
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mechanical switching of magnetic interaction by tweezers-type complex†
Benjamin
Doistau
ab,
Jean-Louis
Cantin
c,
Lise-Marie
Chamoreau
ab,
Valérie
Marvaud
ab,
Bernold
Hasenknopf
*ab and
Guillaume
Vives
*ab
aSorbonne Universités, UPMC Univ Paris 06, UMR 8232, Institut Parisien de Chimie Moléculaire, 4 place Jussieu, 75005, Paris, France. E-mail: bernold.hasenknopf@upmc.fr; guillaume.vives@upmc.fr
bCNRS, UMR 8232, Institut Parisien de Chimie Moléculaire, 4 place Jussieu, 75005, Paris, France
cSorbonne Universités, UPMC Univ Paris 06, INSP, 4 place Jussieu, 75005, Paris, France
First published on 8th July 2015
Abstract
A control of the interaction between two spin centers was achieved by using a mechanical motion in a terpy(Cu–salphen)2 complex. Upon coordination a conformation change and switching from a paramagnetic to an antiferromagnetically coupled system was observed by EPR and SQUID measurements.
The development of the field of molecular machines1 has enabled a fine control of mechanical motion at the molecular scale. Inspired by biological systems or macroscopic counterparts a large variety of molecular machines have been designed by synthetic chemists.2 However, using the motion in these machines to control physical properties at the molecular level remains a challenge. In particular, controlling magnetic interaction is of importance for molecular-scale information processing and memory devices.3 While temperature or light have been used to switch the total spin of a molecule in spin crossover,4 photomagnetic5 or photochromic6 systems, controlling magnetic interactions by a mechanical motion remains largely unexplored. In the latter approach the external stimulus is not focused directly on the electronic state of the site responsible for the magnetic properties but on a remote site leading to a conformational rearrangement that results in modification of magnetic interactions. Thus, the two processes of switching and property change are distinguished, so that a given conformational switch can be combined with different functional units. To the best of our knowledge, only few such examples are reported in the literature. An electronic rearrangement from a diamagnetic to a paramagnetic state was obtained by Herges et al. using a photo-controlled coordination of a tethered pyridine ligand in a Ni-porphyrine based complex.7 Reversible through space control of magnetic interaction has been described by Kaneko et al. using polymer folding8 and by Feringa et al. using a photoswitchable molecular motor functionalized by TEMPO radical moieties.9 Molecular tweezers10 that have already been used for the control of luminescence properties and molecular recognition11 are an alternative architecture to achieve this objective. In particular, we have recently developed switchable molecular tweezers12 based on a terpyridine ligand substituted by metal–salphen complexes that can switch upon metal coordination from a “W” shaped open form to a “U” closed form bringing the two salphen moieties in close spatial proximity (Fig. 1). We propose to use this modular design to control the magnetic interaction between paramagnetic Cu(II)–salphen (d9S = 1/2) complexes via a mechanical motion. This system based on metal coordination is promising as it presents benefits over photochemical switches, giving total conversion and offering thermal stability.
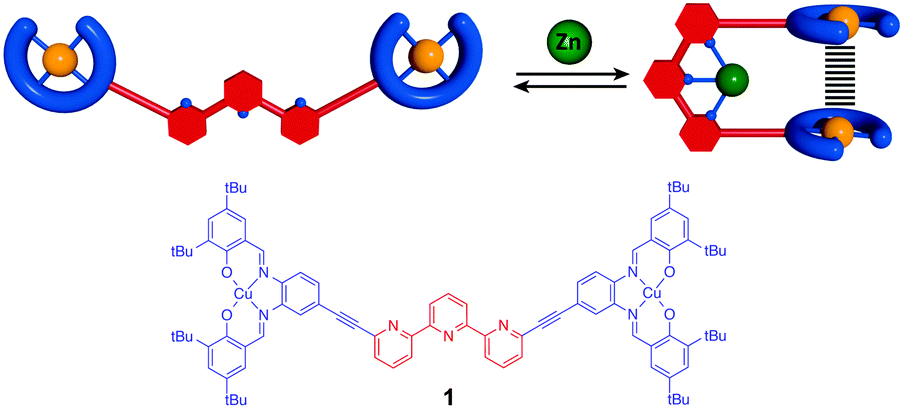 | ||
| Fig. 1 Principle of molecular tweezers 1 mechanical motion. | ||
Herein we describe a dinuclear terpy(Cu–salphen)2 tweezer-type complex composed of a terpyridine ligand as switching unit and two copper salphen spin bearing moieties (Fig. 1). We expect taking advantage of the tunable distance to achieve a reversible control of magnetic interaction between two spins by exchange and dipolar interactions. The synthesis, switching and magnetic studies by EPR and SQUID of the tweezers are reported here.
The tweezers 1 were synthetized in 4 steps following a building block strategy using as a key step a double Sonogashira coupling between alkyne substituted Cu–salphen moieties and 6,6′′ dibromoterpyridine. Complete synthesis and characterizations are detailed in the ESI.† Open tweezers 1 were characterized by mass spectrometry and X-ray diffraction on single crystals obtained by slow solvent evaporation (Fig. 2). Tweezers 1 crystallize in an orthorhombic space group Pbcn with a unit cell of 8831.0(2) Å3 (a = 11.3594(2) Å, b = 40.7108(6) Å, c = 19.0962(3) Å, α = β = γ = 90°). The molecule adopts an almost planar geometry with a small torsion angle between the Cu–salphen planes of 19°. The copper atoms are in a square planar geometry with average Cu–O and Cu–N distances of respectively 1.898(4) and 1.940(4) Å characteristic of Cu–salen complexes.13 The terpyridine adopts an s-trans conformation as expected from the repulsion between the nitrogen lone pairs leading to a large intramolecular Cu–Cu distance of 21.39 Å.
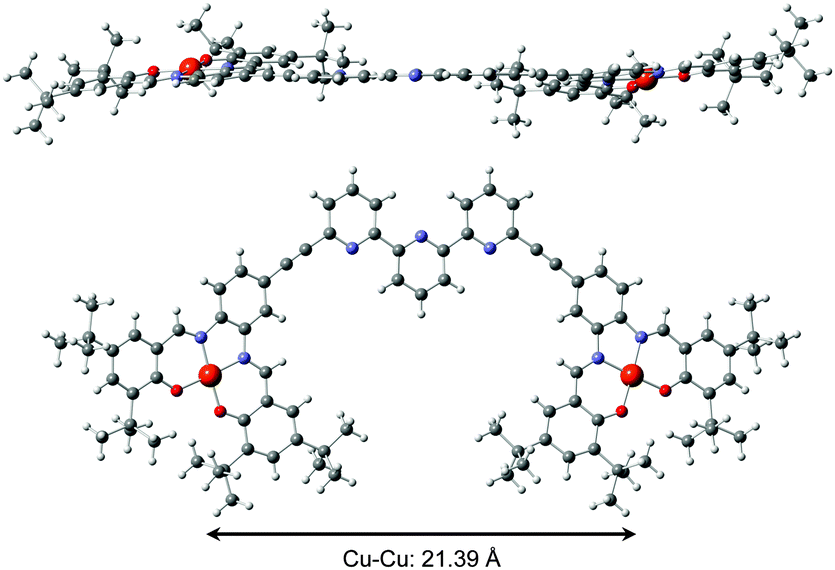 | ||
| Fig. 2 Crystal structure of open tweezers 1 side and top view. | ||
The tweezers mechanical motion of closing and opening was investigated by UV-Visible spectroscopy in solution. Titration of 1 by Zn(ClO4)2 (Fig. 3) showed a single step evolution until addition of one equivalent of zinc with isosbestic points (at 356, 398, and 474 nm), indicating the exclusive formation of the [Zn(1)]2+ complex. Titration with ZnCl2 displayed the same behavior demonstrating no dependency on the counter ions (Fig. S2, ESI†). The 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry of the [Zn(1)]2+ complex was confirmed by mass spectrometry with a signal at m/z 1582.52 corresponding to [Zn(1)Cl]+ (Fig. S4, ESI†). The titration curves were fitted by a 1:1 binding model and revealed a strong binding constant (log K > 8).14
:1 stoichiometry of the [Zn(1)]2+ complex was confirmed by mass spectrometry with a signal at m/z 1582.52 corresponding to [Zn(1)Cl]+ (Fig. S4, ESI†). The titration curves were fitted by a 1:1 binding model and revealed a strong binding constant (log K > 8).14
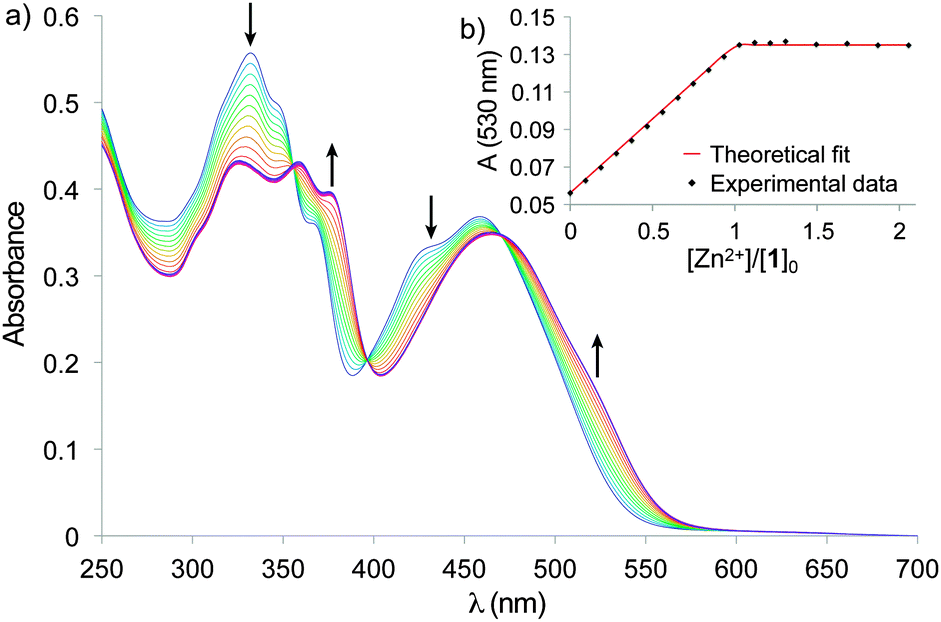 | ||
| Fig. 3 (a) UV-Vis titration of tweezers 1 (5.0 × 10−6 mol L−1) with Zn(ClO4)2 in CHCl3. (b) Absorption at 530 nm and fitting with a 1:1 binding model. | ||
The structure of the closed tweezers was determined in the solid state by X-ray diffraction on single crystal of [Zn(1)]Cl2 complex (Fig. 4). [Zn(1)]Cl2 crystallizes in a monoclinic space group C2/c in a unit cell of 19942.9(14) Å3 (a = 69.071(3) Å, b = 14.7327(6) Å, c = 19.7966(8) Å, α = γ = 90°, β = 98.123(2)°). The molecule adopts a helical folded geometry with the two P and M enantiomers in the crystal as a racemate, like previously observed for platinum analogues.12 Since Zn(II) and Cu(II) have too similar electron density to be unambiguously attributed by XRD, the assignment was confirmed by analysis of the geometry and bond distances. Indeed, the square planar geometry and average M–O and M–N distances in the M–salphen units, of 1.891(3) and 1.938(3) Å respectively, are characteristic of Cu–salphen,13 and very different from Zn–O (1.95 Å) and Zn–N (2.07 Å) distances and pyramidal geometry described in literature for Zn–salphen complexes.15 The zinc is pentacoordinated to the three nitrogen atoms of terpy and two chlorides that complete the coordination sphere. The intramolecular Cu–Cu distance of 4.03 Å is drastically shorter than in the open form and should lead to significant modification in the magnetic interaction between the two unpaired electrons of the copper ions.
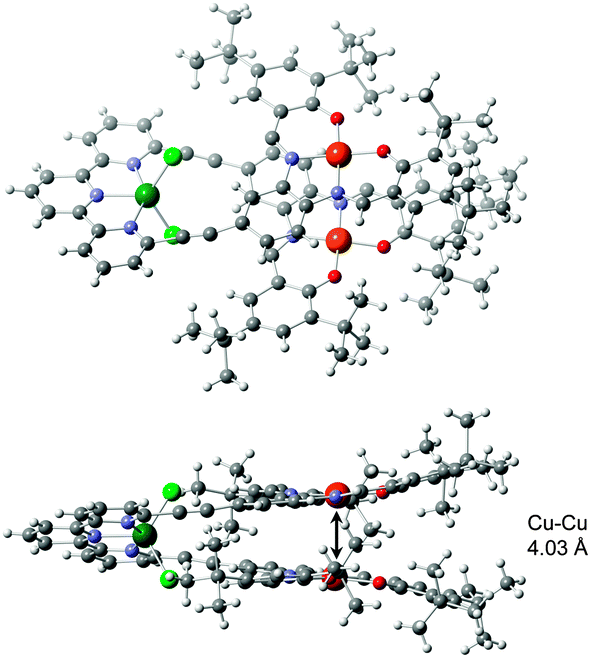 | ||
| Fig. 4 Crystal structure of closed tweezers [Zn(1)]Cl2. | ||
Despite such a high complexation constant with zinc, a reopening was obtained without modification of the Cu complexation by addition of tris(2-aminoethyl)amine (tren) as competitive ligand (Fig. S3, ESI†). Indeed the open tweezers spectrum was recovered after addition of around one equivalent of tren, with the same isosbestic points as for the closing, demonstrating the reversibility of the mechanical motion. The addition of an excess of tren did not lead to copper decoordination, indicating the larger stability of Cu–salphen compared to Zn–terpy or Cu–tren complexes. This is consistent with no observed scrambling between zinc and copper during the tweezers closing.
Having demonstrated the reversible mechanical motion, the metal–metal interactions between the two paramagnetic centers in the open and closed form were first investigated by EPR spectroscopy in frozen solution. Fig. 5a shows the EPR spectrum of the open tweezers 1 in CHCl3, which is characteristic of an electronic spin 1/2 located on a copper atom in an axial symmetry.16 The spectrum is composed of an intense singlet corresponding to the perpendicular transition, and less intense quadruplet corresponding to the parallel transition with a hyperfine coupling with copper (I = 3/2) as determined from simulation (Table 1). This spectrum demonstrates the absence of coupling interactions in the open tweezers 1 which is expected from the large intramolecular Cu–Cu distance observed in the crystal structure.
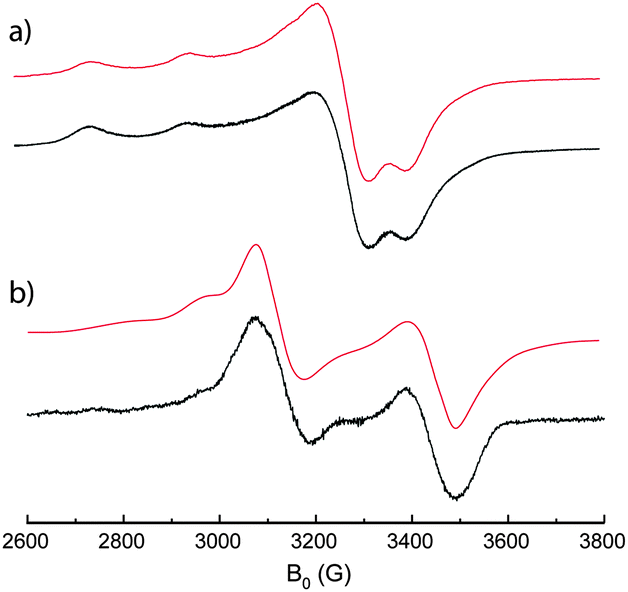 | ||
| Fig. 5 X-band EPR spectrum of (a) open tweezers 1 and (b) closed tweezers [Zn(1)](ClO4)2 in frozen solution of CHCl3 (1.0 × 10−4 mol L−1) at 5 K (black); theoretical fit (red). | ||
| Compound | g ⊥ | g ∥ | Cu | N | D | E | ||
|---|---|---|---|---|---|---|---|---|
| A ⊥ | A ∥ | A ⊥ | A ∥ | |||||
| a In MHz. | ||||||||
| 1 | 2.04 | 2.21 | 10 | 600 | 20 | 110 | — | — |
| [Zn(1)](ClO4)2 | 2.04 | 2.21 | 0 | 500 | 0 | 80 | 880 | 70 |
However, the EPR spectrum of the tweezers closed with Zn2+ is drastically different (Fig. 5b). A doublet-like signal is observed, which is typical for a triplet state in axial geometry as confirmed by the presence of the half-field forbidden transition ΔMs = ±2 (Fig. S5, ESI†). This zero field splitting results from a dipolar coupling interaction between the two 1/2 spins located on each copper(II) ion in close spatial proximity.16b,17 In particular, four transitions are observed (Fig. S5, ESI†) indicating a predominant Zeeman effect (B ≫ hν): a small septuplet corresponding to a parallel transition with hyperfine coupling with the 2 Cu; two unstructured perpendicular transitions; and a small septuplet parallel transition. This spectral analysis confirmed that the tweezers closing results in a through space exchange interaction leading to the formation of two new triplet and singlet levels. Simulation of the spectra permitted the determination of the hyperfine coupling constants (Table 1), as well as the zero field splitting parameter (D = 314.5 G). Since the zero field splitting originates from a dipolar interaction that strongly depends on the spin–spin distance, the D value was utilized to evaluate the distance between the two interacting paramagnetic metal ions (eqn (1) in ESI†).18 The copper–copper intramolecular distance was determined to be ∼4.4 Å, which is in good in agreement with the one measured in the solid state (4.0 Å). In summary the close spatial proximity between the Cu centers generates a S = 1 spin state by through space magnetic coupling showing the interest of this approach to control magnetic interaction by the mechanical motion.
Since the spin ground state and the nature of the exchange coupling interaction is not directly accessible from the EPR spectra, SQUID experiments were carried out (Fig. 6a). The χT versus T curve for open tweezers 1 is characteristic of a paramagnetic system corresponding to non-interacting spins 1/2. χT is constant over a large temperature range, with an experimental value of 0.78 cm3 K mol−1 in agreement with two isolated Cu(II) (theoretical value 0.75 cm3 K mol−1). Even if in the crystal structure the stacking results in a short intermolecular Cu–Cu distance (4.06 Å), the tilted conformation (∼45° dihedral angle) between the two neighboring Cu–salphen might not allow the overlap between the dx2−y2 magnetic orbitals leading to the observed paramagnetic behavior. As the measurement was performed on a powder, the stacking of neighboring complexes might also be negligible. The χT = f(T) data for single crystals of the closed tweezers [Zn(1)]Cl2 shows a decrease starting below 15 K that reveals a weak antiferromagnetic coupling between the metallic centers. Since the intramolecular Cu–Cu distance (4.03 Å) is much shorter than the intermolecular one (9.5 Å) in the crystal structure, this exchange interaction can be attributed to an antiferromagnetic intramolecular interaction in a dinuclear copper complex. In this case, the stacked conformation between the two Cu–salphen allows a better overlap between the magnetic orbitals leading to an antiferromagnetic interaction. The experimental data were fitted by using the Heisenberg-Dirac-Van Vleck Hamiltonian and the exchange coupling value (J) was determined to be −1.4 cm−1. Thus the ground state of the closed tweezers is a singlet and the EPR signal correspond to the excited triplet state thermally populated even at low temperature due to the low J value (Fig. 6b).
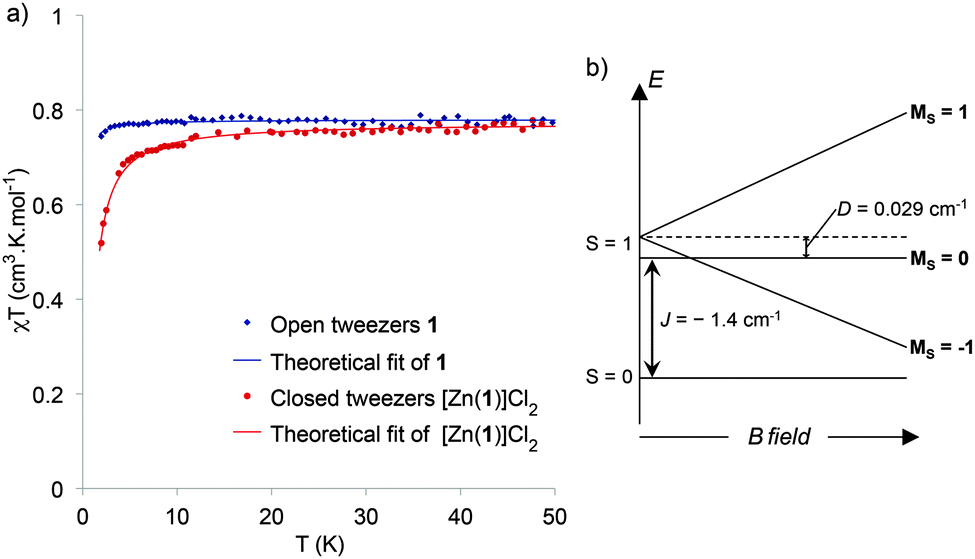 | ||
| Fig. 6 (a) χT = f(T) of open tweezers 1 (blue), and closed tweezers [Zn(1)]Cl2 (red), in the solid state; experimental points, and theoretical fit (plain); (b) energy level diagram of the spin system. | ||
In conclusion, the control of the exchange interaction between two paramagnetic centers was achieved in solution by using a mechanical motion. The choice of a chemical stimulus instead of light or temperature opens the perspective to include this switch in a multicomponent system with chemical messengers. Upon tweezers closing the two isolated 1/2 spins located on each Cu(II) become antiferromagnetically coupled through space leading to a singlet ground state. This system is a rare example of mechano-induced modification of magnetic properties. Conversion between both states is quantitative, and each state is stable at room temperature due to the high binding constant of the terpyridine moiety. These are notable features of such a mechanical switch with a chemical stimulus. Future work will focus on changing the nature of the metal ion and intercalating bridging ligands. This is expected to yield allosteric control of substrate binding and tunable coupling interactions through variation of the bridging ligand.
BD thanks the Ecole Normale Supérieure de Cachan for a PhD Fellowship. Dr Sébastien Blanchard is warmly acknowledged for fruitful discussions. Geoffrey Gontard is thanked for the XRD measurements. Financial support from the ANR Switch (2010-Blan-712) and ANR E-storic (14-CE05-0002) are acknowledged.
Notes and references
- (a) E. R. Kay, D. A. Leigh and F. Zerbetto, Angew. Chem., Int. Ed., 2007, 46, 72–191 CrossRef CAS PubMed; (b) V. Balzani, A. Credi, F. M. Raymo and J. F. Stoddart, Angew. Chem., Int. Ed., 2000, 39, 3348–3391 CrossRef CAS; (c) V. Balzani, M. Venturi and A. Credi, Molecular Devices and Machines: Concepts and Perspectives for the Nanoworld, Wiley-VCH, Weinheim, 2008 Search PubMed.
- (a) R. Eelkema, M. M. Pollard, J. Vicario, N. Katsonis, B. S. Ramon, C. W. M. Bastiaansen, D. J. Broer and B. L. Feringa, Nature, 2006, 440, 163 CrossRef CAS PubMed; (b) G. Vives and J. M. Tour, Acc. Chem. Res., 2009, 42, 473–487 CrossRef CAS PubMed; (c) U. G. E. Perera, F. Ample, H. Kersell, Y. Zhang, G. Vives, J. Echeverria, M. Grisolia, G. Rapenne, C. Joachim and S. W. Hla, Nat. Nanotechnol., 2013, 8, 46–51 CrossRef CAS PubMed; (d) T. Kudernac, N. Ruangsupapichat, M. Parschau, B. Macia, N. Katsonis, S. R. Harutyunyan, K. H. Ernst and B. L. Feringa, Nature, 2011, 479, 208–211 CrossRef CAS PubMed; (e) B. Lewandowski, G. De Bo, J. W. Ward, M. Papmeyer, S. Kuschel, M. J. Aldegunde, P. M. E. Gramlich, D. Heckmann, S. M. Goldup, D. M. D'Souza, A. E. Fernandes and D. A. Leigh, Science, 2013, 339, 189–193 CrossRef CAS PubMed; (f) N. Zigon, A. Guenet, E. Graf and M. W. Hosseini, Chem. Commun., 2013, 49, 3637–3639 RSC.
- G. A. Timco, S. Carretta, F. Troiani, F. Tuna, R. J. Pritchard, C. A. Muryn, E. J. L. McInnes, A. Ghirri, A. Candini, P. Santini, G. Amoretti, M. Affronte and R. E. P. Winpenny, Nat. Nanotechnol., 2009, 4, 173–178 CrossRef CAS PubMed.
- (a) P. Gutlich, Y. Garcia and H. A. Goodwin, Chem. Soc. Rev., 2000, 29, 419–427 RSC; (b) P. Gütlich, A. Hauser and H. Spiering, Angew. Chem., Int. Ed., 1994, 33, 2024–2054 CrossRef PubMed; (c) S. Bonnet, G. Molnár, J. Sanchez Costa, M. A. Siegler, A. L. Spek, A. Bousseksou, W.-T. Fu, P. Gamez and J. Reedijk, Chem. Mater., 2009, 21, 1123–1136 CrossRef CAS.
- (a) O. Sato, Acc. Chem. Res., 2003, 36, 692–700 CrossRef CAS PubMed; (b) S. Ohkoshi and H. Tokoro, Acc. Chem. Res., 2012, 45, 1749–1758 CrossRef CAS PubMed; (c) A. Bleuzen, V. Marvaud, C. Mathoniere, B. Sieklucka and M. Verdaguer, Inorg. Chem., 2009, 48, 3453–3466 CrossRef CAS PubMed; (d) N. Bridonneau, J. Long, J. L. Cantin, J. von Bardeleben, S. Pillet, E. E. Bendeif, D. Aravena, E. Ruiz and V. Marvaud, Chem. Commun., 2015, 51, 8229–8232 RSC.
- (a) M. Irie, T. Fukaminato, K. Matsuda and S. Kobatake, Chem. Rev., 2014, 114, 12174–12277 CAS; (b) K. Matsuda and M. Irie, J. Am. Chem. Soc., 2000, 122, 7195–7201 CrossRef CAS; (c) K. Takayama, K. Matsuda and M. Irie, Chem. – Eur. J., 2003, 9, 5605–5609 CrossRef CAS PubMed; (d) G. Cosquer, B. K. Breedlove and M. Yamashita, Dalton Trans., 2015, 44, 2936–2942 RSC.
- S. Venkataramani, U. Jana, M. Dommaschk, F. D. Sönnichsen, F. Tuczek and R. Herges, Science, 2011, 331, 445–448 CrossRef CAS PubMed.
- T. Kaneko, H. Abe, M. Teraguchi and T. Aoki, Macromolecules, 2013, 46, 2583–2589 CrossRef CAS.
- J. Wang, L. Hou, W. R. Browne and B. L. Feringa, J. Am. Chem. Soc., 2011, 133, 8162–8164 CrossRef CAS PubMed.
- (a) J. Leblond and A. Petitjean, ChemPhysChem, 2011, 12, 1043–1051 CrossRef CAS PubMed; (b) M. Hardouin-Lerouge, P. Hudhomme and M. Salle, Chem. Soc. Rev., 2011, 40, 30–43 RSC; (c) F.-G. Klärner and B. Kahlert, Acc. Chem. Res., 2003, 36, 919–932 CrossRef PubMed; (d) S. Zimmerman, Top. Curr. Chem., 1993, 165, 71–102 CrossRef CAS.
- (a) Y. Tanaka, K. M.-C. Wong and V. W.-W. Yam, Angew. Chem., Int. Ed., 2013, 52, 14117–14120 CrossRef CAS PubMed; (b) M. Barboiu, L. Prodi, M. Montalti, N. Zaccheroni, N. Kyritsakas and J.-M. Lehn, Chem. – Eur. J., 2004, 10, 2953–2959 CrossRef CAS PubMed; (c) J. Leblond, H. Gao, A. Petitjean and J.-C. Leroux, J. Am. Chem. Soc., 2010, 132, 8544–8545 CrossRef CAS PubMed; (d) M. Linke-Schaetzel, C. E. Anson, A. K. Powell, G. Buth, E. Palomares, J. D. Durrant, T. S. Balaban and J.-M. Lehn, Chem. – Eur. J., 2006, 12, 1931–1940 CrossRef CAS PubMed; (e) F. Marchioni, A. Juris, M. Lobert, U. P. Seelbach, B. Kahlert and F.-G. Klarner, New J. Chem., 2005, 29, 780–784 RSC; (f) A. Petitjean and J.-M. Lehn, Inorg. Chim. Acta, 2007, 360, 849–856 CrossRef CAS PubMed.
- (a) B. Doistau, A. Tron, S. A. Denisov, G. Jonusauskas, N. D. McClenaghan, G. Gontard, V. Marvaud, B. Hasenknopf and G. Vives, Chem. – Eur. J., 2014, 20, 15799–15807 CrossRef CAS PubMed; (b) B. Doistau, C. Rossi-Gendron, A. Tron, N. D. McClenaghan, L.-M. Chamoreau, B. Hasenknopf and G. Vives, Dalton Trans., 2015, 44, 8543–8551 RSC.
- (a) F. Thomas, O. Jarjayes, C. Duboc, C. Philouze, E. Saint-Aman and J.-L. Pierre, Dalton Trans., 2004, 2662–2669 RSC; (b) M. J. MacLachlan, M. K. Park and L. K. Thompson, Inorg. Chem., 1996, 35, 5492–5499 CrossRef CAS PubMed.
- P. Thordarson, Chem. Soc. Rev., 2011, 40, 1305–1323 RSC.
- (a) A. L. Singer and D. A. Atwood, Inorg. Chim. Acta, 1998, 277, 157–162 CrossRef CAS; (b) E. C. Escudero-Adán, J. Benet-Buchholz and A. W. Kleij, Chem. – Eur. J., 2009, 15, 4233–4237 CrossRef PubMed; (c) A. Decortes, M. Martinez Belmonte, J. Benet-Buchholz and A. W. Kleij, Chem. Commun., 2010, 46, 4580–4582 RSC.
- (a) K. Butsch, T. Günther, A. Klein, K. Stirnat, A. Berkessel and J. Neudörfl, Inorg. Chim. Acta, 2013, 394, 237–246 CrossRef CAS PubMed; (b) G. Verquin, G. Fontaine, E. Abi-Aad, E. Zhilinskaya, A. Aboukaïs and J.-L. Bernier, J. Photochem. Photobiol., B, 2007, 86, 272–278 CrossRef CAS PubMed; (c) I. Caretti, E. Carter, I. A. Fallis, D. M. Murphy and S. Van Doorslaer, Phys. Chem. Chem. Phys., 2011, 13, 20427–20434 RSC; (d) S. Deshpande, D. Srinivas and P. Ratnasamy, J. Catal., 1999, 188, 261–269 CrossRef CAS; (e) D. M. Murphy, I. Caretti, E. Carter, I. A. Fallis, M. C. Göbel, J. Landon, S. V. Doorslaer and D. J. Willock, Inorg. Chem., 2011, 50, 6944–6955 CrossRef CAS PubMed; (f) M. M. Bhadbhade and D. Srinivas, Inorg. Chem., 1993, 32, 6122–6130 CrossRef CAS.
- (a) G. E. Selyutin, A. A. Shklyaev and A. M. Shul'ga, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1985, 34, 1218–1223 CrossRef; (b) R. Chitta, L. M. Rogers, A. Wanklyn, P. A. Karr, P. K. Kahol, M. E. Zandler and F. D'Souza, Inorg. Chem., 2004, 43, 6969–6978 CrossRef CAS PubMed; (c) C. J. Chang, E. A. Baker, B. J. Pistorio, Y. Deng, Z.-H. Loh, S. E. Miller, S. D. Carpenter and D. G. Nocera, Inorg. Chem., 2002, 41, 3102–3109 CrossRef CAS PubMed.
- (a) M. S. Anders Lund and S. Shimada, Principles and Applications of ESR Spectroscopy, Springer, Netherlands, 2011 Search PubMed; (b) S. S. Eaton, K. M. More, B. M. Sawant and G. R. Eaton, J. Am. Chem. Soc., 1983, 105, 6560–6567 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, spectroscopic, titration and crystallographic data. CCDC 1052163–1052165. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5cc04980f |
| This journal is © The Royal Society of Chemistry 2015 |