
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Direct determination of ionic transference numbers in ionic liquids by electrophoretic NMR†
Martin
Gouverneur
a,
Jakob
Kopp
a,
Leo
van Wüllen
b and
Monika
Schönhoff
*a
aInstitute of Physical Chemistry, University of Muenster, Corrensstraße 28/30, 48419 Münster, Germany. E-mail: schoenho@uni-muenster.de
bInstitute of Physics, Augsburg University, Universitätsstraße 1, 86159 Augsburg, Germany
First published on 27th October 2015
Abstract
Charge transport in ionic liquids is a phenomenon of utmost interest for electrochemical (e.g. battery) applications, but also of high complexity, involving transport of ion pairs, charged clusters and single ions. Molecular understanding is limited due to unknown contributions of cations, anions and clusters to the conductivity. Here, we perform electrophoretic NMR to determine electrophoretic mobilities of cations and anions in seven different ionic liquids. For the first time, mobilities in the range down to 10−10 m2 V−1 s−1 are determined. The ionic transference number, i.e. the fractional contribution of an ionic species to overall conductivity, strongly depends on cation and anion structure and its values show that structurally very similar ionic liquids can have cation- or anion-dominated conductivity. Transference numbers of cations, for example, vary from 40% to 58%. The results further prove the relevance of asymmetric clusters like [CationXAnionY]X−Y, X ≠ Y, for charge transport in ionic liquids.
1. Introduction
Ionic liquids (IL) are of interest in scientific research because of their various applications e.g. as tailored solvents,1,2 catalysts,3–5 or electrolytes in solar cells and batteries.6–9 The properties of IL are tunable by using different combinations of cations and anions.10,11 For choosing a suitable IL for a desired application, the molecular interaction in the IL is essential. The investigation of the microscopic structure, like ion aggregates in IL has thus become very important, especially for their potential use as electrolytes. One of the major topics in the last years was the degree of “ionicity” of IL, which is investigated and discussed in detail in the literature.12,13 The characterization is typically done by diffusion, conductivity and viscosity measurements.14–17 Typically, in IL the ions are not completely dissociated and a significant fraction of ions is forming ion pairs or aggregates.18,19 This can be seen by comparing the molar conductivity with self-diffusion coefficients determined by pulsed-field-gradient-NMR (PFG-NMR) by the Nernst–Einstein equation, which is strictly valid only for dissociated salt ions and their respective diffusion coefficients. In not fully dissociated systems, the Haven-Ratio (HR) is commonly employed as a correction factor,20,21 describing the deviation of conductivity from the fully dissociated case, and thus accounting for correlated motions of non-dissociated salt molecules or other ionic clusters. An alternative description is based on transference numbers, i.e. the fractional contribution of an ion species to the overall conductivity. Transference numbers can be obtained from diffusion coefficients of the respective ionic species, however, again only when correlations of ion motion are neglected. Under this assumption they are often termed apparent transference number, tapp. Employing parameters like HR and tapp, diffusion and conductivity data are often discussed in a simplified model, assuming the existence of simple ion pairs and single ions.21,22 While such descriptions may be reasonable in systems with dilute salt in neutral solvent,23 or salt-in-polymer electrolytes,24 where larger clusters are negligible, they fail in ionic liquids due to the existence of larger aggregates.25To overcome this lack of information, the electrophoretic mobility μ of each ionic species needs to be known in order to calculate transference numbers directly and gain model-free information about charge transport. Until now, most of the reported electrophoretic mobilites are determined only for metal ions like lithium by using non-blocking electrodes.26,27 Such experiments are not feasible for non-metal ions. The method used in this study is electrophoretic NMR (eNMR) which allows the determination of the electrophoretic mobility μ by NMR.28 During a diffusion experiment (PFG-NMR) an electric voltage is applied and the electrophoretic mobility can be determined for each specific type of ion which contains a NMR active nucleus, e.g.1H, 7Li, 19F. Though in principle delivering unique information, a major problem in eNMR is the resistive heating during the measurement, which can cause unwanted convection effects.29 Due to this problem, so far mainly dilute aqueous solutions of low conductivity have been investigated. Hallberg et al. demonstrated eNMR measurements for aqueous salt solutions (0.01 mol L−1).30 Giesecke et al. recently studied salt solutions in water and other neutral solvents, using salt concentrations of 0.002 mol L−1.31 Much earlier, Holz et al. investigated aqueous salt solutions up to 0.3 mol L−1, by stabilizing them against convection in Agar–Agar gel.32 Other approaches to suppress convection are the use of capillaries and convection compensating pulse sequences, which was demonstrated by He et al.33,34 In spite of these technical optimizations, apart from dilute salt solutions, so far only in one case a pure ionic liquid was studied, when Zhang et al. could show that for a highly mobile IL, namely ethyl-methyl-imidazolium (Emim+)BF4−, mobilities on the order of μ ≈ 10−9 m2 V−1 s−1 for cation and anion could be determined by eNMR using glass capillaries.35 In addition, the mobility of an IL cation was determined in a mixture of IL and water.36
In the present work, we employ an improved setup and show here that it is possible to determine mobilities down to 10−10 m2 V−1 s−1 in ionic liquids. With this opportunity, we are able to provide a systematic mobility study of seven different IL. To our knowledge the mobilities presented in this study are the lowest ever measured by electrophoretic NMR. We overcome the heating problem by using capillaries coated with polyimide for easy handling and avoiding electro-osmotic flow. The advantages of our self-built eNMR-insert are fixed wires and electrodes on a rod and a cavity for the capillaries for convenient and easily manageable eNMR measurements. The influence of the most common cation types (imidazolium, pyrrolidinium, ammonium, piperidin-based) in combination with different anions (bis(trifluoromethylsulfonyl)imide, TFSI−; hexafluorophosphate, PF6−; tetrafluoroborate, BF4−) on the electrophoretic mobilities is investigated. Molar conductivities calculated from electrophoretic mobilities are compared with molar conductivities determined by impedance spectroscopy, showing excellent agreement and thus proving artefact-free results. The direct determination of transference numbers shows that the calculation of apparent transference numbers from diffusion experiments does not adequately describe ion transport in ionic liquids. Charge transport in IL is sensitively depending on the molecular structure and the resulting interactions. Small structural variations might change the system from cation-dominated transport to anion-dominated charge transport. Calculation of apparent dissociation values becomes feasible for cations and anions separately, and yields large differences, which is a direct proof for larger, charged ion clusters in the IL. While such clusters had already been documented by spectroscopic methods, now their relevance in charge transport can be quantified.
The herewith achieved availability of eNMR ion mobilities even for viscous ionic liquids opens up interesting perspectives for studies of more complex ionic systems, like Li salts in IL or IL mixtures, which are proposed for optimization of electrolyte materials. In all these systems experimental information about ionic clustering and correlated motions is sparse, but urgently required.
2. Experimental section
Ionic liquids
The ionic liquids 1-ethyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide (EmimTFSI, IOLITEC, 99.5%), 1-butyl-1-methyl pyrrolidiniumbis(trifluoromethylsulfonyl)imide (P14TFSI, IOLITEC, 99.5%), 1-butyl-3-methyl imidazoliumbis(trifluoro-methylsulfonyl)imide (BmimTFSI, IOLITEC, 99%), 1-butyl-1-methylpiperidiniumbis(trifluoromethylsulfonyl)imide (BmPipTFSI, IOLITEC, 99%), butyl trimethylammoniumbis(trifluoromethylsul-fonyl)imide (BMATFSI, IOLITEC, 99%), 1-ethyl-3-methylimidazoliumtetrafluoroborate (EmimBF4, IOLITEC, >98%) and 1-butyl-3-methyl imidazoliumhexafluorophosphate (BmimPF6, IOLITEC, 99%) were used without further purification. All IL were dried at 100 °C at 10−7 bar for 24 hours and stored in a glovebox (dry argon atmosphere).Impedance spectroscopy
Complex impedance spectra of the samples were measured in a frequency range between 100 Hz and 10 MHz by using a Novocontrol α-S high resolution dielectric analyzer equipped with a Quatro cryosystem and controlled by WINDETA software (Novocontrol, Montabaur, Germany). Samples were filled into a coin-shaped sample chamber and sandwiched between two stainless steel electrodes. The temperature was controlled by a preheated flow of dry nitrogen. Impedance measurements were performed at 295 K. The real part of the conductivity σ′ was plotted against the frequency and the conductivity for the direct current case σdc was extrapolated from the conductivity plateau value at low frequencies.NMR experiments
For all NMR measurements a Bruker 400 MHz Avance NMR spectrometer with a gradient probe head (Bruker, diff30) with selective 5 mm radiofrequency inserts for 1H and 19F, respectively, and a maximum gradient strength of 12 T m−1 was used.Diffusion
The self-diffusion coefficient D of the IL was measured at 295 K using pulsed-field-gradient NMR (PFG-NMR),37 employing a stimulated echo pulse sequence. The diffusion coefficient was determined by measuring the decay of the signal intensity I in dependence of the gradient strength g: | (1) |
Electrophoretic NMR
The electrophoretic NMR measurements were carried out at 295 K in the diff30 probehead with a self-built eNMR electrode configuration for 5 mm NMR tubes following the general principle as described by Hallberg et al. and Zhang et al.30,35 (see Scheme S1 in the ESI†). To apply the electric field in the eNMR setup, a self-built pulse generator (Vmax = 1 kV, Imax = 50 mA) was used.The electrodes and wires are fixed on a polyimide (VESPEL) rod, which inhibits distortions by electrode vibration. The rod is hollow in the lower part of the configuration and acts as housing for a bundle of capillaries. A teflon cap is used to fix the NMR tube on the polyimide rod with a sealing ring, which protects the sample from water and air contact. Therefore, even moisture-sensitive samples can be handled. Coated capillaries with polyimide (Polymicro) were used between the electrodes to suppress flow caused by convection in the sample. More details and specifications of the applicability range of this setup will be published soon in a separate paper.38 The pulse program is based on the double stimulated echo pulse sequence, which is used for convection compensation in diffusion experiments.39 The applied electric field is switched in polarity during the echo pulse sequence.34 The pulse sequence is displayed in Scheme S2 (ESI†).
During the experiment the gradient strength g is increased linearly and the drift of the charged ions can be seen by a phase shift in subsequent spectra. The phase shift Φ shows a linear dependence on the gradient strength,
| Φ = γδgΔDriftμEdc, | (2) |
 | (3) |
3. Results and discussion
A. Electrophoretic mobility
As an example for the determination of electrophoretic mobilities, raw data of the phase shift in dependence on gradient strength are shown for BmimTFSI in Fig. 1a and b. Raw data for the other IL can be found in the ESI,† Fig. S1–S6. | ||
| Fig. 1 Phase shift in dependence on the gradient strength of BmimTFSI for (a) cation (1H) and (b) anion (19F). Errors are estimated to ±3°. The red line follows from a linear regression. | ||
The electrophoretic mobilites are determined from a linear fit of phase shift data, as shown in Fig. 1, yielding the slope m, and then calculated employing eqn (3). The results of cation and anion mobilities in seven IL at 295 K are shown in Fig. 2 and are also given in Table S1 in the ESI.† The determined mobilities vary for different IL within one order of magnitude. EmimBF4 and EmimTFSI show the highest mobilities (order of 10−9 m2 V−1 s−1), while the mobilities of BmimPF6 are in the range of 10−10 m2 V−1 s−1. Our results for EmimBF4 are in good agreement with the results from Zhang et al. (μ+Emim,Zhang = 8.9 × 10−10 m2 V−1 s−1,  at 298 K).35 This IL shows the highest anion mobility with
at 298 K).35 This IL shows the highest anion mobility with  . The highest cation mobility (μ+EmimTFSI = 1.3 × 10−9 m2 V−1 s−1) is found for EmimTFSI. The electrophoretic mobilities decrease for the following IL in the order BmimTFSI > P14TFSI > BMATFSI > BmPipTFSI > BmimPF6. Comparing the anion and cation mobilities for one IL, there are also different types of IL which can be identified. For EmimTFSI the cation mobility is higher compared to the anion mobility. For EmimBF4, BmimTFSI and BmimPF6 the anion mobility is higher than the cation mobility. BMATFSI and P14TFSI show similar anion and cation mobilities. The difference between anion and cation mobility in the respective IL is surprising, since in the diffusion experiments all investigated IL in this study show always higher diffusion coefficients for cations than for anions (see Table 1).
. The highest cation mobility (μ+EmimTFSI = 1.3 × 10−9 m2 V−1 s−1) is found for EmimTFSI. The electrophoretic mobilities decrease for the following IL in the order BmimTFSI > P14TFSI > BMATFSI > BmPipTFSI > BmimPF6. Comparing the anion and cation mobilities for one IL, there are also different types of IL which can be identified. For EmimTFSI the cation mobility is higher compared to the anion mobility. For EmimBF4, BmimTFSI and BmimPF6 the anion mobility is higher than the cation mobility. BMATFSI and P14TFSI show similar anion and cation mobilities. The difference between anion and cation mobility in the respective IL is surprising, since in the diffusion experiments all investigated IL in this study show always higher diffusion coefficients for cations than for anions (see Table 1).
| Ionic liquid | EmimBF4 | EmimTFSI | BmimTFSI | P14TFSI | BMATFSI | BmPipTFSI | BmimPF6 |
|---|---|---|---|---|---|---|---|
| D H [10−11 m2 s−1] | 4.2 ± 0.2 | 4.7 ± 0.2 | 2.4 ± 0.1 | 1.5 ± 0.1 | 0.99 ± 0.05 | 0.55 ± 0.03 | 0.55 ± 0.03 |
| D F [10−11 m2 s−1] | 3.4 ± 0.2 | 2.8 ± 0.1 | 1.8 ± 0.1 | 1.25 ± 0.07 | 0.95 ± 0.05 | 0.51 ± 0.03 | 0.43 ± 0.02 |
| t +app | 0.55 ± 0.03 | 0.62 ± 0.03 | 0.57 ± 0.03 | 0.55 ± 0.03 | 0.51 ± 0.03 | 0.52 ± 0.03 | 0.56 ± 0.03 |
| t −app | 0.45 ± 0.02 | 0.38 ± 0.02 | 0.43 ± 0.02 | 0.45 ± 0.02 | 0.49 ± 0.03 | 0.48 ± 0.03 | 0.44 ± 0.02 |
 | ||
| Fig. 2 Electrophoretic mobilities for cations (black squares) and anions (red circles) in seven IL at 295 K. | ||
Comparing the influence of different anions for the imidazolium based cations, the average electrophoretic mobilities of EmimTFSI and EmimBF4 are in the same range, while BmimTFSI to BmimPF6 show a decrease in μave. Some effects can be explained by the more delocalized charge of the TFSI− anion, which weakens the ion–ion interaction in comparison to the hard anion PF6−.40 In addition, the increase of the alkyl chain length of the cation (see Bmim vs. Emim) leads to a decrease in the electrophoretic mobility due to van-der-Waals-interactions. This is in agreement with alkyl-chain induced cation clustering, which was found by 2H NMR,25 and it is furthermore in good correlation with conductivity data of IL based on imidazolium cations with varying alkyl-chain length.41 The variation of the cation type leads to a decrease in the electrophoretic mobilities in the order EmimTFSI > BmimTFSI > P14TFSI > BMATFSI > BmPipTFSI. This result shows the dominating influence of the cation on the electrophoretic ion mobility of both ions in the IL. The imidazolium-based IL show the highest mobility, which can again be explained by their delocalized charge due to their aromatic ring structure.10 For the ammonium- and pyrrolidinium-based IL, the electrophoretic mobility is much lower, which results from their stronger ion–ion interaction caused by the higher local charge density.
B. Impedance spectroscopy
The electrophoretic mobilities determine the respective ionic contribution to the conductivity. Therefore, summing up the electrophoretic mobilities for each ion species should yield the overall conductivity. As mentioned in the introduction, convection artefacts are a serious problem in eNMR measurements and lead to apparent higher electrophoretic mobilities. Therefore, a comparison of the conductivity calculated from the electrophoretic mobilites with the conductivity determined by impedance spectroscopy provides a control of the values of μ. By knowing the density ρ, the molar mass M and the conductivity σdc measured by impedance spectroscopy, one can calculate the molar conductivity of an IL by | (4) |
| ΛeNMR = z·F·(μ+ + μ−) | (5) |
| Ionic liquid | EmimBF4 | EmimTFSI | BmimTFSI | P14TFSI | BMATFSI | BmPipTFSI | BmimPF6 |
|---|---|---|---|---|---|---|---|
| σ dc [mS cm−1] | 13 ± 1 | 8 ± 0.8 | 3.5 ± 0.4 | 2.3 ± 0.2 | 1.5 ± 0.2 | 0.9 ± 0.1 | 1.2 ± 0.1 |
| Λ dc [S cm2 mol−1] | 2.0 ± 0.2 | 2.1 ± 0.2 | 1.0 ± 0.1 | 0.67 ± 0.07 | 0.41 ± 0.04 | 0.29 ± 0.03 | 0.26 ± 0.03 |
 | ||
| Fig. 3 Molar conductivity Λdc determined by impedance spectroscopy (black triangles) compared with the molar conductivity ΛeNMR (blue squares) calculated from electrophoretic mobilities at 295 K. Errors are estimated to ±10%. | ||
C. Transference numbers
The transference numbers can be obtained easily when the electrophoretic mobilities of the cation (μ+) and anion (μ−) of the IL are known. In a simple salt, the cation and anion transference numbers can then be calculated by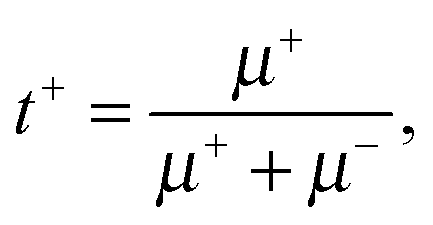 | (6a) |
 | (6b) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :t+) between transference numbers in one IL in this study is about 3:2, which is the case for EmimTFSI, BmimPF6 and BmPipTFSI. These transference numbers are the first reported for a series of IL and can therefore not directly be compared to literature data.
:t+) between transference numbers in one IL in this study is about 3:2, which is the case for EmimTFSI, BmimPF6 and BmPipTFSI. These transference numbers are the first reported for a series of IL and can therefore not directly be compared to literature data.
 | ||
| Fig. 4 Cation transference numbers (t+, black squares); anion transference numbers (t−, red circles). Errors are calculated by error propagation. | ||
However, in the literature it is established to calculate apparent transference numbers t+app from diffusion coefficients by the ratio  , which can give an indication about the fraction of the total current carried by one ion species.21,43,44 Tokuda et al. reported for EmimTFSI, BmimTFSI and BmimPF6 an apparent cationic transference number t+app of 0.63, 0.58 and 0.57, respectively.45,46 These values are in very good agreement with our values of t+app as obtained by diffusion coefficients, being 0.62, 0.57 and 0.56, respectively, for the same compounds (see Table 1). Furthermore, for P14TFSI we had previously determined t+app = 0.53,21 similar to the value of t+app = 0.55 in the present work, see Table 1, which is somewhat larger than t+ = 0.50 for P14TFSI in Fig. 5. Similarly, only for EmimTFSI t+app is similar to t+ determined by eNMR, but for BmimTFSI and BmimPF6t+app is larger than t+. This shows that apparent transference numbers are only an approximate concept and do not reflect the true ionic contributions to conductivity, the reason is ion clustering. The difference in transference numbers for EmimTFSI and BmimTFSI can be explained by the different size of aggregates formed by the cations, see also discussion of mobilities in context with Fig. 2. Imidazolium and pyrrolidinium – based IL show an increase in viscosity and decrease in self-diffusion and molar conductivity with increasing alkyl side chain length,46 comparing IL with the same type of anion. This has been directly observed as reduced ionic rotational diffusion since the cations experience increasing van der Waals forces. The latter act additionally to the electrostatic interaction, which remains independent of alkyl chain length. This leads to enhanced frictional forces in the IL with increasing alkyl chain length.25,46 In the present system this effect should yield significantly lower cation mobility in BmimTFSI compared to EmimTFSI. Apparently the anion mobility is not as strongly influenced by cation clustering as the cation itself, which explains the relatively larger role of the anion in BmimTFSI and the reversal of the transference numbers. Moreover, it can be shown that for BmimTFSI and BmimPF6 the apparent transference numbers45,46 are not agreeing with t+. Therefore, the common practice to calculate transference numbers from diffusion coefficients can give misleading information, because clusters and thus correlated motions of ions are not taken into account.
, which can give an indication about the fraction of the total current carried by one ion species.21,43,44 Tokuda et al. reported for EmimTFSI, BmimTFSI and BmimPF6 an apparent cationic transference number t+app of 0.63, 0.58 and 0.57, respectively.45,46 These values are in very good agreement with our values of t+app as obtained by diffusion coefficients, being 0.62, 0.57 and 0.56, respectively, for the same compounds (see Table 1). Furthermore, for P14TFSI we had previously determined t+app = 0.53,21 similar to the value of t+app = 0.55 in the present work, see Table 1, which is somewhat larger than t+ = 0.50 for P14TFSI in Fig. 5. Similarly, only for EmimTFSI t+app is similar to t+ determined by eNMR, but for BmimTFSI and BmimPF6t+app is larger than t+. This shows that apparent transference numbers are only an approximate concept and do not reflect the true ionic contributions to conductivity, the reason is ion clustering. The difference in transference numbers for EmimTFSI and BmimTFSI can be explained by the different size of aggregates formed by the cations, see also discussion of mobilities in context with Fig. 2. Imidazolium and pyrrolidinium – based IL show an increase in viscosity and decrease in self-diffusion and molar conductivity with increasing alkyl side chain length,46 comparing IL with the same type of anion. This has been directly observed as reduced ionic rotational diffusion since the cations experience increasing van der Waals forces. The latter act additionally to the electrostatic interaction, which remains independent of alkyl chain length. This leads to enhanced frictional forces in the IL with increasing alkyl chain length.25,46 In the present system this effect should yield significantly lower cation mobility in BmimTFSI compared to EmimTFSI. Apparently the anion mobility is not as strongly influenced by cation clustering as the cation itself, which explains the relatively larger role of the anion in BmimTFSI and the reversal of the transference numbers. Moreover, it can be shown that for BmimTFSI and BmimPF6 the apparent transference numbers45,46 are not agreeing with t+. Therefore, the common practice to calculate transference numbers from diffusion coefficients can give misleading information, because clusters and thus correlated motions of ions are not taken into account.
 | ||
| Fig. 5 Effective charge of the IL; black squares: cations (μ+/μ+diff); red circles: anions (μ−/μ−diff); blue triangles: IL dissociation (ΛeNMR/ΛDiff). Errors are calculated by error propagation. | ||
D. Effective charge of ions
By using the Einstein–Smoluchowski equation, one can calculate the electrophoretic mobility by eqn (7) when assuming complete ion dissociation: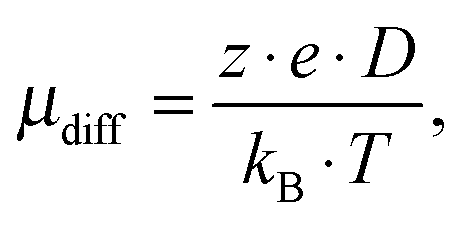 | (7) |
 .31
.31
The effective charge can be interpreted as an apparent dissociation, because it describes the transported charge in relation to the diffusive transport. In the case of single ions, it would be μ = μdiff. Since μdiff includes contributions from uncharged species, the ratio  describes the contribution of transport of dissociated ions or charged aggregates. The ratio
describes the contribution of transport of dissociated ions or charged aggregates. The ratio  is shown in Fig. 5.
is shown in Fig. 5.
The apparent dissociation of the IL then decreases in the order EmimTFSI ≈ EmimBF4 ≈ BmimTFSI ≈ BmPipTFSI ≈ BmimPF6 > P14TFSI ≈ BMATFSI. This result proves the beneficial effect of using charge-delocalized ions like imidazolium-based cations and TFSI− as anion, in contrast to the hard anions BF4− and PF6−, since the tendency is that the soft ions weaken the electrostatic ion interactions and increase the fraction of charged species in the IL. Tokuda et al. found for EmimTFSI, BmimTFSI, BmimPF6 an apparent degree of dissociation via diffusion experiments and impedance spectroscopy of 0.75, 0.61 and 0.68, respectively,47 which agrees with our data, see above. For the IL BmimTFSI, P14TFSI, BMATFSI, BmPipTFSI, BmimPF6 the apparent dissociation of the cation is in the range of 0.5 to 0.6 (Fig. 5). The apparent dissociation of the anion in all these IL is higher. For BmimTFSI and BmimPF6 the apparent dissociation of the anions is even about 100%. In conclusion, it can be proven that in IL the assumption of simple ion pairs and free ions is incorrect, because the apparent degree of dissociation for cation and anion is not equal. The ratio μ/μdiff therefore proves the existence of asymmetric clusters like [CationXAnionY]X−Y (X ≠ Y) in the IL. Due to the overall charge neutrality of the IL, cations and anions have to spend the same fraction of time as neutral pairs. Therefore, the difference in the ratio μ/μdiff for cations and anions is a clear proof that in relation to the expectation from to diffusion results, anion-dominated clusters are drifting faster than cation-dominated ones. By knowing the apparent degrees of dissociation for the cations and anions of the IL, one can judge about the ionicity of the single ions of the IL.
In a molecular picture, these findings imply that the cations preferentially stay in clusters with a high diffusivity but low electrophoretic mobility, while a large fraction of the anions stays in clusters with a high mobility but comparatively low diffusivity, yielding the seeming discrepancy μ+ < μ− and D+ > D−.
We also note that the question of clusters is particularly interesting in systems where ion clustering might be influenced by other molecular constituents of the system. For example, for IL in liquid crystalline structures48 or in polymer gel electrolytes49 ion clustering was discussed as being influenced by structural aspects or direct interactions, respectively. Here, the direct study of ion mobilities will have a large potential to elucidate detailed mechanisms.
4. Conclusions
In this work the cation and anion electrophoretic mobility of seven IL has been measured by eNMR, providing the first systematic comparison of ionic mobilities in different ionic liquids, after overcoming previous technical limitations. The molar conductivity ΛeNMR from electrophoretic mobilities is compared with the molar conductivity Λdc from impedance spectroscopy to prove artefact-free results. The determined cation and anion mobilities show a strong dependence on their chemical structure, which is explained by the strength of the electrostatic interaction depending on the charge delocalization of the ions. The transference numbers derived thereof showed that the ratio t+:t− never exceeds the ratio 3:2, neither is it lower than 2:3, respectively. This implies that in all investigated IL always both ions have a significant contribution to the charge transport. The comparison of the electrophoretic mobilities with mobilities derived from self-diffusion coefficients showed that the apparent cation and anion degree of dissociation 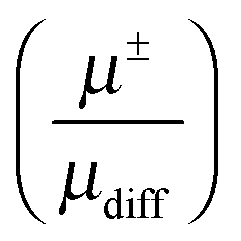 are not equal. This is a direct proof of the existence of asymmetric clusters like [CationXAnionY]X−Y (X ≠ Y), because the assumption that ions exist only in simple ion pairs and as free ions would lead to a similar degree of dissociation for the anion and cation.
are not equal. This is a direct proof of the existence of asymmetric clusters like [CationXAnionY]X−Y (X ≠ Y), because the assumption that ions exist only in simple ion pairs and as free ions would lead to a similar degree of dissociation for the anion and cation.
For the understanding of the interactions between cations and anions in IL it can be therefore concluded that the simple assumption of ion pairs and dissociated single ions is insufficient to describe the ion movement in an electric field. The local structure in IL can be strongly asymmetric with respect to cations and anions. Charge transport in the IL involves to a large extent charged clusters, where in most of the investigated IL the anions move faster than the cations in relation to the expectation from diffusion measurements. EmimTFSI shows highly beneficial properties, as it has the lowest tendency to form large asymmetric clusters, and thus reaches best mobility values. Electrophoretic NMR, now that it is available for comparative studies of ionic liquids, can now be employed to optimize IL, e.g. to identify more “ionic” systems by taking into account the individual ionicity of cations and anions, respectively. It is therefore a unique tool to obtain information about correlated ion motion and about ion clustering, where standard methods providing conductivity, viscosity or even diffusion coefficients fail to provide a detailed molecular picture.
Acknowledgements
We thank J. Kröninger for building the electronic components (pulse generators, filters, etc.) for the eNMR setup, and the collaborators of the mechanical workshop of IPC for providing the mechanical parts to various versions of the setup.Notes and references
- R. D. Rogers and K. R. Seddon, Science, 2003, 302, 792–793 CrossRef PubMed.
- T. Welton, Chem. Rev., 1999, 99, 2071–2083 CrossRef CAS PubMed.
- F. van Rantwijk and R. A. Sheldon, Chem. Rev., 2007, 107, 2757–2785 CrossRef CAS PubMed.
- P. Wasserscheid and W. Keim, Angew. Chem., Int. Ed., 2000, 39, 3772–3789 CrossRef CAS.
- J. S. Wilkes, J. Mol. Catal. A: Chem., 2004, 214, 11–17 CrossRef CAS.
- M. Armand, F. Endres, D. R. MacFarlane, H. Ohno and B. Scrosati, Nat. Mater., 2009, 8, 621–629 CrossRef CAS PubMed.
- M. Galinski, A. Lewandowski and I. Stepniak, Electrochim. Acta, 2006, 51, 5567–5580 CrossRef CAS.
- K. Xu, Chem. Rev., 2004, 104, 4303–4417 CrossRef CAS PubMed.
- S. M. Zakeeruddin and M. Grätzel, Adv. Funct. Mater., 2009, 19, 2187–2202 CrossRef CAS.
- H. Weingärtner, Angew. Chem., Int. Ed., 2008, 47, 654–670 CrossRef PubMed.
- P. Bonhote, A. P. Dias, N. Papageorgiou, K. Kalyanasundaram and M. Grätzel, Inorg. Chem., 1996, 35, 1168–1178 CrossRef CAS PubMed.
- D. R. MacFarlane, M. Forsyth, E. I. Izgorodina, A. P. Abbott, G. Annat and K. Fraser, Phys. Chem. Chem. Phys., 2009, 11, 4962–4967 RSC.
- K. Ueno, H. Tokuda and M. Watanabe, Phys. Chem. Chem. Phys., 2010, 12, 15133–15134 CAS.
- K. Hayamizu, Y. Aihara, S. Arai and C. G. Martinez, J. Phys. Chem. B, 1999, 103, 519–524 CrossRef CAS PubMed.
- K. Hayamizu, S. Tsuzuki, S. Seki, K. Fujii, M. Suenaga and Y. Umebayashi, J. Chem. Phys., 2010, 133, 194505 CrossRef.
- A. Noda, K. Hayamizu and M. Watanabe, J. Phys. Chem. B, 2001, 105, 4603–4610 CrossRef CAS.
- S. Jeremias, M. Kunze, S. Passerini and M. Schönhoff, J. Phys. Chem. B, 2013, 117, 10596–10602 CrossRef CAS.
- F. Castiglione, M. Moreno, G. Raos, A. Famulari, A. Mele, G. B. Appetecchi and S. Passerini, J. Phys. Chem. B, 2009, 113, 10750–10759 CrossRef CAS PubMed.
- H. Weingärtner, Curr. Opin. Colloid Interface Sci., 2013, 18, 183–189 CrossRef.
- G. E. Murch, Solid State Ionics, 1982, 7, 177–198 CrossRef CAS.
- T. Frömling, M. Kunze, M. Schönhoff, J. Sundermeyer and B. Roling, J. Phys. Chem. B, 2008, 112, 12985–12990 CrossRef.
- N. A. Stolwijk and S. Obeidi, Electrochim. Acta, 2009, 54, 1645–1653 CrossRef CAS.
- L. Garrido, A. Mejia, N. Garcia, P. Tiemblo and J. Guzman, J. Phys. Chem. B, 2015, 119, 3097–3103 CrossRef CAS PubMed.
- N. A. Stolwijk, J. Kösters, M. Wiencierz and M. Schönhoff, Electrochim. Acta, 2013, 102, 451–458 CrossRef CAS.
- M. Kunze, S. Jeong, E. Paillard, M. Schönhoff, M. Winter and S. Passerini, Adv. Energy Mater., 2011, 1, 274–281 CrossRef CAS.
- J. Evans, C. A. Vincent and P. G. Bruce, Polymer, 1987, 28, 2324–2328 CrossRef CAS.
- S. Zugmann, M. Fleischmann, M. Amereller, R. M. Gschwind, H. D. Wiemhofer and H. J. Gores, Electrochim. Acta, 2011, 56, 3926–3933 CrossRef CAS.
- M. Holz, Chem. Soc. Rev., 1994, 23, 165–174 RSC.
- M. Holz, O. Lucas and C. Müller, J. Magn. Reson., 1984, 58, 294–305 CAS.
- F. Hallberg, I. Furó, P. V. Yushmanov and P. Stilbs, J. Magn. Reson., 2008, 192, 69–77 CrossRef CAS PubMed.
- M. Giesecke, G. Meriguet, F. Hallberg, Y. Fang, P. Stilbs and I. Furo, Phys. Chem. Chem. Phys., 2015, 17, 3402–3408 RSC.
- M. Holz, C. Müller and A. M. Wachter, J. Magn. Reson., 1986, 69, 108–115 CAS.
- Q. He, Y. Liu, H. Sun and E. Li, J. Magn. Reson., 1999, 141, 355–359 CrossRef CAS.
- Q. He and Z. Wei, J. Magn. Reson., 2001, 150, 126–131 CrossRef CAS.
- Z. Zhang and L. A. Madsen, J. Chem. Phys., 2014, 140, 084204 CrossRef.
- T. J. Simons, P. M. Bayley, Z. Zhang, P. C. Howlett, D. R. MacFarlane, L. A. Madsen and M. Forsyth, J. Phys. Chem. B, 2014, 118, 4895–4905 CrossRef CAS.
- E. O. Stejskal and J. E. Tanner, J. Chem. Phys., 1965, 42, 288–292 CrossRef CAS.
- M. Gouverneur and M. Schönhoff, manuscript in preparation.
- A. Jerschow and N. Müller, J. Magn. Reson., 1997, 125, 372–375 CrossRef CAS.
- J. G. Huddleston, A. E. Visser, W. M. Reichert, H. D. Willauer, G. A. Broker and R. D. Rogers, Green Chem., 2001, 3, 156–164 RSC.
- A. Martinelli, M. Marechal, A. Ostlund and J. Cambedouzou, Phys. Chem. Chem. Phys., 2013, 15, 5510–5517 RSC.
- S. Zhang, X. Lu, Q. Zhou, X. Li, X. Zhang and S. Li, Ionic Liquids – Physicochemical Properties, Elsevier, Amsterdam, 2009 Search PubMed.
- K. R. Harris, J. Phys. Chem. B, 2010, 114, 9572–9577 CrossRef CAS.
- A. L. Rollet and C. Bessada, Annu. Rep. NMR Spectrosc., 2013, 78, 149–207 CrossRef CAS.
- H. Tokuda, K. Hayamizu, K. Ishii, M. Abu Bin Hasan Susan and M. Watanabe, J. Phys. Chem. B, 2004, 108, 16593–16600 CrossRef CAS.
- H. Tokuda, K. Hayamizu, K. Ishii, M. A. B. H. Susan and M. Watanabe, J. Phys. Chem. B, 2005, 109, 6103–6110 CrossRef CAS.
- H. Tokuda, S. Tsuzuki, M. A. B. H. Susan, K. Hayamizu and M. Watanabe, J. Phys. Chem. B, 2006, 110, 19593–19600 CrossRef CAS.
- A. E. Frise, T. Ichikawa, M. Yoshio, H. Ohno, S. V. Dvinskikh, T. Kato and I. Furo, Chem. Commun., 2010, 46, 728–730 RSC.
- M. Gouverneur, S. Jeremias and M. Schönhoff, Electrochim. Acta, 2015, 175, 35–41 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cp05753a |
| This journal is © the Owner Societies 2015 |