
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Eutypellazines A–M, thiodiketopiperazine-type alkaloids from deep sea derived fungus Eutypella sp. MCCC 3A00281†
Siwen Niuab,
Dong Liua,
Zongze Shao*b,
Peter Prokschc and
Wenhan Lin *a
*a
aState Key Laboratory of Natural and Biomimetic Drugs, Peking University, Beijing, 100191, P. R. China. E-mail: whlin@bjmu.edu.cn; Fax: +86-10-82806188
bKey Laboratory of Marine Biogenetic Resources, Third Institute of Oceanography, SOA, Xiamen, 361005, P. R. China
cInstitute of Pharmaceutical Biology and Biotechnology, Heinrich-Heine University, 40225 Duesseldorf, Germany
First published on 3rd July 2017
Abstract
Bioassay in association with the NMR/MS spectroscopic data guided fractionation of the solid fermentation of a deep sea derived fungus Eutypella sp. MCCC 3A00281, resulted in the isolation of 13 new thiodiketopiperazine-type alkaloids, namely eutypellazines A–M (1–13). Their structures were elucidated on the basis of extensive spectroscopic data analysis, including the ECD data, modified Mosher's method, and the Cu-Kα X-ray single-crystal diffraction experiments for the determination of the absolute configurations. An anti-HIV bioassay indicated that compounds 1–12 exhibited inhibitory effects against pNL4.3.Env-.Luc co-transfected 293T cells (HIV-1 model cells) with low cytotoxicity, of which eutypellazine E exerted the highest activity. A preliminary structure–activity relationship was discussed. In addition, eutypellazine J (10) and epicoccin A showed reactivating effects toward latent HIV-1 in J-Lat A2 cells.
1. Introduction
The natural thiodiketopiperazine alkaloids (TDKPs) are a class of unique fungal secondary metabolites, which are characterized by the presence of a diketopiperazine core featuring thiomethyl groups and/or transannular sulfide bridges.1–6 The TDKP derivatives are biogenetically derived from at least one aromatic amino acid (phenylalanine, tryptophan or tyrosine), while the unusual sulfur bridges and thiomethyl units are generated by the mediation of biosynthetic genes such as glutathione S-transferase.7,8 TDKP derivatives are widely distributed in diverse fungal genera, whereas the fungal strains, belonging to the genera of Penicillium,6 Exserohilum,9 and Phoma,10 from marine inhabitation are the rich sources to derive TDKP analogues. So far, an array of TDKPs with diverse scaffolds due to the formation of different amino acids or distinct sulfur bridges has been found from fungal origin, and the structural variety directly affected their bioactivity. The biological activities are mainly focused on cytotoxic,9,11 antibacterial,12,13 antiangiogenic,14 anti-inflammatory,15 and antituberculosis.16 In continuation of our research program aiming the discovery of bioactive metabolites from deep-sea derived microorganisms, the EtOAc extract of the fermentation of a deep-sea derived fungus Eutypella sp. MCCC 3A00281 exhibited inhibitory effect against HIV-1 virus (Table 1). Chromatographic fractionation of the active extract in association with the bioassay and NMR/ESIMS experiments revealed that the active fractions F7 and F8 featured a profile of thiodiketopiperazine-type derivatives. Further chromatographic separation of both F7 and F8 fractions resulted in the isolation of 15 TDKP derivatives, including 13 new analogues namely eutypellazines A–M (1–13) (Fig. 1). In this paper, we intend to report the structure elucidation of new compounds and their anti-HIV-1 activities.| Fractions | c (μg mL−1) | Inhibitory rates (%) |
|---|---|---|
| a EFV (Efavirenz): positive control, bioassay was performed in pNL4.3.Env-.Luc co-transfected 293T cells. | ||
| EtOAc extract | 10 | 46.3 |
| F3 | 10 | 0 |
| F4 | 10 | 9.7 |
| F5 | 10 | 29.4 |
| F6 | 10 | 5.1 |
| F7 | 10 | 99.6 |
| F8 | 10 | 68.6 |
| F9 | 10 | 46.0 |
| F10 | 10 | 18.0 |
| EFV | 0.1 (μM) | 96.22 |
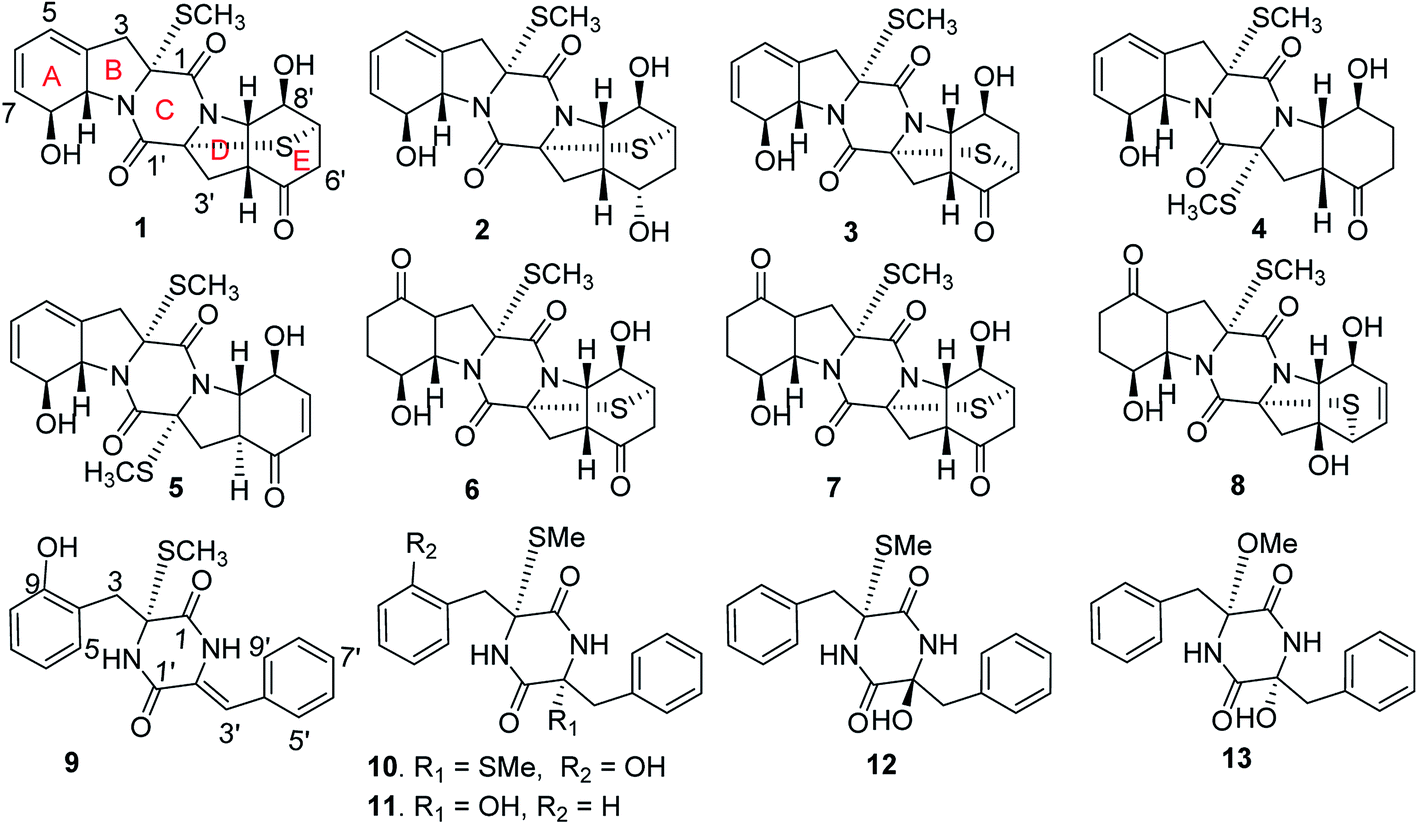 | ||
| Fig. 1 Structures of new compounds. | ||
2. Experimental section
2.1. General procedure
Melting points were measured on X-5 micromelting-point apparatus. IR spectra were determined by a Thermo Nicolet Nexus 470 FT-IR spectrometer. UV spectra were measured on a Cary 300 spectrometer. Optical rotations were obtained from a Rudolph IV Autopol automatic polarimeter at 25 °C. CD spectra were measured on a JASCO J-810 spectropolarimeter. NMR spectra were recorded on Bruker Advance 400 MHz spectrometers. Chemical shifts are expressed in δ (ppm) referenced to the solvent peaks at δH 2.50/δC 39.5 for DMSO-d6 and δH 7.26/δC 77.2 for CDCl3, and coupling constants (J) are in Hz. HRESIMS spectra were obtained from Xevo G2 Q-TOF mass spectrometer (Waters). X-ray data were obtained by a Bruker D8 Advance single crystal X-ray diffractometer using graphite monochromated Cu-Kα radiation. Column chromatography (CC) was performed by silica gel (100–200 and 200–300 mesh, Qingdao Marine Chemistry Co. Ltd. China), ODS gel (50 μm, YMC, Japan) and Sephadex LH-20 (18–110 μm, Amersham Pharmacia Biotech AB, Uppsala, Sweden). The precoated silica gel plates (Merck, Kieselgel 60 F254, 0.25 mm) were used for TLC analysis. HPLC chromatography was performed on a Waters e2695 separation Module coupled with a Waters 2998 photodiode array detector, and a semi-preparative reversed-phased column (YMC-packed C18, 5 μm, 250 mm × 10 mm) was used for purification. 293T cells (ATCC® CRL-3216™) from embryonic kidney of human were provided from American Type Culture Collection (ATCC). J-lat-A2 cells were provided by the Verdin laboratory (Buck Institute, CA), and were generated by transduction of Jurkat T cells (NIH AIDS Reagent Program) with an HIV vector expressing Tat-Flag and GFP under the control of the viral 5′-LTR and IRES positioned in between Tat and GFP (LTR-Tat-Flag-IRES-GFP).2.2. Fungal material and fermentation
The fungus Eutypella sp. MCCC 3A00281 was isolated from the deep sea sediment, collected with TV-multicore from South Atlantic Ocean (GPS 27.90 W, 6.43 S) at the depth of 5610 meters during the Comra 22nd oceanic cruise in June 2011. On the basis of the rRNA analysis, the genus of Eutypella was determined (GeneBank accession number KT366012). The strain was deposited in the Marine Culture Collection of China (MCCC), Third Institute of Oceanography, State Oceanic Administration (SOA), Xiamen, China.The fungal strain MCCC 3A00281 was cultured on the slants of PDA medium at 25 °C for 6 days. The fresh mycelia were cut and inoculated to 45 × 500 mL Erlenmeyer flasks, each flask contains 100 g rice and 140 mL distilled water. After autoclaving at 15 psi for 30 min, these flasks were inoculated and incubated at room temperature for 25 days. The fermented fungal material was extracted with EtOAc for three times, and then concentrated under vacuo to afford crude extract.
2.3. Isolation and purification
The EtOAc extract was tested by the pNL4.3.Env-.Luc co-transfected 293T cells (HIV-1 model cells) showing 46% inhibitory rate in a dose of 10 μg mL−1. The active extract (13 g) was subjected to a silica gel vacuum liquid chromatography (VLC), eluting with a gradient of petroleum ether (PE) and acetone (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0 to 0:1) to yield 10 fractions (F1–F10). Fractions F1 to F4 (sum amount 4.3 g) were detected by the 1H NMR spectra to show the resonances mainly containing fatty acids. Fractions F5 and F6 (sum amount 2.0 g) featured terpenoid resonances in the 1H NMR spectra. The polar fractions F9 and F10 (sum amount 5.4 g) displayed the resonances for polyphenols and saccharides in the 1H NMR spectra. In addition, the 1H NMR and ESIMS spectra of F7 and F8 (sum amount 1.0 g) displayed the metabolite profile of thiodiketopiperazine alkaloids. Bioassay of the fractions F3 to F10 using pNL4.3.Env-.Luc co-transfected 293T cells (HIV-1 model cells) revealed that F7 and F8 possess potential inhibition against HIV-1 virus, of which F7 showed the most active (Table 1). Fraction F7 (0.4 g) was subjected to ODS column chromatography (CC) eluting with a gradient with increasing MeOH in H2O (30% to 100%) to afford 11 subfractions (SF71–SF711). The HPLC fingerprints of SF71–SF76 were overlapped with that of inactive fraction F6. SF77 (112 mg) was separated by the semi-preparative HPLC with a mobile phase of MeCN–H2O (3:2) to obtain 5 (13.4 mg), 10 (2.8 mg), 4 (46.6 mg), and 9 (2.3 mg). SF78 (157 mg) was separated by the semi-preparative HPLC with a mobile phase of MeCN–H2O (7:13) to obtain 13 (15.8 mg) and 12 (4.5 mg). F8 (0.5 g) was fractionated upon an ODS column chromatography using a gradient elution of MeOH in H2O (20–100%) to afford 15 subfractions (SF81–SF815). SF86 (101 mg) was separated by the semi-preparative HPLC (MeCN–H2O, 1:3) to afford epicoccin A (42.3 mg), 6 (28.8 mg), and 7 (14.2 mg), while 8 (2.5 mg) from SF88 (MeCN–H2O = 7:18), 2 (3.5 mg) from SF89 (MeOH/H2O = 43:57), and 1 (47.5 mg) from SF811 (MeCN/H2O = 27:73) were separated by the semi-preparative HPLC chromatography. SF812 (135 mg) was subjected to silica gel CC eluted with CH2Cl2–MeOH (50:1) to yield 3 (12.7 mg), epicoccin I (10.6 mg), and 11 (2.6 mg).
ε) 202 (4.11) and 264 (3.55) nm; CD (MeOH) λmax (Δε) 267 (+5.45), 246 (+7.10), 205 (+20.13) nm; IR (KBr) νmax 3370, 2922, 1713, 1647, 1418, 1324, 1230, 1190 cm−1; 1H and 13C NMR data, see Tables 2 and 4; HRESIMS m/z 421.0887 [M + H]+ (calcd for C19H21N2O5S2, 421.0892).
:0 to 0:1) to yield 10 fractions (F1–F10). Fractions F1 to F4 (sum amount 4.3 g) were detected by the 1H NMR spectra to show the resonances mainly containing fatty acids. Fractions F5 and F6 (sum amount 2.0 g) featured terpenoid resonances in the 1H NMR spectra. The polar fractions F9 and F10 (sum amount 5.4 g) displayed the resonances for polyphenols and saccharides in the 1H NMR spectra. In addition, the 1H NMR and ESIMS spectra of F7 and F8 (sum amount 1.0 g) displayed the metabolite profile of thiodiketopiperazine alkaloids. Bioassay of the fractions F3 to F10 using pNL4.3.Env-.Luc co-transfected 293T cells (HIV-1 model cells) revealed that F7 and F8 possess potential inhibition against HIV-1 virus, of which F7 showed the most active (Table 1). Fraction F7 (0.4 g) was subjected to ODS column chromatography (CC) eluting with a gradient with increasing MeOH in H2O (30% to 100%) to afford 11 subfractions (SF71–SF711). The HPLC fingerprints of SF71–SF76 were overlapped with that of inactive fraction F6. SF77 (112 mg) was separated by the semi-preparative HPLC with a mobile phase of MeCN–H2O (3:2) to obtain 5 (13.4 mg), 10 (2.8 mg), 4 (46.6 mg), and 9 (2.3 mg). SF78 (157 mg) was separated by the semi-preparative HPLC with a mobile phase of MeCN–H2O (7:13) to obtain 13 (15.8 mg) and 12 (4.5 mg). F8 (0.5 g) was fractionated upon an ODS column chromatography using a gradient elution of MeOH in H2O (20–100%) to afford 15 subfractions (SF81–SF815). SF86 (101 mg) was separated by the semi-preparative HPLC (MeCN–H2O, 1:3) to afford epicoccin A (42.3 mg), 6 (28.8 mg), and 7 (14.2 mg), while 8 (2.5 mg) from SF88 (MeCN–H2O = 7:18), 2 (3.5 mg) from SF89 (MeOH/H2O = 43:57), and 1 (47.5 mg) from SF811 (MeCN/H2O = 27:73) were separated by the semi-preparative HPLC chromatography. SF812 (135 mg) was subjected to silica gel CC eluted with CH2Cl2–MeOH (50:1) to yield 3 (12.7 mg), epicoccin I (10.6 mg), and 11 (2.6 mg).
ε) 202 (4.11) and 264 (3.55) nm; CD (MeOH) λmax (Δε) 267 (+5.45), 246 (+7.10), 205 (+20.13) nm; IR (KBr) νmax 3370, 2922, 1713, 1647, 1418, 1324, 1230, 1190 cm−1; 1H and 13C NMR data, see Tables 2 and 4; HRESIMS m/z 421.0887 [M + H]+ (calcd for C19H21N2O5S2, 421.0892).
| Position | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| 3 | 3.03, d (14.0); 3.05, d (14.0) | 3.01, d (14.0); 3.04 d (14.0) | 3.01, d (14.0); 3.05 (14.0) | 3.00, d (14.0); 3.04, d (14.0) | 2.98, d (15.5); 3.06, d (15.5) |
| 5 | 5.98, d (4.5) | 5.97, d (4.5) | 5.98, d (4.5) | 5.96, d (4.5) | 5.99, d (4.5) |
| 6 | 5.91, dd (10.0, 4.5) | 5.90, dd (10.0, 4.5) | 5.91, dd (9.8, 4.5) | 5.89, dd (9.8, 4.5) | 5.91, dd (9.8, 4.5) |
| 7 | 5.62, brd (10.0) | 5.62, d (10.0) | 5.62, brd (9.8) | 5.62, brd (9.8) | 5.65, brd (9.8) |
| 8 | 4.66, brd (13.8) | 4.65, brd (13.9) | 4.60, brd (13.6) | 4.58, brd (13.3) | 4.63, d (13.8) |
| 9 | 4.78, d (13.8) | 4.77, d (13.9) | 4.76, d (13.6) | 4.70, d (13.3) | 4.75, d (13.8) |
| 3′ | 2.76, d (13.1) | 2.59, dd (13.0, 8.5) | 2.88, d (11.6) | 2.31, dd (13.2, 7.8) | 2.29, t (12.7) |
| 2.98, dd (13.1, 8.5) | 2.79, dd (13.0, 1.5) | 3.07, dd (11.6, 7.0) | 2.83, d (13.2) | 2.44, dd (12.7, 5.0) | |
| 4′ | 3.03, dd (11.5, 8.0) | 2.69, dt (8.5, 8.0) | 3.16, t (7.0) | 3.00, dd (8.0, 7.8) | 3.42, dt (13.3, 5.0) |
| 5′ | 4.31, m | ||||
| 6′ | 2.92, dd (18.8, 7.5) | 1.97, dd (14.6, 7.8) | 3.64, d (8.9) | 2.24, ddd (12.0, 6.0, 8.0) | 6.06, dd (10.2, 2.0) |
| 3.12, d (18.8) | 2.74, m | 2.63, dt (12.0, 7.0) | |||
| 7′ | 3.74, dd (7.5, 5.8) | 3.39, dd (8.2, 3.8) | 2.53, dd (15.4, 8.9) | 1.92, m | 6.91, dd (10.2, 1.7) |
| 2.69, dd (15.4, 4.5) | 2.19, m | ||||
| 8′ | 4.03, ddd (5.8, 4.0, 2.5) | 3.85, brdd (4.0, 3.8) | 4.50, t (4.5) | 4.37, brt (4.0) | 4.57, brd (8.7) |
| 9′ | 4.78, dd (8.0, 5.8) | 4.49, ddd (8.0, 4.0) | 4.62, dd (7.0, 4.5) | 4.35, dd (8.0, 4.0) | 3.83, dd (13.3, 8.7) |
| CH3S-2 | 2.24, s | 2.21, s | 2.20, s | 2.12, s | 2.19, s |
| CH3S-2′ | 1.97, s | 2.17, s | |||
| OH-8 | 5.43, d (1.2) | 5.53, br | 5.35, br | 5.31, br | 5.17, d (2.0) |
| OH-5′ | 4.85, d (4.6) | ||||
| OH-8′ | 6.21, d (2.5) | 5.91, br | 5.56, br | 5.37, br | 6.18, br |
ε) 204 (4.01), 265 (3.06) nm; CD (MeOH) λmax (Δε) 264 (+6.78), 245 (+5.45), 220 (+9.58), 201 (+11.38) nm; IR (KBr) νmax 3392, 2920, 1697, 1649, 1416, 1232 cm−1; 1H and 13C NMR data, see Tables 2 and 4; HRESIMS m/z 423.1043 [M + H]+ (calcd for C19H23N2O5S2, 423.1048).ε) 204 (4.18), 266 (3.59) nm; CD (MeOH) λmax (Δε) 307 (+8.45), 248 (−3.19), 213 (+16.65) nm; IR (KBr) νmax 3366, 2921, 1706, 1648, 1418, 1230 cm−1; 1H and 13C NMR data, Tables 2 and 4; HRESIMS m/z 421.0888 [M + H]+ (calcd for C19H21N2O5S2, 421.0892).ε) 205 (4.10), 261 (3.59) nm; CD (MeOH) λmax (Δε) 284 (+1.60), 262 (−3.72), 237 (+4.33), 212 (−8.36) nm; IR (KBr) νmax 3379, 2923, 1706, 1637, 1414, 1328, 1237, 1201 cm−1; 1H and 13C NMR data, see Tables 2 and 4; HRESIMS m/z 437.1201 [M + H]+ (calcd for C20H25N2O5S2, 437.1205).ε) 207 (4.16), 264 (3.55) nm; CD (MeOH) λmax (Δε) 289 (+1.59), 256 (−2.48), 248 (−2.27), 215 (−31.04) nm; IR (KBr) νmax 3351, 2922, 1692, 1639, 1412, 1374, 1262, 1185, 1125 cm−1; 1H and 13C NMR data, see Tables 2 and 4; HRESIMS m/z 435.1049 [M + H]+ (calcd for C20H23N2O5S2, 435.1048).ε) 207 (4.33) nm; IR (KBr) νmax 3396, 2922, 1709, 1655, 1419, 1362, 1324, 1225 cm−1; 1H and 13C NMR data, see Tables 3 and 4; HRESIMS m/z 439.0995 [M + H]+ (calcd for C19H23N2O6S2, 439.0998).
| Position | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 |
|---|---|---|---|---|---|---|---|---|
| 3 | 2.32, dd (13.5, 8.5) | 2.29, dd (13.6, 8.3) | 2.39, dd (13.6, 8.3) | 3.18, d (14.4) | 3.01, d (15.7) | 2.84, d (13.6) | 2.68, d (12.7) | 2.78, d (13.1) |
| 2.86, d (13.5) | 2.85, d (13.6) | 2.76, d (13.6) | 3.46, d (14.4) | 3.09, d (15.7) | 3.21, d (13.6) | 3.27, d (12.7) | 3.09, d (13.1) | |
| 4 | 3.06, t (8.5) | 3.04, dd (8.3, 8.0) | 2.98, t (8.3) | |||||
| 5 | 7.12, d (7.6) | 6.30, dd (7.5, 1.6) | 7.04, d (8.0) | 7.20, d (8.0) | 7.12, d (8.0) | |||
| 6 | 2.26, dt (17.7, 4.6) | 2.25, m | 2.21, dt (17.6, 4.8) | 6.70, t (7.6) | 6.25, t (7.5) | 7.07, t (8.0) | 7.20, t (8.0) | 7.20, t (8.0) |
| 2.59, m | 2.57, m | 2.58, m | ||||||
| 7 | 1.91, m; 2.12, m | 1.90, m; 2.10, m | 1.87, m; 2.03, m | 7.04, t (7.6) | 6.89, t (7.5) | 7.00, t (8.0) | 7.20, t (8.0) | 7.15, t (8.0) |
| 8 | 4.33, br | 4.30, dt (3.8, 2.1) | 4.30, brt (3.4) | 6.81, d (7.6) | 6.67, d (7.5) | 7.07, t (8.0) | 7.20, t (8.0) | 7.20, t (8.0) |
| 9 | 4.39, dd (8.5, 3.8) | 4.39, d (8.0, 3.8) | 4.37, dd (8.3, 3.4) | 7.04, d (8.0) | 7.20, d (8.0) | 7.12, d (8.0) | ||
| 3′ | 2.71, d (13.4) | 2.53, dd (12.8, 8.0) | 2.57, (12.6, 8.0) | 6.62, s | 2.97, d (13.6) | 2.65, d (13.3) | 2.86, d (13.2) | 2.68, d (12.7) |
| 2.91, dd (13.4, 8.0) | 2.74, d (12.8) | 2.68, d (12.6) | 3.48, d (13.6) | 2.90, d (13.3) | 3.32, d (13.2) | 3.27, d (12.7) | ||
| 4′ | 3.00, dd (8.0, 7.9) | 2.67, dt (8.0, 4.0) | ||||||
| 5′ | 4.28, m | 4.17, dd (6.2, 1.5) | 7.47, d (7.6) | 7.14, d (8.0) | 6.89, d (7.4) | 7.08, d (8.0) | 7.20, d (8.0) | |
| 6′ | 2.89, dd (15.7, 5.3) | 1.94, m | 5.98, ddd (10.2, 6.2) | 7.38, t (7.6) | 7.14, t (8.0) | 6.96, t (7.4) | 7.18, t (8.0) | 7.20, t (8.0) |
| 3.09, d (15.7) | 2.72, m | |||||||
| 7′ | 3.70, t (5.3) | 3.35, t (5.0) | 5.76, ddd (10.2, 3.5, 1.5) | 7.29, t (7.6) | 7.17, t (8.0) | 7.04, t (7.4) | 7.20, t (8.0) | 7.20, t (8.0) |
| 8′ | 4.00, brdd (5.3, 4.0) | 3.83, ddd (5.0, 4.0, 3.2) | 4.42, brd (3.5) | 7.38, t (7.6) | 7.14, t (8.0) | 6.96, t (7.4) | 7.18, t (8.0) | 7.20, t (8.0) |
| 9′ | 4.70, dd (7.9, 4.0) | 4.41, dd (8.5, 4.0) | 3.96, brs | 7.47, d (7.6) | 7.14, d (8.0) | 6.89, d (7.4) | 7.08, d (8.0) | 7.20, d (8.0) |
| SMe-2 | 2.04, s | 2.01, s | 1.92, s | 2.20, s | 2.21, s | 2.20, s | 1.19, s | |
| SMe-2′ | 2.30, s | |||||||
| OMe-2 | 2.06, s | |||||||
| OH-8 | 5.40, br | 5.39, d (2.1) | 5.39, br | |||||
| OH-4′ | 6.14, s | 5.76, s | 5.85, s | |||||
| OH-5′ | 4.81, d (4.9) | |||||||
| OH-8′ | 6.20, br | 5.86, d (3.2) | 5.09, br | |||||
| OH-9 | 10.21, s | 9.37, s | ||||||
| NH-1 | 9.84, br s | 9.03, s | 8.68, s | 8.67, s | 8.94, s | |||
| NH-1′ | 8.55, br s | 8.42, s | 8.52, s | 8.58, s | 8.40, s |
| No. | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 159.5, C | 159.5, C | 162.1, C | 165.7, C | 169.2, C | 159.8, C | 159.9, C | 161.4, C | 164.7, C | 166.3, C | 166.0, C | 166.1, C | 165.0, C |
| 2 | 74.7, C | 74.6, C | 74.6, C | 74.0, C | 73.7, C | 72.5, C | 72.4, C | 72.2, C | 67.6, C | 65.2, C | 67.2, C | 68.6, C | 87.4, C |
| 3 | 38.3, CH2 | 38.3, CH2 | 38.1, CH2 | 38.3, CH2 | 38.2, CH2 | 34.2, CH2 | 34.3, CH2 | 34.9, CH2 | 36.6, CH2 | 37.1, CH2 | 43.7, CH2 | 44.4, CH2 | 45.1, CH2 |
| 4 | 134.1, C | 134.2, C | 134.0, C | 134.3, C | 134.0, C | 44.9, CH | 44.9, CH | 44.5, CH | 122.3, C | 122.3, C | 135.2, C | 135.3, C | 134.5, C |
| 5 | 119.6, CH | 119.5, CH | 120.0, CH | 119.4, CH | 119.6, CH | 207.7, C | 207.7, C | 208.2, C | 131.7, CH | 130.0, CH | 130.5, CH | 131.2, CH | 131.1, CH |
| 6 | 123.9, CH | 123.8, CH | 123.9, CH | 123.8, CH | 124.0, CH | 34.5, CH2 | 34.5, CH2 | 34.4, CH2 | 119.5, CH | 119.4, CH | 128.2, CH | 128.3, CH | 128.3, CH |
| 7 | 131.0, CH | 130.9, CH | 131.0, CH | 130.9, CH | 131.4, CH | 26.4, CH2 | 26.4, CH2 | 26.2, CH2 | 128.6, CH | 127.7, CH | 127.1, CH | 127.1, CH | 127.1, CH |
| 8 | 73.9, CH | 74.0, CH | 74.1, CH | 74.2, CH | 74.3, CH | 65.5, CH | 65.9, CH | 64.8, CH | 115.7, CH | 115.2, CH | 128.2, CH | 128.3, CH | 128.3, CH |
| 9 | 69.3, CH | 69.3, CH | 69.2, CH | 68.3, CH | 68.0, CH | 66.5, CH | 66.5, CH | 66.5, CH | 156.0, C | 155.6, C | 130.5, CH | 131.2, CH | 131.1, CH |
| 1′ | 165.9, C | 167.0, C | 165.2, C | 168.1, C | 167.2, C | 164.1, C | 165.1, C | 163.1, C | 160.9, C | 165.6, C | 167.5, C | 167.0, C | 167.6, C |
| 2′ | 70.1, C | 69.2, C | 72.8, C | 71.7, C | 72.8, C | 70.1, C | 69.2, C | 73.4, C | 125.8, C | 65.5, C | 81.9, C | 82.7, C | 82.8, C |
| 3′ | 44.8, CH2 | 43.1, CH2 | 53.3, CH2 | 34.6, CH2 | 31.7, CH2 | 44.6, CH2 | 42.9, CH2 | 54.1, CH2 | 115.3, CH | 43.5, CH2 | 44.8, CH2 | 44.3, CH2 | 44.8, CH2 |
| 4′ | 45.3, CH | 39.1, CH | 46.1, CH | 44.4, CH | 46.4, CH | 45.4, CH | 39.1, CH | 77.5, C | 133.5, C | 135.5, C | 135.1, C | 135.8, C | 135.7, C |
| 5′ | 207.9, C | 61.5, CH | 205.6, C | 207.9, C | 196.6, C | 208.1, C | 61.6, CH | 43.8, CH2 | 129.8, CH | 130.8, CH | 130.4, CH | 131.1, CH | 131.2, CH |
| 6′ | 42.1, CH2 | 37.3, CH2 | 45.7, CH | 34.3, CH2 | 128.6, CH | 42.1, CH2 | 37.3, CH2 | 127.7, CH | 129.2, CH | 128.4, CH | 128.1, CH | 128.3, CH | 128.3, CH |
| 7′ | 41.8, CH | 42.4, CH | 41.5, CH2 | 26.4, CH2 | 152.7, CH | 41.8, CH | 42.4, CH | 128.9, CH | 128.6, CH | 127.2, CH | 126.7, CH | 127.2, CH | 127.1, CH |
| 8′ | 66.1, CH | 68.0, CH | 62.1, CH | 64.1, CH | 72.7, CH | 65.6, CH | 67.5, CH | 62.4, CH | 129.2, CH | 128.4, CH | 128.1, CH | 128.3, CH | 128.3, CH |
| 9′ | 60.3, CH | 57.6, CH | 63.5, CH | 65.5, CH | 69.1, CH | 60.2, CH | 57.6, CH | 66.0, CH | 129.8, CH | 130.8, CH | 130.4, CH | 131.1, CH | 131.2, CH |
| SMe-2 | 14.5, CH3 | 14.5, CH3 | 14.3, CH3 | 14.5, CH3 | 14.6, CH3 | 14.6, CH3 | 14.6, CH3 | 14.2, CH3 | 13.4, CH3 | 13.9, CH3 | 13.5, CH3 | 11.4, CH3 | |
| SMe-2′ | 15.0, CH3 | 15.0, CH3 | 14.0, CH3 | ||||||||||
| MeO-2 | 49.1, CH3 |
ε) 204 (4.19) nm; IR (KBr) νmax 3392, 2923, 1707, 1653, 1420, 1324 cm−1; 1H and 13C NMR data, see Tables 3 and 4; HRESIMS m/z 441.1151 [M + H]+ (calcd for C19H25N2O6S2, 441.1154).ε) 206 (4.16) nm; IR (KBr) νmax 3363, 2923, 1703, 1648, 1414, 1241 cm−1; 1H and 13C NMR data, see Tables 3 and 4; HRESIMS m/z 439.1000 [M + H]+ (calcd for C19H23N2O6S2, 439.0998).ε) 202 (4.09), 300 (3.74) nm; IR (KBr) νmax 3734, 2921, 1670, 1448, 1417, 1375, 1237 cm−1; 1H and 13C NMR data, see Tables 3 and 4; HRESIMS m/z 355.1120 [M + H]+ (calcd for C19H19N2O3S, 355.1116).ε) 203 (4.19), 276 (2.83) nm; IR (KBr) νmax 3180, 2921, 1838, 1663, 1847, 1435, 1369, 1228 cm−1; 1H and 13C NMR data, see Tables 3 and 4; HRESIMS m/z 425.0965 [M + Na]+ (calcd for C20H22N2O3S2Na, 425.0970).ε) 202 (4.11) nm; IR (KBr) νmax 3412, 3292, 3207, 1671, 1495, 1436, 1242, 1109 cm−1; 1H and 13C NMR data, see Tables 3 and 4; HRESIMS m/z 355.1112 [M − H]− (calcd for C19H19N2O3S, 355.1116).ε) 202 (4.01) nm; IR (KBr) νmax 3275, 3204, 1672, 1496, 1453, 1437, 1403, 1307, 1240 cm−1; 1H and 13C NMR data, see Tables 3 and 4; HRESIMS m/z 355.1120 [M − H]− (calcd for C19H19N2O3S, 355.1116).ε) 205 (4.08) nm; IR (KBr) νmax 3430, 3289, 3206, 1668, 1495, 1435, 1405, 1110 cm−1; 1H and 13C NMR data, see Tables 3 and 4; HRESIMS m/z 339.1342 [M − H]− (calcd for C19H19N2O4, 339.1345).2.4. Preparation of (R)-MPA and (S)-MPA esters of 4
Compound 4 (2.0 mg for each) was dissolved in CHCl3 (600 μL) and (R)-MPA (3.0 mg), DMAP (2.8 mg) and DCC (2.5 mg) were added. The mixture was kept at room temperature for 12 h. The reaction products were purified by silica gel CC eluted with PE:acetone (5:1) to get bis-(R)-MPA ester 4a (1.8 mg). In an identical protocol, bis-(S)-MPA ester of 4b (2.1 mg) was obtained from 4.
2.5. X-ray single crystal diffraction
Eutypellazine A (1) was obtained colorless crystal from MeOH/H2O (50:1) using the vapor diffusion method. The monoclinic crystal (0.06 × 0.04 × 0.03) was measured on Bruker D8 Advance single crystal X-ray diffractometer with Cu-Kα radiation at 104.7 K. Crystal data of 1: empirical formula C19H22N2O6S2, M = 438.51; space group P21, unit cell dimensions a = 7.6255 (4) Å, b = 27.2984(13) Å, c = 9.2094(5) Å, α = γ = 90.00°, β = 90.415(5)°, V = 1917.01(17) Å, Z = 4, Dcalcd = 1.1519 mg m−3, μ = 2.886 mm−1, F(000) = 920; a total of 13001 reflections were collected in the range of 6.48° < 2θ < 142.48°, of which 7261 independent reflections [R(int) = 0.0486 (inf-0.9 Å)] were used for the analysis. The structure was solved by the direct methods with the SHELXL-97 program and refined using full-matrix least-squares difference Fourier techniques. The final R indexes [all data] gave R1 = 0.0484, wR2 = 0.1142 and the Flack parameter = −0.004(14). Crystallographic data of 1 have been deposited in the Cambridge Crystallographic Data Center (deposition number CCDC 1416589†).
Eutypellazine E (5) was obtained colorless crystal from MeOH using the vapor diffusion method. The orthorhombic crystal (0.30 × 0.25 × 0.05) was measured on Bruker D8 Advance single crystal X-ray diffractometer with Cu-Kα radiation at 99.9 K. Crystal data of 5: empirical formula C20.21H22.84N2O5.40S2, M = 444.37; space group P212121, unit cell dimensions a = 14.08776(18) Å, b = 15.8916(19) Å, c = 27.9520(4) Å, α = β = γ = 90.00°, V = 6257.82(14) Å, Z = 12, Dcalcd = 1.1415 mg m−3, μ = 2.639 mm−1, F(000) = 2800; a total of 42403 reflections were collected in the range of 6.40° < 2θ < 142.14°, of which 11963 independent reflections [R(int) = 0.0396 (inf-0.9 Å)] were used for the analysis. The structure was solved by the direct methods with the SHELXL-97 program and refined using full-matrix least-squares difference Fourier techniques. The final R indexes [all data] gave R1 = 0.0463, wR2 = 0.1175 and the Flack parameter = 0.015(12). Crystallographic data of 5 have been deposited in the Cambridge Crystallographic Data Center (deposition number CCDC 1416590†).
Upon crystallization from MeOH–H2O (100:1) using the vapor diffusion method, colorless crystals were obtained for eutypellazine F (6). The orthorhombic crystal (0.40 × 0.35 × 0.25) was measured on Bruker D8 Advance single crystal X-ray diffractometer with Cu-Kα radiation at 101.8 K. Crystal data of 6: empirical formula C19H24N2O7S2, M = 456.52; space group P212121, unit cell dimensions a = 9.5541(9) Å, b = 13.1555(17) Å, c = 15.7112(6) Å, α = β = γ = 90.00°, V = 1974.7(3) Å, Z = 4, Dcalcd = 1.536 mg m−3, μ = 2.864 mm−1, F(000) = 960; a total of 6784 reflections were collected in the range of 8.76° < 2θ < 141.64°, of which 3711 independent reflections [R(int) = 0.0199 (inf-0.9 Å)] were used for the analysis. The structure was solved by the direct methods with the SHELXL-97 program and refined using full-matrix least-squares difference Fourier techniques. The final R indexes [all data] gave R1 = 0.0300, wR2 = 0.0772 and the Flack parameter = 0.007(13). Crystallographic data of 6 have been deposited in the Cambridge Crystallographic Data Center (deposition number CCDC 1416591†).
Eutypellazine G (7) was obtained colorless crystal from MeOH–H2O (50:1) using the vapor diffusion method. The monoclinic crystal (0.15 × 0.15 × 0.10) was measured on Bruker D8 Advance single crystal X-ray diffractometer with Cu-Kα radiation at 103.3 K. Crystal data of 7: empirical formula C20H30N2O8S2, M = 490.58; space group P212121, unit cell dimensions a = 9.16165(19) Å, b = 9.5804(2) Å, c = 25.5833(6) Å, α = β = γ = 90.00°, V = 2245.52(9) Å, Z = 4, Dcalcd = 1.451 mg m−3, μ = 2.589 mm−1, F(000) = 1040; a total of 7719 reflections were collected in the range of 6.92° < 2θ < 142.36°, of which 4256 independent reflections [R(int) = 0.0280 (inf-0.9 Å)] were used for the analysis. The structure was solved by the direct methods with the SHELXL-97 program and refined using full-matrix least-squares difference Fourier techniques. The final R indexes [all data] gave R1 = 0.0414, wR2 = 0.1015 and the Flack parameter = −0.003(18). Crystallographic data of 7 have been deposited in the Cambridge Crystallographic Data Center (deposition number CCDC 1416592†).
Eutypellazine H (8) was obtained colorless crystal from MeOH using the vapor diffusion method. The orthorhombic crystal (0.60 × 0.25 × 0.25) was measured on Bruker D8 Advance single crystal X-ray diffractometer with Cu-Kα radiation at 102.4 K. Crystal data of 8: empirical formula C19H22N2O6S2, M = 438.51; space group P212121, unit cell dimensions a = 8.27744(17) Å, b = 10.60684(20) Å, c = 21.3739(4) Å, α = β = γ = 90.00°, V = 1876.57(6) Å, Z = 4, Dcalcd = 1.552 mg m−3, μ = 2.948 mm−1, F(000) = 920; a total of 7441 reflections were collected in the range of 8.28° < 2θ < 142.24°, of which 3517 independent reflections [R(int) = 0.0238 (inf-0.9 Å)] were used for the analysis. The structure was solved by the direct methods with the SHELXL-97 program and refined using full-matrix least-squares difference Fourier techniques. The final R indexes [all data] gave R1 = 0.0330, wR2 = 0.0827 and the Flack parameter = −0.008(13). Crystallographic data of 8 have been deposited in the Cambridge Crystallographic Data Center (deposition number CCDC 1416593†).
2.6. Anti-HIV bioassay
293T cells were cultured at 37 °C in a 5% CO2 humidified atmosphere and split twice a week. The vector pNL4.3-Luc was generated by cloning the luciferase gene (Thermo Fisher Scientific) in the HIV-1 proviral clone pNL4.3. Plasmid pNL4.3-Ren was generated by cloning the renilla gene (Promega) in the Luc site of pNL4.3-Luc (NIH AIDS Reagent Program). Infectious supernatants were obtained from Ca3(PO4)2 transfection on 293T cells of plasmid pNL4.3-Ren. These supernatants were used to infect cells in the presence or absence of the compounds to be evaluated. Anti-HIV activity quantification was performed 48 h postinfection. Briefly, cells were lysed with 100 μL of buffer. Relative luminescence units (RLUs) were obtained in a luminometer (Berthold Detection Systems) after the addition of substrate to cell extracts. Viability was performed in parallel treated cells with the same concentrations of compound. After 48 h, cell viability was evaluated with the CellTiter Glo (Promega) assay system following the manufacturer's specifications. Inhibitory concentrations 50% (IC50) and cytotoxic concentrations 50% (CC50) were calculated using GraphPad Prism software.2.7. In vitro latent HIV reactivating assay
For flow cytometry-bases screening, the J-Lat A2 cell line containing an integrated HIV-1 long terminal repeat (LTR) luciferase reporter construct but expressing no Tat was used. The J-Lat A2 cells were grown in Dulbecco's modification of eagle's medium (DMEM) (Thermo Fisher Scientific) supplemented with 10% fetal bovine serum (Aleken Biologicals), 2 mM L-glutamine (Invitrogen), 4500 mg mL−1 glucose, and antibiotic solution (0.2% kanamycin, 0.2% streptomycin, and 0.12% penicillin) (Sigma Aldrich) at 37 °C under 5% CO2 atmosphere. The DMEM replaced every date until cells grew to 90% confluency from explants and then using Tyrisin (Invitrogen) to dissociate them. These cultures of J-Lat A2 cells were then dispensed into 24-well plates at 1 × 105 cells per mL per well 24 h prior to the test compounds treatment and incubated at 37 °C under 5% CO2 condition. The test compounds (100 μM for each), two positive controls prostratin (5 μM), and SAHA (2.5 μM), and negative control DMSO (10 μL) were added to per well. After 48 h incubation, cells were rinsed and resuspended by phosphate-buffered saline (PBS) (Thermo Fisher Scientific) and then the flow cytometry was used to detect EGFP-positive cells. Cytotoxicities of all compounds were measured by the Promega CellTiter kit (CellTiter-Glo® Reagent) following the manufacturer's instructions in 96-well plates incubated with test compounds at 37 °C for 48 h.3. Results and discussion
3.1. Structure elucidation of new compounds
The EtOAc extract of the fermentation broth of Eutypella sp. MCCC 3A00281 was examined by the HPLC-ESIMS, which featured an array of TDKP-based derivatives. Detailed chromatographic separation including semipreparative HPLC purification resulted in the isolation of 15 TDKP analogues including 13 new compounds (Fig. 1).Eutypellazine A (1) was isolated as white monoclinic crystals. Its molecular formula was established as C19H20N2O5S2 by the HRESIMS (m/z 421.0887 [M + H]+) and NMR data. The IR absorptions at 3370, 1713 and 1648 cm−1 suggested the presence of hydroxy and carbonyl functionalities. The 1H and 13C NMR data (Tables 1 and 2) were characteristic of a diketopiperazine-based derivative, while analyses of 1H–1H COSY, HSQC and HSBC data revealed the presence of a 6/5/6/5/6-membered pentacyclic diketopiperazine skeleton, structurally related to the coexisted epicoccin I.15 The spin system coupled the protons from the olefinic proton H-5 (δH 5.98) to H-9 (δH 4.78), while the HMBC correlations from H-9 to C-2 (δC 74.7), C-3 (δC 38.3), C-4 (δC 134.1), and C-5 (δC 119.6) and from H-5 to C-3 and C-9 (δC 69.3) assigned a dihydroindoline for rings A–B, in which a hydroxy substitution at C-8 (δC 73.9) was deduced by the COSY relationship between H-8 (δH 4.66) and a D2O exchangeable proton OH-8 (δH 5.43). Extensive analyses of the 2D NMR data uncovered rings D–E to be a perhydroindole, in which the location of a ketone group at C-5′ (δC 207.9) and a hydroxy group at C-8′ (δC 66.1) was evident from the HMBC correlations from C-5′ to H2-3′, H-4′, and H2-6′ and the COSY relationship between H-8′ (δH 4.03) and OH-8′ (δH 6.21). In addition, the chemical shifts of a methyl group at δH 2.24 (3H, s)/δC 14.5 were characteristic of a thiomethyl group, which was located at C-2 on the basis of the HMBC correlation between the methyl protons and C-2. The second sulfur element was bonded across C-2′ (δC 70.1) and C-7′ (δC 41.8) to form a thioether bridge on the basis of the HMBC correlation between H-7′ (δH 3.74) and C-2′.
The relative configurations of 1 were established by the coupling constants and the NOE data. The JH-8/H-9 value (13.8 Hz) in association with the NOE interaction between OH-8 and H-9 was indicative of trans orientation of H-8 toward H-9. The observation of the NOE correlations from CH3S (δH 2.24) to H-8 and H-8′, and from H-9′ to OH-8′ and H-4′ assigned the same face of thiomethyl group as H-8 and H-8′, while the cis fusion of rings D and E was deduced by the NOE interactions from H-4′ and H-9′ to OH-8′. The thioether orientation in opposite face to H-9′ was due to the NOE interaction between OH-8′ and H-7′ (Fig. 2). The absolute configurations of the stereogenic centers were determined by the X-ray diffraction experiment, while the Flack parameter (−0.004(14)) using Cu-Kα reflection measurement unambiguously determined 2R, 8S, 9S, 2′R, 4′R, 7′R, 8′R, and 9′S configurations, respectively (Fig. 3).
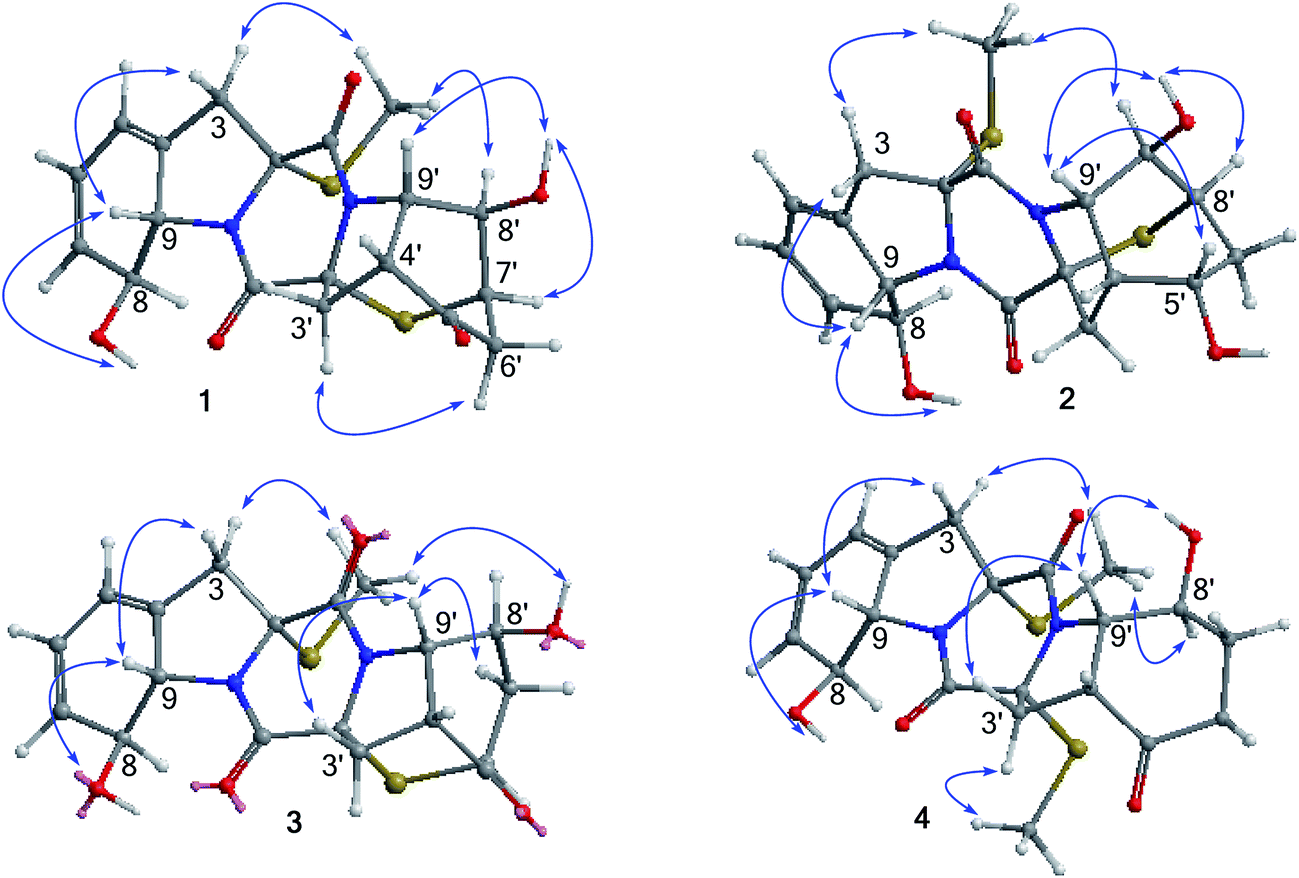 | ||
| Fig. 2 Key NOE correlations of 1–4. | ||
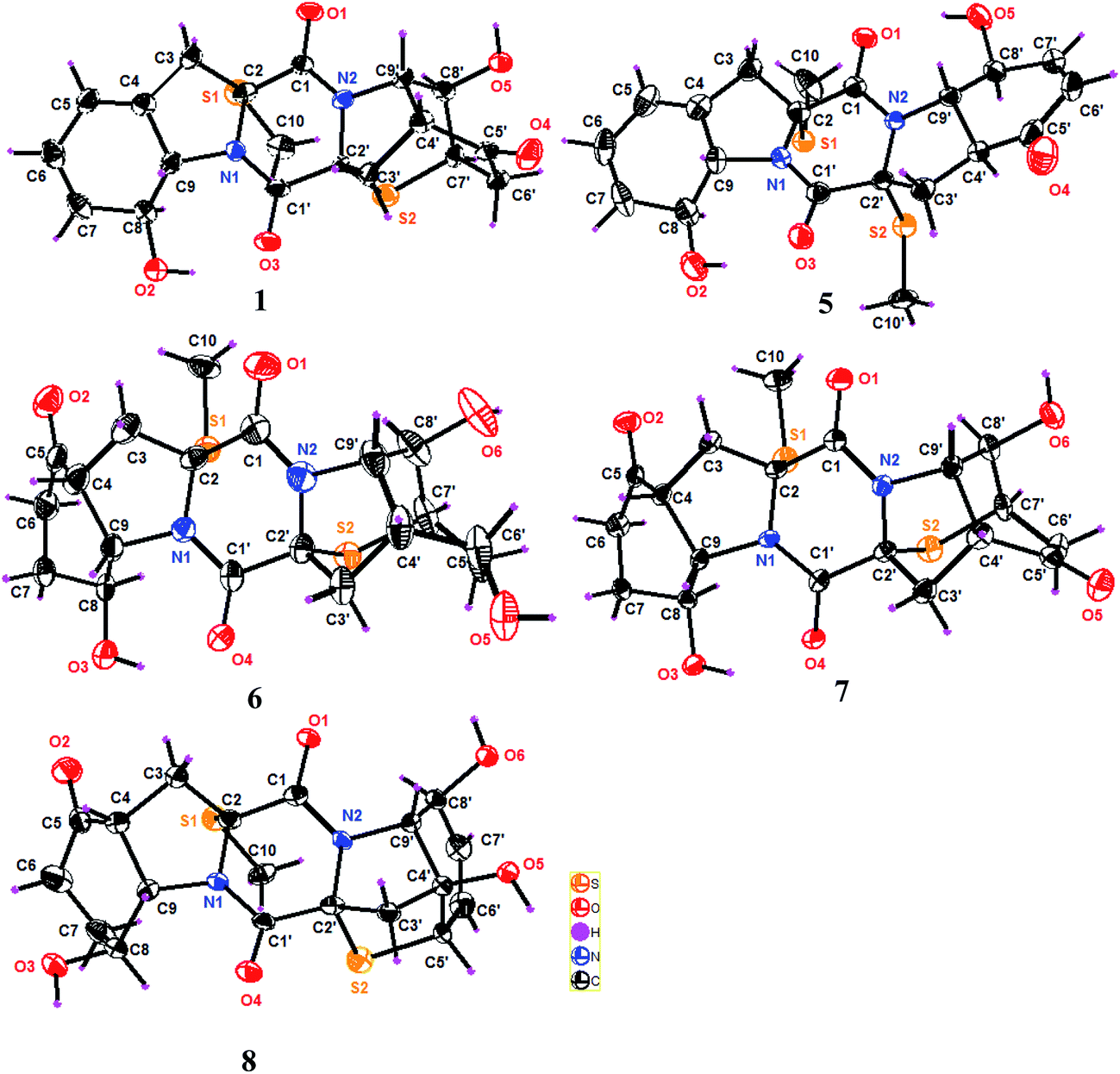 | ||
| Fig. 3 ORTEP plots of the X-ray crystal structures of 1 and 5–8. | ||
Analyses of the 2D NMR data revealed the structure of eutypellazine B (2) closely related to 1. The distinction was observed in ring E, where a ketone at C-5′ of 1 to be replaced by a hydroxy group, as recognized by the COSY relationship between the D2O exchangeable proton at δH 4.85 (OH) and H-5′ (δH 4.31), in addition to the HMBC correlations from the OH proton to C-4′, C-5′ (δC 61.5), and C-6′. Comparison of the NOE data and coupling constants indicated that both 2 and 1 share the same relative configurations in rings A–C, whereas the NOE interaction between H-5′/H-9′ and OH-5′/H-8′ (Fig. 2) assigned the opposite face of OH-5′ and OH-8′. Considering the absolute configuration established for 1 by X-ray data, the similar ECD data such as the positive Cotton effects at 230 and 270 nm which reflected the orientation of the thiomethyl group and thioether assumed both 2 and 1 sharing the same absolute configurations. Thus, the stereogenic center C-5′ in 2 was suggested to have the S configuration.
Eutypellazine C (3) has the same molecular formula as that of 1, as determined by the HRESIMS and NMR data. The 2D NMR data indicated that the partial structure of 3 regarding rings A–C was identical to that of 1, while rings D and E presented as a perhydroindoline unit related to that of 1. The COSY and HMQC data conducted H-6′ (δH 3.64) to be a methine proton instead of H-7′ ring E of 1. This assignment was evident from the COSY relationship from H2-7′ (δH 2.53, 2.69) to H-6′ and H-8′ (δH 4.50). The HMBC correlation between H-6′ and C-2′ confirmed the connection of a thioether bond across C-2′ and C-6′. The closely similar NOE data (Fig. 2) and coupling constants allowed the assignment of the relative configurations of 3 to be the same as those of 1. The positive Cotton effects at 220 and 307 nm and the negative Cotton effect at 255 nm were attributed to 2R configuration, whereas the positive Cotton effect at 270 nm was induced by the π → π* transition of conjugated hexadiene.17 Thus, the remaining stereogenic centers of 3 were assigned as 2R, 8S, 9S, 2′R, 4′R, 6′S, 8′S, and 9′S, respectively.
The molecular formula of eutypellazine D (4) was established as C20H24N2O5S2 by the HRESIMS (m/z 437.1201 [M + H]+) and NMR data. Comparison of the NMR (Tables 2 and 4) and ESIMS data indicated that the structure of 4 closely related to ent-epicoccin G.15 The distinction was attributed to ring A, where the 2D NMR data assigned a 8-hydroxycyclohexadiene with the same moiety as that of 1. Based on the modified Mosher's method,18 compound 4 was esterified by the (R)- and (S)-MPA to form MPA esters 4a and 4b. Calculation of the chemical shift values (ΔδRS = δR − δS) resulted in a S configuration for C-8 and C-8′ (Fig. 4). In combination with the NOE interactions (Fig. 2), the absolute configurations of the remaining chiral centers were determined. These assignments were also supported by the negative Cotton effect at 262 nm, which was in agreement with 2R/2′R configurations for TDKPs bearing two S-methyl groups.17
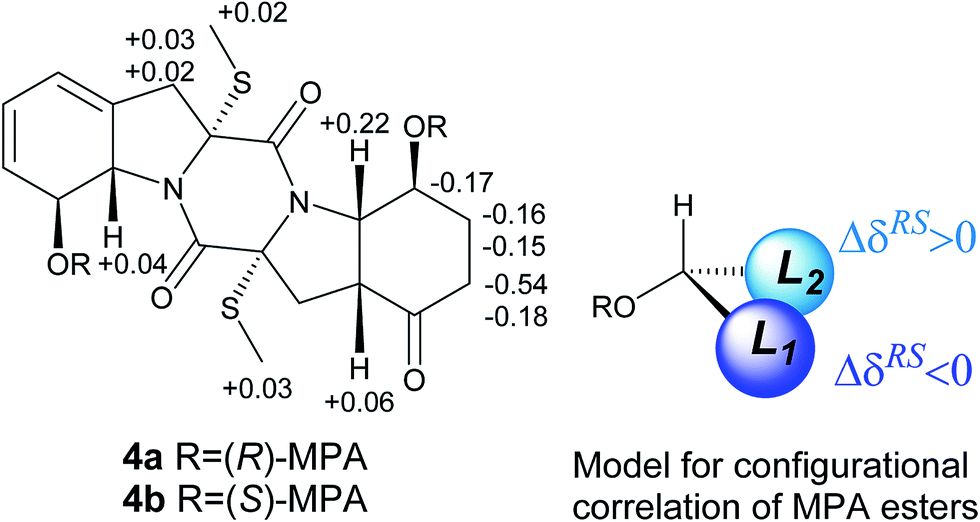 | ||
| Fig. 4 ΔδRS (δR − δS) values of the MPA esters of 4 in CDCl3. | ||
Analyses of the 2D NMR data revealed that eutypellazine E (5) was a 6′,7′-dehydrogenated analogue of 4. This assignment was evident from the similar NMR data of both 4 and 5, with the exception of the presence of two olefinic protons at δH 6.06 (H-6′) and δH 6.91 (H-7′) in addition to the HMBC correlations from H-6′ and H-7′ to C-5′ and C-8′. The NOE relationships revealed the same relative configuration in rings A–C of both 4 and 5. However, the NOE interactions between H-9′/OH-8′ and H-4′/H-8, in association with JH-4′/H-9′ value (13.3 Hz) conducted a trans fusion of rings D and E. This assignment was further supported by the X-ray single crystal diffraction data using Flack parameter (0.015(12)) (Fig. 3), which deduced the absolute configurations to be 2R, 8S, 9S, 2′R, 4′S, 8′S, and 9′S, respectively.
Eutypellazine F (6) has a molecular formula of C19H22N2O6S2 as determined by the HRESIMS and NMR data. Comparison of the NMR data in association with the 2D NMR data resulted in the partial structure regarding rings C–E to be the same as that of 1. The distinction was recognized in ring A, where the location of a ketone group at C-5 was based on the COSY relationships of the spin system from H2-3 to H-9, and the HMBC correlations from C-5 to H2-3, H-4, H2-6, H2-7, and H-9. The relative configurations of 6 were determined by the NOE relationships, while the absolute configurations were determined by the single crystal X-ray diffraction data using the Flack parameter (0.007(13)) as obtained by Cu-Kα diffraction to assign 2R, 4R, 8S, 9S, 2′R, 4′R, 7′R, 8′R, and 9′S, respectively (Fig. 3).
Comparison of the NMR data (Tables 3 and 4) indicated that the partial structure of rings A–C in eutypellazine G (7) is the same as that of 6, while the second partial structure in rings C-E of 7 was identical to that of 2. The absolute configurations of 7 were unequivocally assigned as 2R, 4R, 8S, 9S, 2′R, 4′R, 5′S, 7′R, 8′R, and 9′S, respectively, on the basis of the Flack parameter −0.003(18), which was obtained by the X-ray Cu-Kα crystallographic experiment.
The 2D NMR data assigned eutypellazine H (8) to be a thiodiketopiperazine with the partial structure of rings A–C being the same as that of 7, whereas the structure of rings C-E agreed with that of epicoccin I.15 The absolute configurations 8 were determined by the single crystal X-ray crystallographic data with the Flack parameter −0.008(13), as obtained by the X-ray Cu-Kα experiment, indicating 2R, 4R, 8S, 9S, 2′R, 4′S, 5′S, 8′S, and 9′R, respectively.
The molecular formula of eutypellazine I (9) was established as C19H18N2O3S on the basis of the HRESIMS (m/z 355.1120, [M + H]+) and NMR data. The 1H and 13C NMR data (Tables 3 and 4) of 9 were closely related to those of coexisted emethacin A,19 whereas the aromatic ring A presented a phenolic proton and an ABCD spin system instead of the mono-substituted aromatic ring of the known counterpart. The aromatic spin system among H-5 (δH 7.12, d, J = 7.3 Hz), H-6 (δH 6.70, t, J = 7.3 Hz), H-7 (δH 7.04, t, J = 7.5 Hz), and H-8 (δH 6.80, d, J = 8.0 Hz), in association with the HMBC correlations from H2-3 to C-9 (δC 156.0), clarified 9 to be a 9-hydroxyemethacin A. This assignment was supported by the negative sign and the similar value of the specific rotation of both 9 and emethacin A.
Analyses of the NMR and HRESIMS data conducted eutypellazine J (10) to be a 9-hydroxyemethacin B, while the distinction was attributed to the aromatic ring A where an ABCD spin system among H-5 (δH 6.30), H-6 (δH 6.25), H-7 (δH 6.89), and H-8 (δH 6.67) and the HMBC correlations from H2-3 (δH 3.01, 3.09) to C-4 (δC 122.3), C-5 (δC 130.0), and C-9 (δC 155.6) were observed in the 1H–1H COSY and HMBC spectra. The similar magnitude and the same sign of the specific rotation of 10 ([α]25D −128, MeOH) and emethacin B ([α]25D −168, CHCl3)19 assumed 10 possessing 2R/2′R configurations.
Comparison of the NMR data (Tables 3 and 4) indicated the structure of eutypellazine K (11) to be closely related to emethacin B. The distinction was found by the absence of a thiomethyl group and the deshielded C-2′ (δC 81.9) in the NMR spectra of 11. In addition, a D2O exchangeable proton (δH 6.60, s) showed the HMBC correlations with C-1′ (δC 167.5), C-2′, and C-3′ (δC 44.8), confirming C-2′ of 11 to be substituted by a hydroxy group to replace a thiomethyl group of emethacin B. The NOE interaction between CH3S and OH-2′ assigned the spatial approximation of both functional groups. In addition, the negative specific rotation of 11 ([α]25D −165, MeOH) which was contributed by the chiral centers at C-2 and C-2′ and was comparable to that of 10, suggested 2R/2′R configurations.
Eutypellazine L (12) was determined to have a planer structure to be the same as that of 11, based on the 2D NMR and HRESIMS data. The distinction was observed by the deshielded C-2′ (δC 82.7) and the lower magnitude of the specific rotation ([α]25D −80) in comparison with those of 11. Since the hydroxylated and methoxylated diatretol with 2S and 2′S configurations exhibited positive specific rotation ([α]25D +42, MeOH),20 the lowering value of the specific rotation of 11 was derived by the 2′S contribution.
Eutypellazine M (13) has a molecular formula of C19H20N2O4 as provided by the HRESIMS (m/z 339.1342 [M − H]−) and NMR data. Analyses of the NMR data revealed that 13 structurally related to 11 with the exception of the substitution at C-2, in which a methoxy group (δH 2.06/δC 49.1) instead of a thiomethyl group was recognized to position at C-2. This assignment was supported by the HMBC correlation between the methoxy protons and C-2 (δC 87.4). The remarkable shielded protons of MeO (δH 2.06) was due to the location of the MeO group under the shielded zone of the nucleus, while the NOE interaction between MeO and OH-2′ (δH 5.85) clarified the same orientation of both MeO and OH-2′. Thus, the negative specific rotation of 13 ([α]25D −72) was in agreement with 2R configuration.
The known compounds were determined as epicoccin I15 and epicoccin A,6 on the basis of comparison their spectra data with those reported in the literatures.
3.2. Anti-HIV assay
The isolated compounds were tested for their inhibitory effects against human immunodeficiency virus type 1 (HIV-1) replication. Anti-HIV screening was performed by the pNL4.3.Env-.Luc co-transfected 293T cells. Before bioassay, all compounds were tested for their cytotoxicity toward 293T cells, while the tested compounds showed low cytotoxicity with CC50 > 100 μM. Compounds in a single dose of 20 μM showing more than 50% of HIV-1 inhibition were subjected to IC50 measurement. As shown in Table 5, most of the tested compounds exerted inhibitory effects, while compound 5 showed the most inhibitory effect. A preliminary structure–activity relationship study revealed that the analogues with thiomethyl group at C-2/C-2′ (4 and 5) showed more active than those with sulfide bridge (1–3, 6–8) in the pentacyclic thiodiketopiperazines. Comparison of the inhibitory effect between 4 and 5 revealed a double bond at C-6′/C-7′ in 5 enhancing the activity. In regard to compounds 9–13, the analogues with thiomethyl group at C-2/C-2′ (10) showed more effect than those with hydroxyl substitution (11–12), whereas the analogue with methoxy/hydroxyl substitution at C-2/C-2′ dramatically reduced the activity.| Compounds | IC50 + SD (μM) | CC (μM) |
|---|---|---|
| a IC50 (inhibitory concentration 50%) and CC50 (cytotoxic concentration 50%). | ||
| 1 | 14.8 + 1.2 | >100 |
| 2 | 11.5 + 0.8 | >100 |
| 3 | 10.7 + 1.3 | >100 |
| 4 | 8.7 + 0.5 | >100 |
| 5 | 3.2 + 0.4 | >100 |
| 6 | 16.6 + 0.5 | >100 |
| 7 | 18.2 + 1.3 | >100 |
| 8 | 13.3 + 0.6 | >100 |
| 9 | 6.7 + 2.1 | >100 |
| 10 | 4.9 + 1.1 | >100 |
| 11 | 5.8 + 0.7 | >100 |
| 12 | 5.9 + 0.9 | >100 |
| 13 | >20 | >100 |
| EFV | 0.1 | >100 |
In addition, compound 10 and epicoccin A showed the reactivation on latent HIV-1 transcription with dose-dependent manner, whereas the remaining compounds exerted inactive in a dose of 100 μM. As shown in Fig. 5, compound 10 and epicoccin A showed the reactivation activities at 80 μM, which were comparable to the positive controls prostratin (5 μM) and SAHA (2.5 μM). Latent HIV reservoirs are the primary hurdle to eradicate human immunodeficiency virus by the highly active antiretroviral therapy (HAART), because the residual provirus harbored in cellular reservoirs quickly rebound when treatment is interrupted.21,22 One promising strategy to expunge HIV-1 infection is to reactive latent viral reservoirs in combination with HAART.23,24 Thus, finding new latency reactivating agents with noncytotoxic, clinically effective treatment of HIV infections is urgently needed.
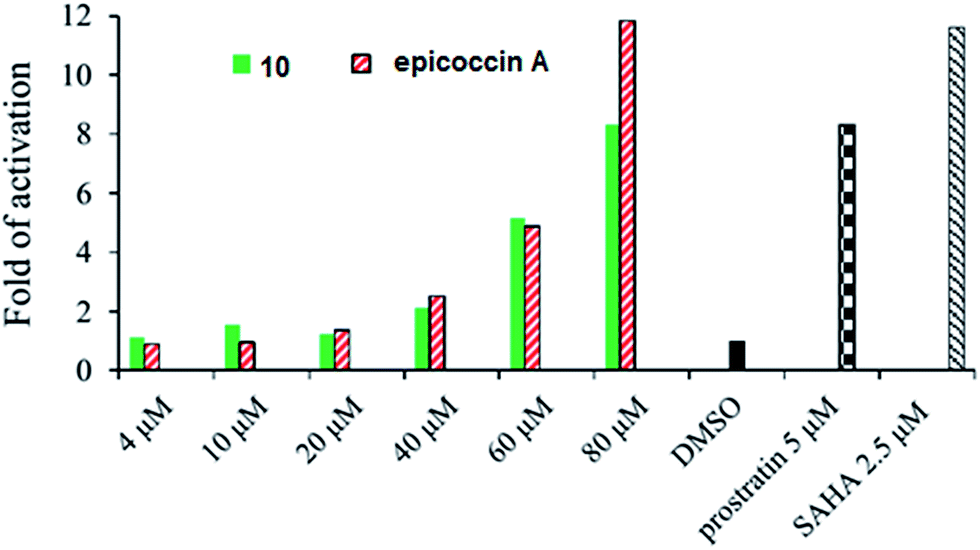 | ||
| Fig. 5 Reactivating effects of 10 and epicoccin A on latent HIV-1 expression in J-Lat A2 cell. Positive control: prostratin and SAHA; negative control: DMSO. The compounds were detected by flow cytometry for the EGFP-positive cells. The activity detected in cells treated with DMSO was set to 1 and the values shown in figure were mean from two independent experiments. | ||
4. Conclusion
Previous investigation of Eutypella fungi inhabited in terrestrial and marine habitats have uncovered diverse terpenoids25–28 and cytosporin-related compounds.29,30 This is the first report of thiodiketopiperazine-type alkaloids to be derived from Eutypella sp., suggesting the presence of diverse biogenetic synthetic pathways in a fungal strain and the distinct synthetic pathway to be activated from a fungal strain inhabited in different location. In addition, the present work not only enriched the numbers of new analogues in thiodiketopiperazine family, also extended the pharmaceutical usage of thiodiketopiperazines to anti-HIV activities. The potent anti-HIV effects of the new compounds suggested that they may serve as new scaffolds for structural modification to be developed as a group of new anti-HIV candidates. The reactivation on latent HIV-1 expression induced by 10 and epicoccin A suggested that both compounds may be applied as new latency reactivating agents, which are rarely found from natural products.Acknowledgements
This work was supported by the grants of National High Technology and Science 973 program (2015CB755906), and NSFC (81630089, 41376127).References
- R. W. Timothy and M. W. Robert, Nat. Prod. Rep., 2014, 31, 1376–1404 RSC.
- G. C. Horace, H. Karst, H. L. Robert and H. A. Byron, Agric. Biol. Chem., 1991, 55, 2037–2042 Search PubMed.
- Y. Zhang, S. Liu, Y. Che and X. Liu, J. Nat. Prod., 2007, 70, 1522–1525 CrossRef CAS PubMed.
- H. Guo, B. Sun, H. Gao, X. Chen, S. Liu, X. Yao, X. Liu and Y. Che, J. Nat. Prod., 2009, 72, 2115–2119 CrossRef CAS PubMed.
- L. Meng, X. Li, C. Lv, C. Huang and B. Wang, J. Nat. Prod., 2014, 77, 1921–1927 CrossRef CAS PubMed.
- Y. Sun, K. Takada, Y. Takemoto, M. Yoshida, Y. Nogi, S. Okada and S. Matsunaga, J. Nat. Prod., 2012, 75, 111–114 CrossRef CAS PubMed.
- C. Guo, H. Yeh, Y. Chiang, J. F. Sanchez, S. Chang, K. S. Bruno and C. C. C. Wang, J. Am. Chem. Soc., 2013, 135, 7205–7213 CrossRef CAS PubMed.
- H. S. Daniel, H. Andreas, H. Thorsten, A. B. Axel and H. Christian, J. Am. Chem. Soc., 2014, 136, 11674–11679 CrossRef PubMed.
- R. Tan, P. R. Jensen, P. G. Williams and W. Fenical, J. Nat. Prod., 2004, 67, 1374–1382 CrossRef CAS PubMed.
- F. Kong, Y. Wang, P. Liu, T. Dong and W. Zhu, J. Nat. Prod., 2014, 77, 132–137 CrossRef CAS PubMed.
- C. Takahashi, Y. Takai, Y. Kimura, A. Numata, N. Shigematsu and H. Tanaka, Phytochemistry, 1995, 38, 155–158 CrossRef CAS PubMed.
- L. H. Meng, P. Zhang, X. M. Li and B. G. Wang, Mar. Drugs, 2015, 13, 276–287 CrossRef CAS PubMed.
- M. Saka, S. Palasarn, P. Rachtawee, S. Vimuttipong and P. Kongsaeree, Org. Lett., 2005, 7, 2257–2260 CrossRef PubMed.
- H. J. Lee, J. H. Lee, B. Y. Hwang, H. S. Kim and J. J. Lee, Arch. Pharmacal Res., 2001, 25, 397–401 CrossRef.
- J. Wang, G. Ding, L. Fang, J. Dai, S. Yu, Y. Wang, X. Chen, S. Ma, J. Qu, S. Xu and D. Du, J. Nat. Prod., 2010, 73, 1240–1249 CrossRef CAS PubMed.
- J. Wang, W. He, X. Qin, X. Wei, X. Tian, L. Liao, S. Liao, B. Yang, Z. Tu, B. Chen, F. Wang, X. Zhou and Y. Liu, RSC Adv., 2015, 5, 68736–68742 RSC.
- J. Wang, N. Jiang, J. Ma, S. Yu, R. Tan, J. Dai, Y. Si, G. Ding, S. Ma, J. Qu, L. Fang and D. Du, Tetrahedron, 2013, 69, 1195–1201 CrossRef CAS.
- F. Freire, F. Calderon, J. M. Seco, A. Fernandez-Mayoralas, E. Quinoa and R. Riguera, J. Org. Chem., 2007, 72, 2297–2301 CrossRef CAS PubMed.
- N. Kawahara, K. Nozawa, S. Nakajima, M. Yamazaki and K. Kawai, Heterocycles, 1989, 29, 397–402 CrossRef CAS.
- A. Alberto, C. Silvia, N. Gianluca, V. M. Stefano and V. P. Orso, Liebigs Ann., 1996, 1875–1877 Search PubMed.
- B. León, G. Navarro, B. J. Dickey, G. Stepan, A. Tsai, G. S. Jones, M. E. Morales, T. Barnes, S. Ahmadyar, M. Tsiang, R. Geleziunas, T. Cihlar, N. Pagratis, Y. Tian, H. Yu and R. G. Linington, Org. Lett., 2015, 17, 262–265 CrossRef PubMed.
- A. Jordan, D. Bisgrove and E. Verdin, EMBO J., 2003, 22, 1868–1877 CrossRef CAS PubMed.
- E. J. Mejia, S. T. Loveridge, G. Stepan, A. Tsai, G. S. Jones, T. Barnes, K. N. White, M. Drašković, K. Tenney, M. Tsiang, R. Geleziunas, T. Cihlar, N. Pagratis, Y. Tian, H. Yu and P. Crews, J. Nat. Prod., 2014, 77, 618–624 CrossRef CAS PubMed.
- Z. Li, J. Guo, Y. Wu and Q. Zhou, Nucleic Acids Res., 2013, 41, 277–287 CrossRef CAS PubMed.
- W. Pongcharoen, V. Rukachaisirikul, S. Phongpaichit, N. Rungjindamai and J. Sakayaroj, J. Nat. Prod., 2006, 69, 856–858 CrossRef CAS PubMed.
- L. Sun, D. Li, M. Tao, Y. Chen, F. Dan and W. Zhang, Mar. Drugs, 2012, 10, 539–550 CrossRef CAS PubMed.
- M. Isaka, S. Palasarn, W. Prathumpai and P. Laksanacharoen, Chem. Pharm. Bull., 2011, 59, 1157–1159 CrossRef CAS PubMed.
- X. Lu, J. Liu, X. Liu, Y. Gao, J. Zhang, B. Jiao and H. Zheng, J. Antibiot., 2014, 67, 171–174 CrossRef CAS PubMed.
- M. Isaka, S. Palasarn, S. Lapanun, R. Chanthaket, N. Boonyuen and S. Lumyong, J. Nat. Prod., 2009, 72, 1720–1722 CrossRef CAS PubMed.
- M. L. Ciavatta, M. P. Lopez-Gresa, M. Gavagnin, R. Nicoletti, E. Manzo, E. Mollo, Y. Guo and G. Cimino, Tetrahedron, 2008, 64, 5365–5369 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1416589–1416593. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra05774a |
| This journal is © The Royal Society of Chemistry 2017 |