
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn vivo evaluation of small-molecule thermoresponsive anticancer drugs potentiated by hyperthermia
Catherine M.
Clavel
,
Patrycja
Nowak-Sliwinska
,
Emilia
Păunescu
,
Arjan W.
Griffioen
and
Paul J.
Dyson
*
Institut des Sciences et Ingénierie Chimiques, Ecole Polytechnique Fédérale de Lausanne (EPFL), CH-1015 Lausanne, Switzerland. E-mail: paul.dyson@epfl.ch; Fax: +41 21 693 97 80; Tel: +41 21 693 98 54
First published on 17th March 2015
Abstract
Hyperthermia used as an adjuvant with chemotherapy is highly promising in the treatment of certain cancers. Currently, the small molecule drugs used in combination with hyperthermia were not designed for this application. Herein, we report the evaluation of a chlorambucil and a ruthenium compound modified with a long fluorous chain, which exhibit thermoresponsive activity in colorectal adenocarcinoma xenografts in athymic mice in combination with mild hyperthermia (42 °C). Intraperitoneal injection of the derivatives followed by local hyperthermia showed a synergistic tumor growth reduction by 79% and 90% for the chlorambucil and ruthenium-based derivatives, respectively, with the latter exhibiting a higher synergy in combination with hyperthermia compared to the monotherapies. Histological analysis shows that both derivatives in combination with hyperthermia significantly decrease the number of proliferating tumor cells.
Introduction
The most widely applied anticancer drugs tend to exhibit severe side-effects1 and, consequently, considerable efforts have been directed towards the design of more selective anticancer drugs or treatment protocols that reduce side-effects and improve the quality of life of the patient, both during and after chemotherapy. It has been shown that combining chemotherapy with regional mild hyperthermia (applying heat locally to raise the tumor temperature between 40 and 42 °C), can sensitize tumor tissue to anticancer agents resulting in improved local control, treatment efficacy and overall survival prolongation.2,3 A promising approach used to enhance drug action when combined with hyperthermia is to employ drugs that are activated only under hyperthermia conditions.4,5Attaching known drugs to thermoresponsive macromolecules has led to hyperthermia treatments that are considerably more effective and selective than the application of the drug alone.6–11
For example, doxorubicin has been extensively studied in liposomal formulations designed for thermoresponsive release,11,12 which also benefit from preferential accumulation in solid tumors due to the enhanced permeability and retention effect that relies on microvascular hyperpermeability to macromolecules.13–15 One such formulation termed ThermoDox® is based on a low temperature sensitive liposome containing doxorubicin, which upon heating to 42 °C releases the doxorubicin.16,17 ThermoDox® is currently in phase III clinical trials in combination with radiofrequency ablation for the treatment of hepatocellular carcinoma.18
Hyperthermia used as an adjuvant with chemotherapy, but also with radiotherapy19,20 or their combination, also shows promise in cancer treatment. A triple therapy comprising radiotherapy, hyperthermia and chemotherapy, employing temozolomide (with liposomal doxorubicin included in resistant cases), led to enhanced survival rates in a glioblastoma clinical trial.21 In this study over 50% of patients showed prolonged survival to 26 months whereas the median survival following surgery is usually less than 4 months, which slightly increases with radiotherapy.22,23 Chlorambucil has also been encapsulated in a biodegradable and thermoresponsive micellar system based on triblock copolymers that increase water solubility and control its release near the tumor.24,25
Methods to transform anticancer drugs such as doxorubicin, chlorambucil, cisplatin and other compounds into thermoresponsive agents focus mainly on the use of macromolecules, notably liposomal formulations, or magnetic nanoparticles that encapsulate the drug and deliver it at the tumor site under hyperthermia.26–31 Nevertheless, replacing macromolecules with low molecular weight thermosensitive drugs remains an attractive alternative option due, in part, to the variability and complexity of nanoscale medicines.
We have recently shown that perfluorinated chains covalently bound to small molecule drugs give rise to thermoresponsive solubility, i.e. low solubility that rapidly increases with small increases in temperature, with the change in solubility being considerably greater than that of the parent drug. A modified chlorambucil derivative 132 and a monofunctional RAPTA-C like ruthenium(II) compound 233 (see Fig. 1) are essentially non-cytotoxic at 37 °C and highly cytotoxic to cancer cells when activated by a 2 hour hyperthermia signal (41.5 °C). In this study, compounds 1 and 2 were evaluated in a pre-clinical model in combination with mild hyperthermia validating their synergy in cancer treatment in vivo.
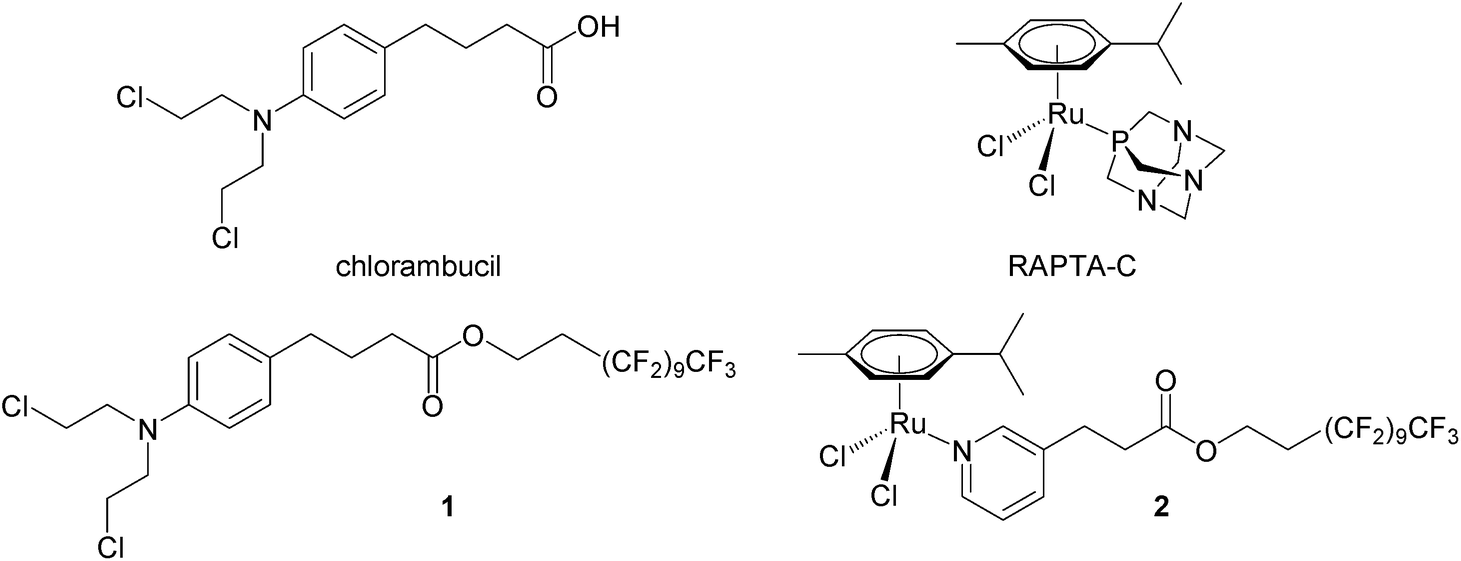 | ||
| Fig. 1 Chemical structures of chlorambucil, RAPTA-C and the thermoactive derivatives 1 and 2. | ||
Results and discussion
Human adenocarcinoma LS174T cells were implanted subcutaneously on the hind leg of athymic mice, as local mild hyperthermia can easily be applied at this site, and were allowed to grow until a palpable tumor mass was observed. The compounds (1 or 2) were injected intraperitoneally (i.p.) at a dose of 12.5 mg per kg per day in a 10% DMSO solution in sterile saline (0.9% NaCl) with control groups receiving a 10% DMSO saline solution. In parallel monotherapy groups with hyperthermia treatment only (30 minutes at 42 °C) or 1 or 2 only were studied. Dual therapy was performed by injection of the compound followed by hyperthermia treatment with a drug-hyperthermia interval of a maximum of 15 minutes. The rapid induction of hyperthermia following drug injection was shown to be optimal for several clinically approved drugs, e.g. cisplatin, melphalan and carboplatin.34–37Tumor growth was measured daily starting at the first day of treatment (day 6, following inoculation on day 0). The median tumor volume of each group for 1 is shown in Fig. 2.
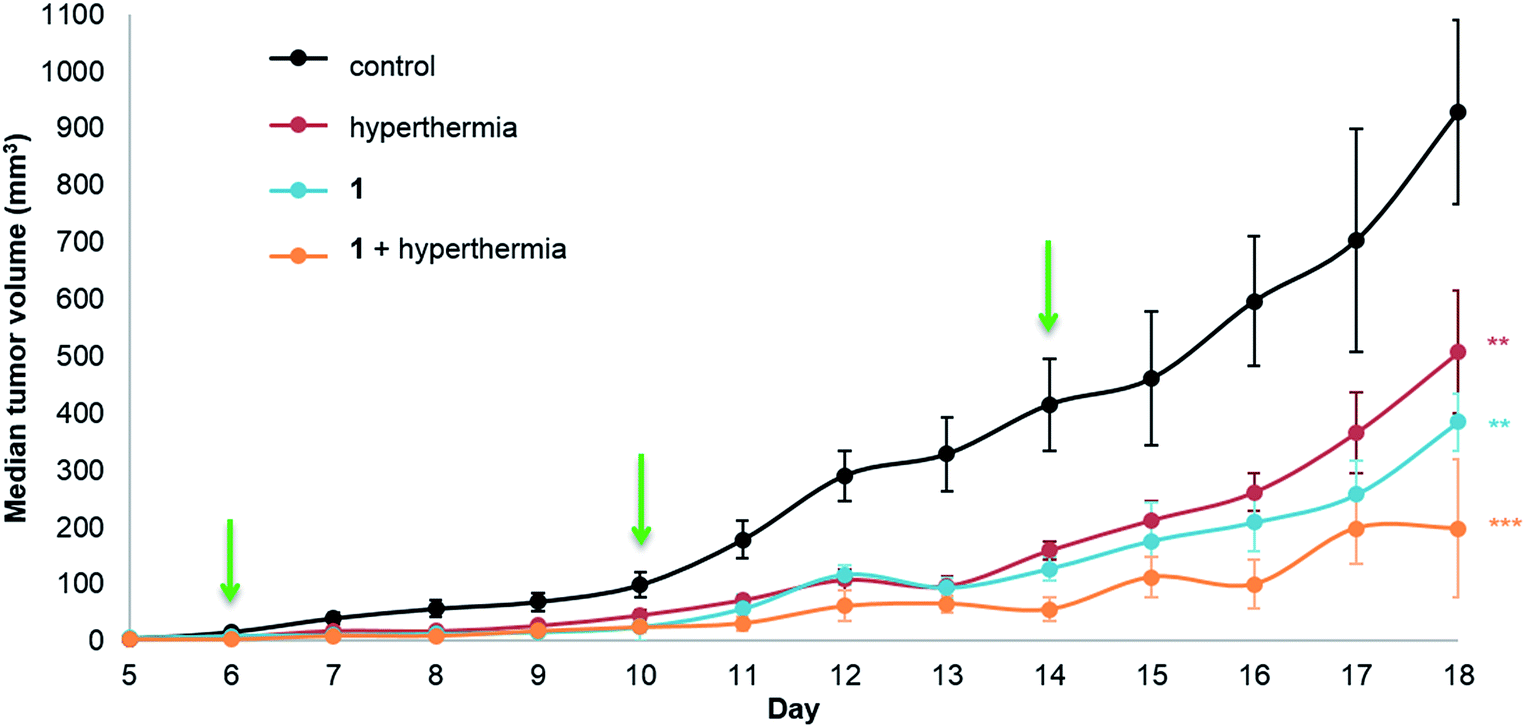 | ||
| Fig. 2 LS174T adenocarcinoma growth inhibition in athymic mice. Mice were treated with 1 (12.5 mg kg−1, 300 μL i.p.) given every 4 days (indicated by the green arrows) with or without mild hyperthermia (42 °C for 30 minutes) within 15 minutes following drug injection. The control group animals were treated with 10% DMSO in sterile saline (0.9% NaCl) also given every 4 days. Results are expressed as medians ± the standard error of the mean, SEM (n = 5 to 8 mice/group, ANOVA, **p < 0.01, ***p < 0.001), efficacy DE1,H = 0.787. Tumor volume (mm3) was calculated using the formula: volume = width2 × length × 0.5. | ||
Significant tumor inhibition from day 7 to 18 is observed (based on ANOVA testing). In addition, post ad-hoc statistical analysis using the one-sided pairwise t-test indicates that 1 alone (p = 0.00184), hyperthermia alone (p = 0.00610) and the duotherapy of 1 + hyperthermia (p = 3.2 × 10−4) significantly reduce tumor growth compared to the control group. Indeed, on the last day of the experiment, duotherapy gives a drug efficacy of 79% versus 59% (for 1 alone) and 45% (for hyperthermia alone), with the 1 + hyperthermia duotherapy acting synergistically on tumor growth reduction following the fractional product, DE1,H = 0.787 (see Experimental). It should be noted that hyperthermia alone has previously been shown to reduce tumor size in patients,38 as cancer cells are more sensitive than normal cells to mild hyperthermia (41–43 °C).39,40
The same in vivo model was also used to evaluate in duotherapy 2 with hyperthermia on tumor growth reduction (Fig. 3) using the protocol described for 1. ANOVA testing confirmed significant tumor growth reduction and post ad hoc statistical analysis using the one-sided pairwise t-test gave significant differences in tumor growth for 2 alone (p = 0.0013), hyperthermia alone (30 minutes at 42 °C, p = 0.0058) and 2 + hyperthermia (p = 7.4 × 10−5) compared to the control group. Moreover, the tumor volume in the mice undergoing duotherapy is significantly different from hyperthermia alone and from 2 given as a monotherapy (*p-values < 0.05). Compound 2 inhibited tumor growth by 66%, whereas the duotherapy of 2 + hyperthermia resulted in tumor growth inhibition of 90%, with the combination displaying strong synergistic effects (DE2,H = 0.899).
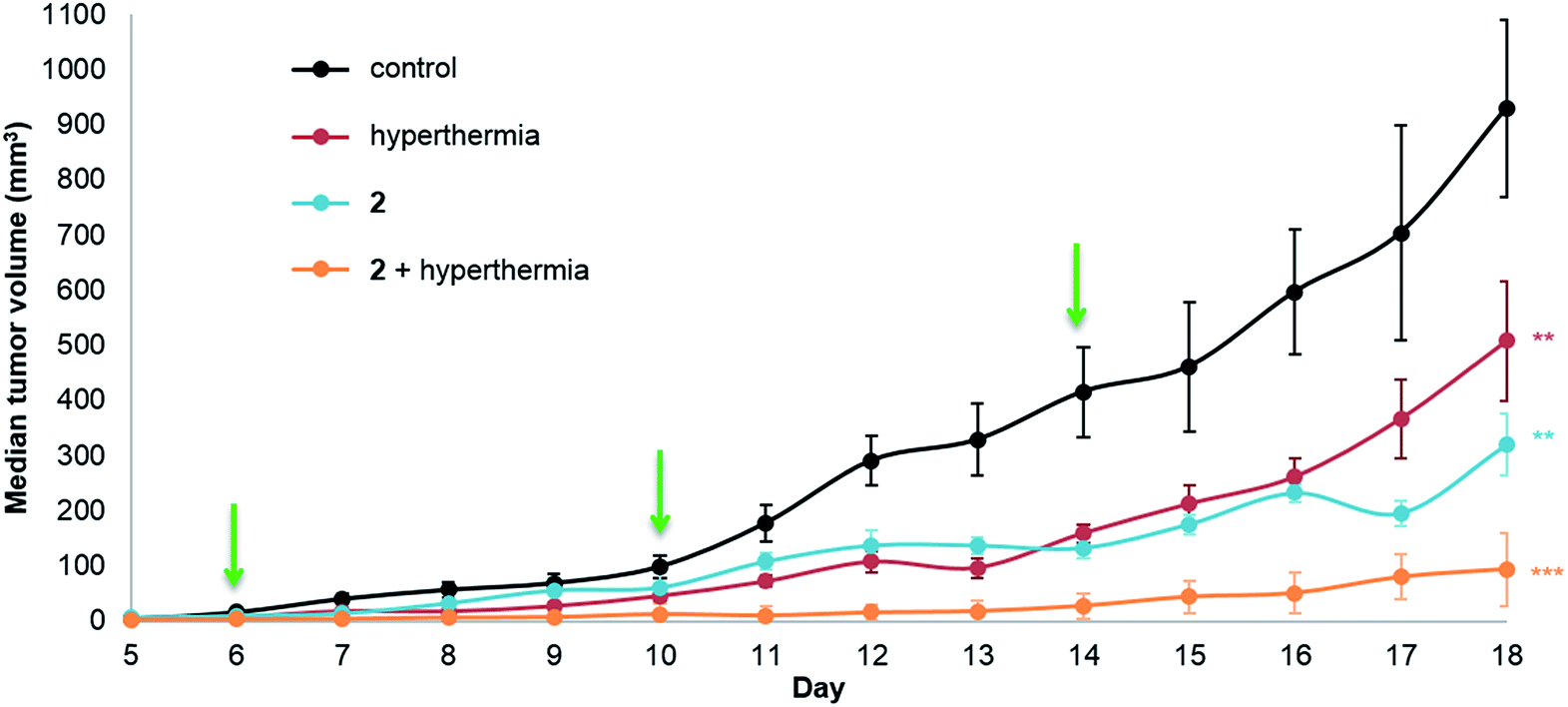 | ||
| Fig. 3 LS174T adenocarcinoma growth inhibition in athymic mice. Mice were treated with 2 (12.5 mg kg−1, 300 μL i.p.) given every 4 days (indicated by green arrows) with or without mild hyperthermia (42 °C for 30 minutes) administered intraperitoneally. The vehicle control was treated with three doses of 10% DMSO in sterile saline (0.9% NaCl) also given every 4 days. Values plotted are medians ± the standard error of the mean, SEM (n = 5–8 mice per group, ANOVA, **p < 0.01, ***p < 0.001), efficacy DE2,H = 0.899. | ||
In order to detect proliferating cells, treated tumor tissue sections from the various studies were stained with the nuclear proliferation marker Ki67 (Fig. 4). Hyperthermia by itself showed an anticancer effect, decreasing the number of proliferating cells by 33% (p = 0.0462). The application of 1 or 2 both significantly reduces the number of proliferating cells (28%, p = 0.041 and 37%, p = 0.0465, respectively). Duotherapy of 1 + hyperthermia resulted in a 84% decrease of proliferating cells (p = 0.0037), whereas 2 + hyperthermia inhibited proliferating tumor cells by 91%, i.e. inducing an enhanced state of quiescence. This cellular quiescence, or proliferation arrest, presumably represents a homogeneous state induced by diverse anti-mitogenic signals.41 Previously, it was shown that RAPTA-C treated mice bearing LS174T tumors leads to similar effects,42 however, RAPTA-C was administered daily at 100 mg kg−1, an 8-fold higher dose than 2 (12.5 mg kg−1 given every 4 days). Hyperthermia combined with known chemotherapy agents has previously been shown to lead to some necrosis,43 which was also observed with 1 and 2 (Fig. 5). However, with these specially-designed thermoactive compounds, antiproliferative activity is efficiently triggered by a short mild hyperthermia signal without apparently affecting the surrounding tissues, therefore providing enhanced selectivity.
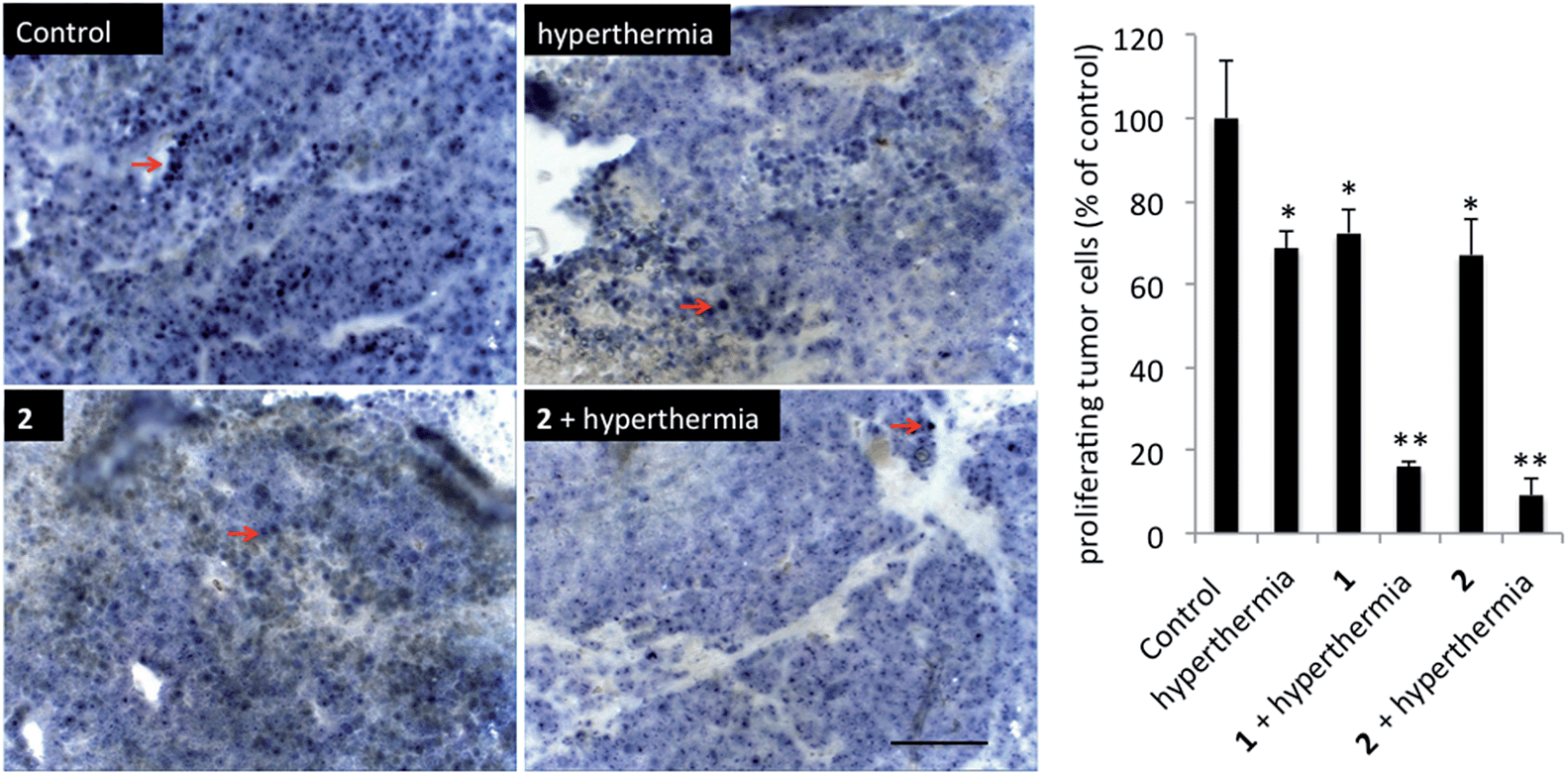 | ||
| Fig. 4 Histochemical analysis of tumors stained for proliferating cells (Ki67). Representative sections of tumors of the different treatment groups are shown. Ki67 + nuclei are indicated with red arrows. Scale bar = 100 μm (left). Quantification of proliferating tumor cells per treatment group, errors are shown as means ± the standard error of the mean (right). | ||
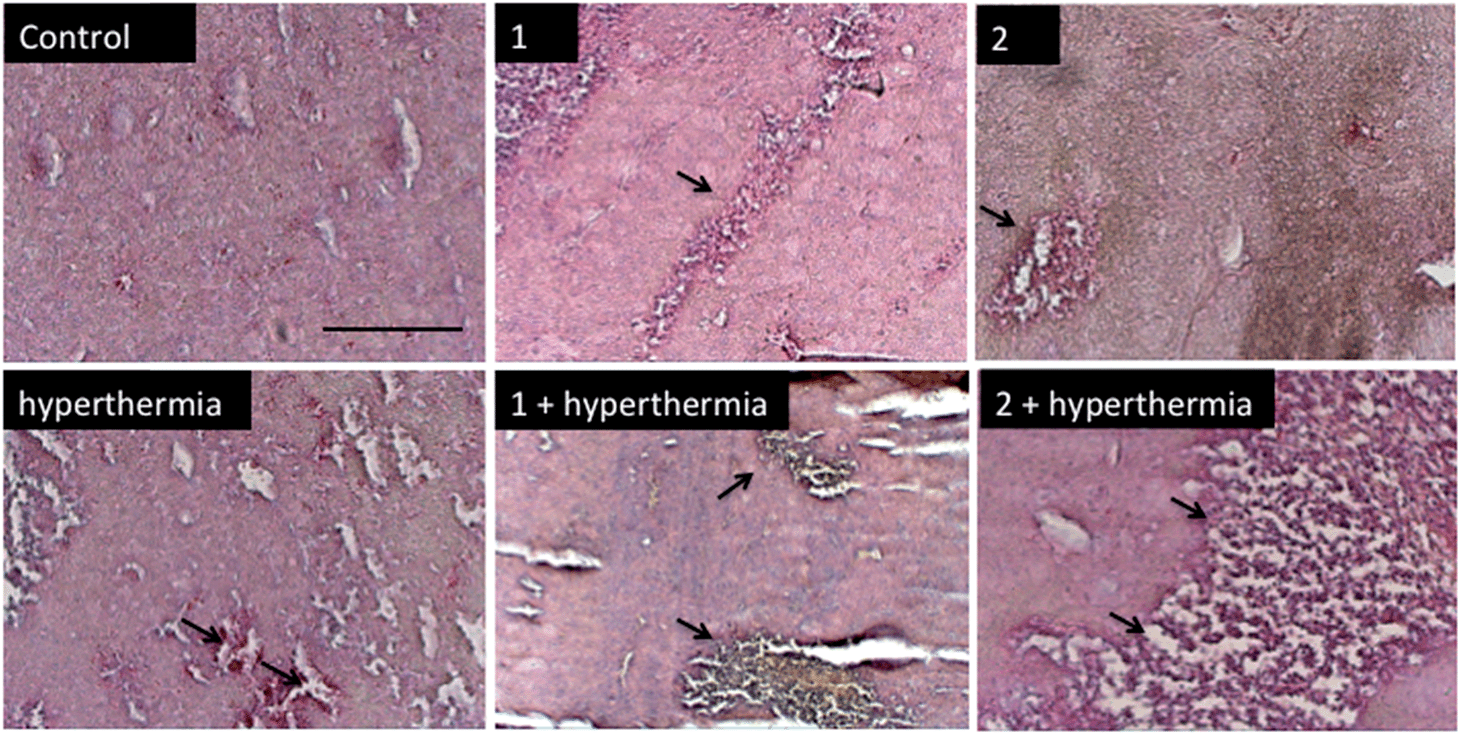 | ||
| Fig. 5 Histochemical analysis of tumor necrosis using hematoxylin and eosin (H&E) staining. Representative sections of tumors from different treatment groups are shown. Tumor necrosis is observed in the hyperthermia group and is extensive in the 2 + hyperthermia group (indicated by the black arrows). Scale bar = 100 μm. | ||
Treatment toxicity and side-effects of the monotherapies and duotherapy were assessed by monitoring the relative bodyweight of the mice on daily basis. At the end of the experiment the average body weight of the mice was not significantly different between all the treatment groups. At a behavioral level no unusual changes were observed including no change in food and water intake throughout the experiment period. Moreover, acute induction of inflammation of the tumors was not observed. Since ruthenium is not naturally present in living organisms, the distribution of ruthenium in tissues was determined after sacrificing mice treated with either 2 alone or in duotherapy. Organs were removed and dried, digested in nitric acid, and their ruthenium content determined by ICP-MS (Fig. 6).
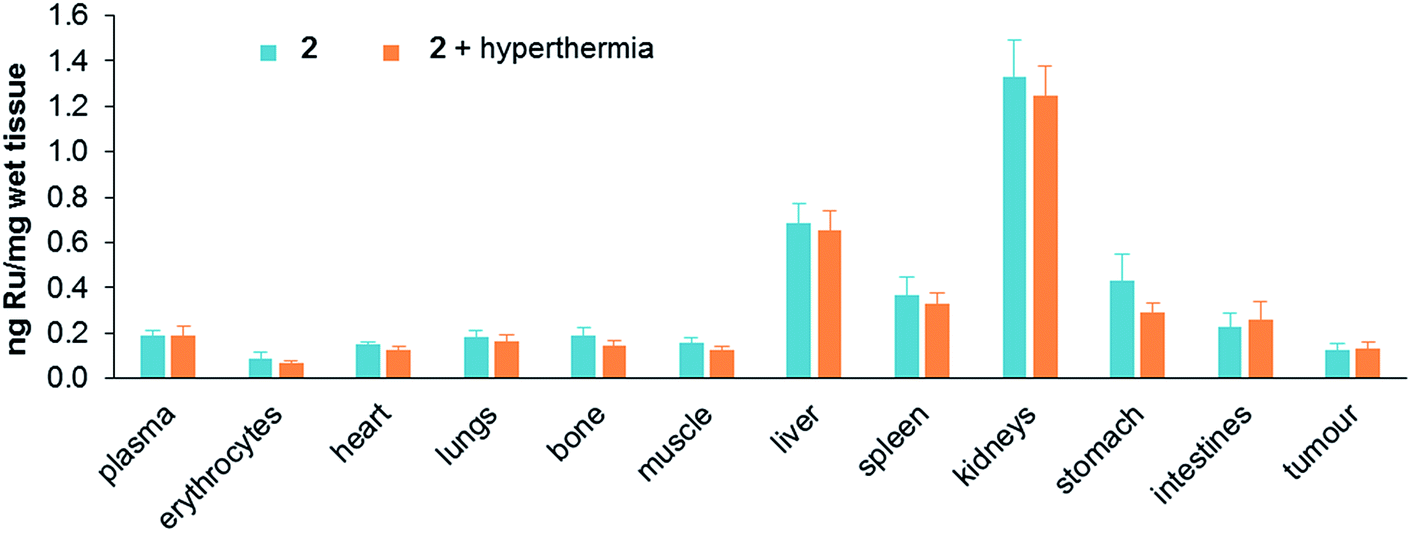 | ||
| Fig. 6 Ruthenium content in tissues (ng mg−1 of wet tissue) after mouse sacrifice for 2 applied as a monotherapy and 2 used in duotherapy with hyperthermia. Values are means ± SEM. | ||
Ruthenium accumulates in the kidneys, an organ associated with penetration and elimination of drugs, similar to that observed for the antimetastatic ruthenium(III) drug NAMI-A, which accumulates in kidneys in a dose-dependent manner,44 and in RAPTA-C-treated mice.42 Ruthenium also accumulates in the liver, but two fold less than in kidneys, followed by the stomach and spleen. The concentration of ruthenium in the lungs is low, in contrast to NAMI-A, which exhibits equivalent concentrations in lungs, kidneys and liver.45 Notably, duotherapy with hyperthermia does not significantly affect the ruthenium distribution in the organs.
Conclusions
We have validated the concept of using small molecule thermoresponsive compounds in combination with hyperthermia to inhibit tumor growth. Two very different classes of drugs with perfluorinated chains were studied, with the perfluorinated chain endowing the compounds with thermomorphic properties. Compound 1, which is derived from the alkylating agent chlorambucil, and 2, which is structurally related to a compound that selectively targets histone proteins in chromatin,46,47 exhibit a synergistic tumor growth reduction in duotherapy with local hyperthermia, with 2 showing a higher synergistic level with hyperthermia than 1, and resulting in a 90% reduction of tumor growth. Attempts were not made to elucidate the biomolecular targets of 1 and 2, however, it is not unreasonable to assume that 1, like chlorambucil, targets DNA (since the fluorous chain can be lost following cellular uptake), and that 2 may behave in the same way as other bifunctional ruthenium(II)-arene compounds.47,50 Notably, duotherapy decreases the number of proliferating cancer cells and enhances tumor necrosis in the absence of systemic damage to the surround tissues and organs. These findings not only validate the strategy, but also indicate more generally that drug modification with perfluorinated chains may enhance tumor growth inhibition efficacy without inducing additional side-effects, and could be applied to other drugs currently used in the clinic to treat cancer.Experimental
Compounds 1 and 2 were prepared as described previously.32,33 Compounds were dissolved in DMSO (Sigma-Aldrich) and diluted to a final concentration of 12.5 mg kg−1 in sterile saline (0.9% NaCl) prior i.p. injections. The control group animals were treated with 10% DMSO in sterile saline (0.9% NaCl) also given every 4 days.Cell implantation in athymic mice and treatment protocol
Experiments in 8 week old Swiss female athymic mice purchased from Charles River (Orleans, France) were carried out according to a protocol approved by the Committee for Animal Experiments for the Canton of Vaud, Switzerland (license 2772). LS174T colorectal adenocarcinoma cells (Cell Line Service GmbH, Eppelheim, Germany) were cultured in DMEM culture medium (Gibco, Carlsbad, USA) supplemented with 10% heat-inactivated fetal calf serum and 1% antibiotics (Sigma-Aldrich, St. Luis, USA). Mice were injected subcutaneously in the hind left leg with 5 × 105 LS174T cells resuspended in culture medium. When tumors reached a size of 5–6 mm in diameter, i.e. on day 6, mice received 12.5 mg kg−1 of either 1 or 2 in 10% DMSO in saline as 300 μL i.p. injection with or without hyperthermia (30 minutes at 42 °C, drug-heat interval of <15 minutes). Commercially available heating pads (Thermopad, 10200034 type 226, Solis AG – Switzerland, 30 × 40 cm heated pad, with 100 watt output and 6 temperature settings) to locally heat a subcutaneous tumor in the hind leg were used. To minimize the stress due to hyperthermia treatment immobilization anesthesia was carried out under 2.5% isoflurane and kept under 1.5% of isoflurane for the duration of the experiment with a humidified isofluorane system (Rothhacher GmbH, Bern, Switzerland). This treatment was repeated following the q4d3 schedule, i.e. a total of 3 doses at 4 day intervals. Tumor dimensions and body weight were measured daily. Tumor volumes (mm3) were calculated using the following formula volume = width2 × length × 0.5. At the end of the experiment, when the control tumor size reached 1000 mm3 the mice were euthanized and the tumors resected and fixed in formalin solution for immunohistochemistry. Blood, part of tumors and organs were snap-frozen for ruthenium distribution measurements.Immunohistochemistry
Tumor sections (5 μm) were blocked with 5% BSA in PBS and incubated with primary antibodies against Ki-67 (mouse anti-human Ki-67; 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :200, clone 1A4, Dako, Gostrup, Denmark). This step was followed by the addition of alkaline phosphatase conjugated polymer Mach 2 (Biocare Medical, Concord, USA) and visualized by Fast Blue BB/Naphthol-AS-MX-Phosphate, resulting in a blue/purple-colored precipitate. Hematoxylin and eosin staining was routinely preformed. Briefly, sections were deparaffinized in xylene I, II and III, washed and stained in hematoxylin (Harris Hematoxylin, Sigma) followed by eosin staining. In the next step they were dehydrated by washing in ethanol and mounted with DPX mountant and coverslip. Sections were imaged using Olympus microscope IX81 connected to Leica 200 scientific camera.
:200, clone 1A4, Dako, Gostrup, Denmark). This step was followed by the addition of alkaline phosphatase conjugated polymer Mach 2 (Biocare Medical, Concord, USA) and visualized by Fast Blue BB/Naphthol-AS-MX-Phosphate, resulting in a blue/purple-colored precipitate. Hematoxylin and eosin staining was routinely preformed. Briefly, sections were deparaffinized in xylene I, II and III, washed and stained in hematoxylin (Harris Hematoxylin, Sigma) followed by eosin staining. In the next step they were dehydrated by washing in ethanol and mounted with DPX mountant and coverslip. Sections were imaged using Olympus microscope IX81 connected to Leica 200 scientific camera.
Drug distribution measurements
Tissues of interest were collected, weight and dried until their weight remained constant. Samples were digested in concentrated nitric acid (1 mL) for 2 days. Samples were completed to a total volume of 8 mL with water. Indium was added as an internal standard at a concentration of 0.5 ppb. Determination of the metal content was achieved using an Elan DRC II ICP-MS instrument (Perkin-Elmer, Switzerland) equipped with a Meinhard nebulizer and a cyclonic spray chamber. The ICP-MS instrument was tuned using a solution containing 1 ppb of the following elements: Mg, In, Ce, Ba, Pb and U. External standards were prepared gravimetrically in an identical matrix to the samples with single element standards obtained from CPI International (Amsterdam, The Netherlands). Values were normalized per mg of wet tissue.Drug efficacy and synergy calculation
Drug efficacy (DE), or tumor regression, was calculated from the tumor growth of the treated group (T) and the control group (C) using the relation: DE = 1 − T/C. A synergy between the drug and hyperthermia was calculated with the fractional product method (for independent inhibitors).48 Limited to the final dose-point, the isobologram resulting from the Chou–Talalay combination index method cannot be built since it needs two points for each monotherapy. Nevertheless, the fractional product gives a first insight at the synergistic level between 1 and hyperthermia with the formula: f1,H = f1 × f1,H, where f1,H, f1, and f1,H are fractions of the total activity unaffected by the combination of 1 + hyperthermia, 1 and hyperthermia, respectively. Using drug efficacy (tumor reduction) values from the tumor growth curves: f1 × fH = (1 − DE1) (1 − DEH) = 0.226. Therefore, the combination 1 with hyperthermia was considered as synergistic if: DE1,H > 1 − 0.226 = 0.774, which was the case as DE1,H = 0.787. In the same way, for a synergistic effect between 2 with hyperthermia the value must be higher than: 1 − (1 − DE2)(1 − DEH) = 0.812, where DE2 = 0.656 and DEH = 0.454, which was the case as DE2,H = 0.899.Toxicity evaluation
To monitor the toxicity during the treatment the mice were observed daily in terms of relevant indexes such as body weight, appearance, provoked and natural behavior change and food and water intake.Statistical analysis
Tumor growth values are given as median values ± standard error of the mean. Statistical analysis was conducted using the R software (version 3.1.0).49 ANOVA tests were used to assess the significance of results and were accompanied by one-sided pairwise t-test ad-hoc analysis. *p < 0.05, **p < 0.01, ***p < 0.001.Acknowledgements
We thank the Swiss National Science Foundation and EPFL for financial support and Dr Andrea Weiss for maintenance of the LS174T cells.References
- T. Henne and D. Schmhl, Cancer Treat. Rev., 1985, 12, 77–94 CrossRef CAS.
- R. D. Issels, L. H. Lindner, J. Verweij, P. Wust, P. Reichardt, B. C. Schem, S. Abdel-Rahman, S. Daugaard, C. Salat, C. M. Wendtner, Z. Vujaskovic, R. Wessalowski, K. W. Jauch, H. R. Durr, F. Ploner, A. Baur-Melnyk, U. Mansmann, W. Hiddemann, J. Y. Blay and P. Hohenberger, Lancet Oncol., 2010, 11, 561–570 CrossRef CAS.
- K. Pietzner, R. B. Schmuck, C. Fotopoulou, J. Gellermann, F. Ismaeel, C. H. Cho, M. Kalden and J. Sehouli, Anticancer Res., 2011, 31, 2675–2677 CAS.
- R. D. Issels, Eur. J. Cancer, 2008, 44, 2546–2554 CrossRef CAS PubMed.
- A. Chilkoti, M. R. Dreher, D. E. Meyer and D. Raucher, Adv. Drug Deliver. Rev., 2002, 54, 613–630 CrossRef CAS.
- L. Li, T. L. ten Hagen, D. Schipper, T. M. Wijnberg, G. C. van Rhoon, A. M. Eggermont, L. H. Lindner and G. A. Koning, J. Controlled Release, 2010, 143, 274–279 CrossRef CAS PubMed.
- D. Needham, G. Anyarambhatla, G. Kong and M. W. Dewhirst, Cancer Res., 2000, 60, 1197–1201 CAS.
- L. H. Lindner, M. E. Eichhorn, H. Eibl, N. Teichert, M. Schmitt-Sody, R. D. Issels and M. Dellian, Clin. Cancer Res., 2004, 10, 2168–2178 CrossRef CAS.
- A. M. Ponce, Z. Vujaskovic, F. Yuan, D. Needham and M. W. Dewhirst, Int. J. Hyperthermia, 2006, 22, 205–213 CrossRef CAS PubMed.
- S. Unezaki, K. Maruyama, N. Takahashi, M. Koyama, T. Yuda, A. Suginaka and M. Iwatsuru, Pharm. Res., 1994, 11, 1180–1185 CrossRef CAS.
- G. Kong, G. Anyarambhatla, W. P. Petros, R. D. Braun, O. M. Colvin, D. Needham and M. W. Dewhirst, Cancer Res., 2000, 60, 6950–6957 CAS.
- Y. Min, C.-Q. Mao, S. Chen, G. Ma, J. Wang and Y. Liu, Angew. Chem., Int. Ed., 2012, 51, 6742–6747 CrossRef CAS PubMed.
- H. Maeda, H. Nakamura and J. Fang, Adv. Drug Deliver. Rev., 2013, 65, 71–79 CrossRef CAS PubMed.
- H. Maeda, Bioconjugate Chem., 2010, 21, 797–802 CrossRef CAS PubMed.
- A. K. Iyer, G. Khaled, J. Fang and H. Maeda, Drug Discovery Today, 2006, 11, 812–818 CrossRef CAS PubMed.
- J. K. Mills and D. Needham, Methods Enzymol., 2004, 387, 82–113 CAS.
- M. L. Hauck, S. M. LaRue, W. P. Petros, J. M. Poulson, D. Yu, I. Spasojevic, A. F. Pruitt, A. Klein, B. Case, D. E. Thrall, D. Needham and M. W. Dewhirst, Clin. Cancer Res., 2006, 12, 4004–4010 CrossRef CAS PubMed.
- D. Needham, in Biomaterials for cancer therapeutics : diagnosis, prevention, and therapy, ed. K. Park, Woodhead Publishing Series in Biomaterials, Oxford, 2013, ch. 12, vol. 66, pp. 270–348 Search PubMed.
- M. Franckena, Int. J. Hyperthermia, 2012, 28, 543–548 CrossRef PubMed.
- T. M. Zagar, J. R. Oleson, Z. Vujaskovic, M. W. Dewhirst, O. I. Craciunescu, K. L. Blackwell, L. R. Prosnitz and E. L. Jones, Int. J. Hyperthermia, 2010, 26, 618–624 CrossRef PubMed.
- G. Baronzio, G. Parmar, M. De Santis and A. Gramaglia, in Cancer Treatment – Conventional and Innovative Approaches, ed. L. Rangel, InTech, 2013, ch. 13, pp. 285–296 Search PubMed.
- K. Nakagawa, Y. Aoki, T. Fujimaki, M. Tago, A. Terahara, K. Karasawa, K. Sakata, Y. Sasaki, M. Matsutani and A. Akanuma, Int. J. Radiat. Oncol., Biol., Phys., 1998, 40, 1141–1149 CrossRef CAS.
- P. J. Miller, R. S. Hassanein, P. G. Giri, B. F. Kimler, P. O'Boynick and R. G. Evans, Int. J. Radiat. Oncol., Biol., Phys., 1990, 19, 275–280 CrossRef CAS.
- V. San Miguel, A. J. Limer, D. M. Haddleton, F. Catalina and C. Peinado, Eur. Polym. J., 2008, 44, 3853–3863 CrossRef CAS PubMed.
- J. Woo, G. N. C. Chiu, G. Karlsson, E. Wasan, L. Ickenstein, K. Edwards and M. B. Bally, Int. J. Pharm., 2008, 349, 38–46 CrossRef CAS PubMed.
- K. Kakinuma, R. Tanaka, H. Takahashi, Y. Sekihara, M. Watanabe and M. Kuroki, Int. J. Hyperthermia, 1996, 12, 157–165 CrossRef CAS.
- R. T. P. Poon and N. Borys, Expert Opin. Pharmacother., 2009, 10, 333–343 CrossRef CAS PubMed.
- N. Andhariya, B. Chudasama, R. V. Mehta and R. V. Upadhyay, J. Nanopart. Res., 2011, 13, 1677–1688 CrossRef CAS.
- K. Iga, N. Hamaguchi, Y. Igari, Y. Ogawa, K. Gotoh, K. Ootsu, H. Toguchi and T. Shimamoto, J. Pharmacol. Exp. Ther., 1991, 257, 1203–1207 CAS.
- A. A. Petryk, A. J. Giustini, R. E. Gottesman, P. A. Kaufman and P. J. Hoopes, Int. J. Hyperthermia, 2013, 29, 845–851 CrossRef CAS PubMed.
- M. P. Alvarez-Berrios, A. Castillo, J. Mendez, O. Soto, C. Rinaldi and M. Torres-Lugo, Int. J. Nanomed., 2013, 8, 1003–1013 Search PubMed.
- C. M. Clavel, O. Zava, F. Schmitt, B. H. Kenzaoui, A. A. Nazarov, L. Juillerat-Jeanneret and P. J. Dyson, Angew. Chem., Int. Ed., 2011, 50, 7124–7127 CrossRef CAS PubMed.
- C. M. Clavel, E. Păunescu, P. Nowak-Sliwinska and P. J. Dyson, Chem. Sci., 2014, 5, 1097–1101 RSC.
- J. B. Marmor, Cancer Res., 1979, 39, 2269–2276 CAS.
- O. Dahl and O. Mella, Anticancer Res., 1982, 2, 359–364 CAS.
- J. Overgaard, Int. J. Radiat. Oncol., Biol., Phys., 1989, 16, 535–549 CrossRef CAS.
- P. L. Ausmus, A. V. Wilke and D. L. Frazier, Cancer Res., 1992, 52, 4965–4968 CAS.
- R. Cavaliere, E. C. Ciocatto, B. C. Giovanella, C. Heidelberger, R. O. Johnson, M. Margottini, B. Mondovi, G. Moricca and A. Rossi-Fanelli, Cancer, 1967, 20, 1351–1381 CrossRef CAS.
- K. Overgaard and J. Overgaard, Eur. J. Cancer, 1972, 8, 65–78 CrossRef CAS.
- R. T. Pettigrew, J. M. Galt, C. M. Ludgate and A. N. Smith, Br. Med. J., 1974, 4, 679–682 CrossRef CAS.
- H. A. Coller, L. Sang and J. M. Roberts, PLoS Biol., 2006, 4, e83 Search PubMed.
- A. Weiss, B. H. Berndsen, M. Dubois, M. Müller, R. Schibli, A. W. Griffioen, P. J. Dyson and P. Nowak-Sliwinska, Chem. Sci., 2014, 5, 4742–4748 RSC.
- W. Nie, X. L. Ma, Y. X. Sang, Y. L. Li, X. Gao, G. C. Xu, G. B. Shen, H. S. Shi, X. X. Liu, F. T. Wang and Y. Q. Wei, Clin. Exp. Med., 2014, 14, 203–213 CrossRef CAS PubMed.
- S. Zorzet, A. Sorc, C. Casarsa, M. Cocchietto and G. Sava, Met.-Based Drugs, 2001, 8, 1–7 CrossRef CAS PubMed.
- M. Cocchietto and G. Sava, Pharmacol. Toxicol., 2000, 87, 193–197 CAS.
- B. Wu, M. S. Ong, M. Groessl, Z. Adhireksan, C. G. Hartinger, P. J. Dyson and C. A. Davey, Chem.–Eur. J., 2011, 17, 3562–3566 CrossRef CAS PubMed.
- Z. Adhireksan, G. E. Davey, P. Campomanes, M. Groessl, C. M. Clavel, H. J. Yu, A. A. Nazarov, C. H. F. Yeo, W. H. Ang, P. Droge, U. Rothlisberger, P. J. Dyson and C. A. Davey, Nat. Commun., 2014, 5, 3462 Search PubMed.
- A. Ocana, E. Amir, C. Yeung, B. Seruga and I. F. Tannock, Ann. Oncol., 2012, 23, 2161–2166 CrossRef CAS PubMed.
- R Core Team, R Foundation for Statistical Computing, Vienna, Austria, 3.1.0 edn, 2014 Search PubMed.
- M. V. Babak, S. M. Meier, K. Huber, J. Reynisson, A. A. Legin, M. A. Jakupec, A. Roller, A. Stukalov, M. Gridling, K. L. Bennett, J. Colinge, P. J. Dyson, G. Superti-Furga, B. K. Keppler and C. G. Hartinger, Chem. Sci., 2015, 6, 2449–2456 RSC.
| This journal is © The Royal Society of Chemistry 2015 |