
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn vivo visible light-triggered drug release from an implanted depot†
Carl-Johan
Carling
ab,
Mathieu L.
Viger
ab,
Viet Anh
Nguyen Huu
bc,
Arnold V.
Garcia
ab and
Adah
Almutairi
*abcd
aSkaggs School of Pharmacy and Pharmaceutical Sciences, University of California, San Diego, 9500 Gilman Dr., La Jolla, California 92093, USA. E-mail: aalmutairi@ucsd.edu; Tel: +1-858-246-0871
bKACST-UCSD Center for Excellence in Nanomedicine, University of California, San Diego, 9500 Gilman Dr., La Jolla, California 92093, USA
cDepartment of NanoEngineering, University of California, San Diego, 9500 Gilman Dr., La Jolla, California 92093, USA
dDepartment of Materials Science and Engineering, University of California, San Diego, 9500 Gilman Dr., La Jolla, California 92093, USA
First published on 6th October 2014
Abstract
Controlling chemistry in space and time has offered scientists and engineers powerful tools for research and technology. For example, on-demand photo-triggered activation of neurotransmitters has revolutionized neuroscience. Non-invasive control of the availability of bioactive molecules in living organisms will undoubtedly lead to major advances; however, this requires the development of photosystems that efficiently respond to regions of the electromagnetic spectrum that innocuously penetrate tissue. To this end, we have developed a polymer that photochemically degrades upon absorption of one photon of visible light and demonstrated its potential for medical applications. Particles formulated from this polymer release molecular cargo in vitro and in vivo upon irradiation with blue visible light through a photoexpansile swelling mechanism.
Introduction
Controlling chemistry in space and time with light has proven enormously useful in biological research. For example, optogenetics is considered the breakthrough of the decade and has revolutionized neuroscience.1 However, current means of controlling cellular processes using light require tedious synthetic or genetic development of tools specific to each target. On the other hand, light-responsive nanocarriers2 can be used to control the activity of any encapsulated bioactive molecule, including proteins and nucleic acids, without the need for chemical modification. Such versatile tools have enormous potential to advance fundamental understanding of physiological, developmental, and disease processes. Externally triggered release of drugs from nanomaterials may also have exciting applications in the treatment of disease, as it could allow precise coordination with biological rhythms or disease activity. Since particles can be injected within the tissue of interest and remain localized in a depot-like manner, the pharmacokinetic profile of the encapsulated drug can be altered. Such an approach would minimize injections and harmful side effects while maximizing therapeutic outcome.Light-responsive technologies useful in vivo must be highly efficient and capable of responding to electromagnetic radiation that can innocuously penetrate into tissue. To date most photochemical systems do not meet such physiological requirements, as the majority rely on UV light activation,2,3 which is both damaging to cells and does not appreciably penetrate tissues. Systems making use of near-infrared (NIR) laser light by multi-photon approaches, which include two-photon excitation4 and upconverted UV light,5 have been developed to sidestep these limitations. Unfortunately, achieving sufficient photocleavage of polymer to induce release from nanoparticles by these approaches currently requires extended irradiation at high laser powers, which could lead to heat-induced damage of biological tissues.6
The development of one-photon visible light-responsive carriers4c,h,i,7 represents an attractive alternative to both one-photon UV- and two-photon NIR-responsive carriers for in vivo applications because it is several orders of magnitude more efficient than two-photon excitation,8 requiring much shorter irradiation times at lower powers, and is less harmful to cells than UV light.
Inspired by recent developments in the photo-uncaging field, we synthesized a polymer that degrades upon one-photon absorption of blue visible light (polymer 1, Fig. 1, Scheme S1†) by incorporating a recently developed red-shifted photocage developed by Donato et al.8 and used it to formulate particles encapsulating various payloads. Advantages of this photocage over existing systems3 such as coumarin, nitroveratryls, ruthenium complexes and perylene are the intense visible light absorption up to 500 nm and photo-reactivity in hydrophobic environments, a crucial requirement as most polymer-based delivery vehicles have a hydrophobic interior.
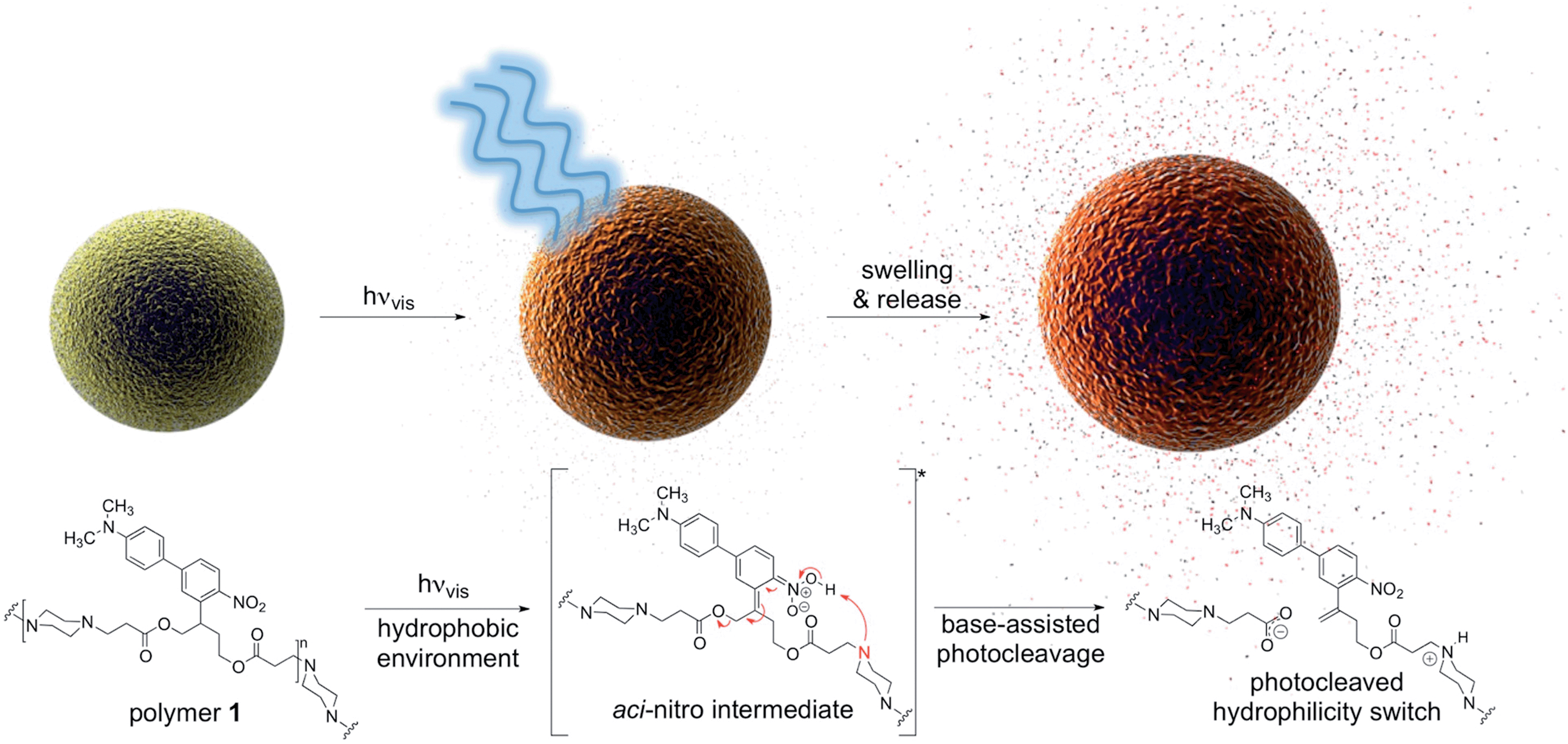 | ||
| Fig. 1 Visible light irradiation of particles composed of polymer 1 in aqueous media induces swelling and release of molecular cargo. Tertiary amines within the polymer backbone assist deprotonation of the aci-nitro intermediate in the hydrophobic particle microenvironment, leading to β-elimination and photocleavage in favour of photorearrangement. | ||
The results presented in this study highlight the practical utility of one-photon visible light photochemistry for applications in dense, non-transparent mammalian tissues. In an in vivo mouse model we demonstrate light-triggered drug release from a subcutaneously implanted polymer 1 particle depot with blue visible light.
Results and discussion
Design, light response and degradation mechanism
In this work, the propyloxy chain of the previously reported ANBP8 (2-(4′-(N,N-dimethylamino)-4-nitro-[1,1′-biphenyl]-3-yl)propyl carbonyl) photocage was elongated to yield the butane-diol derivative 2-(4′-N-dimethylamino-4-nitro-[1,1′-biphenyl]-3-yl)butane-1,4-diyl dicarbonyl (ANBB, Fig. 1 and 2, Scheme S1†). This modification facilitates polymerization and enhances the kinetics of photodegradation by placing the photolabile bond directly in the polymer backbone (Fig. 1).4i,9 Like ANBP, ANBB is photochemically active in hydrophobic environments. As previously reported, the photochemistry of this class of photoresponsive molecules is strongly dependent on solvent and basicity.10 In water, the photo-induced aci-nitro intermediate (Fig. 1) is deprotonated, causing the compound to undergo photocleavage by a β-elimination pathway to yield a nitro-alkene derivative.8 However, in hydrophobic non-basic environments, these molecules photorearrange to a hydroxy-nitroso isomer10 (Fig. 2). To increase the release efficiency from corresponding particles and potentially enhance metabolism/excretion of the photodegraded carrier material, we reasoned that the introduction of basic tertiary amines in the polymer 1 backbone would assist deprotonation of the photo-induced aci-nitro intermediate in the hydrophobic particle microenvironment in favour of photorearrangement (Fig. 1, 2c and d). To inform the design of our polymer, we examined the photoreactions of model compounds 2 (no tertiary amines) and 3 (tertiary amines) in hydrophobic solvent to mimic the particle microenvironment (Fig. 2, Scheme S1†). As revealed by HPLC-MS, the non-basic compound 2 yielded only the photostable photorearranged nitroso-hydroxy isomer 2a upon irradiation with visible light (λex = 400–500 nm) in CH2Cl2 (Fig. 2a and b), whereas the basic compound 3 underwent a much more complex photoreaction (Fig. 2c and d). Analysis of the photoreaction of compound 3 revealed that the major reaction product was indeed the photocleaved nitro-alkene derivative 3a, which was isolated and identified by 1H NMR spectroscopy (Fig. S1†) and high-resolution mass spectroscopy. However, compound 3 was also observed to directly generate the photorearranged hydroxy-nitroso minor product 3b, alongside a photocleaved minor product 3c upon irradiation (Fig. 2c and d). Furthermore, as has been previously observed with similar compounds,10 the photocleaved nitro-alkene product 3a is not photostable and undergoes multiple subsequent photochemical transformations upon prolonged irradiation (Fig. 2c and d). Despite considerable efforts, secondary photoproducts (3c, 3d, 3e and minor products) could not be identified. The observed photochemical behavior of compound 3 confirms that the introduction of tertiary amines indeed promotes photocleavage in hydrophobic environments, such as the interior of a polymeric particle.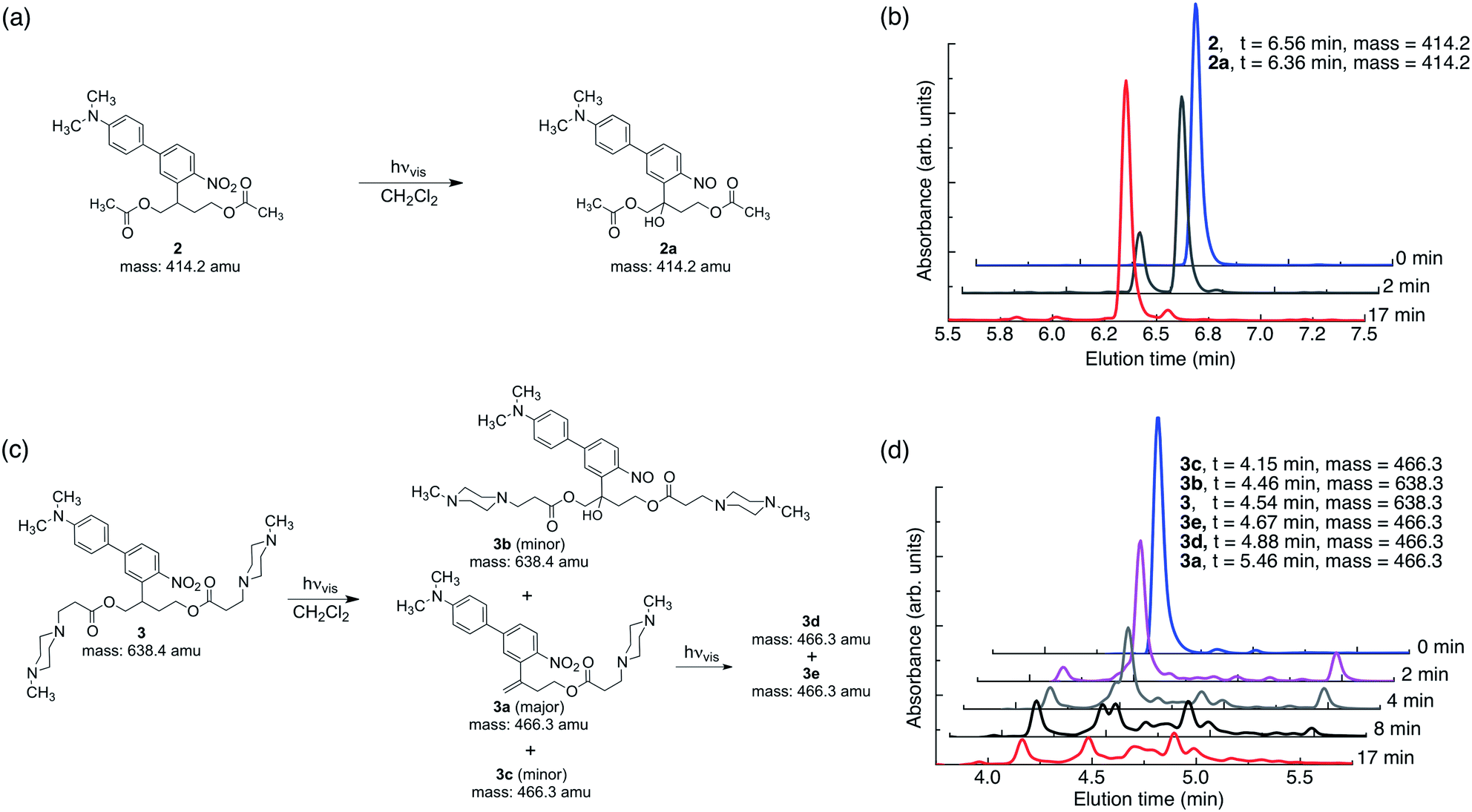 | ||
| Fig. 2 Tertiary amines modulate the photochemistry of ANBB in hydrophobic environments. (a and b) Compound 2 cleanly reacts upon irradiation with visible light (λex = 400–500 nm, 0.18 W, 0.21 W cm−2) in CH2Cl2 (6 × 10−5 M) to yield photorearranged hydroxy-nitroso product 2a. (c and d) Compound 3 has more complex photochemistry and yields photocleaved 3a (major product), photorearranged 3b (minor product) and photocleaved 3c (minor product) upon irradiation with visible light in CH2Cl2. 3a is not photostable and reacts further upon prolonged irradiation to yield major photoproducts 3d and 3e. The exact mass calculated (2, 2a, 3, 3a and 3c) and measured (2, 2a, 3a, 3b, 3c, 3d and 3e (mass of protons subtracted)) is indicated. | ||
The photoresponsive polymer 1 (Mw = 31.7 kDa, PDI = 2.7) was synthesized in five steps at 17% overall yield starting from a previously reported compound8 (see ESI and Scheme S1† for synthetic details). Upon irradiation of polymer 1 in CH2Cl2 with visible light (λex = 400–500 nm), the long-wavelength absorption peak at λmax = 398 nm decreases as a new absorption peak at λmax = 458 nm increases, resulting in a visually recognizable color change from yellow to orange (Fig. 3a inset). The substantial increase in the GPC retention time of polymer 1 after irradiation in CH2Cl2 indicates amine-assisted photocleavage upon irradiation and the formation of smaller segments (Fig. 3b). The greater solubility of the photoproducts of polymer 1 in methanol and in water containing 1% w/v Pluronic® F127 further indicates photodegradation into smaller, more polar segments (Fig. S2†).
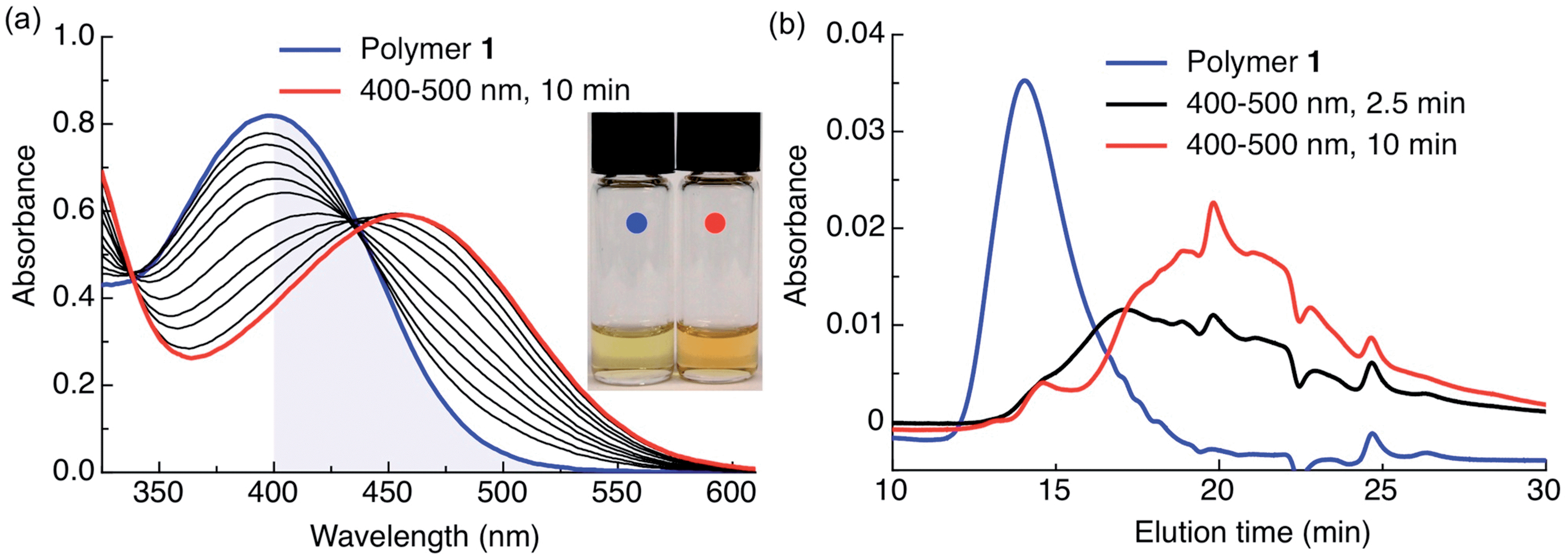 | ||
| Fig. 3 Polymer 1 photodegrades upon irradiation with visible light in hydrophobic environments. (a) Spectral changes in the UV-Vis spectrum of polymer 1 irradiated in CH2Cl2 (0.04 mg mL−1, 0.5 mL) with visible light (λex = 400–500 nm, 0.18 W, 0.21 W cm−2). The blue shaded area highlights visible light absorption. Inset: color change following complete photoconversion. (b) GPC chromatogram of polymer 1 before (blue trace, Mw = 31.7 kDa, PDI = 2.7, PMMA standard) and after irradiation in CH2Cl2 (0.04 mg mL−1, 0.5 mL) with visible light (λex = 400–500 nm, 0.18 W, 0.21 W cm−2) for 2.5 min (black trace) and 10 min (red trace). Absorbance sampled at 300 nm; peak at 24 min is the toluene internal standard. | ||
HPLC-MS analysis of the methanol-soluble photodegraded material revealed a complex mixture of photoproducts, including peaks with masses corresponding to the smallest photocleaved products anticipated (Fig. S3†). We also studied the photochemistry of polymer 1 in CDCl3 by 1H NMR spectroscopy. Peaks corresponding to the photocleaved nitro-alkene product were observed upon irradiation by comparing the spectrum of irradiated polymer 1 to that of 3a (Fig. S1†). However, since the photocleaved nitro-alkene product was quickly consumed by secondary photoreactions and the product precipitated (likely because of high polarity), line broadening and decreased peak heights thwarted detailed analysis.
Attenuation of visible light by dermal tissue does not prevent polymer photocleavage
To test the practical utility of one-photon visible light driven photochemistry for in vivo applications, we examined the efficiency of photochemistry after passage of light through hairless mouse skin (thickness: ∼0.45 mm) sandwiched between two glass slides (Fig. 4a, S4†). Polymer 1 solutions were irradiated with and without this tissue filter in the beam path and the photoreaction kinetics were measured by UV-Vis absorption spectroscopy (Fig. 4b). Under direct blue light excitation (λex = 400–500 nm, 0.18 W), the photoreaction reached 100% completion after only 10 min (Fig. 4b, open circles), whereas under skin-attenuated irradiation, the photoreaction reached 22% completion within the same irradiation time (Fig. 4b, solid circles). As the optical power reaching the sample after passage through the glass/skin tissue filter was attenuated by 82% (glass/skin = 0.03 W, skin alone = 0.06 W, Table S1†), this reduction indicates a linear correlation between optical power and reactivity of one-photon absorption, since the rate constant and reaction completion were reduced by 79% and 78%, respectively. These results thus highlight the utility of delivery systems that respond efficiently to one-photon visible light.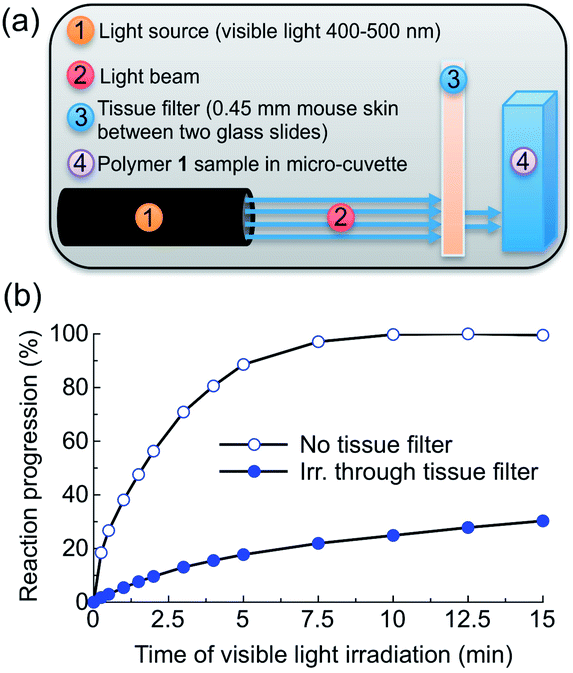 | ||
| Fig. 4 Visible light triggers polymer 1 photochemistry after passage through mouse skin. (a) Experimental setup. (b) Reaction progression of CH2Cl2 solutions of polymer 1 (0.04 mg mL−1, 0.5 mL) as they are irradiated directly (open circles) or through mouse skin (solid circles) with visible light (λex = 400–500 nm, 0.18 W, 0.21 W cm−2). The photoreactions were monitored by UV-Vis spectroscopy; the skin-attenuated reaction was further irradiated directly to fully react the polymer. Reaction progression (RP) was calculated by the following formula: RP = ((A − Amin)/(Amax − Amin)) × 100%, where A is the absorption at 490 nm, Amin the absorbance at 0 min, and Amax the maximum absorbance after complete photoreaction. | ||
Formulation of Nile red-loaded polymeric particles and photo-induced release
As our ultimate goal is light-triggered drug delivery, we next examined release of cargo in vitro using Nile red as a model drug. As the traditional method of using Nile red to follow release by measuring fluorescence quenching cannot distinguish between an increase in particle hydrophilicity and cargo release, we employed a distinct approach. So that release of dye would correspond with an increase in fluorescence, we sought to encapsulate a high concentration of dye to quench its fluorescence inside the particle. For this purpose, polymer 1 was formulated into particles by electrospray11 (see ESI† for Experimental details). This convenient formulation method produces dense, solid polymer particles in a one-step process with unrivalled payload encapsulation efficiencies, a highly desirable feature for therapeutic applications. Representative scanning electron microscopic (SEM) images of the particles are shown in Fig. S5.† We studied the kinetics of light-induced release of Nile red from polymer 1 particles (P-1-NR, size: 1.1 ± 0.3 μm (SEM), dye loading: 5.6% w/w) by directly monitoring changes in fluorescence intensity of the aqueous particle suspensions containing 1% w/v Pluronic® F127 upon irradiation with visible light. When P-1-NR was irradiated with blue visible light, a substantial increase in fluorescence intensity, indicative of photo-triggered release, was observed (Fig. 5a, solid circles). In contrast, the fluorescence intensity did not change significantly for particles not exposed to visible light (Fig. 5a, open circles). We further quantitatively measured Nile red released from particles into solution by measuring the fluorescence of the supernatant following centrifugation. We found that while only 2.0 ± 0.7% Nile red was released from non-irradiated controls, samples irradiated for 90 min released 57 ± 5% of the encapsulated Nile red, which corresponds to 1.3 ± 0.1 μg (Fig. 5b). Light-triggered release from the P-1-NR particles can also be followed with the naked eye, as the supernatant of centrifuged irradiated samples becomes pink, whereas non-irradiated controls display colourless supernatant as the dye is well retained in the particles without light stimulation (Fig. 5c). We also investigated whether release of Nile red continues after shorter irradiation times, but we did not observe continued release under these conditions.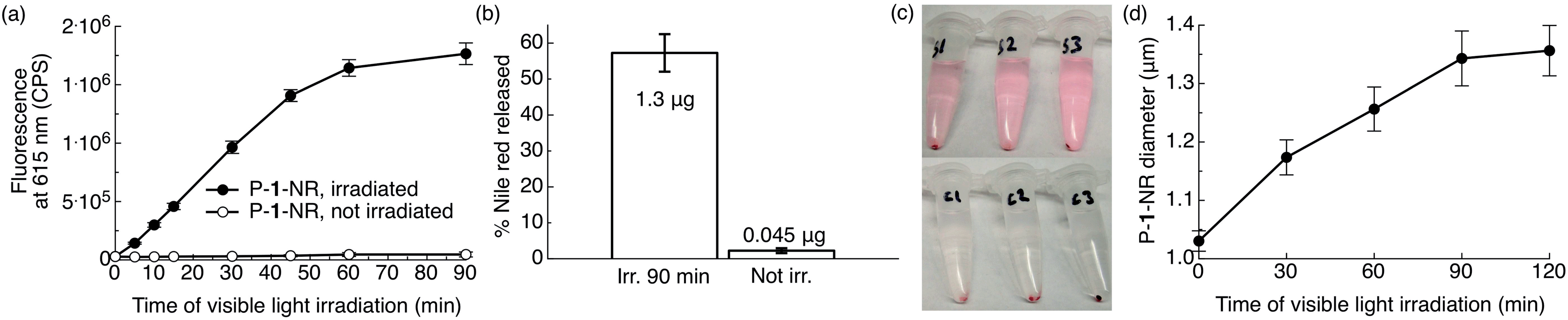 | ||
| Fig. 5 Photo-triggered release of Nile red from photoexpansile polymer 1 particles (P-1-NR). P-1-NR (0.08 mg mL−1, 0.5 mL, total amount Nile red per sample: 2.25 μg) in PBS (1×, pH = 7.4) containing 1% w/v Pluronic® F127 were irradiated in ambient temperature with visible light (λex = 400–500 nm, 0.18 W, 0.21 W cm−2) or kept in the dark for the same time. (a) Changes in fluorescence intensity at 615 nm (n = 3). (b) Relative and actual amount of Nile red released from the same samples after 90 min of visible light irradiation (see ESI† for calculation). (c) Photographs illustrating Nile red (pink) release from the samples/controls in (a and b) after 90 min of irradiation and after centrifugation (S1–3 = irradiated samples, C1–3 = non-irradiated controls kept in the dark). (d) Changes in particle diameter upon irradiation with visible light (Multisizer; n = 3). | ||
The kinetics of P-1-NR particles' photolysis were measured by UV-Vis spectroscopy. We observed photochemical changes in P-1-NR particles over 60 min of irradiation, which matches well with the Nile red release kinetics (Fig. 5a, S6a and b†). When further comparing the absorption and rate of photolysis between polymer 1, P-1-NR and empty polymer 1 particles (P-1), significant differences were observed. First, the absorbance of P-1-NR (λmax = 475 nm) and P-1 (λmax = 438 nm) was red-shifted compared to polymer 1 in CH2Cl2 (λmax = 398 nm). Second, photolysis rates differed considerably, that is, photolysis was complete at 10, 15 and 60 min, for polymer 1, P-1 and P-1-NR respectively (Fig. 4b, S6†). Although the particles absorb more visible light than polymer 1 in CH2Cl2, it appears that particle formulation decreases the photochemical efficiency of polymer 1. This decrease may result from water quenching and/or close proximity of the photocages in the particle, leading to self-quenching. Furthermore, it appears that Nile red reduces the rate of photolysis, either due to inner filter effects or energy transfer from the photocages to Nile red molecules.
To examine the physical changes underlying release, the morphology, size, and structure of the particles before and after irradiation were studied by particle size analyser (Multisizer™) and SEM. Upon prolonged irradiation, we observed a progressive increase in particle diameter from 1.03 ± 0.02 to 1.35 ± 0.04 μm after 90 min of irradiation (Fig. 5d). This expansion likely results from hydrophobic-to-hydrophilic photoswitching within the particles, which leads to influx of water. Interestingly, we did not observe evidence of particle degradation or disruption by SEM (Fig. S5†), which may indicate ionic and hydrogen bonding between the photodegraded segments and/or insufficient water solubility of the photoproducts. Based on these observations, release appears to occur through increased hydrophilicity and fluidity of the particles upon amine-assisted photocleavage and photorearrangement of the polymer.
In vivo photorelease of dexamethasone from implanted depot
On-demand one-photon visible light-triggered release of biologically active agents from an implanted depot would allow local self-administration while minimizing invasive injections and systemic side effects. Furthermore, a particle depot alters the pharmacokinetic behaviour of the encapsulated cargo, allowing it to remain localized at the injection site for much longer than small molecules. Upon irradiation the cargo is released locally and on demand.To assess the viability of the technology in animals, we examined the effect of subcutaneous light-triggered release of dexamethasone (Dex), a well-characterized anti-inflammatory drug,12 on carrageenan-induced hind paw inflammation, a favoured model to assess the anti-inflammatory properties of potential therapeutic agents.13
Carrageenan, a linear sulfated polysaccharide that triggers the release of inflammatory and proinflammatory mediators, was injected into the left hind footpad of the mouse; the right hind footpad was injected with Dulbecco's phosphate buffered saline (DPBS, 25 μL) as an internal control. Using electrospray, we encapsulated Dex in photoresponsive polymer 1 particles (P-1-Dex, Dex loading: 1% w/w, size by SEM: 0.42 ± 0.07 μm, hydrodynamic size by dynamic light scattering (DLS): 0.95 ± 0.5 μm, Fig. S7†) along with the NIR fluorescent probe IR780 (0.1% w/w) to allow in vivo monitoring of the depot distribution in real time (Fig. 6a and b, S8†).
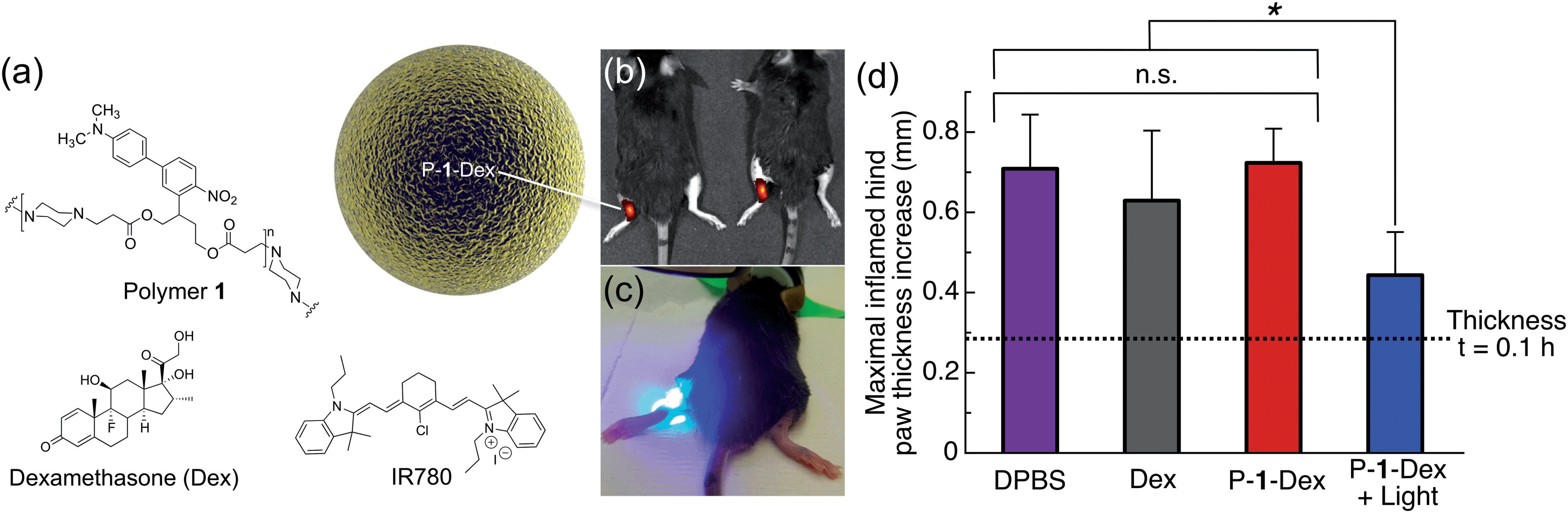 | ||
| Fig. 6 One-photon visible light photo-release of dexamethasone from an implanted depot reduces carrageenan-induced hind paw inflammation in mice. (a) Composition of P-1-Dex particle depot. (b) Representative IVIS image of mice implanted with NIR fluorescent P-1-Dex depot before irradiation. (c) Photograph illustrating irradiation of the P-1-Dex depot with blue visible light (λex = 400–500 nm, 0.18 W, 0.21 W cm−2, 432 J h−1). (d) Maximal inflamed hind paw thickness increase at t = 5 h after carrageenan injection. The P-1-Dex dose was 200 μg containing 2 μg Dex; free Dex dose was 2 μg; all mice were injected with a total of 25 μL of solution. n = 3 for all groups except DPBS, for which n = 6. Drug depots were injected in the left hind hock 1 h before inflammation was induced with carrageenan (2% w/v in 25 μL DPBS) in the left hind footpad. Hind paw thickness was measured using a caliper (see Fig. S9†) and the results are plotted relative to the non-inflamed right hind paw (injected at the same time with DPBS). Error bars are standard error of mean; *p = 0.02, n.s. = not statistically significant. | ||
One hour prior to induction of inflammation by carrageenan, P-1-Dex depots were injected subcutaneously in the left hind hock. Depots were then irradiated at the injection site with blue light for 40 minutes between hind paw thickness measurements (Fig. 6c, S9†). IR780 labelling allowed tracking of the depot location by an in vivo imaging system (IVIS) before and after irradiation of the particles (Fig. 6b, S8†). Controls included non-irradiated P-1-Dex depot, DPBS with and without light exposure, free Dex, empty particles (P-1) and pre-irradiated P-1 particles (Fig. S10†). The thickness of hind paws of mice injected with DPBS (Fig. 6d, S10†) increased from 0.28 ± 0.1 mm (t = 0.1 h after carrageenan injection) to a maximum of 0.71 ± 0.1 mm thicker than untreated paws (t = 5 h), while swelling in those treated with free Dex (Fig. 6d) was slightly but insignificantly (p = 0.5) lower, with a maximal difference in thickness of 0.63 ± 0.2 mm greater than control paws (t = 5 h). In contrast, irradiation of the P-1-Dex particle depot (Fig. 6d), led to considerably and significantly (p = 0.02) less swelling at maximal inflammation, i.e., 0.44 ± 0.1 mm (t = 5 h). We hypothesize that the lower therapeutic efficacy associated with free Dex compared to P-1-Dex + light results from the rapid diffusion of free Dex into the systemic circulation. In comparison, the sustained light-triggered release of Dex from the implanted P-1-Dex depot makes it more available over time because the depot remains stationary during the experiment, as monitored by IVIS (Fig. S8†).
As we did not observe any statistically significant reduction in inflammation associated with the non-irradiated P-1-Dex group (t = 5 h, paw thickness = 0.72 ± 0.08 mm, p = 0.9) (Fig. 6d), we can assume that Dex stays well retained within the drug carrier. Treatment with light alone, empty P-1 particles, or pre-irradiated P-1 particles resulted in similar paw inflammation as in mice given DPBS (Fig. S10†), ruling out therapeutic effects by either light or the carrier material.
We observed no gross changes to the color or texture of the skin following irradiation (Fig. S11†). However, we observed a slight swelling (oedema) of the thigh in a few cases (Fig. S12†) that appeared to result from prolonged irradiation, which resorbed the day after light exposure. Furthermore, no signs of adverse effects from the carrier material in non-irradiated mice were apparent (Fig. S11†), in agreement with the cytotoxicity data (MTT assay) obtained for empty P-1 particles, which showed excellent tolerance by Raw 264.7 mouse macrophage cells at concentrations up to 400 μg mL−1 (400 μg mL−1 = 115 ± 7% viability) (Fig. S13a†). However, irradiated P-1 particles were less tolerated above concentrations of 100 μg mL−1 (400 μg mL−1 = 65 ± 30% viability) (Fig. S13a†). The free polymer 1 before and after irradiation was not as well tolerated as particles, which demonstrates that particle formulation mitigates polymer toxicity (Fig. S13b†). In conclusion, we demonstrated on-demand one-photon visible light-triggered release of an anti-inflammatory agent in vivo, which reduced local hind paw inflammation more efficiently than free Dex (Fig. 6d). Greater therapeutic effects might be possible with more hydrophobic drugs, which would allow higher loading efficiency.
Conclusions
This report introduces a polymer that degrades upon one-photon absorption of blue visible light, which we show enables in vivo photorelease of dexamethasone from a subcutaneously implanted depot of polymeric particles. Due to the altered pharmacokinetics of the implanted depot, compared to the free drug, the photoreleased Dex lowered inflammation with greater efficiency than the free drug, which rapidly diffused from the site of injection. The release mechanism appears to involve expansion of the polymeric matrix by photoinduced photodegradation and hydrophilicity switching, as demonstrated by size measurements.The in vivo model used in this work demonstrates the practical utility of one-photon visible light for on-demand subcutaneous release of biological effectors. Visible light-responsive polymers and materials could become an important tool to enable light-mediated release in a multitude of animal research models.
Materials that respond to long-wavelength one-photon visible light, which is more suited for deeper tissue in vivo photorelease applications because of lower absorption of these wavelengths relative to the blue light used here, are currently under development.
Acknowledgements
The authors gratefully acknowledge the NIH New Innovator Award (DP 2OD006499) and King Abdulaziz City for Science and Technology (through the KACST-UCSD Center for Excellence in Nanomedicine and Engineering) for funding. NMR spectra were acquired at UCSD Skaggs School of Pharmacy and Pharmaceutical Sciences NMR facility. High-resolution mass spectroscopic data was measured at the UCSD Biomolecular and Proteomics Mass Spectrometry Facility. The authors are grateful to Dr Jacques Lux, Jessica Moore, Marlene Arredondo, Jason Olejniczak, Dr Caroline de Gracia Lux, Dr Amy Moore, Dr Shivanjali Joshi-Barr and Dr Brendan Duggan for insightful and technical help.Notes and references
- K. Deisseroth, Nat. Methods, 2011, 8, 26 CrossRef CAS PubMed.
- N. Fomina, J. Sankaranarayanan and A. Almutairi, Adv. Drug Delivery Rev., 2012, 64, 1005 CrossRef CAS PubMed; J. F. Gohy and Y. Zhao, Chem. Soc. Rev., 2013, 42, 7117 RSC; G. Liu, W. Liu and C.-M. Dong, Polym. Chem., 2013, 4, 3431 RSC; G. Pasparakis, T. Manouras, P. Argitis and M. Vamvakaki, Macromol. Rapid Commun., 2012, 33, 183 CrossRef PubMed; Y. Zhao, Macromolecules, 2012, 45, 3647 CrossRef; R. Tong and D. S. Kohane, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2012, 4, 638 CrossRef PubMed.
- P. Klan, T. Solomek, C. G. Bochet, A. Blanc, R. Givens, M. Rubina, V. Popik, A. Kostikov and J. Wirz, Chem. Rev., 2013, 113, 119 CrossRef CAS PubMed.
- (a) A. P. Goodwin, J. L. Mynar, Y. Z. Ma, G. R. Fleming and J. M. J. Frechet, J. Am. Chem. Soc., 2005, 127, 9952 CrossRef CAS PubMed; (b) N. Fomina, C. McFearin, M. Sermsakdi, O. Edigin and A. Almutairi, J. Am. Chem. Soc., 2010, 132, 9540 CrossRef CAS PubMed; (c) Q. N. Lin, Q. Huang, C. Y. Li, C. Y. Bao, Z. Z. Liu, F. Y. Li and L. Y. Zhu, J. Am. Chem. Soc., 2010, 132, 10645 CrossRef CAS PubMed; (d) S. Kumar, J.-F. Allard, D. Morris, Y. L. Dory, M. Lepage and Y. Zhao, J. Mater. Chem., 2012, 22, 7252 RSC; (e) G.-Y. Liu, C.-J. Chen, D.-D. Li, S.-S. Wang and J. Ji, J. Mater. Chem., 2012, 22, 16865 RSC; (f) J. Cao, S. Huang, Y. Chen, S. Li, X. Li, D. Deng, Z. Qian, L. Tang and Y. Gu, Biomaterials, 2013, 34, 6272 CrossRef CAS PubMed; (g) W. Ji, N. Li, D. Chen, X. Qi, W. Sha, Y. Jiao, Q. Xu and J. Lu, J. Mater. Chem. B, 2013, 1, 5942 RSC; (h) Q. Lin, C. Bao, Y. Yang, Q. Liang, D. Zhang, S. Cheng and L. Zhu, Adv. Mater., 2013, 25, 1981 CrossRef CAS PubMed; (i) J. Olejniczak, J. Sankaranarayanan, M. L. Viger and A. Almutairi, ACS Macro Lett., 2013, 2, 683 CrossRef CAS PubMed; (j) C. de Gracia Lux, C. L. McFearin, S. Joshi-Barr, J. Sankaranarayanan, N. Fomina and A. Almutairi, ACS Macro Lett., 2012, 1, 922 CrossRef CAS PubMed.
- C.-J. Carling, F. Nourmohammadian, J.-C. Boyer and N. R. Branda, Angew. Chem., 2010, 49, 3782 CrossRef CAS PubMed; B. Yan, J.-C. Boyer, N. R. Branda and Y. Zhao, J. Am. Chem. Soc., 2011, 133, 19714 CrossRef PubMed; B. Yan, J.-C. Boyer, D. Habault, N. R. Branda and Y. Zhao, J. Am. Chem. Soc., 2012, 134, 16558 CrossRef PubMed; Y. Yang, Q. Shao, R. Deng, C. Wang, X. Teng, K. Cheng, Z. Cheng, L. Huang, Z. Liu, X. Liu and B. Xing, Angew. Chem., 2012, 51, 3125 CrossRef PubMed; M. L. Viger, M. Grossman, N. Fomina and A. Almutairi, Adv. Mater., 2013, 27, 3733 CrossRef PubMed; Y. Yang, F. Liu, X. Liu and B. Xing, Nanoscale, 2013, 5, 231 RSC; M. K. G. Jayakumar, N. M. Idris and Y. P. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 8483 CrossRef PubMed.
- M. L. Denton, M. S. Foltz, L. E. Estlack, D. J. Stolarski, G. D. Noojin, R. J. Thomas, D. Eikum and B. A. Rockwell, Invest. Ophthalmol. Visual Sci., 2006, 47, 3065 Search PubMed; W. Watanabe, N. Arakawa, S. Matsunaga, T. Higashi, K. Fukui, K. Isobe and K. Itoh, Opt. Express, 2004, 12, 4203 CrossRef.
- N. Z. Knezevic, B. G. Trewyn and V. S. Lin, Chem. Commun., 2011, 47, 2817 RSC; E. S. Shibu, K. Ono, S. Sugino, A. Nishioka, A. Yasuda, Y. Shigeri, S.-I. Wakida, M. Sawada and V. Biju, ACS Nano, 2013, 11, 9851 CrossRef CAS PubMed; M. Frasconi, Z. C. Liu, J. Y. Lei, Y. L. Wu, E. Strekalova, D. Malin, M. W. Ambrogio, X. Q. Chen, Y. Y. Botros, V. L. Cryns, J. P. Sauvage and J. F. Stoddart, J. Am. Chem. Soc., 2013, 135, 11603 CrossRef PubMed.
- L. Donato, A. Mourot, C. M. Davenport, C. Herbivo, D. Warther, J. Leonard, F. Bolze, J. F. Nicoud, R. H. Kramer, M. Goeldner and A. Specht, Angew. Chem., 2012, 51, 1840 CrossRef CAS PubMed.
- S. Sun, E. A. Chamsaz and A. Joy, ACS Macro Lett., 2012, 1, 1184 CrossRef CAS; C. Lv, Z. Wang, P. Wang and X. Tang, Int. J. Mol. Sci., 2012, 13, 16387 CrossRef PubMed; C. Lv, Z. Wang, P. Wang and X. Tang, Langmuir, 2012, 28, 9387 CrossRef PubMed; D. Han, X. Tong and Y. Zhao, Langmuir, 2012, 28, 2327 CrossRef PubMed; M. A. Azagarsamy, D. L. Alge, S. J. Radhakrishnan, M. W. Tibbitt and K. S. Anseth, Biomacromolecules, 2012, 13, 2219 CrossRef PubMed.
- S. Walbert, W. Pfleiderer and U. E. Steiner, Helv. Chim. Acta, 2001, 84, 1601 CrossRef CAS; D. Woll, J. Smirnova, M. Galetskaya, T. Prykota, J. Buhler, K. P. Stengele, W. Pfleiderer and U. E. Steiner, Chem.–Eur. J., 2008, 14, 6490 CrossRef PubMed.
- Y. X. Xu and M. A. Hanna, Int. J. Pharm., 2006, 320, 30 CrossRef CAS PubMed; A. Jaworek, Powder Technol., 2007, 176, 18 CrossRef PubMed; S. Chakraborty, I. C. Liao, A. Adler and K. W. Leong, Adv. Drug Delivery Rev., 2009, 61, 1043 CrossRef PubMed.
- S. Tsurufuji, K. Sugio and F. Takemasa, Nature, 1979, 280, 408 CrossRef CAS.
- C. J. Morris, Methods Mol. Biol., 2003, 225, 115 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and synthetic details and additional graphs, chromatograms, photographs and micrographs. See DOI: 10.1039/c4sc02651a |
| This journal is © The Royal Society of Chemistry 2015 |