
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Self-assembling characteristics of amphiphilic zwitterionic brush random copolymers at the air–water interface
Jonghyun Kima,
Jin Chul Kimab,
Minh Dinh Phan c,
Heesoo Kim*d,
Kwanwoo Shin*c and
Moonhor Ree*a
c,
Heesoo Kim*d,
Kwanwoo Shin*c and
Moonhor Ree*a
aDepartment of Chemistry, Division of Advanced Materials Science, Polymer Research Institute, Pohang University of Science and Technology, Pohang 37673, Korea. E-mail: ree@postech.edu; Fax: +82-54-279-3399; Tel: +82-54-279-2120
bResearch Centre for Green Fine Chemicals, Chemical Convergence Research Division, Korea Research Institute of Chemical Technology, Ulsan 44412, Republic of Korea
cDepartment of Chemistry, Program of Integrated Biotechnology, Sogang University, Seoul 04107, Republic of Korea. E-mail: kwshin@sogang.ac.kr; Fax: +82-2-701-0967; Tel: +82-2-705-8441
dDepartment of Microbiology, Dongguk University College of Medicine, Gyeongju 38066, Korea. E-mail: hskim@dongguk.ac.kr; Fax: +82-54-770-2447; Tel: +82-54-770-2417
First published on 16th February 2017
Abstract
Self-assembling characteristics of a series of amphiphilic zwitterionic brush random copolymers, poly(oxy(11-(3-sulfonylpropyltrimethylglycinyl)undecylesterthiomethyl)ethylene-co-oxy(n-do-decylthiomethyl)ethylene)s (PECH-DMAPSm where m is the mol% of DMAPS end group), were investigated at an air–water interface by using surface pressure–area isotherms, infrared spectroscopy, and X-ray reflectivity analysis. Interestingly the random polymers (m: 20–60 mol%) always formed Langmuir monolayer structures only rather than any other structures, regardless of the surface pressures. The Langmuir monolayers possessed enhanced lateral ordering together with conformational changes of the backbone and bristles through increasing the surface pressure. The monolayer structures were basically composed of a hydrophobic bristle phase in the air side and a hydrophilic backbone and bristle phase in the water side. The unique, highly ordered Langmuir monolayer structures could be realized by positive, cooperative efforts of several factors such as the compositional balance of hydrophobic and hydrophilic zwitterionic bristles, the air- and water-induced segregations of the bristles and backbone, the lateral ordering capabilities of the alkyl groups and the alkylenyl linkers in the bristles under the assistance of surface pressure, and the relatively strong anchoring power of DMAPS end groups in the water side using their high water affinity due to the hydrophilic zwitterionic nature. Overall, PECH-DMAPSm revealed a very unique self-assembling behavior at the air–water interface, always producing the Langmuir monolayer structure.
Introduction
Biomimicking polymers have gained great attention from academia and industry because of their potential applications as biomaterials in various fields of biomedical science, tissue culture, pharmaceutics, and drug delivery.1,2 Therefore, much effort has been given to develop biomimicking polymers; as a result, a number of biomimicking polymers were reported.1,3–15 They can be classified into two major families: biomolecule-containing polymer brushes or self-assembled monolayers (SAMs) tethered to substrate surfaces1,3–7 and biomolecule-containing graft, comb-like, random and block copolymers.13–15 However, the first family has some serious drawbacks, as follows. Much care is needed for the selection of substrates in the aspect of biocompatibility and reliability. The substrates employed require surface functionalization or activation before tethering of polymers or surface-induced polymerization and SAM formation. The tethering approach of polymers to substrate surfaces is limited by steric hindrance, causing incomplete surface coverage. The SAM approach possesses structural defects due to the non-uniformity and the chemical instability. Therefore, the first family of materials is generally restricted to in vitro applications. In the case of the second family, there are also some disadvantages. A number of reaction steps are generally necessary to synthesize biomolecule-containing polymers. Thus, the development of synthetic schemes with high yields is required. And, it is not assured that the biomolecule groups are exposed to the surface, because detailed structural information has not been given. Their structures and functionalities were mainly investigated in bulk state or fiber form. In applications they are highly demanded to use as a thin film rather than a bulk state or fiber form. Moreover, they very often elicit inflammatory responses, including foreign body responses and fibrous encapsulation,1,2 indicating that their biocompatibility is needed to improve more. Therefore, the development of polymeric biomaterials with high performances still remains in the exploration stage.Recently we have developed a new strategy to develop high performance polymeric biomaterials with significantly enhanced biocompatibility. Our strategy stands on the molecular design and synthesis of chemically well-defined brush (i.e., comb-like) polymers bearing proper biomolecules at the bristle ends which can enable to favorably self-assemble as an organized structure, providing biomolecule-enriched surface. We have successfully demonstrated this strategy and, indeed, newly introduced several polymeric biomaterial systems.16–27 Their thin film morphologies were well investigated, confirming that they self-assembled favorably as horizontally-oriented multibilayer structures with fully extended chain conformation, always providing biomolecule-rich film surface. In fact, these biomaterials can be used in aqueous solutions including buffers or in contact with water or buffers, in addition to the film and bulk state. Thus, it is necessary to investigate their self-assembly behaviors at an air–water interface or in water and buffer solutions.
In this study, we investigated Langmuir film formation characteristics of amphiphilic zwitterionic brush random copolymers, poly(oxy(11-(3-sulfonylpropyltrimethylglycinyl)-undecylesterthiomethyl)ethylene-co-oxy(n-dodecylthiometh-yl)ethylene)s (PECH-DMAPSm, where m is the mol% of DMAPS (i.e., sulfobetaine) end group) at an air–water interface using surface pressure–area isotherm, infrared spectroscopy, and X-ray reflectivity analysis. PECH-DMAPS100 was found to dissolve in water, failed to form Langmuir film. In contrast, the other brush random polymers (m: 20–60 mol%) nicely demonstrated to always form only Langmuir monolayer structure, regardless of the surface pressures. The Langmuir monolayer films were further improved their structural ordering by increasing the surface pressure. These interesting monolayer formation behaviors could be understood with taking into account all possible chemical, physical, and thermodynamic factors.
Experimental
Materials
A series of well-defined PECH-DMAPSm polymers were prepared, according to the synthetic method described in the literature.23 Poly(epichlorohydrin) (PECH) was synthesized as a base polymer from epichlorohydrin by cationic ring-opening polymerization. The obtained PECH polymer was determined to have a weight-average molecular weight![[M with combining macron]](https://www.rsc.org/images/entities/i_char_004d_0304.gif) w of 38
w of 38![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 and n of 22700 by using gel permeation chromatography calibrated with polystyrene standards. A portion of the PECH polymer was reacted with sodium 11-hydroxyundecylthiolate and further reacted with 1,3-propanesultone, producing PECH-DMAPSm (Fig. 1). The PECH-DMAPSm polymers were identified by proton and carbon nuclear magnetic resonance spectroscopy analysis. The PECH-DMAPSm polymers were thermally stable up to 190–215 °C (= Td, degradation temperature) in nitrogen atmosphere, depending on the compositions. A glass transition temperature Tg was observed over the temperature range −33 °C to −25 °C, whereas a melting temperature Tm was detected over the temperature range 0 °C to 30 °C. Higher DMAPS content in the brush polymer lowered Td and Tg but increased Tm.
500 and n of 22700 by using gel permeation chromatography calibrated with polystyrene standards. A portion of the PECH polymer was reacted with sodium 11-hydroxyundecylthiolate and further reacted with 1,3-propanesultone, producing PECH-DMAPSm (Fig. 1). The PECH-DMAPSm polymers were identified by proton and carbon nuclear magnetic resonance spectroscopy analysis. The PECH-DMAPSm polymers were thermally stable up to 190–215 °C (= Td, degradation temperature) in nitrogen atmosphere, depending on the compositions. A glass transition temperature Tg was observed over the temperature range −33 °C to −25 °C, whereas a melting temperature Tm was detected over the temperature range 0 °C to 30 °C. Higher DMAPS content in the brush polymer lowered Td and Tg but increased Tm.
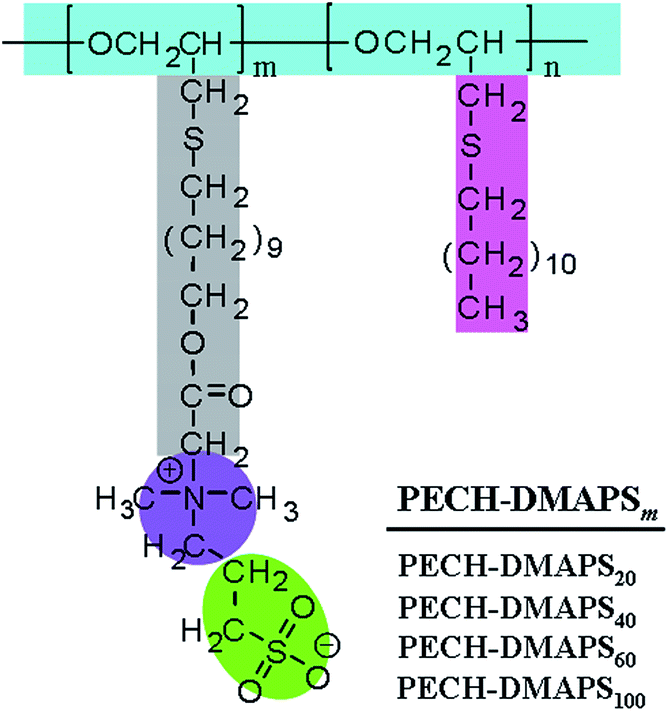 | ||
| Fig. 1 Chemical structures of the PECH-DMAPSm polymers used in this study. | ||
Langmuir film formation
Each brush polymer sample was dissolved in chloroform (analytical grade, Sigma-Aldrich, St. Louis, MO, USA), giving polymer solution with a concentration of 0.001 wt%. The obtained polymer solutions were filtered through polytetrafluoroethylene (PTFE) membrane microfilters with a pore size of 0.2 μm. A Langmuir–Blodgett (LB) trough system (model 302M, Nima, Coventry, England) was used for the preparation of Langmuir films at air–water interface. The trough was filled with deionized water and then maintained at room temperature. Each brush polymer solution was spread onto the air–water interface by using a Hamilton microsyringe. The compression was started around 30 min after spreading in order to allow the evaporation of the used chloroform solvent. The surface pressure–area (π–A) isotherm was recorded at a compression rate of 10 cm2 min−1 in a symmetric mode with an initial area of 245 cm2.XR measurement
Taking into account the measured π–A isotherms, in situ specular XR measurements were carried out at chosen surface pressures during Langmuir film formation process, using a Bruker diffractometer (model D8, Bruker AXS, Germany) with a vertical goniometer, which allows the study of a liquid surface without disturbing a LB trough during measurements.28,29 The LB trough was enclosed in a plexiglass box with Kapton windows, and the incoming and reflected beams were detected through the Kapton-covered windows. The LB trough setup was placed on an active vibration table platform (NANO 30, Halcyonics, Goettingen, Germany). An anode X-ray generator was used to produce CuKα radiation (λ = 1.542 Å), followed by paralleling the incident beam with a Göbel mirror (model GM III, Incoatec, Geesthacht, Germany). Two slits prior to the sample defined the incident beam size and reduced the vertical divergence. The measured XR data were quantitatively analyzed with a recursive formula given by the Parratt dynamic theory.30,31IR measurement
Attenuated total reflection Fourier transform infrared (ATR-FTIR) spectroscopy measurements were conducted on LB films that were prepared at various surface pressures. All IR measurements were obtained by recording 256 scans with a resolution of 2 cm−1 using FTIR spectrometers (model Cary 640, Agilent Technologies, Santa Clara, CA, USA; model UATR, Perkin-Elmer, Waltham, MA, USA) with a Peltier-cooled DLaTGS detector. A ZnSe ATR crystal was purchased from the Pike Technologies (Madison, WI, USA) and used for ATR-FTIR measurements. The ZnSe crystal was dipped in water on the LB trough and then Langmuir film was prepared on water surface. The barrier in the LB trough was moved with a compression rate of 10 cm2 min−1. When surface pressure was reached at a target pressure, the ZnSe crystal was pulled up with a rate of 2 mm min−1 while the barrier was compressed to maintain the target pressure. The LB film deposited onto the ZnSe crystal was loaded onto the ATR-FTIR cell after evaporation of the residual water and then IR spectroscopic measurements were conducted. In addition, the polymers in bulk states were examined by IR spectroscopy.Results and discussions
π–A isotherms were successfully measured at the air–water interface for PECH-DMAPS20, PECH-DMAPS40, and PECH-DMAPS60. However, π–A isotherm could not be measured for PECH-DMAPS100 because of its good solubility in water. Representatives of the measured π–A isotherm curves are shown in Fig. 2. For the PECH-DMAPS20 molecules spread on the water surface, the surface pressure π begins to rise up when the surface area reaches to 9148 Å2 per molecule. Then, with decreasing area, the π value initially increases very slowly until around 0.5 mN m−1 and changes to a rapidly rising mode. The π value continues such rapid rise, reaching to around 30 mN m−1. Thereafter, the π value turns to increase slowly with decreasing surface area until 33 mN m−1 and again changes to a rising mode, ultimately reaching to 36 mN m−1. A similar mode of π–A isotherm curve is observed for PECH-DMAPS60. However, the π value starts to rise at the surface area of 9647 Å2 per molecule, which is slightly larger than that of PECH-DMAPS20. The rising mode of π is changed at 1, 23, and 33 mN m−1. PECH-DMAPS40 shows somewhat different π–A isotherm curve from those of PECH-DMAPS20 and PECH-DMAPS60. The π value begins to rise at the surface area of 9723 Å2 per molecule, which is slightly larger than that of PECH-DMAPS60 but much larger than that of PECH-DMAPS20. The rising mode of π is changed at 1 and 28 mN m−1.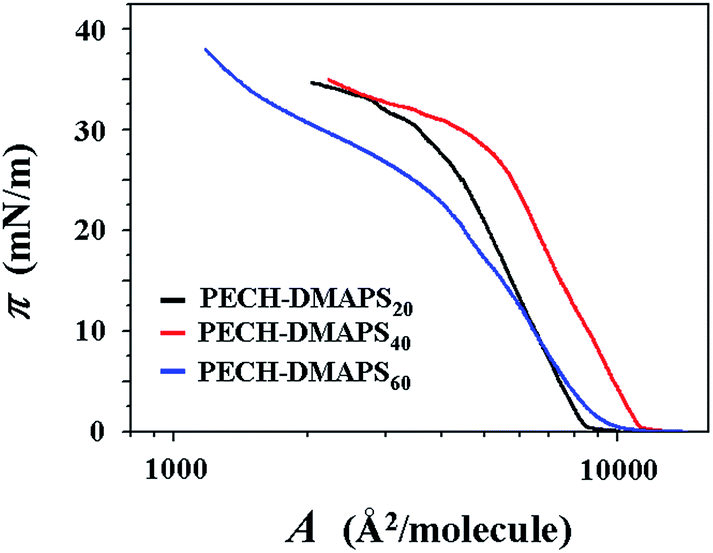 | ||
| Fig. 2 Representative surface pressure–area (π–A) isotherms of the amphiphilic zwitterionic brush random polymers at the air–water interface. | ||
With the π–A isotherms above, Langmuir films were sampled out at chosen π values and transformed onto ZnSe prisms for IR spectroscopy analysis. In addition, IR spectroscopy analysis was extended for the brush polymers in solid states. Representatives of the measured IR spectra are shown in Fig. 3. In general, the methyl (CH3) group of alkyl-containing compounds is known to reveal asymmetric stretching vibration peak around 2955 cm−1 and symmetric vibration peak around 2875 cm−1.32–37 And their methylenyl (CH2) units are also known to exhibit asymmetric stretching vibration peak around 2920 cm−1 and symmetric vibration peak around 2850 cm−1.32–37 For both cases, the asymmetric vibration peak is stronger in intensity than the symmetric vibration peak. The CH2 stretching peaks are much stronger in intensity than the CH3 stretching bands. On the other hand, the methyl (N–CH3) group of zwitterionic sulfobetaine-containing compounds has been reported to show asymmetric stretching vibration peak around 3020 cm−1 and symmetric vibration peak around 2854 cm−1; in general both the peaks are known to be very weak and broad, perhaps due to their low absorptivities.34,35 Taking these facts into account, the characteristic C–H stretching vibration peaks observed for the PECH-DMAPSm polymers in Langmuir films or bulk states of this study could be assigned.
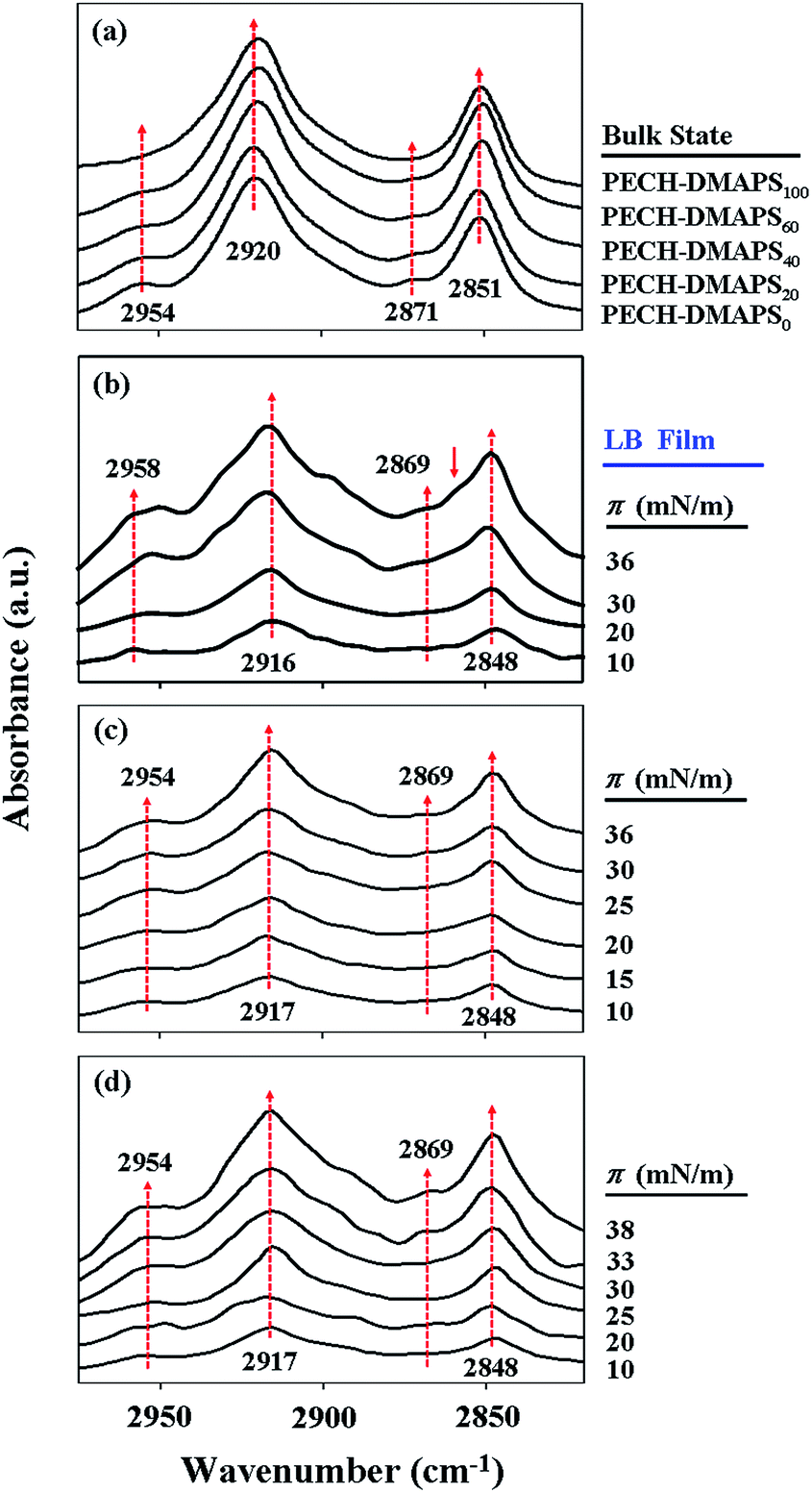 | ||
| Fig. 3 Representative IR spectra measured for the brush polymers in bulk states as well as the Langmuir–Blodgett (LB) films prepared at various surface pressures: (a) brush polymers in bulk states; (b) PECH-DMAPS20; (c) PECH-DMAPS40; (d) PECH-DMAPS60. The LB films were prepared on ZnSe prisms. | ||
As shown in Fig. 3a, PECH-DMAPS0 in bulk state reveals CH3 stretching peaks at 2954 and 2871 cm−1 and CH2 stretching peaks at 2920 and 2851 cm−1. PECH-DMAPS100 in bulk state also exhibits CH2 stretching peaks at 2920 and 2851 cm−1, which are very similar to those of PECH-DMAPS0; but the symmetric vibration peak of the methyl groups (N–CH3) is not discernible clearly because of its very and broad nature. All brush random copolymers in bulk states show IR spectra which resemble that of PECH-DMAPS0.
For the PECH-DMAPS20 film formed at π = 10 mN m−1, the CH2 asymmetric and symmetric stretching vibrations are observed in slightly lower frequencies (2916 and 2848 cm−1 respectively) (Fig. 3b), suggesting that the bristles are in ordered states. However, the CH3 asymmetric vibration peak of n-dodecylthiomethyl bristles weakly appears in slightly higher frequency (2958 cm−1). Furthermore, the symmetric CH3 vibration peak could not be discernible. These observations inform that the n-dodecylthiomethyl bristles are in a less ordered state rather than in a well-ordered state. The results collectively indicate that in the Langmuir film the bristles are present in a relatively low degree of ordered state.
As the surface pressure π is increased, the CH2 asymmetric and symmetric stretching frequencies are varied slightly (Fig. 3b). In fact, the CH2 stretching bands of alkyl compounds are known to vary very little in frequency; for example, they shift only 1 cm−1 as the alkyl packing density changes by a factor of 1000.34 And both the hydrophobic and the hydrophilic bristles possess CH2 units. The CH2 units in the hydrophobic bristle may be less associated with water molecules, leading to a red shift in the stretching frequencies. This red shift effect may compete with a blue shift caused by the CH2 units in the hydrophilic bristle that may have more opportunity to associate with water molecules. Due to these characteristics, in this study it is hard to see significant variations in the CH2 stretching frequencies with increasing surface pressure.
In contrast, the CH3 asymmetric stretching band shows red shifts as the π value is increased; in addition, the CH3 symmetric stretching band becomes discernible in the films formed with higher surface pressures. These results indicate that the packing order of the n-dodecylthiomethyl bristles is enhanced with increasing surface pressure. Moreover, a shoulder peak is additionally observed at 2860 cm−1 in the films formed with π = 30 and 36 mN m−1, suggesting that the disordered and ordered DMAPS bristles associated with water molecules coexist in the films.
Overall, these results inform that both the hydrophobic and the hydrophilic bristles of PECH-DMAPS20 tend to pack together laterally under increasing surface pressure, forming ordered states. Such ordered states result from segregations of the hydrophobic and the hydrophilic bristles at the air–water interface which are induced by surface pressure in the film formation process.
Similar trends are observed in the Langmuir film formation processes of PECH-DMAPS40 and PECH-DMAPS60 (Fig. 3c and d). It is noted that for the film formed with π = 10 mN m−1 the CH3 asymmetric vibration peak of n-dodecylthiomethyl bristles appears at 2954 cm−1 rather than at 2958 cm−1, indicating that the hydrophobic bristles have already formed an ordered state at the air–water interface even under such low surface pressure. The symmetric CH3 vibration peak is discernible in the films formed with high surface pressures. But, in these cases a shoulder peak at 2860 cm−1 could not be discernible clearly because of its weak and broad nature.
Overall, the IR analysis results inform that the hydrophobic n-dodecylthiomethyl bristles, as well as the hydrophilic DMAPS bristles undergo lateral packing in the Langmuir film formation process of PECH-DMAPSm; their packing orders are enhanced with increasing surface pressure. Such packing orderings are associated with segregations of the hydrophobic and the hydrophilic bristles at the air–water interface under surface pressure.
With the π–A isotherms and IR spectroscopy analysis results discussed above, XR measurements were conducted during Langmuir layer formation processes of the PECH-DMAPSm polymers. Fig. 4 shows representatives of the measured XR data. To quantitatively analyze the measured XR data, all possible structural models were considered and tested. As a result, structural models consisted of two to four sublayers were found to be most suitable for analyzing the XR data, depending on the surface pressure and the chemical composition of the random copolymer. All XR data were successfully analyzed using a recursive formula based on the Parratt dynamic theory.30,31 The analysis results are displayed in Fig. 5 and 6. From the analysis results, morphological structures are further proposed for the Langmuir layers formed at various conditions, as shown in Fig. 7–9.
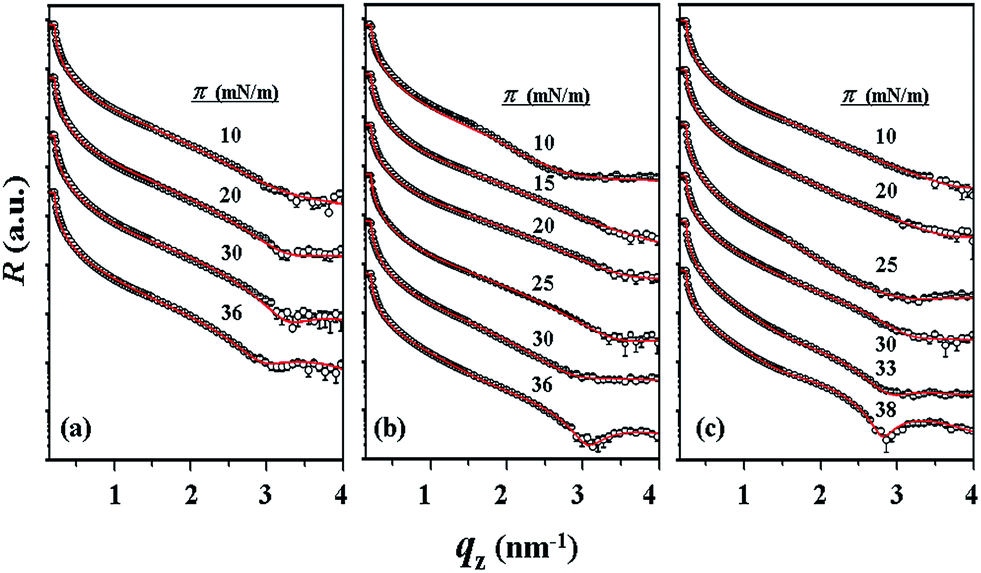 | ||
| Fig. 4 Representative specular X-ray reflectivity R profiles measured for the Langmuir films formed on the air–water interface under various surface pressures: (a) PECH-DMAPS20; (b) PECH-DMAPS40; (c) PECH-DMAPS60. qz is the magnitude of the scattering vector along the direction of the film thickness, which is defined by qz = (4π/λ)sinθi where λ is the wavelength of the X-ray beam and θi is the grazing incidence angle. The symbols are the measured data and the solid lines represent the best fit curves. | ||
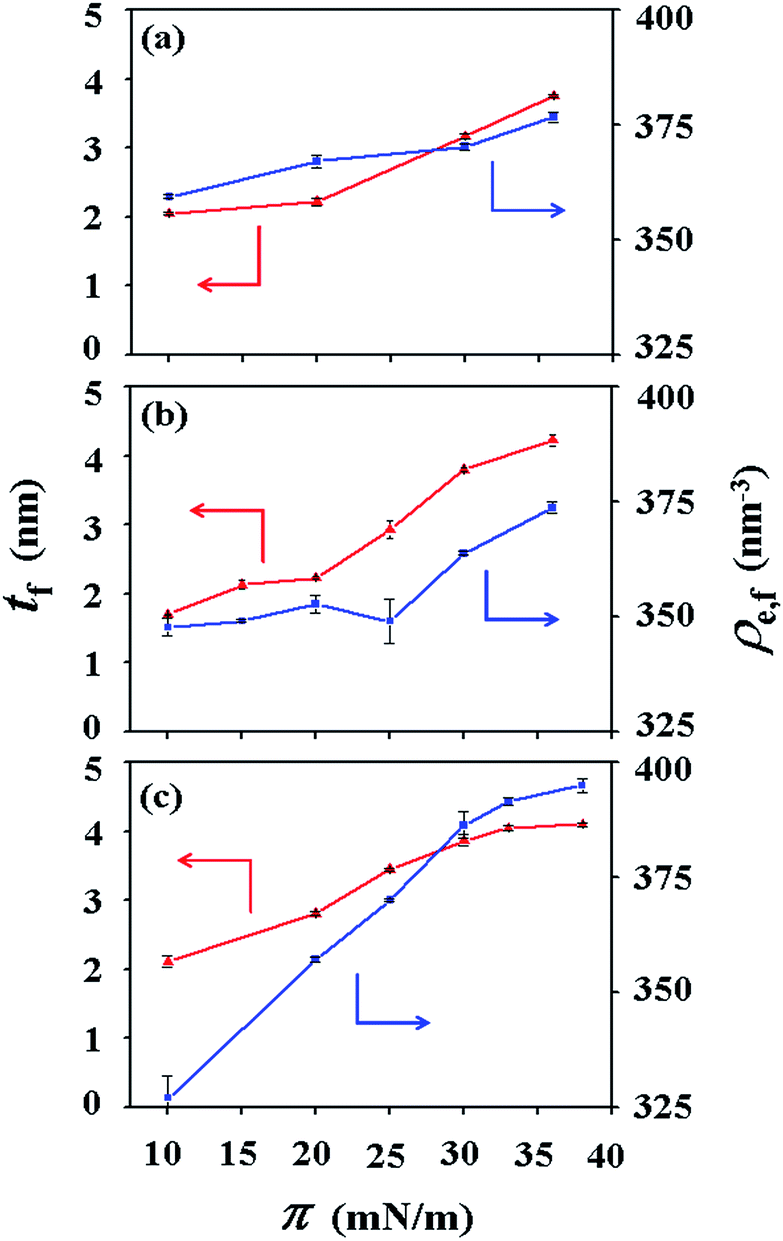 | ||
| Fig. 5 Thickness tf and electron density ρe,f variations of the Langmuir film prepared on the air–water interface as a function of surface pressure: (a) PECH-DMAPS20; (b) PECH-DMAPS40; (c) PECH-DMAPS60. | ||
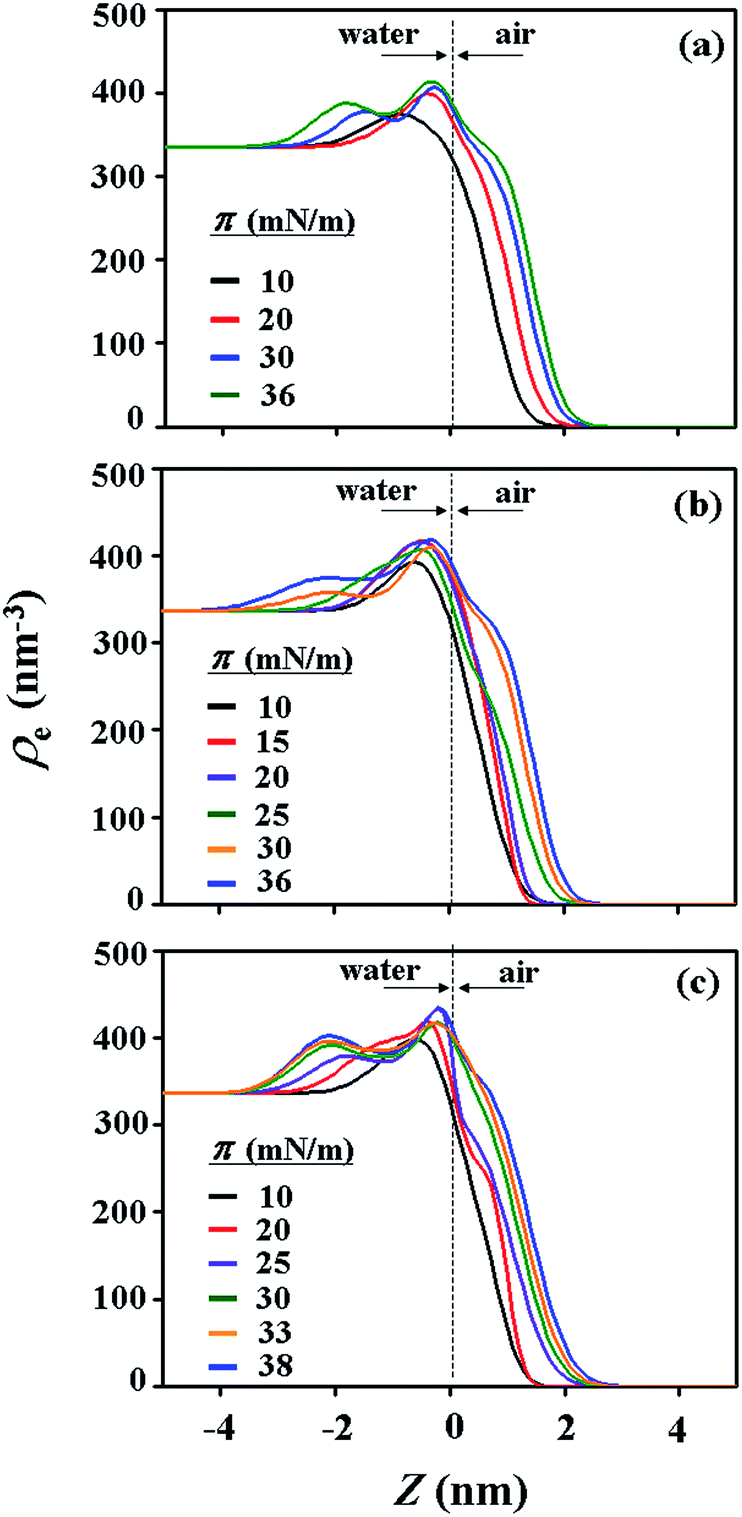 | ||
| Fig. 6 Electron density ρe profiles as a function of the film thickness, for the films prepared at the air–water interface under various surface pressures: (a) PECH-DMAPS20; (b) PECH-DMAPS40; (c) PECH-DMAPS60. | ||
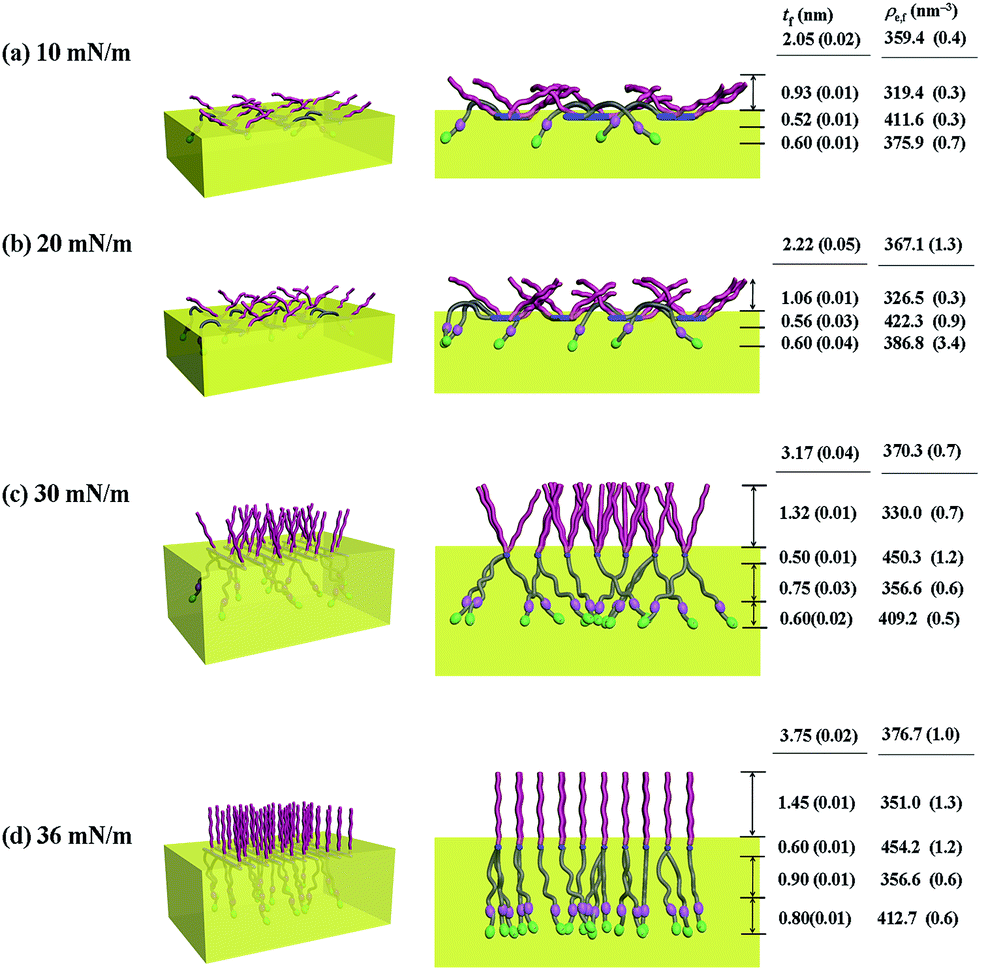 | ||
| Fig. 7 Molecular models of the Langmuir films of PECH-DMAPS20 prepared at the air–water interface under various surface pressures: (left) view with a tilt angle from the top; (right) front view along the polymer backbone. | ||
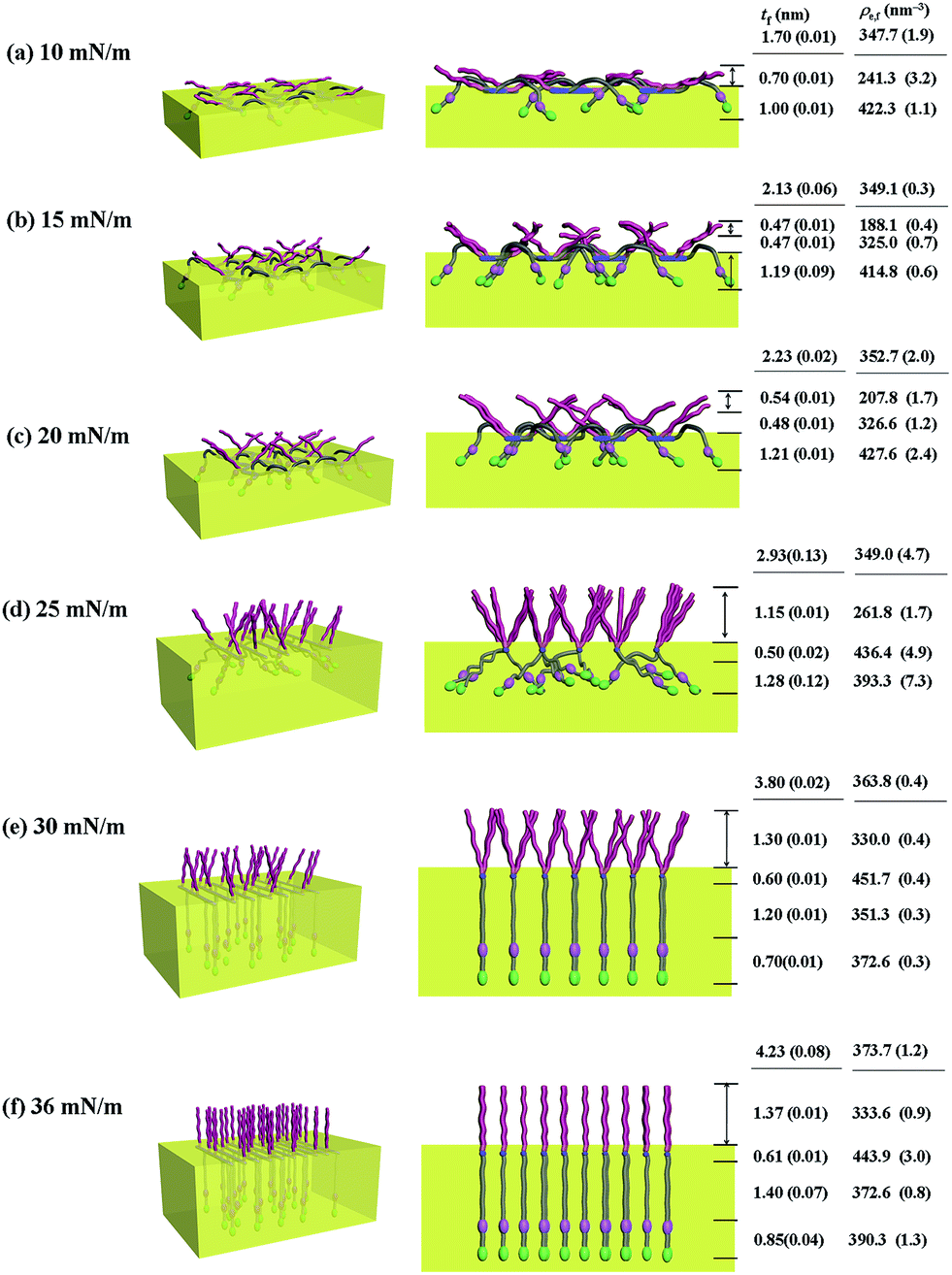 | ||
| Fig. 8 Molecular models of the Langmuir films of PECH-DMAPS40 prepared at the air–water interface under various surface pressures: (left) view with a tilt angle from the top; (right) front view along the polymer backbone. | ||
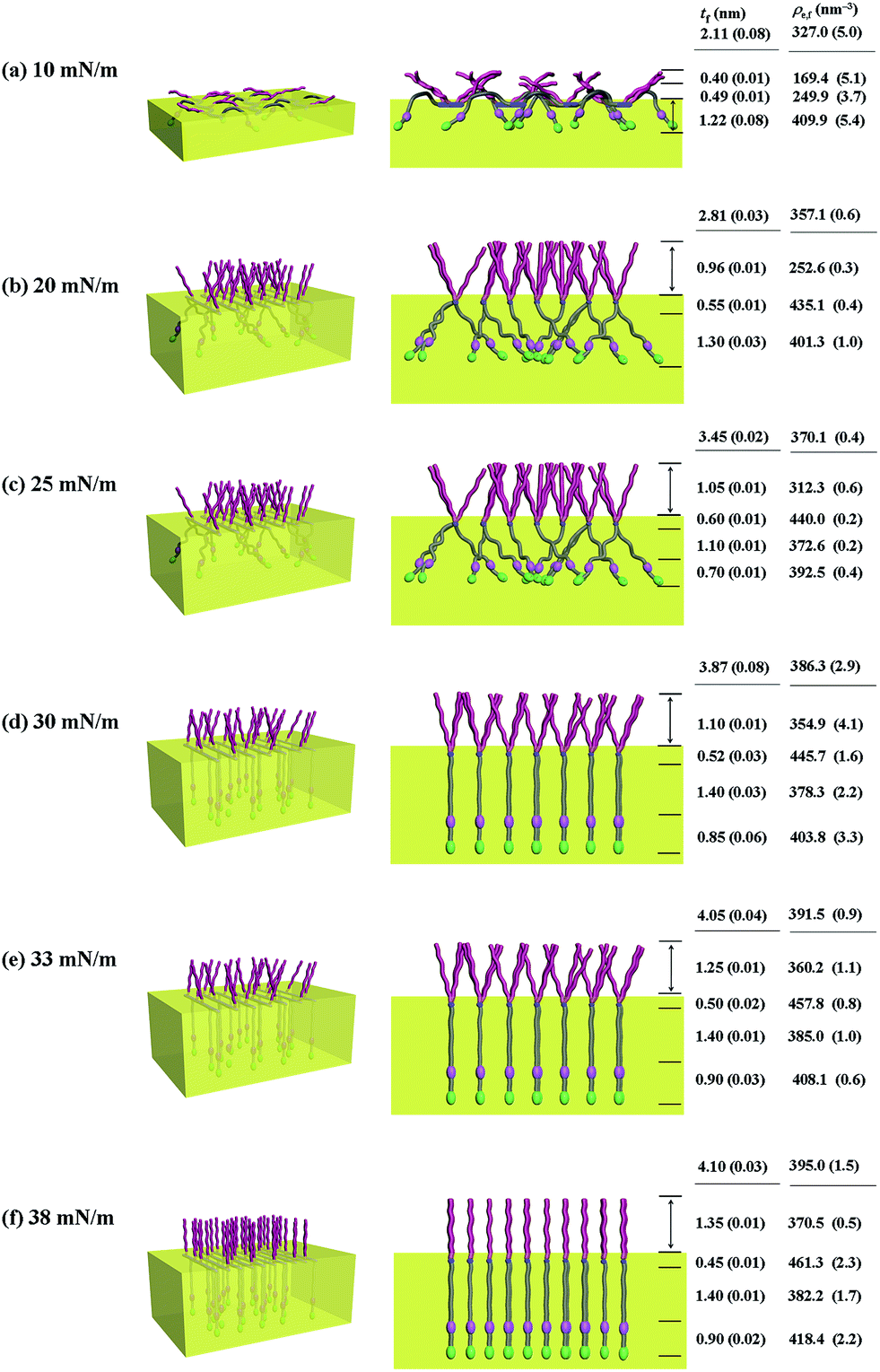 | ||
| Fig. 9 Molecular models of the Langmuir films of PECH-DMAPS60 prepared at the air–water interface under various surface pressures: (left) view with a tilt angle from the top; (right) front view along the polymer backbone. | ||
For PECH-DMAPS20, the Langmuir film is formed with a thickness tf of 2.05 nm and an electron density ρe,f of 359.4 nm−3 under π = 10 mN m−1. The film consists of three sublayers: the first sublayer (t1 = 0.60 nm and ρe,1 = 375.9 nm−3), the second sublayer (t2 = 0.52 nm and ρe,2 = 411.6 nm−3), and the third sublayer (t3 = 0.93 nm and ρe,3 = 319.4 nm−3). The overall film thickness tf is slightly larger than the length (1.80 nm) of the fully extended hydrophobic bristle but much smaller than that (2.84 nm) of the fully extended hydrophilic bristle; here the bristle lengths were estimated by performing molecular simulations using the Cerius2 software package. And the ρe,1 and ρe,2 values are larger than that (336.2 nm−3) of water layer; but the ρe,3 value is lower than that of water layer. These results collectively indicate that a Langmuir monolayer is formed at π = 10 mN m−1, where the bristles might be tilted with certain angles with respect to the backbone and/or be in less extended conformation; the first and second sublayers are immersed in the water side while the third sublayer is present in the air side. Considering the polymer chemical structure, the first sublayer is composed mainly with the hydrophilic, zwitterionic DMAPS moieties while the second sublayer consists of the hydrophilic backbones and the linker parts of the bristles. The third sublayer is made of the hydrophobic n-dodecylthiomethyl bristles in the air side.
The Langmuir film is found to undergo structural changes including thickening and densification by increasing surface pressure π. The film is slightly thickened and densified at π = 20 mN m−1. The film is further thickened and densified at π = 30 mN m−1, consequently causing structural change that the three-sublayer structure is transformed to a four-sublayer structure. The four-sublayer structure continues thickening and densification by increasing π up to 36 mN m−1. The ultimately obtained film is characterized by a set of structural parameters: tf = 3.75 nm (t1 = 0.80 nm, t2 = 0.90 nm, t3 = 0.60 nm, and t4 = 1.45 nm) and ρe,f = 376.7 nm−3. The tf value is somewhat smaller than the sum (4.64 nm) of the lengths of the hydrophobic bristle fully extended upward and the hydrophilic, zwitterionic DMAPS bristle fully extended downward. Moreover, the t4 value is slightly smaller than the length (1.80 nm) of the fully extended hydrophobic bristle; in fact, the sum of t4 and a half of t3 (this part is made of the inner portion of the hydrophobic bristle) is almost same with the length of the fully extended hydrophobic bristle. And the fourth sublayer reveals the lowest ρe value, compared to those of the other sublayers. This result is an indication that no or less water molecules are associated with the fourth sublayer. In comparison, the sum of t1, t2, and a half of t3 (this part is made of the inner portion of the hydrophilic bristle), which is originated from the hydrophilic bristle, is much smaller than the length (2.84 nm) of the fully extended hydrophilic bristle. These results collectively inform that in the Langmuir film formed at π = 36 mN m−1, the fourth sublayer is composed of the hydrophobic bristles in nearly extended conformation and positioned in the air side while the first and second sublayers including roughly a half of the third sublayer are consisted of the DMAPS bristles in less extended conformation and present in the water side. The lateral packing of such the nearly-extended hydrophobic bristles is further supported by the lateral packing of the hydrophilic polymer backbones being extended under surface pressure as high as 36 mN m−1.
Similar Langmuir monolayer formation behaviors are observed for PECH-DMAPS40 and PECH-DMAPS60. However, the ultimately formed Langmuir films are relatively thicker, compared to the PECH-DMAPS20 film. The films formed with the highest surface pressures are found to have a thickness slightly smaller than the sum of the lengths of the fully extended hydrophobic bristle upward and the fully extended hydrophilic bristle downward. These results are evidenced that the hydrophobic and hydrophilic bristles, which are segregated into the air and the water side respectively, are in nearly-extended conformations. And the Langmuir films are identified to be composed of four sublayers rather than three sublayers.
The above π–A isotherm, IR spectroscopy and XR analysis results collectively provide the following insights into the correlations of chemical structure, self-assembly and Langmuir film morphology in PECH-DMAPSm, an amphiphilic, zwitterionic random copolymer system, at the air–water interface.
Firstly, the PECH-DMAPSm polymer bearing higher mol% of DMAPS tends to cover a larger area of water surface. Such the water surface coverage reaches to a maximum as the DMAPS content approaches to around 40 mol%. These suggest that the hydrophilic, zwitterionic DMAPS bristles, as well as the hydrophilic backbones make important role in the spread of PECH-DMAPSm at the water surface via their favorable interactions with or solvations in water while the hydrophobic n-dodecylthiomethyl bristles contribute to the Langmuir monolayer formation and its stability against the complete solvation (i.e., dissolution) of the polymer in water. However, Langmuir films could not be made from PECH-DMAPS100 because it is completely soluble in water. This fact indicates that the solvation power in water of the hydrophilic, zwitterionic DMAPS bristle with the aid of the hydrophilic backbone is relatively stronger than the stabilization power of the n-dodecylthiomethyl bristle for Langmuir monolayer. Therefore, a Langmuir film could be formed at the air–water interface from PECH-DMAPSm containing <100 mol% DMAPS in which the structural stabilization power of the hydrophobic bristle is balanced with the water-solvation power of the hydrophilic, zwitterionic bristle in a certain level.
Secondly, once a Langmuir film of PECH-DMAPSm is formed, it could undergo structural changes by increasing surface pressure, revealing highly ordered monolayer structure. The highly ordered monolayer structure could be achieved by thickening and densification through the conformational changes of the backbone and bristles to nearly or fully extended forms and their lateral packing under increasing surface pressure. Such conformational changes and lateral packing could be driven by the hydrophobic bristles, the alkylenyl linkers in the hydrophilic bristles, and the backbone chain. In particular, the alkylenyl linkers of the hydrophilic bristles present in the water side could get a certain degree of stabilization (i.e., enthalpy gain) by the lateral packing with the aid of their hydrophobicity against the solvation with water molecules. This could make a significant contribution to enhance the ordering and stability of the Langmuir film. Such enthalpy gain could be more feasible for the hydrophobic alkyl groups in the air side.
Thirdly, at the water surface all PECH-DMAPSm polymers (DMAPS: 20–60 mol%) formed only monolayers rather than bilayer or multilayers. These interesting, unique monolayer formation behaviors might be attributed to the relatively high solvation power of DMAPS moieties in the random copolymers under the presence of the hydrophobic bristles.
Lastly, the Langmuir monolayer structure formations are the evidences that the bristles of PECH-DMAPSm favorably underwent segregation, forming a hydrophobic bristle phase and a hydrophilic bristle phase. The segregation behavior is remarkable in regard to the random copolymeric nature. In general, such kind of segregation is very rare or not easily happened for random copolymers in bulk or film state. Even though the PECH-DMAPSm polymers are random copolymers, such the segregation could be driven at the air–water interface by the selective interactions of the bristles with water molecules and air, leading to Langmuir monolayer formation.
Conclusions
A series of amphiphilic, zwitterionic brush random copolymers, PECH-DMAPSm, was investigated at the air–water interface in the aspect of their self-assembling behavior by using π–A isotherm, IR spectroscopy, and XR analyses. These analyses collectively provide insights about the self-assembling behavior of the random copolymer system as follows. PECH-DMAPS20, PECH-DMAPS40, and PECH-DMAPS60 successfully demonstrated the formation of Langmuir layer at the air–water interface, whereas PECH-DMAPS100 failed to make Langmuir film because of its high solubility in water. The observed Langmuir films might result from the dual functionalities of PECH-DMAPSm, namely its favorable and unfavorable interactions with water molecules in a balanced manner. The favorable interaction with water could be driven by both the hydrophilic backbone and the hydrophilic, zwitterionic DMAPS groups at the bristle ends while the unfavorable interaction with water could be attributed to the hydrophobic n-dodecylthiomethyl bristles. Therefore, the composition of the bristles plays a key role in the Langmuir film formation. Interestingly, PECH-DMAPSm always forms only Langmuir monolayer film, regardless of the surface pressure in the film formation process. Instead, the Langmuir monolayer undergoes to enhance the lateral packing order in both the hydrophobic bristle phase and the hydrophilic bristle and backbone phase. In particular, the hydrophobic linkers of the bristles could make a significant contribution to the overall lateral packing of the bristles although they are present in the water side. That is, the hydrophobic linkers could maximize enthalpy gains thermodynamically through the lateral packing with the aid of increasing surface pressure, consequently making positive contribution to enhance the packing order of Langmuir film structure. Furthermore, the zwitterionic DMAPS moieties may have made significant contribution to hold the hydrophilic bristle and backbone phase in the waterside by using their high water affinity (or water salvation power), therefore keeping only Langmuir monolayer structure. Overall, PECH-DMAPSm revealed very unique self-assembling behavior at the air–water interface, always producing Langmuir monolayer structure.Acknowledgements
This study was supported by the National Research Foundation of Korea (Haeksim Program 2015R1A2A2A2A01005642).Notes and references
- J. E. Raynor, J. R. Capadona, D. M. Collard, T. A. Petrie and A. J. García, Biointerphases, 2009, 4, FA3 CrossRef CAS PubMed.
- J. M. Anderson, Annu. Rev. Mater. Res., 2001, 31, 81 CrossRef CAS.
- B. Zhao and W. J. Brittain, Prog. Polym. Sci., 2000, 25, 677 CrossRef CAS.
- D. Bozukova, C. Pagnoulle, M.-C. De Pauw-Gillet, S. Desbief, R. Lazzaroni, N. Ruth, R. Jerome and C. Jerome, Biomacromolecules, 2007, 8, 2379 CrossRef CAS PubMed.
- H. Ma, D. Li, X. Sheng, B. Zhao and A. Chilkoti, Langmuir, 2006, 22, 3751 CrossRef CAS PubMed.
- Z. Zhang, T. Chao, S. Chen and S. Jiang, Langmuir, 2006, 22, 10072 CrossRef CAS PubMed.
- E. Ostuni, R. G. Chapman, M. N. Liang, G. Meluleni, G. Pier, D. E. Ingber and G. M. Whitesides, Langmuir, 2001, 17, 6336 CrossRef CAS.
- R. G. Chapman, E. Ostuni, M. N. Liang, G. Meluleni, E. Kim, L. Yan, G. Pier, H. S. Warren and G. M. Whitesides, Langmuir, 2001, 17, 1225 CrossRef CAS.
- S. Chen, J. Zheng, L. Li and S. Jiang, J. Am. Chem. Soc., 2005, 127, 14473 CrossRef CAS PubMed.
- Z. Zhang, S. Chen, Y. Chang and S. Jiang, J. Phys. Chem. B, 2006, 110, 10799 CrossRef CAS PubMed.
- W. Feng, J. L. Brash and S. Zhu, Biomaterials, 2006, 27, 847 CrossRef CAS PubMed.
- J. Zheng, L. Li, S. Chen and S. Jiang, Langmuir, 2004, 20, 8931 CrossRef CAS PubMed.
- H. Kitano, T. Mori, Y. Takeuchi, S. Tada, M. Gemmei-Ide, Y. Yokoyama and M. Tanaka, Macromol. Biosci., 2005, 5, 314 CrossRef CAS PubMed.
- Y. Chang, S. Chen, Z. Zhang and S. Jiang, Langmuir, 2006, 22, 2222 CrossRef CAS PubMed.
- A. B. Lowe, M. Vamvakaki, M. A. Wassall, L. Wong, N. C. Billingham, S. P. Armes and A. W. Lloyd, J. Biomed. Mater. Res., 2000, 52, 88 CrossRef CAS PubMed.
- G. Kim, S. Park, J. Jung, K. Heo, J. Yoon, H. Kim, I. J. Kim, J. R. Kim, J. I. Lee and M. Ree, Adv. Funct. Mater., 2009, 19, 1631 CrossRef CAS.
- G. Kim, L. Y. Hong, J. Jung, D.-P. Kim, H. Kim, I. J. Kim, J. R. Kim and M. Ree, Biomaterials, 2010, 31, 2517 CrossRef CAS PubMed.
- G. Kim, Y. Rho, S. Park, H. Kim, S. Son, H. Kim, I. J. Kim, J. R. Kim, W. J. Kim and M. Ree, Biomaterials, 2010, 31, 3816 CrossRef CAS PubMed.
- J. Jung, J. C. Kim, Y. Rho, M. Kim, H. Kim and M. Ree, ACS Appl. Mater. Interfaces, 2011, 3, 2655 CAS.
- J. C. Kim, J. Jung, Y. Rho, M. Kim, W. Kwon, H. Kim, I. J. Kim, J. R. Kim and M. Ree, Biomacromolecules, 2011, 12, 2822 CrossRef CAS PubMed.
- Y. Rho, J. Min, J. Yoon, B. Ahn, S. Jung, K. Kim, P. Shah, J.-S. Lee and M. Ree, NPG Asia Mater., 2012, 4, e29 CrossRef.
- J. C. Kim, M. Kim, J. Jung, H. Kim, I. J. Kim, J. R. Kim and M. Ree, Macromol. Res., 2012, 20, 746 CrossRef CAS.
- M. Kim, J. C. Kim, Y. Rho, J. Jung, W. Kwon, H. Kim and M. Ree, J. Mater. Chem., 2012, 22, 19418 RSC.
- J. C. Kim, Y. Rho, G. Kim, M. Kim, H. Kim, I. J. Kim, J. R. Kim and M. Ree, Polym. Chem., 2013, 4, 2260 RSC.
- J. Jung, H. Kim and M. Ree, Soft Matter, 2014, 10, 701 RSC.
- J. C. Kim, M. Kim, J. Jung, J.-S. Lee, B. J. Ree, H. Kim, I. J. Kim, J. R. Kim and M. Ree, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 1151 CrossRef CAS.
- K. Kwon, C. Kim, J. Lee, H. Kim and M. Ree, Biomacromolecules, 2016, 17, 974 CrossRef CAS PubMed.
- I. Kuzmenko, H. Rapaport, K. Kjaer, J. Als-Nielsen, I. Weissbuch, M. Lahav and L. Leiserowitz, Chem. Rev., 2001, 101, 1659 CrossRef CAS PubMed.
- M. Tolan, X-ray Scattering from Soft Matter Thin Films, Springer-Verlag Berlin, Heidelberg, 1999 Search PubMed.
- L. G. Parratt, Phys. Rev., 1954, 95, 359 CrossRef.
- J. Bolze, M. Ree, H. S. Youn, S. H. Chu and K. Char, Langmuir, 2001, 17, 6683 CrossRef CAS.
- M. D. Porter, T. B. Bright, D. L. Allara and C. E. D. Chidsey, J. Am. Chem. Soc., 1987, 109, 3559 CrossRef CAS.
- M. R. Linford and C. E. D. Chidsey, J. Am. Chem. Soc., 1993, 115, 12631 CrossRef CAS.
- R. B. Viana, A. B. F. da Silva and A. S. Pimentel, Adv. Phys. Chem., 2012, 2012, 903272 Search PubMed.
- K.-S. S. Kung and K. F. Hayes, Langmuir, 1993, 9, 263 CrossRef CAS.
- Y.-L. Su, J. Wang and H.-Z. Liu, J. Phys. Chem. B, 2002, 106, 11823 CrossRef CAS.
- E. E. Dormidontova, Macromolecules, 2002, 35, 987 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |