
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Molecular glues for manipulating enzymes: trypsin inhibition by benzamidine-conjugated molecular glues†
Rina
Mogaki
a,
Kou
Okuro
*a and
Takuzo
Aida
*ab
aDepartment of Chemistry and Biotechnology, School of Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan. E-mail: okuro@macro.t.u-tokyo.ac.jp; aida@macro.t.u-tokyo.ac.jp; Tel: +81-3-5841-7251
bRIKEN Center for Emergent Matter Science, 2-1 Hirosawa, Wako, Saitama 351-0198, Japan
First published on 18th March 2015
Abstract
Water-soluble bioadhesive polymers bearing multiple guanidinium ion (Gu+) pendants at their side-chain termini (Gluen–BA, n = 10 and 29) that were conjugated with benzamidine (BA) as a trypsin inhibitor were developed. The Gluen–BA molecules are supposed to adhere to oxyanionic regions of the trypsin surface, even in buffer, via a multivalent Gu+/oxyanion salt-bridge interaction, such that their BA group properly blocks the substrate-binding site. In fact, Glue10–BA and Glue29–BA exhibited 35- and 200-fold higher affinities for trypsin, respectively, than a BA derivative without the glue moiety (TEG–BA). Most importantly, Glue10–BA inhibited the protease activity of trypsin 13-fold more than TEG–BA. In sharp contrast, mGlue27–BA, which bears 27 Gu+ units along the main chain and has a 5-fold higher affinity than TEG–BA for trypsin, was inferior even to TEG–BA for trypsin inhibition.
Introduction
If the behaviours of enzymes are manipulated by noncovalent interactions,1–4 one may possibly alter their functions and eventually control related biological events. In this context, one ambitious goal might be to noncovalently operate enzymes such that they perform different functions from their original tasks. In early studies, for instance, amphiphilic molecules have been utilized to introduce enzymes to non-aqueous media in order to expand the range of substrates.5 Nevertheless, from a pharmacological viewpoint, noncovalent enhancement or attenuation of certain enzymatic activities6–9 is a highly important and challenging subject. As a proof-of-concept study, we developed a benzamidine (BA) derivative appended with a particular bioadhesive polymer, i.e., molecular glue (Gluen–BA, Fig. 1), which bears at its side-chain termini multiple guanidinium ion (Gu+) pendants that can be salt-bridged with oxyanionic groups on target protein surfaces. BA is known to inhibit the protease activity of trypsin by blocking its substrate-binding site (Fig. 2a).10 In proximity to this binding site, trypsin has oxyanionic regions11 (blue-coloured) that allow the glue moiety of Gluen–BA to adhere (Fig. 2a). Hence, we envisioned that Gluen–BA could inhibit the protease activity of trypsin much more than a BA derivative without the glue moiety such as TEG–BA (Fig. 1), if the adhesion of the glue moiety (Gluen) does not hamper the appropriate BA positioning toward the active site (Fig. 2b).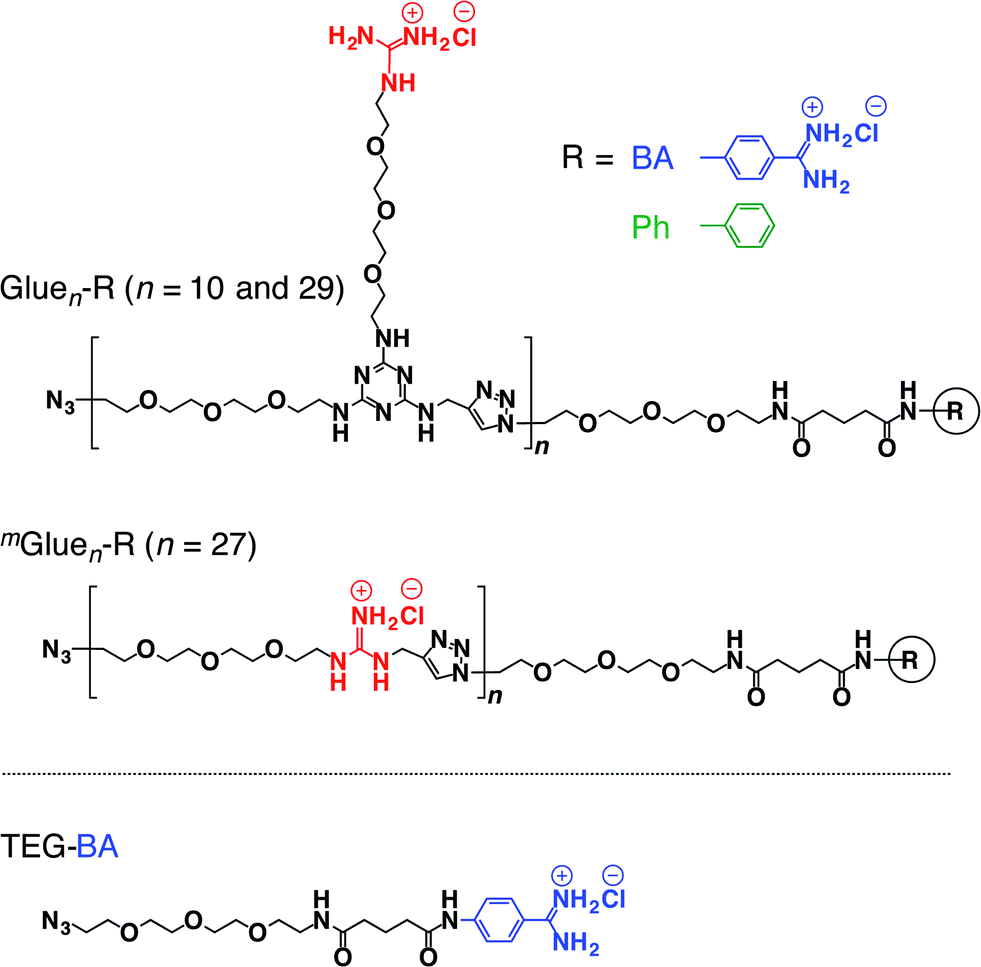 | ||
| Fig. 1 Schematic structures of bioadhesive molecular glues Gluen–R (n = 10 and 29) and mGluen–R (n = 27) conjugated with benzamidine (R = BA) as a trypsin inhibitor, and those of the reference molecular glue Gluen–Ph without an inhibitory terminus and TEGylated benzamidine (TEG–BA) without the glue moiety. | ||
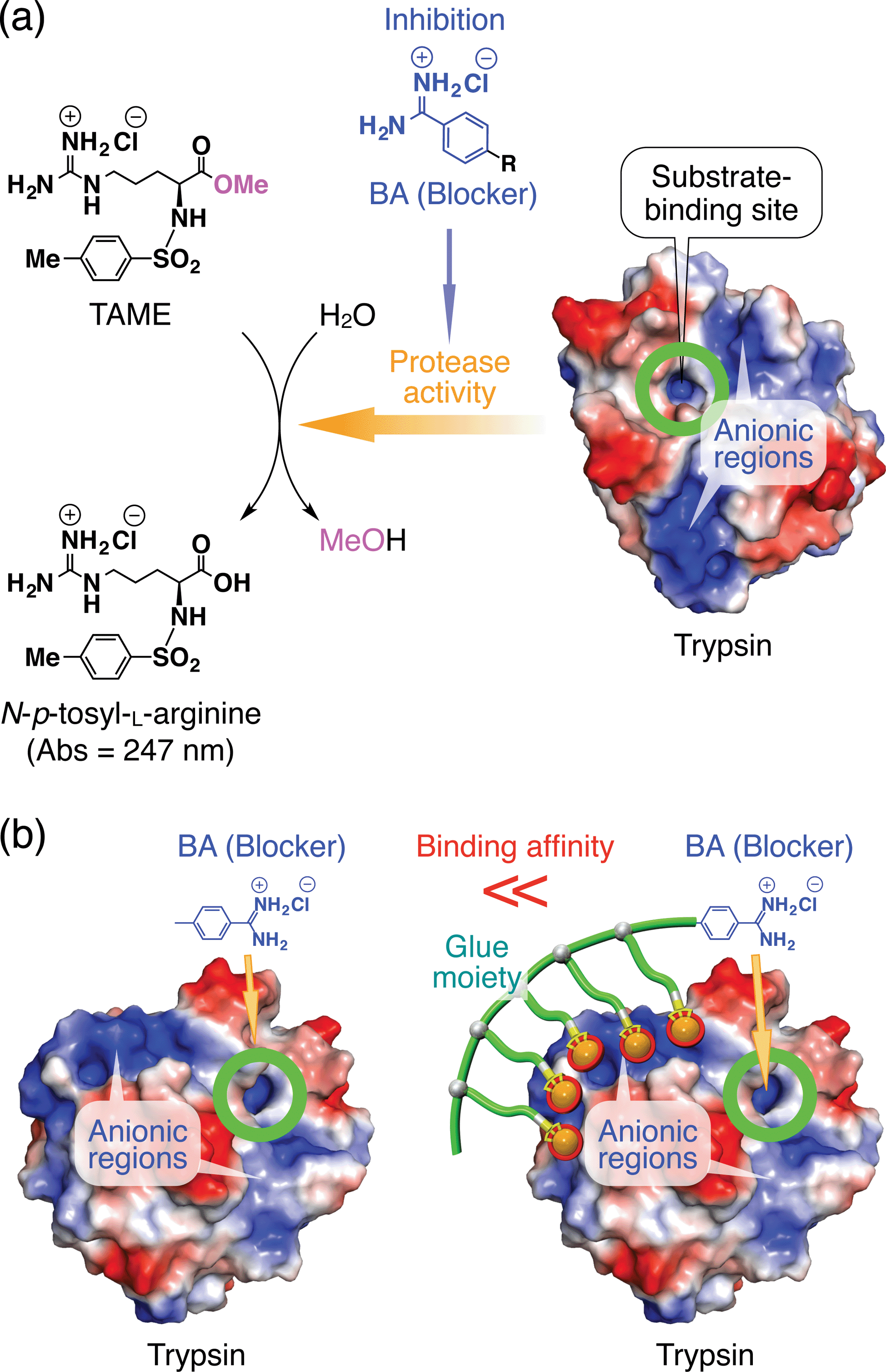 | ||
| Fig. 2 (a) Schematic illustration of the trypsin-catalyzed hydrolysis of N-p-tosyl-L-arginine methyl ester (TAME) to N-p-tosyl-L-arginine, exhibiting its characteristic absorption at 247 nm.10,11 Benzamidine (BA) derivatives are known to bind to the substrate-binding site of trypsin and inhibit its enzymatic activity.10 Trypsin has oxyanionic regions on its surface (blue-coloured) in proximity to the substrate-binding site, which allow the glue moiety of Gluen–BA to adhere. (b) Schematic representation of how the inhibitory effect of the blocking unit BA on the protease activity of trypsin is enhanced by conjugation with molecular glue. | ||
We have developed a series of dendritic molecular glues that bear multiple guanidinium ion (Gu+) pendants in the periphery of their water-soluble dendritic scaffolds.12–16 Such dendritic molecular glues tightly adhere to proteins,12–14 phospholipid membranes15 and clay nanosheets16 in aqueous media via the formation of multiple salt bridges between their Gu+ pendants and oxyanionic groups located on those targets. Most interesting along the line of this study was the observation that the photomechanical motion of an azobenzene-cored molecular glue can be transmitted to a phospholipid vesicular membrane via salt-bridge interactions and can consequently modulate its transmembrane ion permeation.15 This finding motivated us to extend the scope of the present study more to bio-related applications, i.e., noncovalent manipulation of enzymes. Recently, we confirmed that non-dendritic, linear polymers bearing side-chain Gu+ pendants17 are readily accessible alternatives to our prototype dendritic molecular glues. Hence, in the present study, we designed linear Gluen–BA with short (n = 10) and long (n = 29) glue moieties (Fig. 1). In addition to TEG–BA as a reference, we also prepared mGluen–BA (n = 27, Fig. 1) carrying 27 Gu+ units along the polymer main chain. As highlighted in this article, Glue10–BA inhibited the trypsin activity much more than TEG–BA (Fig. 1) without the glue moiety, whereas mGlue27–BA (Fig. 1) was inferior to TEG–BA despite the fact that it carries 27 Gu+ units and has a higher affinity than TEG–BA for trypsin.
Results and discussion
Gluen–BA was synthesized using a “click” reaction18–23 between TEG–BA and a three-armed monomer containing Gu+, azide and alkyne moieties. The reaction mixture was subjected to preparative size exclusion chromatography to allow fractionation of Glue10–BA and Glue29–BA. mGluen–BA (average n = 27, Fig. 1) was synthesized in a similar fashion by polymerizing a Gu+-containing linear monomer with terminal azide and alkyne groups. The average molecular weights of Gluen–BA and mGluen–BA were estimated by 1H NMR spectroscopy and static light scattering (SLS) analysis (Table S1†).We first investigated the effect of conjugation of molecular glues to BA on the binding affinity for trypsin. Trypsin is known to alter its conformation upon interaction with metal ions,24 polymers25 and proteins,26 resulting in circular dichroism (CD) spectral changes. Upon mixing with Glue10–BA, trypsin also changed its CD spectrum. As shown in Fig. S10c,† the CD intensity of trypsin (5 μM) at 237 nm decreased upon titration with Glue10–BA (0–7 μM) at 25 °C in Tris–HCl buffer (50 mM Tris–HCl, 10 mM CaCl2, pH 8.0). According to the reported method,27,28 we estimated the association constant (Kassoc) of Glue10–BA with trypsin to be 5.5 × 105 M−1 by fitting the fractions of bound trypsin (Fig. 3, red) to the Hill equation.27,28 As expected, Glue29–BA bearing a larger number (29) of Gu+ pendants exhibited a significantly higher Kassoc value of 3.2 × 106 M−1 (Fig. 3, brown and S10b†), reflecting an important role of multivalency. In sharp contrast, when TEG–BA without the glue moiety was used in the titration, the CD spectral change of trypsin was too small to detect unless the concentration range of TEG–BA for the titration was extended to 200 μM (Fig. 3, blue and S10a†). Accordingly, the Kassoc value was estimated to be 1.6 × 104 M−1, which is 35- and 200-fold lower than those observed for Glue10–BA and Glue29–BA, respectively. We also found that Glue10–Ph without BA (Fig. 1) binds to trypsin (Fig. S9†) with a Kassoc value (2.8 × 105 M−1; Fig. 3, green and S11a†) that is comparable to that of Glue10–BA, indicating that the glue moiety predominantly contributes to the binding affinity of Glue10–BA. Notably, the Kassoc value of mGlue27–BA containing 27 Gu+ units along the main chain (8.2 × 104 M−1; Fig. 3, purple and S11b†) was 40-fold lower than that of Glue29–BA possessing an almost comparable number of Gu+ pendants, and even 6.7-fold lower than that of Glue10–BA. As previously reported,12 the poor binding behaviour of mGlue27–BA is most likely due to a presumably small conformational flexibility of its in-chain Gu+ units compared with that of the Gu+ units at the side-chain termini in Gluen–BA.
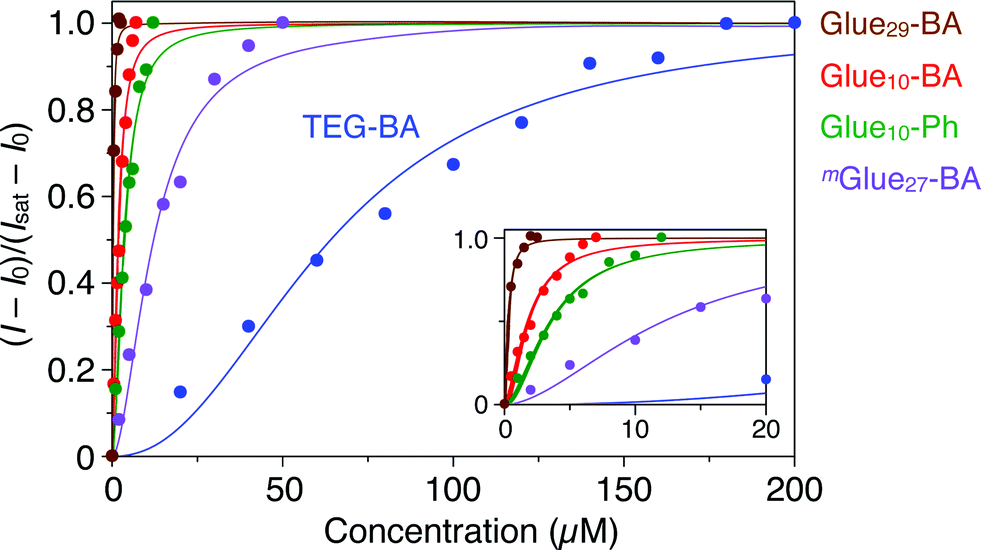 | ||
| Fig. 3 Circular dichroism (CD) spectral titration profiles of trypsin (5 μM) at 237 nm with molecular glues Glue29–BA (brown; 0–2.5 μM), Glue10–BA (red; 0–7 μM), Glue10–Ph (green; 0–12 μM) and mGlue27–BA (purple; 0–50 μM), together with reference TEG–BA (blue; 0–200 μM), at 25 °C in Tris–HCl buffer (50 mM Tris–HCl, 10 mM CaCl2, pH 8.0). The fractions of bound trypsin were calculated from (I − I0)/(Isat − I0), where I0, I and Isat represent the CD intensities before titration, observed with titrants and at the saturation point, respectively. | ||
Trypsin hydrolyses peptide linkages at the carboxyl side of lysine and arginine residues.29 As already described in Fig. 2a, this protease activity is inhibited by BA.10 Considering the exceptionally high affinity of Glue29–BA for trypsin, we expected that this BA-appended molecular glue might be the best inhibitor among those listed in Fig. 1. However, as observed by dynamic light scattering (DLS; Fig. S12†), trypsin/Glue29–BA, in contrast with other complexes such as trypsin/Glue10–BA and trypsin/Glue10–Ph, tends to form large aggregates (>200 nm), most likely due to the formation of physical crosslinks between its excessively long glue moiety and trypsin. Therefore, we conducted inhibitory assay experiments using Glue10–BA and mGlue27–BA, along with Glue10–Ph and TEG–BA as references, but did not use Glue29–BA. Nevertheless, we found that upon conjugation with Glue10, BA’s inhibitory effect was considerably enhanced. As shown in Fig. 4 (black), when N-p-tosyl-L-arginine methyl ester (TAME, 1 mM) as a substrate was mixed with trypsin (20 nM) at 25 °C in Tris–HCl buffer (50 mM Tris–HCl, 10 mM CaCl2, pH 8.0), TAME was hydrolysed to N-p-tosyl-L-arginine (Fig. 2a), exhibiting an increase in its characteristic absorption at 247 nm.10 However, when 10 μM of Glue10–BA was added to the reaction system, the hydrolysis of TAME was considerably decelerated (Fig. 4, red). Although TEG–BA did not exhibit detectable inhibition at 10 μM (Fig. 4, blue), Glue10–BA explicitly inhibited the trypsin activity even at 2.5 μM (Fig. 4, orange). As shown in Fig. 5, the hydrolytic activity of trypsin was evaluated using the pseudo-first order reaction kinetics, and normalized to that of untreated trypsin (20 nM). The sigmoidal profile, obtained for the case with TEG–BA in Fig. 5 (blue), allowed estimation of the half-maximal inhibitory concentration (IC50) of TEG–BA as 79 μM. Notably, Glue10–BA exhibited a 13-fold greater inhibitory effect (IC50 = 6.2 μM; Fig. 5, red) than TEG–BA. In sharp contrast, when Glue10–Ph was used in place of Glue10–BA, no inhibition of the trypsin activity was observed (Fig. 5, green) even when [Glue10–Ph] was higher than 20 μM. Hence, the adhesion of the glue moiety does not hamper the enzymatic activity of trypsin, but primarily contributes to the stabilization of the BA/trypsin complex. As previously described, the binding affinity of mGlue27–BA for trypsin is only 15% of that of Glue10–BA, but still 5-fold higher than that of TEG–BA. However, mGlue27–BA exhibited a lower inhibitory effect than TEG–BA under identical conditions, and minimally inhibited the hydrolytic activity of trypsin (Fig. 5, purple). We presume that the poor conformational flexibility of the in-chain Gu+ units in the glue moiety hinders the ability of the conjugated BA terminus to properly block the substrate-binding site of trypsin. To rationalize the concept of blocker-appended molecular glues for pharmacological applications, this issue should be taken into consideration.
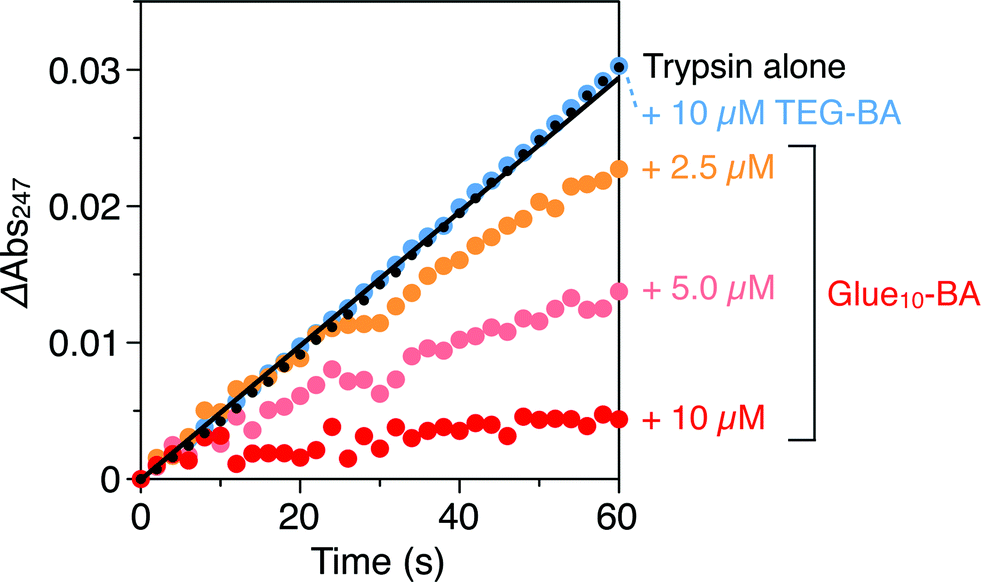 | ||
| Fig. 4 Absorption spectral changes at 247 nm of Tris–HCl buffer (50 mM Tris–HCl, 10 mM CaCl2, pH 8.0) solutions of a mixture of TAME (1 mM) and trypsin (20 nM) in the absence (black) and presence of 2.5 (orange), 5.0 (pink) and 10 μM (red) of Glue10–BA, and in the presence of 10 μM TEG–BA (blue). | ||
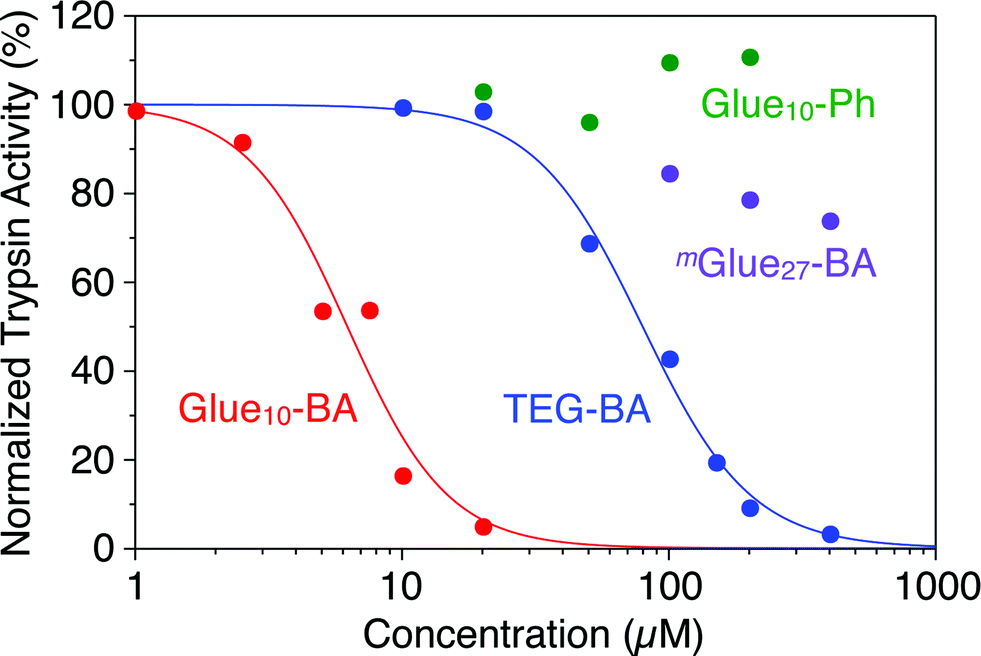 | ||
| Fig. 5 Hydrolytic activities of trypsin (20 nM), as estimated from the rates of absorption increase at 247 nm, normalized to that of untreated trypsin, at 25 °C in Tris–HCl buffer (50 mM Tris–HCl, 10 mM CaCl2, pH 8.0) containing TAME (1 mM) in the presence of TEG–BA (blue), Glue10–BA (red), Glue10–Ph (green) and mGlue27–BA (purple). | ||
Conclusions
Through a comparative inhibition study on the protease activity of trypsin using Gluen–BA, mGluen–BA and TEG–BA (Fig. 1) as potential trypsin inhibitors, we demonstrated that an active-site blocker such as BA efficiently inhibits the trypsin activity when its conjugated glue moiety (Gluen) can hold the blocker stably onto the active site through adhesion to a proximal oxyanionic region (Fig. 2b). Of particular interest is the obviously smaller inhibitory effect of mGlue27–BA compared to TEG–BA, despite the fact that mGlue27–BA has a 5-fold higher affinity than TEG–BA for trypsin. The incorporation of a mechanism to respond to biological or physical stimuli for controlling the operation of the blocker unit is an interesting subject worthy of further investigation.Methods
Trypsin activity assay
To a Tris–HCl buffer (50 mM Tris–HCl, 10 mM CaCl2, pH 8.0) solution of trypsin (20 nM) was added a Tris–HCl buffer solution of TEG–BA, and the mixture was incubated at 25 °C for 1 min. Then, to the resultant solution was added a Tris–HCl buffer solution of N-p-tosyl-L-arginine methyl ester hydrochloride (TAME, final concentration 1 mM; Fig. 2a), and the absorption intensity at 247 nm (ref. 10) was traced over a period of 1 min. The trypsin activity was determined using pseudo-first order reaction kinetics and normalized to that of untreated trypsin. The trypsin activities in the presence of Gluen–BA, mGluen–BA and Gluen–Ph were likewise evaluated.Acknowledgements
This work was partially supported by a JSPS Grant-in-Aid for Young Scientists (B) (26810046) to K.O. R.M. thanks JSPS for Program for Leading Graduate Schools (GPLLI) financial support. We appreciate Prof. T. Nagamune and Dr K. Minamihata (The University of Tokyo) for zeta potential measurements.Notes and references
- M. A. Kostiainen, O. Kasyutich, J. J. L. M. Cornelissen and R. J. M. Nolte, Nat. Chem., 2010, 2, 394 CrossRef CAS PubMed.
- N. M. Matsumoto, P. Prabhakaran, L. H. Rome and H. D. Maynard, ACS Nano, 2013, 7, 867 CrossRef CAS PubMed.
- T. H. Nguyen, S.-H. Kim, C. G. Decker, D. Y. Wong, J. A. Loo and H. D. Maynard, Nat. Chem., 2013, 5, 221 CrossRef CAS PubMed.
- D. A. Torres, M. Garzoni, A. V. Subrahmanyam, G. M. Pavan and S. Thayumanavan, J. Am. Chem. Soc., 2014, 136, 5385 CrossRef PubMed.
- P. L. Luisi, Angew. Chem., Int. Ed. Engl., 1985, 24, 439 CrossRef.
- B. S. Sandanaraj, D. R. Vutukuri, J. M. Simard, A. Klaikherd, R. Hong, V. M. Rotello and S. Thayumanavan, J. Am. Chem. Soc., 2005, 127, 10693 CrossRef CAS PubMed.
- R. Roy, B. S. Sandanaraj, A. Klaikherd and S. Thayumanavan, Langmuir, 2006, 22, 7695 CrossRef CAS PubMed.
- P. De, M. Li, S. R. Gondi and B. S. Sumerlin, J. Am. Chem. Soc., 2008, 130, 11288 CrossRef CAS PubMed.
- A. J. Keefe and S. Jiang, Nat. Chem., 2012, 4, 59 CrossRef CAS PubMed.
- D. R. Corey and M. A. Phillips, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 4106 CrossRef CAS.
- Image of 1S85 from: T. R. Transue, J. M. Krahn, S. A. Gabel, E. F. DeRose and R. E. London, Biochemistry, 2004, 43, 2829 CrossRef CAS PubMed , created with PyMol 1.3.
- K. Okuro, K. Kinbara, K. Tsumoto, N. Ishii and T. Aida, J. Am. Chem. Soc., 2009, 131, 1626 CrossRef CAS PubMed.
- K. Okuro, K. Kinbara, K. Takeda, Y. Inoue, A. Ishijima and T. Aida, Angew. Chem., Int. Ed., 2010, 49, 3030 CrossRef CAS PubMed.
- N. Uchida, K. Okuro, Y. Niitani, X. Ling, T. Ariga, M. Tomishige and T. Aida, J. Am. Chem. Soc., 2013, 135, 4684 CrossRef CAS PubMed.
- Y. Suzuki, K. Okuro, T. Takeuchi and T. Aida, J. Am. Chem. Soc., 2012, 134, 15273 CrossRef CAS PubMed.
- Q. Wang, J. L. Mynar, M. Yoshida, E. Lee, M. Lee, K. Okuro, K. Kinbara and T. Aida, Nature, 2010, 463, 339 CrossRef CAS PubMed.
- S. Tamesue, M. Ohtani, K. Yamada, Y. Ishida, J. M. Spruell, N. A. Lynd, C. J. Hawker and T. Aida, J. Am. Chem. Soc., 2013, 135, 15650 CrossRef CAS PubMed.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004 CrossRef CAS.
- D. D. Díaz, S. Punna, P. Holzer, A. K. McPherson, K. B. Sharpless, V. V. Fokin and M. G. Finn, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 4392 CrossRef.
- R. Roux, L. Sallet, P. Alcouffe, S. Chambert, N. Sintes-Zydowicz, E. Fleury and J. Bernard, ACS Macro Lett., 2012, 1, 1074 CrossRef CAS.
- T. Gong, B. J. Adzima, N. H. Baker and C. N. Bowman, Adv. Mater., 2013, 25, 2024 CrossRef CAS PubMed.
- A. A. Accurso, M. Delaney, J. O'Brien, H. Kim, P. M. Iovine, D. D. Díaz and M. G. Finn, Chem.–Eur. J., 2014, 20, 10710 CrossRef CAS PubMed.
- B. Sandmann, B. Happ, M. D. Hager, J. Vitz, E. Rettler, P. Burtscher, N. Moszner and U. S. Schubert, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 239 CrossRef CAS.
- L. Yang, Z. Gao, Y. Cao, R. Xing and X. Zhang, ChemBioChem, 2005, 6, 1191 CrossRef CAS PubMed.
- H. L. Luessen, J. C. Verhoef, G. Borchard, C.-M. Lehr, A. G. de Boer and H. E. Junginger, Pharm. Res., 1995, 12, 1293 CrossRef CAS.
- H. Huang and M. Zhao, Eur. Food Res. Technol., 2007, 227, 361 CrossRef PubMed.
- D. Hebenstreit and F. Ferreira, Allergy, 2005, 60, 1208 CrossRef CAS PubMed.
- N. J. Greenfield, Nat. Protoc., 2007, 1, 2733 CrossRef PubMed.
- K. A. Walsh, Methods Enzymol., 1970, 19, 41 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of TEG–BA, Gluen–BA, mGluen–BA and Gluen–Ph; 1H NMR, 13C NMR, MALDI-TOF MS, electronic absorption, and CD spectra; zeta potential distributions; SLS plots; DLS histograms; and related experimental procedures. See DOI: 10.1039/c5sc00524h |
| This journal is © The Royal Society of Chemistry 2015 |