layer was formed on a gold-coated sensor in order to investigate the propensity for the CMP NPs to disrupt lipid membranes.48,50,54 After a stable baseline was achieved by rinsing the sensor with Buffer C, a 0.05 g L−1 CF-DOPC vesicle suspension was flowed through the chamber for ca. 30 min. The CF-DOPC vesicles deposited on the gold surface to form a SVL.50,54 The CF dye outside the vesicles was removed by rinsing the SVL with Buffer C for at least 2 h. Following the rinsing stage, a NaCl solution (150 mM NaCl, phosphate buffer, pH 7.4) was introduced into the chamber to achieve a stable baseline. Next, a CMP NP suspension prepared in the same solution chemistry was introduced into the chamber for 40 min. At the end of each experiment, the vesicles were exposed to a 32 mM Triton X-100 solution, a membrane solubilizer,55 to induce the complete rupture of the deposited vesicles. The fluorescence of the outflow was measured using a spectrofluorometer (Horiba Jobin Yvon, France) at an excitation wavelength of 490 nm and an emission wavelength of 517 nm.Results and discussion
Electrokinetic properties of CMP NPs
The EPMs of the CMP NPs are presented in Fig. 1a as a function of NaCl concentration in phosphate buffer at pH 7.4. As shown in the figure, all four NPs carried negative charge in the presence of phosphate. The magnitude of the EPMs for all four NPs decreased with an increase in NaCl concentration due to the screening of surface charge.56 In our recent work,7 we have measured the pH of zero point of charge (pHZPC) of c-SiO2, f-SiO2, CeO2, and Al2O3 NPs in the absence of phosphate to be below 2, 2, 8, and 10, respectively. These values are consistent with the pHZPC values of silica (ca. 2),53,57 CeO2 (6.0–8.5), and γ-Al2O3 (7.6–9.3)58,59 reported in the literature. The negative charge of SiO2 NPs at pH 7.4 in the presence of phosphate is consistent with the pHZPC of silica. Interestingly, in the presence of phosphate, the CeO2 and Al2O3 NPs carried considerable negative charge at pH 7.4.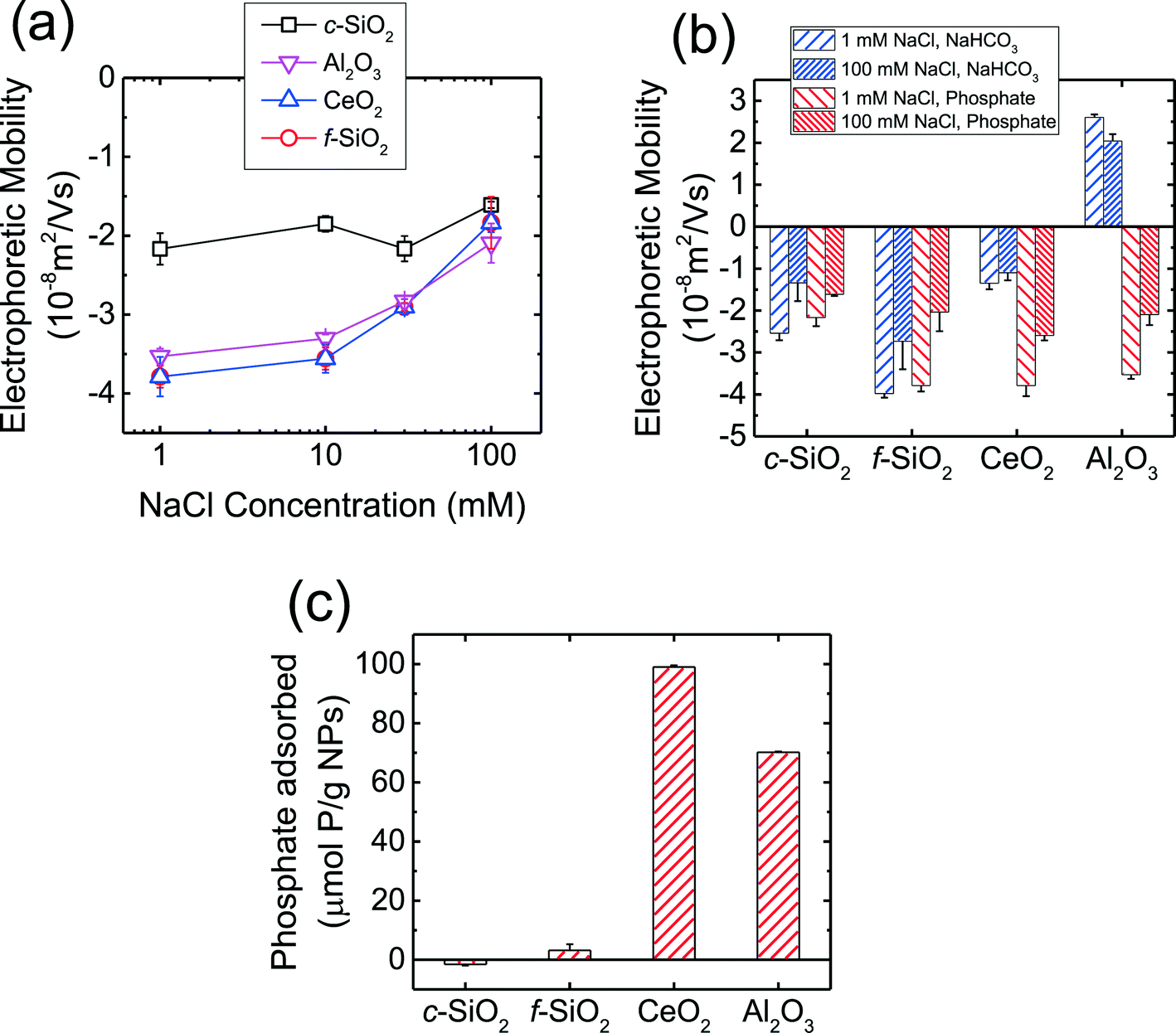 | ||
| Fig. 1 (a) EPMs of CMP NPs over a wide range of NaCl concentrations at pH 7.4 in phosphate buffer. (b) EPMs of CMP NPs at 1 and 100 mM NaCl and pH 7.4 in phosphate and bicarbonate buffer. The data in phosphate buffer was reproduced from Fig. 1a for comparison. (c) Phosphate adsorption by CMP NPs within a time duration of 10 min. The adsorption experiments were conducted at an initial phosphate concentration of 100 μM, 1 g L−1 NPs, 1 mM NaCl, and pH 7.4. Error bars represent standard deviations. | ||
The EPMs of the four CMP NPs were further measured in 1 and 100 mM NaCl in bicarbonate buffer (i.e., in the absence of phosphate) at pH 7.4 and the EPMs in the absence and presence of phosphate are compared in Fig. 1b. In bicarbonate buffer, the c-SiO2 and f-SiO2 NPs were negatively charged, while the CeO2 and Al2O3 NPs carried slight negative charge and considerable positive charge, respectively. These results were consistent with the pHZPC values for SiO2, CeO2, and Al2O3 that had been reported in the literature.53,57–59 For both c-SiO2 and f-SiO2 NPs, the EPMs in the two buffers at the same NaCl concentration was not statistically different (two tailed t-test, significant level of 0.05). In contrast, for CeO2 and Al2O3 NPs, their EPMs in the two buffers were significantly different. This discrepancy is likely due to the adsorption of the phosphate ions on the surface of CeO2 and Al2O3 NPs. In order to test this hypothesis, phosphate adsorption experiments were further conducted for the four NPs (Fig. 1c). The amount of phosphate adsorbed on CeO2 and Al2O3 NPs over a time period of 10 min (expressed as μmol phosphate per g NPs) was one to two orders of magnitude higher than that on f-SiO2 NPs (no adsorption detected on c-SiO2 NPs). Since the densities of CeO2 (ca. 7.3 g cm−3)60 and Al2O3 (ca. 3.9 g cm−3)61 are both higher than that of amorphous SiO2 (ca. 2.2 g cm−3),62 a more pronounced difference in phosphate adsorption on CeO2 and Al2O3 NPs versus SiO2 NPs would be expected if the amount of adsorbed phosphate is expressed on a particle number basis. Therefore, the significant change in the EPMs of CeO2 and Al2O3 NPs was very likely due to the adsorption of phosphate on the NPs.
At pH 7.4, phosphate (pKa1 = 2.1, pKa2 = 7.2, pKa3 = 11.9) mainly exists in the form of monovalent (H2PO4−) and divalent (HPO42−) phosphate ions (calculated to be 39 and 61%, respectively).53 As shown in Fig. 1b, the Al2O3 NPs underwent charge reversal in the presence of phosphate. It has been shown through 31P NMR measurements that phosphate can adsorb to γ-Al2O3via both outer- and inner-sphere complexation.63 Outer-sphere complexation occurs through the electrostatic attraction between the positively charged –AlOH2+ groups and negatively charged phosphate ions,63 whereas inner-sphere complexation takes place through direct bond formation between the Al atom and the oxygen donor atoms in the phosphate ion.63,64 Hunter65 reported that inner-spherically adsorbed ions have a higher propensity to reverse the sign of surface charge, whereas indifferent ions can only screen the surface charge. Therefore, inner-sphere complexation of phosphate is expected to be the primary mechanism for the reversal of surface charge of Al2O3 NPs. For CeO2 NPs which already carried a negative charge at pH 7.4 (Fig. 1b), the enhancement of negative surface charge in the presence of phosphate, as observed elsewhere,66,67 was likely a result of the inner-sphere complexation between phosphate ions and the CeO2 NPs.65 For both SiO2 NPs, the surface charges in the presence and absence of phosphate were not significantly different (Fig. 1b), consistent with the negligible adsorption of phosphate on the SiO2 NPs compared to that on CeO2 and Al2O3 NPs (Fig. 1c).
Stability of c-SiO2 and f-SiO2 NPs at pH 7.4
The aggregation profiles of the four CMP NPs at 150 mM NaCl and pH 7.4 in the presence and absence of phosphate are presented in Fig. S1 of the ESI.† This NaCl concentration was chosen because the ionic strength in physiological fluids is ca. 150 mM.19 The hydrodynamic diameter of c-SiO2 and f-SiO2 NPs did not change significantly within a duration of 20 min, whereas CeO2 and Al2O3 NPs underwent appreciable aggregation. This observation shows that both SiO2 NPs were considerably more stable than CeO2 and Al2O3 NPs at pH 7.4. Since phosphate ions do not adsorb favorably on both NPs, as discussed in the previous section, they are not expected to be involved in the mechanisms for the elevated stability of the SiO2 NPs.In order to elucidate the mechanisms for the high stability of SiO2 NPs at pH 7.4, aggregation experiments were further carried out at pH 2.0 (adjusted with 1 M HCl), which is the reported pHZPC of SiO2.53,57 Thus, at pH 2.0, the electric double layer (EDL) repulsion between the SiO2 NPs was expected to be largely eliminated. As shown in Fig. 2a, the f-SiO2 NPs underwent considerable aggregation at pH 2.0 compared to at pH 7.4 (both in the presence and absence of phosphate). It can therefore be concluded that the elevated stability of f-SiO2 NPs at pH 7.4 was due to the high surface charge that was not completely screened even at 150 mM NaCl.
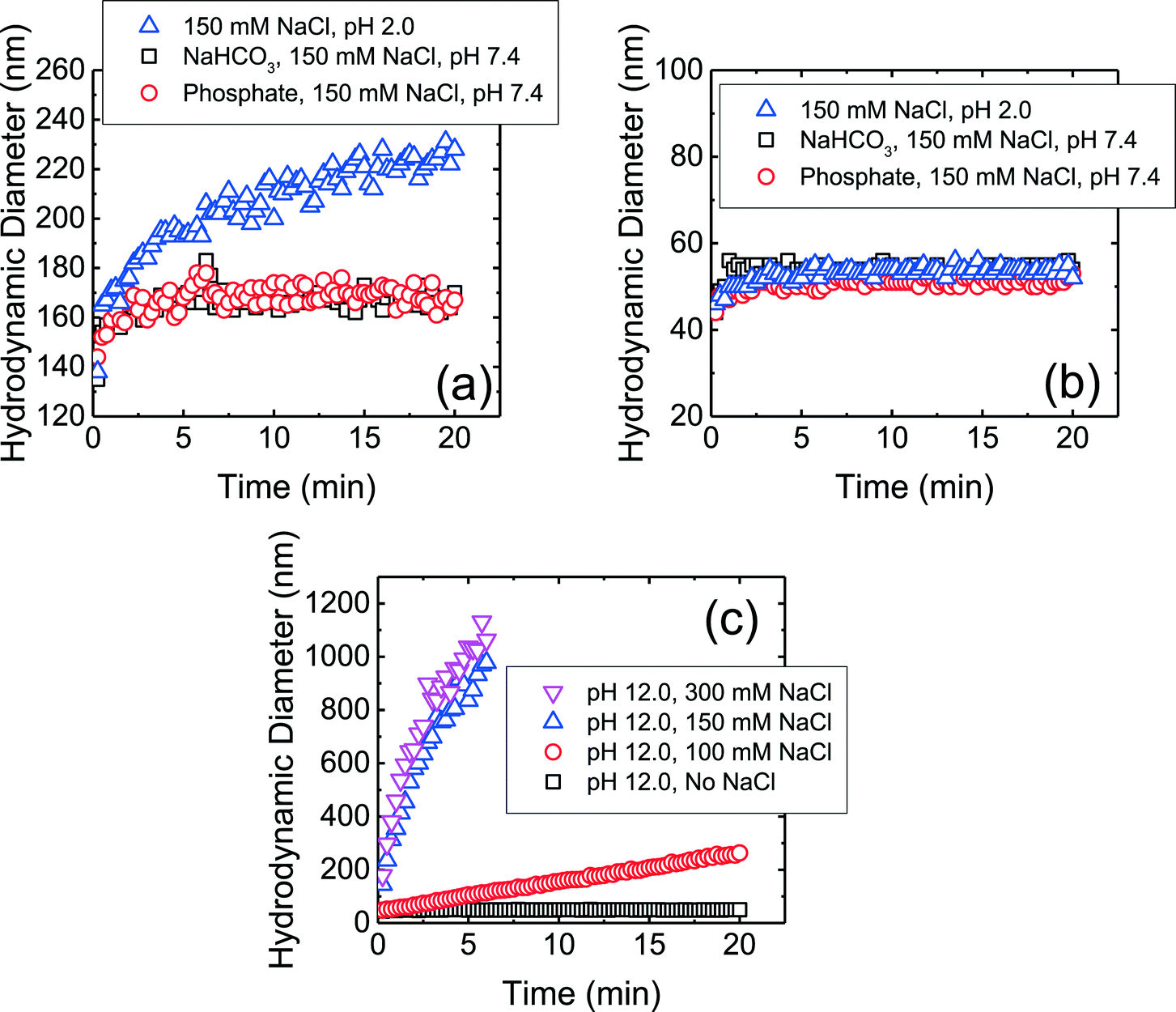 | ||
| Fig. 2 Aggregation profiles of (a) f-SiO2 NPs at 150 mM NaCl at pH 7.4 (in phosphate and bicarbonate buffer) and pH 2.0 (pH adjusted with 1 M HCl), (b) c-SiO2 NPs at 150 mM NaCl at pH 7.4 (in phosphate and bicarbonate buffer) and pH 2.0 (pH adjusted with 1 M HCl), and (c) c-SiO2 NPs at pH 12 (pH adjusted with 1 M NaOH) at varying NaCl concentrations. | ||
In contrast to f-SO2 NPs, c-SiO2 NPs were stable at pH 2.0 (Fig. 2b). Additional aggregation experiments of c-SiO2 NPs were carried out at pH 1.0 and, similar to at pH 2.0 and 7.4, no aggregation was observed (data not shown). The high stability of c-SiO2 colloids at ca. pH 2 has been reported by different researchers.68–70 The presence of a repulsive force between silica surfaces at short separation distances, in addition to EDL repulsion, has been detected through force measurements using atomic force microscopy and surface force apparatus.71,72 Israelachvili and co-workers72,73 proposed that a hairy layer of protruding silanol and silicic acid groups on silica surface was the origin of the repulsive force between SiO2 colloids and also their stability in water. Borkovec and co-workers70 reported that SiO2 particles underwent fast aggregation at pH 12 which they attributed to the dissolution of the hairy layers at high pH conditions. The aggregation behavior of c-SiO2 at pH 12 is presented in Fig. 2c. In the absence of NaCl at pH 12, c-SiO2 was stable, likely due to the EDL repulsion between the NPs. With the increase in NaCl concentration, the aggregation rates of the NPs increased and fast aggregation was achieved at 150 mM NaCl. This result supports the hypothesis that the presence of hairy layer on the surface of c-SiO2, which was dissolved at pH 12, contributed to the remarkable stability of the c-SiO2 NPs at pH 7.4. Zhang et al.14 investigated the surface silanol densities of both c-SiO2 and f-SiO2 through Fourier transform infrared spectroscopy and found that f-SiO2 had a lower total silanol content (ca. 2.8 OH per nm2) than c-SiO2 (ca. 4.5 OH per nm2). It is thus expected that the protruding hairy silanol and silicic acid groups were less abundant on f-SiO2 NPs than on c-SiO2 NPs. Therefore, the repulsive force caused by the hairy layers is likely to play a more important role in the stability of c-SiO2 than that of f-SiO2. We do not, however, rule out the possibility of other mechanisms for the stability of c-SiO2 NPs, such as hydration force resulting from the hydrogen bonding network between silanol groups.74
Phosphate adsorption enhanced the stability of CeO2 and Al2O3 NPs
The aggregation kinetics of CeO2 and Al2O3 NPs were investigated over a wide range of NaCl concentrations in the presence of phosphate and the attachment efficiencies are presented in Fig. 3. At NaCl concentrations of ca. 10–90 mM, the attachment efficiencies of both CeO2 and Al2O3 NPs increased with increasing NaCl concentrations (reaction-limited regime) as a result of the screening of their surface charge. At NaCl concentrations higher than 90 mM, a further increase in the salt concentration did not result in any substantial increase in the attachment efficiencies (diffusion-controlled regime) since the energy barrier to aggregation was eliminated due to charge screening under these conditions. The critical coagulation concentrations (CCCs) that demarcated the reaction- and diffusion-limited regimes were ca. 90 mM for both CeO2 and Al2O3 NPs in the presence of phosphate.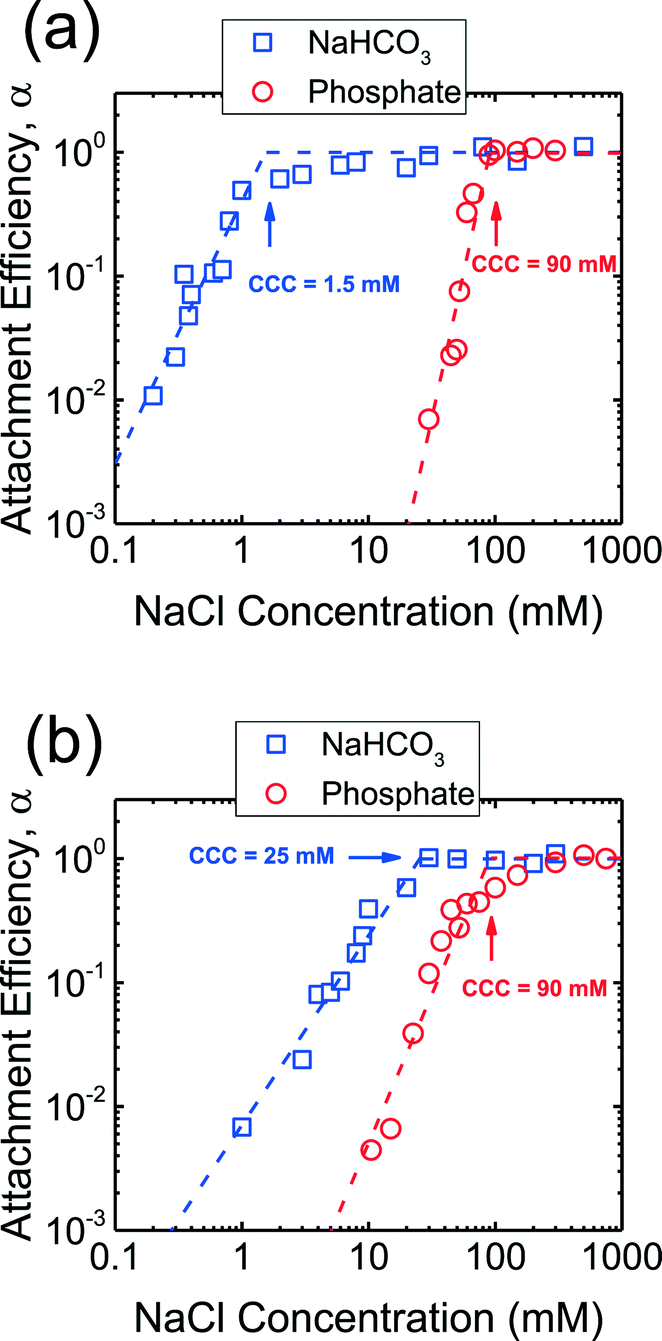 | ||
| Fig. 3 Aggregation attachment efficiencies of (a) CeO2 and (b) Al2O3 NPs as a function of NaCl concentration at pH 7.4 in phosphate and bicarbonate buffer. The dashed lines are extrapolated from the favorable and unfavorable aggregation regimes, and their intersections yield the CCCs. | ||
In addition, aggregation experiments were carried out at pH 7.4 in the absence of phosphate in order to elucidate the role of phosphate adsorption on the stability of CeO2 and Al2O3 NPs (Fig. 3). Reaction- and diffusion-limited regimes were also observed for both CeO2 and Al2O3 NPs in bicarbonate buffer, indicating that the aggregation kinetics were controlled by electrostatic interactions in the absence of phosphate. The CCC values were determined to be ca. 2 mM for CeO2 (Fig. 3a) and 25 mM for Al2O3 NPs (Fig. 3b), both considerably lower than their respective CCCs in the presence of phosphate. This comparison implies that the stability of CeO2 and Al2O3 NPs was enhanced as a result of the adsorption of phosphate on the NP surface. As shown in Fig. 1b, the adsorption of phosphate resulted in a significant increase in the magnitude of the surface charge of CeO2 NPs, thereby increasing the energy barrier to NP aggregation. For Al2O3 NPs, the surface charge was reversed in the presence of phosphate and, additionally, the magnitude of the surface charge was increased (as shown in the increase in the magnitude of EPM in Fig. 1b), thus raising the EDL repulsion between the NPs. Because the increase in the magnitude of surface charge due to the adsorption of phosphate was smaller for Al2O3 than for CeO2 (Fig. 1b), the rise in CCC in the presence of phosphate was less pronounced for Al2O3 than for CeO2 NPs (Fig. 3).
Deposition of CMP NPs on DOPC SLBs in the presence of phosphate
The deposition of the CMP NPs on DOPC SLBs was investigated in order to probe the propensity for CMP NPs to attach to lipid membranes. In our previous work, the DOPC bilayers were determined to be negatively charged at pH 7.23 Fig. S2 of the ESI† presents the rates of frequency shift during the deposition of CMP NPs on the SLBs in the presence of phosphate. When the NaCl concentration was raised from 1 to 30 mM, the deposition rates of c-SiO2 and f-SiO2 NPs on SLBs increased due to the reduction of the EDL repulsion between the NPs and SLBs. In contrast, no deposition of CeO2 and Al2O3 NPs on SLBs was detected over the NaCl concentration range of 1–100 mM.By normalizing the deposition rates of CMP NPs on SLBs to the transport-limited deposition rates on PLL (presented in Fig. S3 of the ESI†), we obtained the deposition attachment efficiencies of the four NPs on SLBs, as presented in Fig. 4. For the two SiO2 NPs, the attachment efficiencies increased when the NaCl concentration was increased from 1 to 100 mM. At NaCl concentrations higher than 100 mM, the attachment efficiency did not increase with a further increase in NaCl concentration, most likely due to the complete screening of surface charges under these high ionic strength conditions. Interestingly, for both CeO2 and Al2O3 NPs, the attachment efficiency did not reach unity even at 300 mM NaCl.
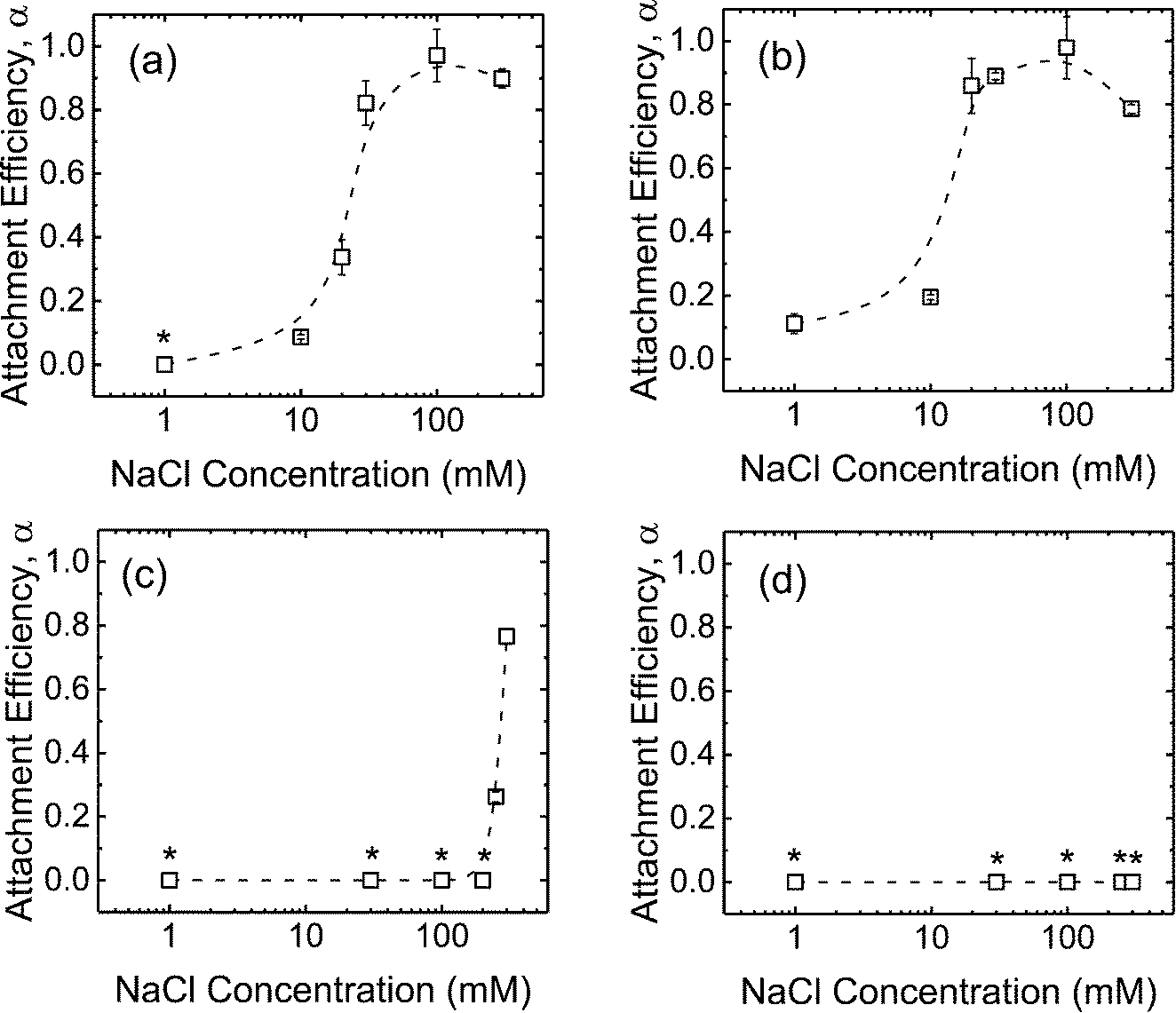 | ||
| Fig. 4 Deposition attachment efficiencies of (a) c-SiO2, (b) f-SiO2, (c) CeO2, and (d) Al2O3 NPs on DOPC SLBs at pH 7.4 in phosphate buffer. Error bars represent standard deviations. The lines are meant to guide the eye. Asterisks (*) indicate that no frequency shift was detected on SLBs. | ||
Comparing deposition kinetics of CMP NPs on DOPC SLBs in the presence and absence of phosphate
In order to obtain further insights into the low propensity for CeO2 and Al2O3 NPs to deposit on SLBs in the presence of phosphate, additional deposition experiments on SLBs were performed in bicarbonate buffer. As shown in Fig. 5a and b, both c-SiO2 and f-SiO2 NPs had similar propensities to attach to SLBs in phosphate and bicarbonate buffers, owing to their similar surface charge in the two buffers (Fig. 1b).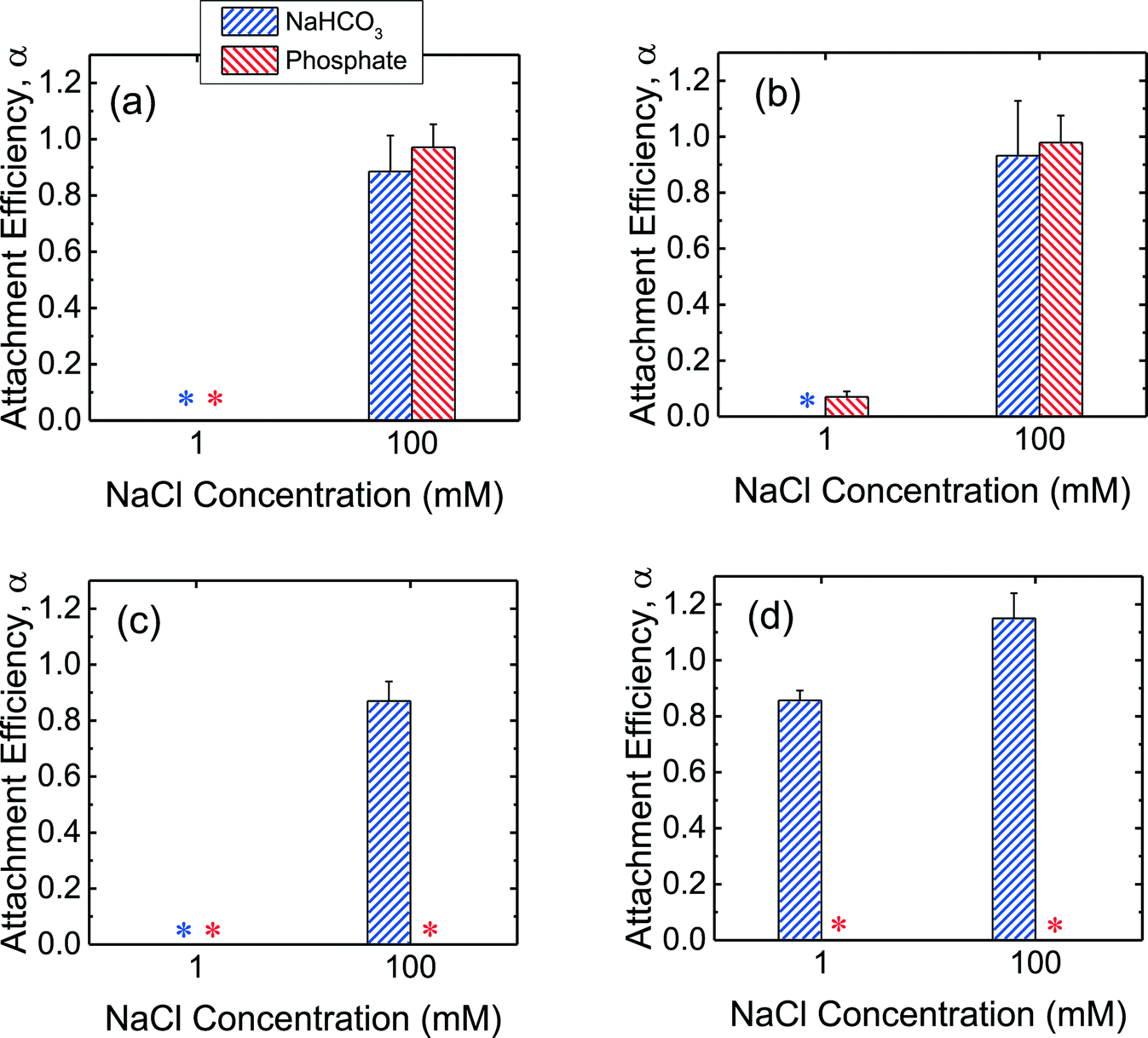 | ||
| Fig. 5 Deposition attachment efficiencies of (a) c-SiO2, (b) f-SiO2, (c) CeO2, and (d) Al2O3 NPs on DOPC SLBs at pH 7.4 in phosphate and bicarbonate buffer. Error bars represent standard deviations. The data in phosphate buffer was reproduced from Fig. 4 for comparison. Asterisks (*) indicate that the attachment efficiencies were 0 (i.e., no frequency shift was detected on SLBs). | ||
For the CeO2 NPs, the deposition attachment efficiency in bicarbonate buffer increased from zero to close to unity when the NaCl concentration was raised from 1 to 100 mM (Fig. 5c) due to the reduction in the EDL repulsion between the NPs and SLBs. In the presence of phosphate, however, the deposition attachment efficiencies of CeO2 NPs on SLBs were zero at both 1 and 100 mM NaCl. According to the EPM measurements (Fig. 1b), additional negative charge was imparted to the CeO2 NPs through phosphate adsorption. Thus, the EDL repulsion between the CeO2 NPs and SLBs may still be strong enough even at 100 mM NaCl to inhibit the deposition of the NPs.
For the deposition of Al2O3 NPs on SLBs, a more pronounced contrast in attachment efficiency was observed in phosphate and bicarbonate buffers (Fig. 5d). In bicarbonate buffer, Al2O3 was positively charged at both 1 and 100 mM NaCl (Fig. 1b). The attractive EDL and vdW interactions between Al2O3 NPs and SLBs resulted in close-to-unity attachment efficiencies at both 1 and 100 mM NaCl in bicarbonate buffer. In the presence of phosphate, however, the attachment of Al2O3 NPs on SLBs was not detected at both 1 and 100 mM NaCl due to the negative charge of Al2O3 NPs imparted by phosphate adsorption (Fig. 1b). The distinctly different deposition behaviors of both CeO2 and Al2O3 NPs on SLBs in the presence and absence of phosphate shows that it is imperative to account for the influence of phosphate adsorption in the assessment of NP association with cell membranes.
No disruption of vesicles by CMP NPs detected
The attachment of NPs on cell membranes has been reported in some studies75,76 to result in the damage of the membranes and the leakage of intracellular contents. Dye leakage experiments have been employed in several studies on the disruption of cell membranes by silica,31 gold,30 TiO2,32 and graphene oxide54 NPs. Herein, the disruption of the supported vesicles upon the attachment of CMP NPs was investigated by QCM-D coupled with fluorescence detection measurements. This technique enables us to simultaneously monitor the deposition of CMP NPs on the supported vesicles using QCM-D and detect the disruption of the vesicles by measuring the dye leakage from the vesicles.54 These experiments were only performed in phosphate buffer in the context of cell membrane disruption in biological systems. Since no attachment of CeO2 and Al2O3 NPs on DOPC bilayers was expected to take place at 150 mM NaCl (i.e., ionic strength of physiological fluids), as shown in Fig. 4c and d, these experiments were only conducted for both SiO2 NPs.The frequency and dissipation shifts during the formation of a SVL and the deposition of f-SiO2 NPs on the SVL are presented in Fig. 6a. Fig. 6b and c show the frequency and dissipation shifts and the dye concentration in the outflow of QCM-D, respectively, during the deposition of f-SiO2 NPs on the SVL. The experimental protocol involves three stages: 1) deposition of dye-encapsulated vesicles on a gold-coated sensor to form a SVL (26–57 min, only in Fig. 6a); 2) removal of unadsorbed vesicles with buffer rinse (57–190 min); and 3) deposition of NPs on the SVL at 150 mM NaCl (213–257 min). The fluorescence intensity of the outflow was monitored from close to the end of stage 2 onwards (from 176 min). In a typical experiment, four QCM-D chambers were employed. In two of the chambers, deposition experiments were performed with a suspension of CMP NPs prepared in 150 mM NaCl. In the other two chambers, control experiments were conducted using the same procedure as the deposition experiments, except that a 150 mM NaCl solution was used in lieu of the NP suspension.
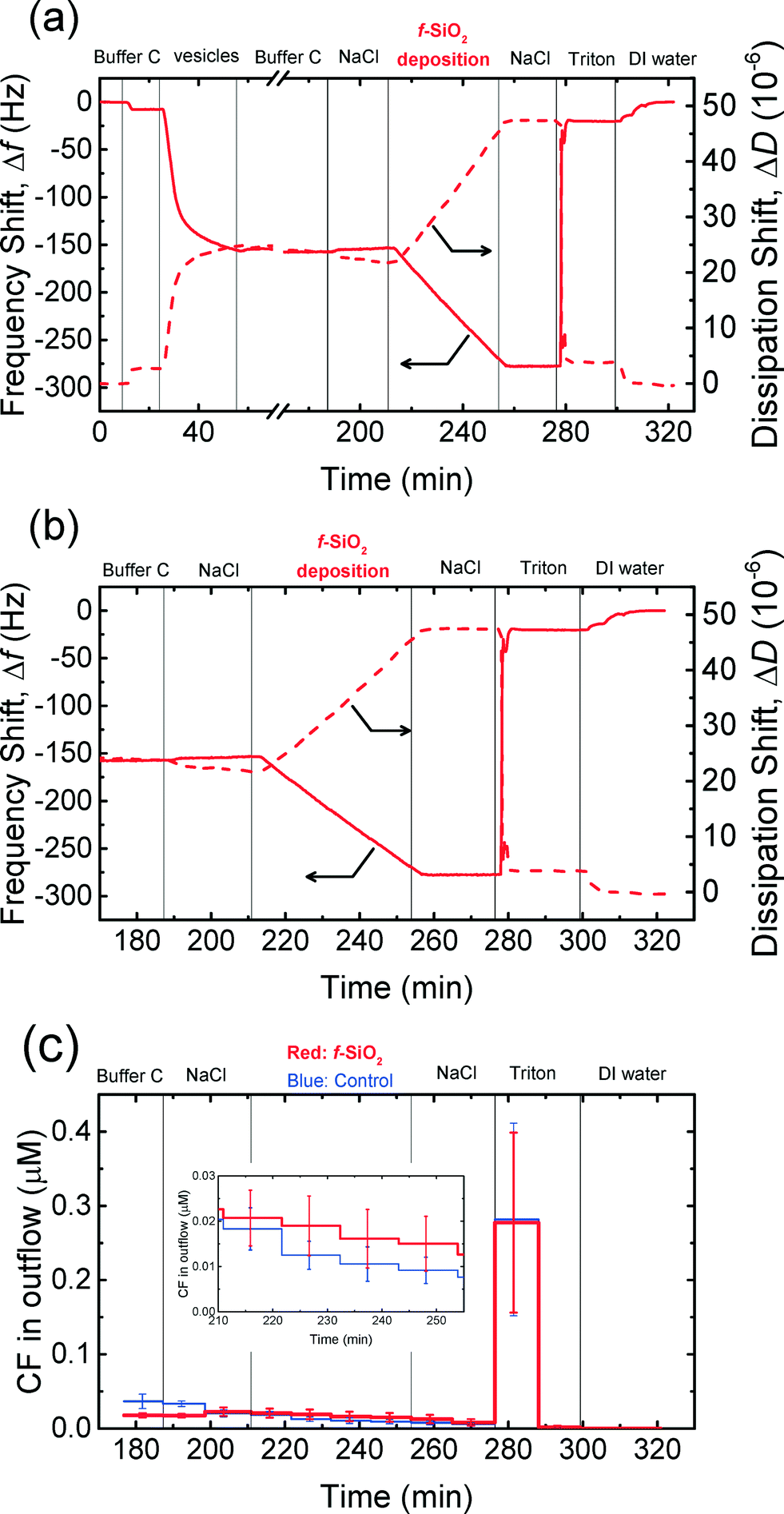 | ||
| Fig. 6 (a) Frequency and dissipation shifts during the formation of a SVL (26–57 min), the deposition of f-SiO2 NPs on the SVL at 150 mM NaCl and pH 7.4 (213–257 min), and the rupture of the vesicles when they were exposed to 32 mM Triton X-100 (277–301 min). (b) Frequency and dissipation shifts when supported vesicles were exposed to f-SiO2 NPs (in 150 mM NaCl), 150 mM NaCl, and 32 mM Triton X-100. (c) Release of CF dye from supported vesicles upon exposure to f-SiO2 NPs (in 150 mM NaCl), 150 mM NaCl, and 32 mM Triton X-100. Insert: Close-up of the release of dye in the f-SiO2 NP deposition stage. | ||
As shown in Fig. 6a and b, f-SiO2 NPs deposited on the SVL continuously without eliciting significant rupture of the vesicles as evidenced from the monotonic decrease in frequency and increase in dissipation shifts (213–257 min). At the same time, no significant difference in dye leakage concentration was observed between the deposition and control experiments (inset of Fig. 6c, two-tailed t test, significance level of 0.05). As a positive control, the exposure of the vesicles to Triton (276 min) resulted in precipitous frequency and dissipation shifts (Fig. 6b), as well as a dramatic increase in fluorescence intensity (Fig. 6c), indicating that the vesicles had been ruptured by the surfactant. Hence, it can be inferred that the deposition of f-SiO2 NPs on the SVL did not induce appreciable disruption of the lipid membrane. Similar experiments were performed for c-SiO2 NPs and no significant dye leakage was observed during the deposition of c-SiO2 NPs on the vesicles (Fig. S4†). These results demonstrate that both of SiO2 NPs have a low propensity to disrupt DOPC bilayers at 150 mM NaCl despite their high affinity to associate with the model membranes under these conditions.
Conclusions
In this study, the aggregation behavior of four CMP NPs and their interactions with model cell membranes at neutral pH conditions were investigated. One important aspect of this study is to examine the influence of phosphate, a key component of biological fluids, on the surface charge and colloidal stability of the NPs, as well as on the nanoparticle–membrane interactions. The surface charge of both c-SiO2 and f-SiO2 NPs was not appreciably altered by phosphate as phosphate did not adsorb favorably on the NP surface. As a result, both SiO2 NPs had similar aggregation and deposition kinetics on SLBs in the presence and absence of phosphate. Conversely, the CeO2 NPs became more negatively charged while the Al2O3 NPs underwent charge reversal (positive to negative) in the presence of phosphate, both likely due to the adsorption of phosphate on the NPs through inner-sphere complexation. The enhancement of surface charge in the presence of phosphate led to an increase in the stability of CeO2 and Al2O3 NPs and decreased the NPs' propensity to attach to SLBs. These result imply that the nonspecific interactions between NPs and cell membranes and potentially the biological effects of NPs may be controlled by phosphate. Thus, the role of phosphate needs to be accounted for in the assessment of the stability and cellular interactions of NPs. Additionally, the deposition of both c-SiO2 and f-SiO2 NPs on supported vesicles did not result in detectable disruption of the vesicles. Future studies that include a systematic variation of membrane properties (e.g., lipid composition and incorporation of proteins in membranes) and CMP NP properties (e.g., sizes and shapes) will enable additional mechanistic insights into the interactions between the nanoparticles and cell membranes. The effects of other major constituents in biological fluids (e.g., proteins) should also be investigated.Acknowledgements
This work is supported by the Semiconductor Research Corporation (award 425-MC-2001, project 425.041) and National Science Foundation (CBET-1133559). We acknowledge Cabot Inc. for providing the CMP slurries used in this study, which are currently available through Dr. Farhang Shadman (University of Arizona). X. L. acknowledges funding support from the Gary Anderson Fellowship. We thank Dr. Kalina Hristova from the Department of Materials Science and Engineering at Johns Hopkins University for the use of the fluorescence spectrophotometer.References
- J. M. Steigerwald, S. P. Murarka and R. J. Gutmann, Chemical mechanical planarization of microelectronic materials, John Wiley & Sons, 2008 Search PubMed.
- M. Krishnan, J. W. Nalaskowski and L. M. Cook, Chem. Rev., 2010, 110, 178–204 CrossRef CAS PubMed.
- C. L. Lai and S. H. Lin, Chemosphere, 2004, 54, 235–242 CrossRef CAS PubMed.
- S. H. Lin and C. R. Yang, J. Hazard. Mater., 2004, 108, 103–109 CrossRef CAS PubMed.
- W. Den and C. P. Huang, Colloids Surf., A, 2005, 254, 81–89 CrossRef CAS.
- Y. Li, Microelectronic applications of chemical mechanical planarization, John Wiley & Sons, 2007 Search PubMed.
- D. Speed, P. Westerhoff, R. Sierra-Alvarez, R. Draper, P. Pantano, S. Aravamudhan, K. L. Chen, K. Hristovski, P. Herckes, X. Y. Bi, Y. Yang, C. Zeng, L. Otero-Gonzalez, C. Mikoryak, B. A. Wilson, K. Kosaraju, M. Tarannum, S. Crawford, P. Yi, X. Liu, S. V. Babu, M. Moinpour, J. Ranville, M. Montano, C. Corredor, J. Posner and F. Shadman, Environ. Sci.: Nano, 2015, 2, 227–244 RSC.
- L. K. Limbach, R. Bereiter, E. Mueller, R. Krebs, R. Gaelli and W. J. Stark, Environ. Sci. Technol., 2008, 42, 5828–5833 CrossRef CAS PubMed.
- M. N. Shepard and S. Brenner, Ann. Occup. Hyg., 2014, 58, 251–265 CrossRef CAS PubMed.
- S. A. Brenner and N. M. Neu-Baker, J. Chem. Health Saf., 2014, 22, 10–19 CrossRef.
- H. E. Bergna and W. O. Roberts, Colloidal silica: fundamentals and applications, CRC Press, 2005 Search PubMed.
- S. E. Pratsinis, Prog. Energy Combust. Sci., 1998, 24, 197–219 CrossRef CAS.
- P. B. Zantye, A. Kumar and A. K. Sikder, Mater. Sci. Eng., R, 2004, 45, 89–220 CrossRef.
- H. Y. Zhang, D. R. Dunphy, X. M. Jiang, H. Meng, B. B. Sun, D. Tarn, M. Xue, X. Wang, S. J. Lin, Z. X. Ji, R. B. Li, F. L. Garcia, J. Yang, M. L. Kirk, T. Xia, J. I. Zink, A. Nel and C. J. Brinker, J. Am. Chem. Soc., 2012, 134, 15790–15804 CrossRef CAS PubMed.
- W. S. Lin, Y. W. Huang, X. D. Zhou and Y. F. Ma, Int. J. Toxicol., 2006, 25, 451–457 CrossRef CAS PubMed.
- M. G. Song, J. H. Lee, Y. G. Lee and J. H. Koo, J. Colloid Interface Sci., 2006, 300, 603–611 CrossRef CAS PubMed.
- L. Otero-Gonzalez, R. Sierra-Alvarez, S. Boitano and J. A. Field, Environ. Sci. Technol., 2012, 46, 10271–10278 CAS.
- M. P. Vinardell, A. Sorde, J. Diaz, T. Baccarin and M. Mitjans, J. Nanopart. Res., 2015, 17 Search PubMed.
- A. E. Nel, L. Madler, D. Velegol, T. Xia, E. M. V. Hoek, P. Somasundaran, F. Klaessig, V. Castranova and M. Thompson, Nat. Mater., 2009, 8, 543–557 CrossRef CAS PubMed.
- K. L. Chen and G. D. Bothun, Environ. Sci. Technol., 2014, 48, 873–880 CrossRef CAS PubMed.
- K. Yang and Y. Q. Ma, Nat. Nanotechnol., 2010, 5, 579–583 CrossRef CAS PubMed.
- J. M. Troiano, L. L. Olenick, T. R. Kuech, E. S. Melby, D. H. Hu, S. E. Lohse, A. C. Mensch, M. Dogangun, A. M. Vartanian, M. D. Torelli, E. Ehimiaghe, S. R. Walter, L. Fu, C. R. Anderton, Z. H. Zhu, H. F. Wang, G. Orr, C. J. Murphy, R. J. Hamers, J. A. Pedersen and F. M. Geiger, J. Phys. Chem. C, 2015, 119, 534–546 CAS.
- P. Yi and K. L. Chen, Environ. Sci. Technol., 2013, 47, 5711–5719 CrossRef CAS PubMed.
- X. F. Zhang and S. H. Yang, Langmuir, 2011, 27, 2528–2535 CrossRef CAS PubMed.
- B. Jing and Y. Zhu, J. Am. Chem. Soc., 2011, 133, 10983–10989 CrossRef CAS PubMed.
- R. Frost, G. E. Jonsson, D. Chakarov, S. Svedhem and B. Kasemo, Nano Lett., 2012, 12, 3356–3362 CrossRef CAS PubMed.
- K. H. Jacobson, I. L. Gunsolus, T. R. Kuech, J. M. Troiano, E. S. Melby, S. E. Lohse, D. Hu, W. B. Chrisler, C. J. Murphy, G. Orr, F. M. Geiger, C. L. Haynes and J. A. Pedersen, Environ. Sci. Technol., 2015, 49, 10642–10650 CrossRef CAS PubMed.
- P. R. Leroueil, S. A. Berry, K. Duthie, G. Han, V. M. Rotello, D. Q. McNerny, J. R. Baker, B. G. Orr and M. M. B. Holl, Nano Lett., 2008, 8, 420–424 CrossRef CAS PubMed.
- A. Lesniak, A. Salvati, M. J. Santos-Martinez, M. W. Radomski, K. A. Dawson and C. Åberg, J. Am. Chem. Soc., 2013, 135, 1438–1444 CrossRef CAS PubMed.
- C. M. Goodman, C. D. McCusker, T. Yilmaz and V. M. Rotello, Bioconjugate Chem., 2004, 15, 897–900 CrossRef CAS PubMed.
- H. Pera, J. M. Kleijn and F. A. M. Leermakers, Chem. Lett., 2012, 41, 1322–1324 CrossRef CAS.
- B. Y. Moghadam, W. C. Hou, C. Corredor, P. Westerhoff and J. D. Posner, Langmuir, 2012, 28, 16318–16326 CrossRef CAS PubMed.
- S. L. Tobey and E. V. Anslyn, Org. Lett., 2003, 5, 2029–2031 CrossRef CAS PubMed.
- A. Albanese and W. C. W. Chan, ACS Nano, 2011, 5, 5478–5489 CrossRef CAS PubMed.
- X. S. Liu, Y. J. Chen, H. Li, N. Huang, Q. Jin, K. F. Ren and J. Ji, ACS Nano, 2013, 7, 6244–6257 CrossRef CAS PubMed.
- T. Masui, K. Fujiwara, K. Machida, G. Adachi, T. Sakata and H. Mori, Chem. Mater., 1997, 9, 2197–2204 CrossRef CAS.
- K. M. Parida, A. C. Pradhan, J. Das and N. Sahu, Mater. Chem. Phys., 2009, 113, 244–248 CrossRef CAS.
- J. Murphy and J. P. Riley, Anal. Chim. Acta, 1962, 27, 31–36 CrossRef CAS.
- K. A. Huynh and K. L. Chen, Environ. Sci. Technol., 2011, 45, 5564–5571 CrossRef CAS PubMed.
- P. Yi and K. L. Chen, Langmuir, 2011, 27, 3588–3599 CrossRef CAS PubMed.
- K. L. Chen, S. E. Mylon and M. Elimelech, Environ. Sci. Technol., 2006, 40, 1516–1523 CrossRef CAS PubMed.
- K. L. Chen and M. Elimelech, Langmuir, 2006, 22, 10994–11001 CrossRef CAS PubMed.
- L. L. Hoekstra, R. Vreeker and W. G. M. Agterof, J. Colloid Interface Sci., 1992, 151, 17–25 CrossRef CAS.
- N. B. Saleh, L. D. Pfefferle and M. Elimelech, Environ. Sci. Technol., 2008, 42, 7963–7969 CrossRef CAS PubMed.
- I. Chowdhury, M. C. Duch, N. D. Mansukhani, M. C. Hersam and D. Bouchard, Environ. Sci. Technol., 2013, 47, 6288–6296 CrossRef CAS PubMed.
- H. Holthoff, S. U. Egelhaaf, M. Borkovec, P. Schurtenberger and H. Sticher, Langmuir, 1996, 12, 5541–5549 CrossRef CAS.
- N. J. Cho, C. W. Frank, B. Kasemo and F. Höök, Nat. Protoc., 2010, 5, 1096–1106 CrossRef CAS PubMed.
- P. Yi and K. L. Chen, Environ. Sci. Technol., 2013, 47, 5711–5719 CrossRef CAS PubMed.
- X. L. Qu, P. J. J. Alvarez and Q. L. Li, Environ. Sci. Technol., 2012, 46, 13455–13462 CrossRef CAS PubMed.
- C. A. Keller and B. Kasemo, Biophys. J., 1998, 75, 1397–1402 CrossRef CAS PubMed.
- K. L. Chen and M. Elimelech, Environ. Sci. Technol., 2008, 42, 7607–7614 CrossRef CAS PubMed.
- A. J. de Kerchove and M. Elimelech, Macromolecules, 2006, 39, 6558–6564 CrossRef CAS.
- W. Stumm and J. J. Morgan, Aquatic chemistry: an introduction emphasizing chemical equilibria in natural waters, John Wiley, 1981 Search PubMed.
- X. Liu and K. L. Chen, Langmuir, 2015 DOI:10.1021/acs.langmuir.5b02414.
- C. A. Schnaitman, J. Bacteriol., 1971, 108, 545–552 CAS.
- M. Elimelech, J. Gregory, X. Jia and R. A. Williams, Particle Deposition and Aggregation: Measurement, Modelling and Simulation, Butterworth-Heinemann, Oxford, England, 1995 Search PubMed.
- T. Hiemstra, P. Venema and W. H. VanRiemsdijk, J. Colloid Interface Sci., 1996, 184, 680–692 CrossRef CAS PubMed.
- M. Kosmulski, J. Colloid Interface Sci., 2002, 253, 77–87 CrossRef CAS PubMed.
- M. Kosmulski, J. Colloid Interface Sci., 2011, 353, 1–15 CrossRef CAS PubMed.
- S. Tsunekawa, T. Fukuda and A. Kasuya, J. Appl. Phys., 2000, 87, 1318–1321 CrossRef CAS.
- G. J. Zhang, J. F. Yang and T. Ohji, J. Am. Ceram. Soc., 2001, 84, 1395–1397 CrossRef CAS.
- S. W. Wang, M. Di Ventra, S. G. Kim and S. T. Pantelides, Phys. Rev. Lett., 2001, 86, 5946–5949 CrossRef CAS PubMed.
- B. B. Johnson, A. V. Ivanov, O. N. Antzutkin and W. Forsling, Langmuir, 2002, 18, 1104–1111 CrossRef CAS.
- E. Laiti, P. Persson and L. O. Ohman, Langmuir, 1996, 12, 2969–2975 CrossRef CAS.
- R. J. Hunter, Zeta potential in colloid science: principles and applications, Academic press, 2013 Search PubMed.
- K. Van Hoecke, J. T. K. Quik, J. Mankiewicz-Boczek, K. A. C. De Schamphelaere, A. Elsaesser, P. Van der Meeren, C. Barnes, G. McKerr, C. V. Howard, D. Van De Meent, K. Rydzynski, K. A. Dawson, A. Salvati, A. Lesniak, I. Lynch, G. Silversmit, B. De Samber, L. Vincze and C. R. Janssen, Environ. Sci. Technol., 2009, 43, 4537–4546 CrossRef CAS PubMed.
- G. Cornelis, B. Ryan, M. J. McLaughlin, J. K. Kirby, D. Beak and D. Chittleborough, Environ. Sci. Technol., 2011, 45, 2777–2782 CrossRef CAS PubMed.
- K. Higashitani, M. Kondo and S. Hatade, J. Colloid Interface Sci., 1991, 142, 204–213 CrossRef CAS.
- J. Depasse, J. Colloid Interface Sci., 1999, 220, 174–176 CrossRef CAS PubMed.
- M. Kobayashi, F. Juillerat, P. Galletto, P. Bowen and M. Borkovec, Langmuir, 2005, 21, 5761–5769 CrossRef CAS PubMed.
- W. A. Ducker, T. J. Senden and R. M. Pashley, Nature, 1991, 353, 239–241 CrossRef CAS.
- G. Vigil, Z. H. Xu, S. Steinberg and J. Israelachvili, J. Colloid Interface Sci., 1994, 165, 367–385 CrossRef CAS.
- J. Israelachvili and H. Wennerstrom, Nature, 1996, 379, 219–225 CrossRef CAS PubMed.
- J. P. Chapel, Langmuir, 1994, 10, 4237–4243 CrossRef CAS.
- Y. S. Lin and C. L. Haynes, J. Am. Chem. Soc., 2010, 132, 4834–4842 CrossRef CAS PubMed.
- T. Yu, A. Malugin and H. Ghandehari, ACS Nano, 2011, 5, 5717–5728 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5en00176e |
| This journal is © The Royal Society of Chemistry 2016 |