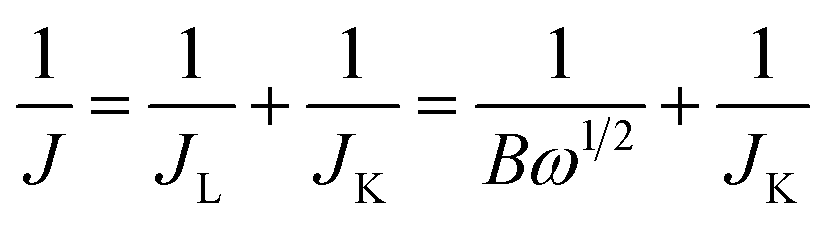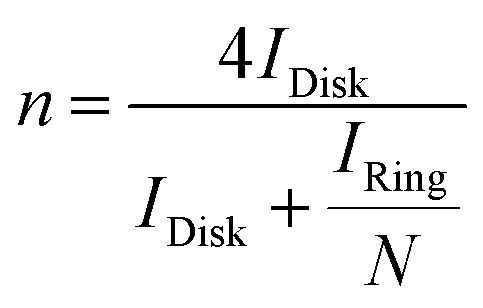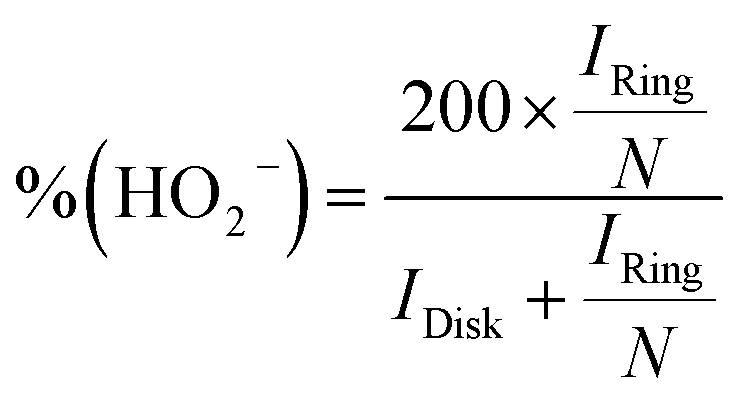DOI:
10.1039/C5RA02027A
(Paper)
RSC Adv., 2015,
5, 27953-27963
Sulfur-doped porous carbon nanosheets as high performance electrocatalysts for PhotoFuelCells†
Received
2nd February 2015
, Accepted 12th March 2015
First published on 12th March 2015
Abstract
Sulfur-doped porous carbon nanosheets have been prepared by pyrolysis of graphene-coupled conjugated microporous polymers under an inert atmosphere. The obtained carbon nanosheets exhibited large specific surface areas up to 642 m2 g−1 and high sulfur weight content up to 7.11%. These highly porous carbon nanosheets have been studied as metal-free oxygen reduction electrocatalysts in alkaline environments and they were found to undergo oxygen reduction via a major 4-electron transfer pathway. They were then examined as substitutes for Pt–carbon electrocatalysts in PhotoFuelCells functioning in the presence of ethanol as a model fuel. It has been shown that sulfur doped porous carbon nanosheets yield functional cells with approximately the same characteristics as those employing Pt–carbon electrocatalysts, therefore, they mark a new class of metal-free catalysts.
1. Introduction
Conversion of solar energy simultaneously with chemical energy and electricity is achieved by means of photoelectrochemical cells. A standard configuration for a photoelectrochemical cell involves a photoanode electrode carrying an n-type semiconductor photocatalyst, a counter electrode carrying an electrocatalyst and an electrolyte. Light is absorbed by the photocatalyst generating electron–hole pairs. Electrons are guided through an external circuit to the cathode (counter) electrode, where they take part in reduction reactions while holes are consumed through oxidation reactions. In regenerative solar cells, the electrolyte involves a redox couple, which is reduced at the counter electrode and oxidized at the photoanode electrode thus refilling holes and converting photon energy into electricity. Those cells act as photovoltaic devices. The present work will not deal with such cells but it will focus on photoelectrochemical procedures where both oxidation and reduction reactions involve consumption or production of chemical substances. In that case, holes are consumed by oxidizing an organic or inorganic substance, which acts as sacrificial agent. Electrons are consumed by some standard reduction reactions, for example, water, oxygen or CO2 reduction. A less common configuration for a photoelectrochemical cell involves a photocathode electrode carrying a p-type photocatalyst while the anode electrode carries an oxidation electrocatalyst. The present work will deal with a standard cell employing a photoanode and n-type semiconductor photocatalyst. In both types of cells, the energy of photons and the chemical energy of the sacrificial agent, i.e. of the “fuel”, are converted into electricity and/or stored in a useful form of chemical energy, for example, hydrogen. For this reason, photoelectrochemical cells operating by consumption of a fuel are called PhotoFuelCells (PFCs).1,2
The origins of photoelectrochemical cells can be traced back to the findings of Alexander-Edmond Becquerel in 1839, who reported for the first time the photovoltaic (in reality, the photoelectrochemical) conversion of light into electricity.3 However, a work by Fujishima and Honda4 in 1972 describing water splitting by using a TiO2 photoanode has set the foundations of modern photoelectrochemistry. After more than 40 years, the conception of a photoelectrochemical cell remains the same but the new materials and procedures employed for their construction has kept the field alive with ever increasing popularity.5–7 Photoanode electrodes are usually made by depositing nanoparticulate titania (np-TiO2) on transparent conductive glasses like Fluorine-doped Tin Oxide (FTO). Since titania absorbs only UV light, it is combined with appropriate sensitizers. The choice of sensitizers is very limited in the case of PhotoFuelCells since the sensitized photoanode must preserve its oxidative capacity towards the fuel. Thus the only applicable sensitizers in the presence of organic fuels are a few medium band gap semiconductor quantum dots like CdS and ZnSe.8,9 A second major component of PhotoFuelCells is the counter electrode, which carries the electrocatalyst. Nanoparticulate Pt is acknowledged as the best electrocatalyst for both oxidation and reduction reactions. There are two reasons for this property. Pt can be easily obtained in fine nanoparticles and it has the highest work function thus becoming an electron sink, which can exchange charges with the electrolyte. However, Pt is rare and costly and this discourages massive applications. In addition, Pt suffers from migration and aggregation that progressively decreases its efficiency. Alternative materials employed as electrocatalysts should have high specific surface area and be good conductors in order to facilitate transfer of charges. Various doped carbon nanostructures as well as conductive polymers have been employed for this purpose.10–13 In the present work we studied a series of novel 2D sulfur-doped porous carbon sheets (Scheme 1), which combine the properties of both and we tested their applicability as oxygen reduction electrocatalysts (ORE).
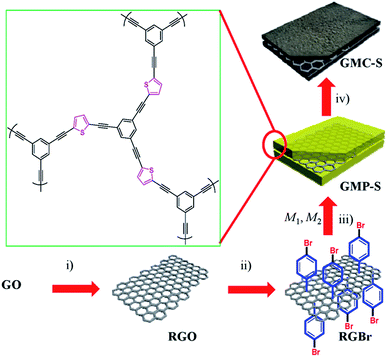 |
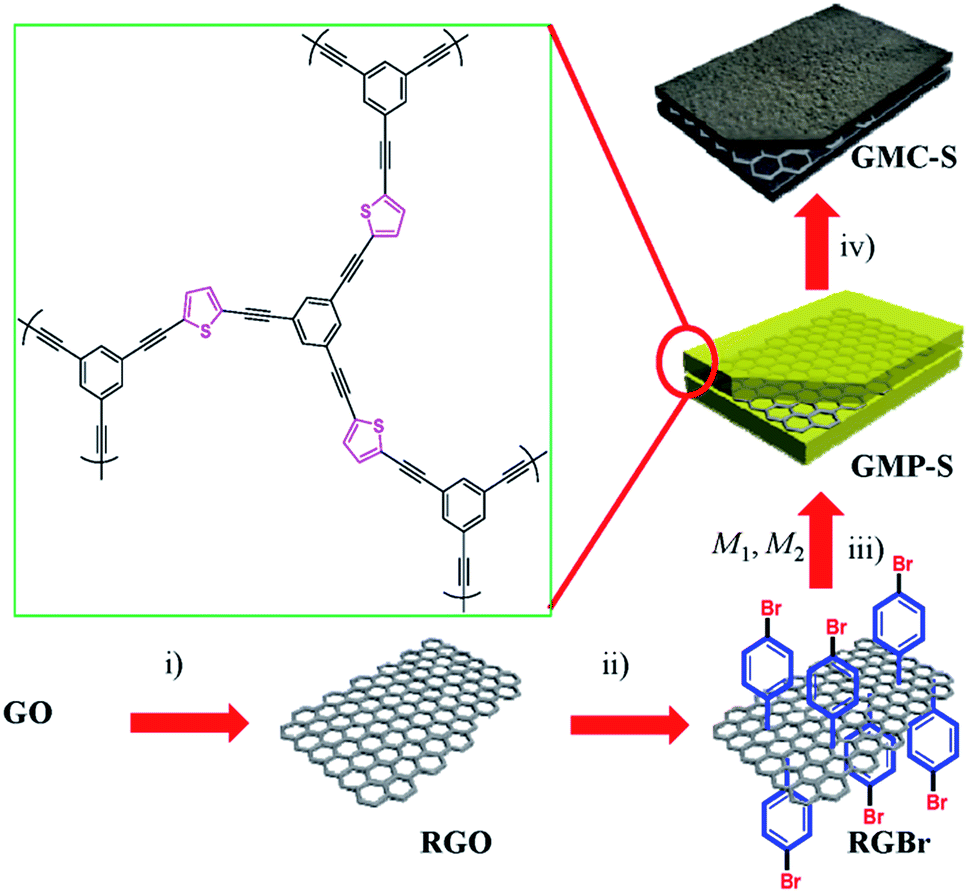
| | Scheme 1 Preparation of GMP-S and GMC-S. (i) Sodium dodecylbenzenesulfonate, N2H4·H2O, 100 °C, 8 h; (ii) 4-bromobenzenediazonium tetrafluoroborate, 0 °C to RT, 2 h; (iii) Ar, Pd[(PPh3)4], CuI, Et3N, DMF, 80 °C, 3 days (M1, 1,3,5-triethynylbenzene; M2, 2,5-dibromothiophene); (iv) Ar, RT to 700, 800 and 900 °C, 5 °C min−1, 2 h. GO and RGO stand for graphene oxide and reduced graphene oxide, respectively. | |
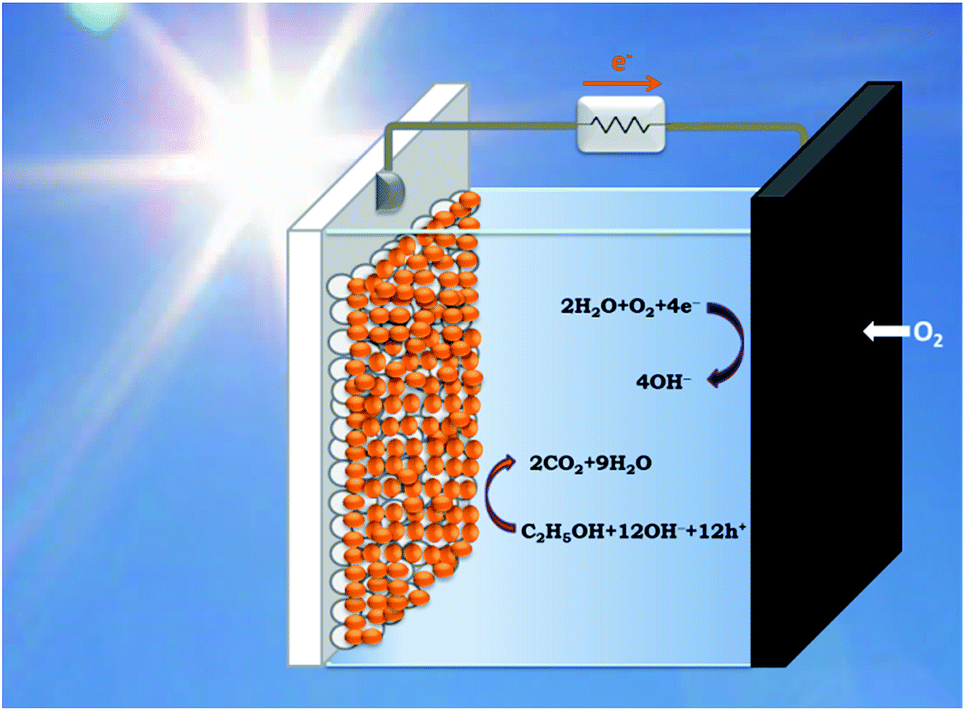
PhotoFuelCells can be used either for production of electricity or for production of hydrogen.14 The present work exclusively focuses on the production of electricity. In that case, the photoanode oxidizes the fuel while the cathode functions by oxygen reduction.8 Therefore the cell works in the presence of oxygen. In order to facilitate continuous supply of oxygen, the counter electrode is usually made of an “air-breathing” material using, for example, carbon cloth. Fig. 1 illustrates such a cell. It is seen that the photoanode plays the role of reactor window while the counter electrode seals the reactor on the opposite side. In order to avoid water leak through the carbon cloth, a hydrophobic layer is applied, which provides this property. Carbon cloth will be thus employed as material for counter electrode also in the present case. The currently studied materials are hydrophobic graphene-based sulfur-doped porous carbon nanosheets (GMC-S, Scheme 1),15 which provide necessary hydrophobicity to prevent water leak when applied on carbon cloth electrodes. For the synthesis of GMC-S, graphene-based 2D sandwich-like conjugated microporous polymer sheets were firstly prepared utilizing thiophene-based monomer polymerized with 1,3,5-triethynylbenzene monomer on the graphene surface via the metal-catalyzed Hagihara–Sonogashira coupling reaction. Then, the as-prepared GMP-S was pyrolyzed at 700, 800 and 900 °C for 2 h under an argon atmosphere, affording 2D porous carbon nanosheets (denoted GMC-S700, GMC-S800 and GMC-S900, respectively). The resultant GMP-S and GMC-S exhibit large aspect ratio and specific surface area as high as 888 m2 g−1 and 462–642 m2 g−1 respectively. The as-prepared sulfur-doped porous carbon nanosheets are very promising as new generation, low cost, oxygen reduction electrocatalysts.
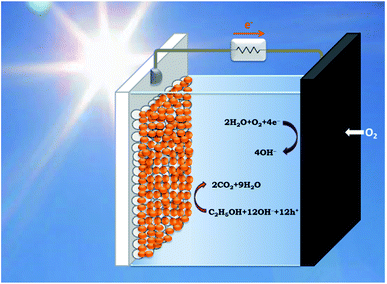 |
| | Fig. 1 Schematic illustration of a PhotoFuelCell operating with a sensitized nanoparticulate titania photoanode and an oxygen-reducing cathode in the presence of ethanol as model fuel. | |
2. Experimental
Fabrication of the sulfur-doped carbon nanosheets
The fabrication of sulfur-doped porous carbon nanosheets is illustrated in Scheme 1 and detailed in the Results and discussion section. All reactions were carried out under nitrogen atmosphere by standard Schlenk techniques. All chemicals were purchased from Aldrich, Aladdin Reagent Inc. and Titan Sci. Inc. and used as received, unless otherwise noted.
SEM measurements were performed on a FEI Sirion-200 field emission scanning electron microscope. Transmission electron microscopy (TEM) characterizations were conducted using a JEM-2100 (JEOL Ltd., Japan) with an accelerating voltage of 200 KV. X-ray photoelectron spectroscopy (XPS) experiments were carried out on a AXIS Ultra DLD system from Kratos with Al Kα radiation as X-ray source for radiation. The Brunauer–Emment–Teller (BET) specific surface area was measured on Autosorb-iQ-C surface area and porosimetry analyzer (Quantachrome Instruments, USA) based on N2 adsorption. The pore size distributions were calculated based on non-local density functional theory (NLDFT) method using the adsorption isotherms.
Cyclic voltammetry (CV) and rotating disk electrode (RDE) measurements
The working electrode was prepared by loading a catalyst sample film of 0.60 mg cm−2 onto a glassy carbon electrode.16 First, the ink was prepared by dispersing 10 mg of catalyst (GMCs) in 500 μL of 1 wt% Nafion ethanol solution, and then sonicated for at least 30 min to form a homogeneous dispersion. Next, 6 μL of the catalyst ink was loaded onto a glassy carbon electrode with a diameter of 5 mm (0.6 mg cm−2). Pt/C ink was prepared by dispersing 4 mg of Pt/C (20 wt% Pt in carbon black) in 1 mL of ethanol with 35 μL of 5 wt% Nafion solution (40 wt% of Nafion to catalyst ratio), and then 5 μL of Pt/C ink was loaded onto a glassy carbon electrode. The ink was dried slowly in air, and the drying condition was adjusted by trial and error until a uniform catalyst distribution across the electrode surface was obtained.
Electrochemical measurements of cyclic voltammetry, rotating disk electrode (RDE) and rotating ring-disk electrode (RRDE) were performed by a basic bipotentiostat (Pine Research Instrumentation, USA) with a three-electrode cell system. A rotating glass carbon disk and platinum ring electrode (Pine Research Instrumentation, USA) after loading the electrocatalyst was used as the working electrode, an Ag/AgCl (KCl, 3 M) electrode as the reference electrode, and a Pt wire as the counter electrode. The electrochemical experiments were conducted in O2 saturated 0.1 M KOH and 0.5 M NaOH electrolyte for the oxygen reduction reaction. The potential range was cyclically scanned between −1.0 and +0 V at a scan rate of 100 mV s−1 at the room temperature after purging O2 gas for 30 min. RDE measurements were conducted at different rotating speeds from 225 to 2500 rpm.
The transferred electron number (n) per oxygen molecule in the ORR process at electrode can be calculated by the Koutechy–Levich (K–L) eqn (1)–(3):
| | 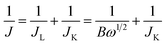 | (1) |
| | | B = 0.62nFC0(D0)2/3ν−1/6 | (2) |
JK and
JL are the kinetic- and diffusion-limiting current densities,
μ is the angular velocity of the rotating electrode (
ω = 2π
N,
N is the linear rotation speed),
F is the Faraday constant (
F = 96
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif)
485 C mol
−1),
C0 is the concentration of O
2,
D0 is the diffusion coefficient of O
2,
ν is the kinematic viscosity of the electrolyte, and
k is the electron transfer rate constant. Since the electrolyte is O
2 saturated 0.5 M NaOH,
C0,
D0 and
v are constant as 0.92 × 10
−3 M, 1.93 × 10
−5 cm
2 s
−1, and 0.01 cm
2 s
−1, respectively.
17
Rotating ring-disk electrode (RRDE) measurement
Catalyst inks and electrodes were prepared by the same method as described above. The disk electrode was scanned cathodically at a rate of 5 mV s−1, and the ring potential was constant at 0.5 V vs. Ag/AgCl.
Based on the RRDE result, the HO2− (%) and the electron transfer number (n) were determined by the following equations:
| | 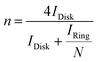 | (4) |
| | 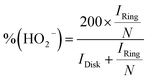 | (5) |
where
Idisk is disk current,
Iring is ring current, and
N is current collection efficiency of the Pt ring, which was provided as 0.37 by manufacture. The glassy carbon electrode with a diameter of 5.61 mm was used in this experiment.
Preparation of np-TiO2 films and deposition of CdS by the SILAR method
Nanoparticulate titania (np-TiO2) films were deposited on FTO transparent electrodes by the following procedure. A FTO glass was cut in the appropriate dimensions and was carefully cleaned first with soap and then by sonication in isopropanol, water and acetone. A thin layer of compact titania was first sprayed over a patterned area by using 0.2 mol L−1 diisopropoxytitanium bis(acetylacetonate) solution in ethanol and was calcined at 500 °C. Deposition of this bottom compact layer is a common practice with nanocrystalline titania photoanodes, since it enhances attachment of the top thick film, prevents short circuits and facilitates electron flow towards the electrode. On the top of this compact film, we applied a titania paste made of P25 nanoparticles by doctor blading. The film was calcined up to 550 °C at a rate of 20 °C min−1. The final thickness of the film, as measured by SEM, was approximately 10 μm. The geometrical area of the film varied according to the particular application. CdS was deposited by Successive Ionic Layer Adsorption and Reaction (SILAR).8,9,18 10 SILAR cycles were applied by using Cd(NO3)2 as Cd2+ and Na2S as S2− precursor. In all cases, we used 0.1 mol L−1 aqueous solutions for both cations and anions. After SILAR deposition and final washing with tripled distilled water, the films were dried in an oven at 100 °C and were ready for use.
Construction of the counter electrode
Carbon cloth was used as counter electrode as already mentioned above. For reasons of comparison, we have used an electrode made by a standard procedure, which was functionalized with carbon black and Pt, similar to previous publications.8,9,19 A hydrophobic layer was first applied as follows. 0.246 g of carbon black was mixed with 8 mL of distilled water by vigorous mixing (about 2400 r.p.m.) until it became a viscous paste. This paste was further mixed with 0.088 mL polytetrafluorethylene (Teflon 60 wt% dispersion in water) and then applied on a carbon cloth cut in the necessary dimensions. This has been achieved by first spreading the paste with a spatula, preheating for 30 min at 80 °C and finally heating for a few minutes in an oven at 340 °C. Subsequently, the catalytic layer was prepared as follows: 1 g of Pt–carbon black electrocatalyst (30% on Vulcan XC72) was mixed with 8 g of Nafion perfluorinated resin (5 wt% solution in lower aliphatic alcohols and water) and 15 g of a solution made of 7.5 g H2O and 7.5 g isopropanol. The mixture was ultrasonically homogenized and then applied on the previously prepared carbon cloth bearing carbon black. The electrode was then heated at 80 °C for 30 min and the procedure was repeated as many times as necessary to load about 0.5 mg of Pt cm−2. The thus prepared Pt–carbon black/Carbon-Cloth (Pt/CC) electrode was ready for use. Its dimensions were 10 cm2 (3.0 cm × 3.3 cm). The above porous carbon cloth electrode allows contact with air while the deposited hydrophobic materials prevent water leak.
The as-synthesized sulfur doped porous carbon electrocatalyst was deposited by the following procedure. 10 mg of the porous carbon (GMC-S700, GMP-S800 or GMC-S900) was mixed with 1.3 mL of isopropanol and 0.5 mL of water, first by magnetic stirring and then under sonication. Then we added 0.1 mL of the above referenced polytetrafluoroethylene, under mild stirring. The mixture was applied on the carbon cloth with a spatula and was annealed at 340 °C. The porous carbon nanosheet on carbon cloth is abbreviated GMP-S/CC. The quantity of the active component was about 5 mg spread over an area of 10 cm2 (3.0 cm × 3.3 cm).
Construction of the PhotoFuelCell
The PhotoFuelCell apparatus was made of Plexiglas designed to accommodate photoanode and counter electrode, which were set up in the configuration presented by Fig. 1. The distance between the two electrodes was 5 mm. This cell also allows incorporation of a thin hydrogen reference electrode. The latter was a home-made construction. A thin glass tube was sealed on one end by a silicon sealant allowing the introduction of a Pt wire. The other end was closed but it carried a thin hole on its side. The tube was first filled by using a syringe with the same electrolyte as the one used to fill the cell. Then it was rapidly vertically immersed into the electrolyte of the cell taking care to avoid any air coming in. For this reason the side hole must be small. Then the Pt wire was connected to a potentiostat providing about −4 V potential. At this high negative potential, hydrogen is produced by water electrolysis. Hydrogen bubbles move up inside the tube and accumulate themselves at the top of the tube pushing electrolyte down. The hydrogen is thus trapped between the electrolyte and the sealed top of the tube. When half of the Pt wire was in hydrogen atmosphere and the other half in contact with the electrolyte electrolysis was terminated. The thus obtained hydrogen reference electrode was ready for use. It remained immersed in the electrolyte for the whole period of current–voltage measurements in 3-electrode configuration. The cell was filled with an aqueous alkaline electrolyte made of 0.5 mol L−1 NaOH, either alone or in the presence of 5 vol% EtOH. For the formation of hydrogen in the reference electrode we used only alkaline electrolyte without ethanol. For measurements in 2-electrode configuration hydrogen electrode was taken out from the cell. Illumination was made with a xenon lamp providing 100 mW cm−2 simulated solar radiation at the position on the photocatalyst. Current–voltage curves were traced with the help of an Autolab PGSTAT128N potentiostat.
3. Results and discussion
The strategy for the synthesis of 2D graphene-based conjugated microporous polymer sheet (GMP-S) is presented in Scheme 1. First, hydrazine-hydrate-reduced graphene oxide sheet (RGO)20 was functionalized by p-bromobenzene in aqueous conditions using the diazonium salt. The obtained bromo-functionalized RGO (RGBr) was highly dispersed in various organic solvents, such as dimethylformamide (DMF, 1.0 mg mL−1), chloroform, and toluene. Next, thiophene-based halogen monomers (M2) were mixed with alkyne monomer (M1: 1,3,5-triethynylbenzene) and RGBr in dry DMF, and then sealed and reacted under Hagihara–Sonogashira reaction conditions. After precipitation, a black solid was collected and purified by Soxhlet extraction. Finally, 2D sandwich-like GMP, denoted as GMP-S for thiophene monomer was obtained after vacuum drying. Based on inductively coupled plasma mass spectrometry (ICP-MS), it was found that only 0.64 wt% residual Pd remained in the polymer matrix. The highly dispersible RGBr serves as a 2D template for in situ polymerization of conjugated microporous polymer on its surfaces. That came with effective separating of individual graphene layers and preventing the aggregation problems between them.
The morphology and microstructure of as-prepared GMP was investigated by means of transmission electron microscopy (TEM) and scanning electron microscopy (SEM). As shown in Fig. 2a and b, many free-standing sheets with morphology similar to that of graphene and with sizes ranging from 200 nm to several micrometers were observed. In addition, these porous polymer sheets exhibited wrinkles and flexible features that stand in contrast to the rigid inorganic porous silica sheets21 reported previously. No free porous polymer particles or naked graphene sheets were identified by either the TEM or SEM visualizations. This result suggests that, most of the monomers have been polymerized on the surface of graphene.22
 |
| | Fig. 2 Typical SEM (a) and TEM (b) images of GMP. | |
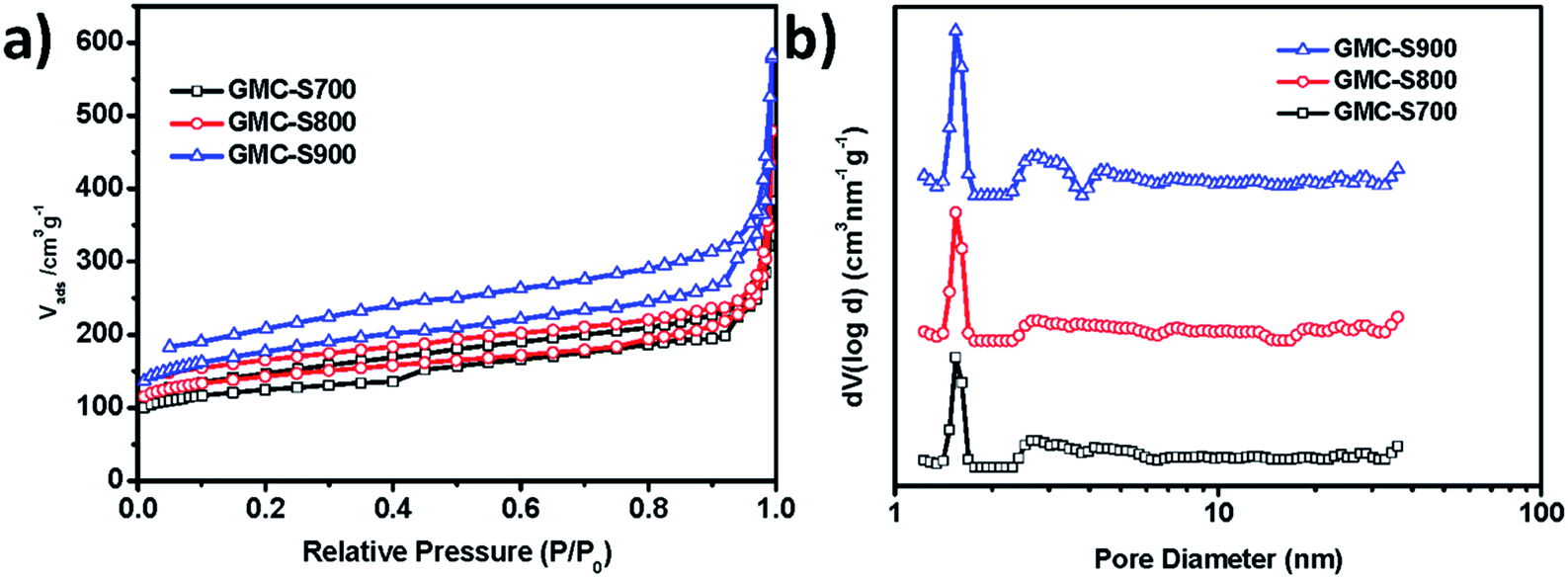
Conjugated microporous polymer is a type of carbon-rich precursor23 that can integrate heteroatoms,24,25 metals, and metal oxides26 into the carbon framework for various applications, such as energy storage and electrochemical catalysis. Thermogravimetric analysis (TGA, Fig. S1†) revealed that GMP-S can feasibly be transformed into carbon materials with a high carbon yield (81%). Therefore, S-doped 2D porous carbons, denoted as GMC-S, was generated by the direct pyrolysis of GMP-S at 700, 800 and 900 °C for 2 h under an argon atmosphere (Scheme 1). Based on the nitrogen physisorption results (Fig. 3 and Table 1), it is to be noted that GMC-S-X (X = 700, 800 and 900 °C) maintain the porous feature with a high BET surface area of 462, 530, and 642 m2 g−1 respectively. According to the NL-DFT method, the pore size distribution of these porous carbon nanosheets were calculated based on their adsorption isotherms (Fig. 3a). There are two main kinds of pores in these materials, which locate at ∼1.7 nm and 2.5–6 nm respectively. This result indicates that these porous carbon nanosheets are typical micro- (<2 nm) and meso- (2–50 nm) porous nanostructures. Thereby, the present approach is a highly efficient method for the preparation of 2D hierarchical porous carbon nanosheets without using any inorganic templates. The difference in the surface area for GMCs with respect to the pyrolysis temperatures is probably due to the different degradation degree of GMP-S and recombination of fragments under the carbonization conditions.23
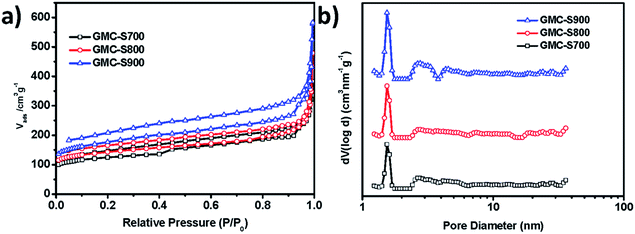 |
| | Fig. 3 (a) Nitrogen adsorption–desorption isotherms of GMC-S-X (X: 700, 800, 900) at 77.3 K; (b) pore-size distribution calculated by non-localized density functional theory (NLDFT). | |
Table 1 Physical properties for sulfur-doped porous carbon nanosheets
| Samples |
S
BET
a/m2 g−1 |
S
Langmuir
b/m2 g−1 |
V
micro
c/cm3 g−1 |
S
micro
d/m2 g−1 |
V
Tot
e/cm3 g−1 |
D
av
f/nm |
|
Surface area calculated from the N2 adsorption isotherm using the BET method and Langmuir method, respectively.
Surface area calculated from the N2 adsorption isotherm using the BET method and Langmuir method, respectively.
Micropore volume.
Micropore surface area.
Total pore volume at p/p0 = 0.99.
Average pore size based on the adsorption isotherm.
|
| GMC-S-700 |
462 |
514 |
0.077 |
212 |
0.59 |
5.10 |
| GMC-S-800 |
530 |
590 |
0.176 |
407 |
0.74 |
5.59 |
| GMC-S-900 |
642 |
750 |
0.209 |
462 |
0.89 |
5.57 |
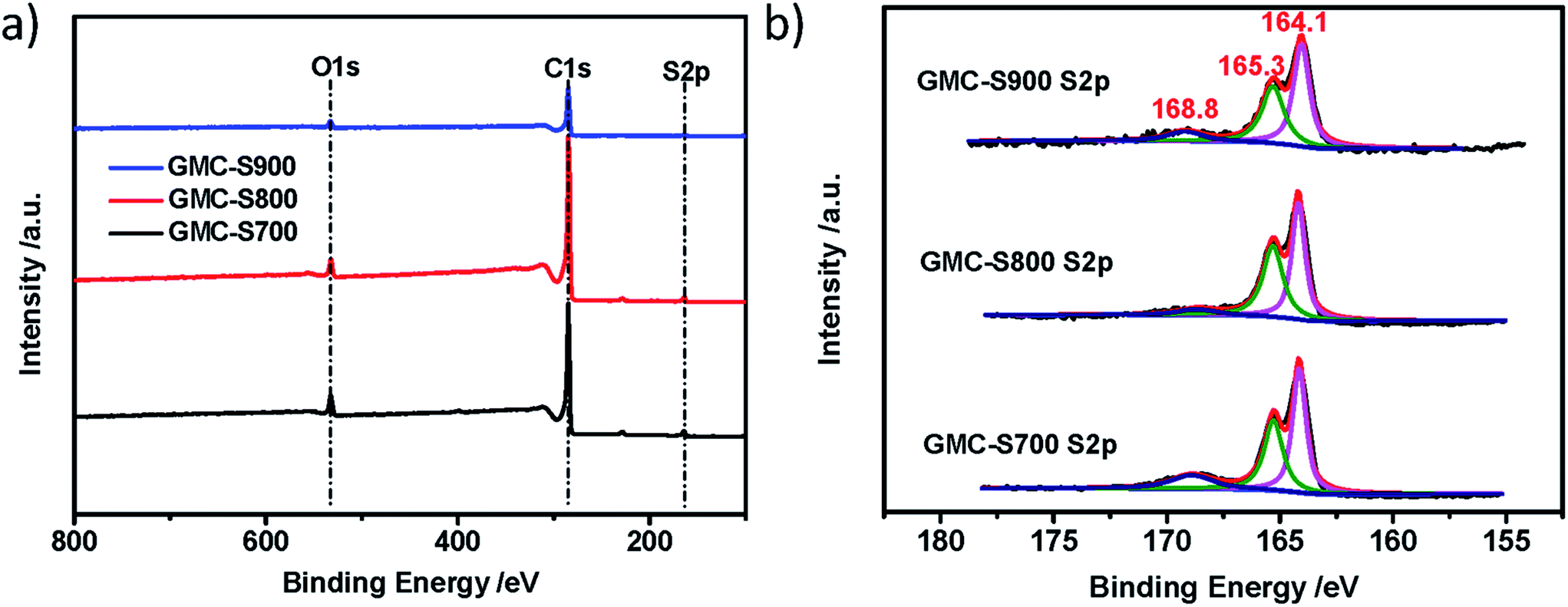
The chemical nature of GMCs was further investigated by X-ray photoelectron spectroscopy (XPS) measurement and elemental analysis. The obtained GMCs show similar chemical compositions: carbon, oxygen and sulfur (Fig. 4a). Interestingly, the S 2p spectra of GMC-S can be fitted by almost the same sulfur components.27 The weight contents of sulfur in GMC-S700, GMC-S800 and GMC-S900 are 7.11%, 5.02% and 4.74%, respectively. The major contributions at binding energies of around 164.1 and 165.3 eV can be attributed to the sulfur species of the C–Sn–C (n = 1 or 2) bonds and conjugated –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S– bonds28 (which should be of the thiophene type), respectively. Another minor contribution at the higher binding energy of around 168.8 eV can be assigned to the oxidized (–SOn–) sulfur moieties in which the oxygen may originate from the residual oxygen groups in the RGO. ICP-MS result indicated only <0.038 wt% residual Pd in these porous carbons. This strategy thus realizes a feasible way to build up 2D porous carbons with a high sulfur doping content.
S– bonds28 (which should be of the thiophene type), respectively. Another minor contribution at the higher binding energy of around 168.8 eV can be assigned to the oxidized (–SOn–) sulfur moieties in which the oxygen may originate from the residual oxygen groups in the RGO. ICP-MS result indicated only <0.038 wt% residual Pd in these porous carbons. This strategy thus realizes a feasible way to build up 2D porous carbons with a high sulfur doping content.
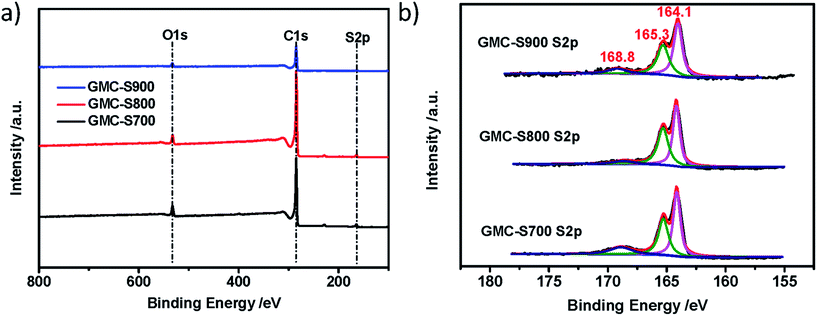 |
| | Fig. 4 XPS survey (a) and S 2p core level (b) spectra of GMC-SX (X = 700, 800, 900 °C). | |
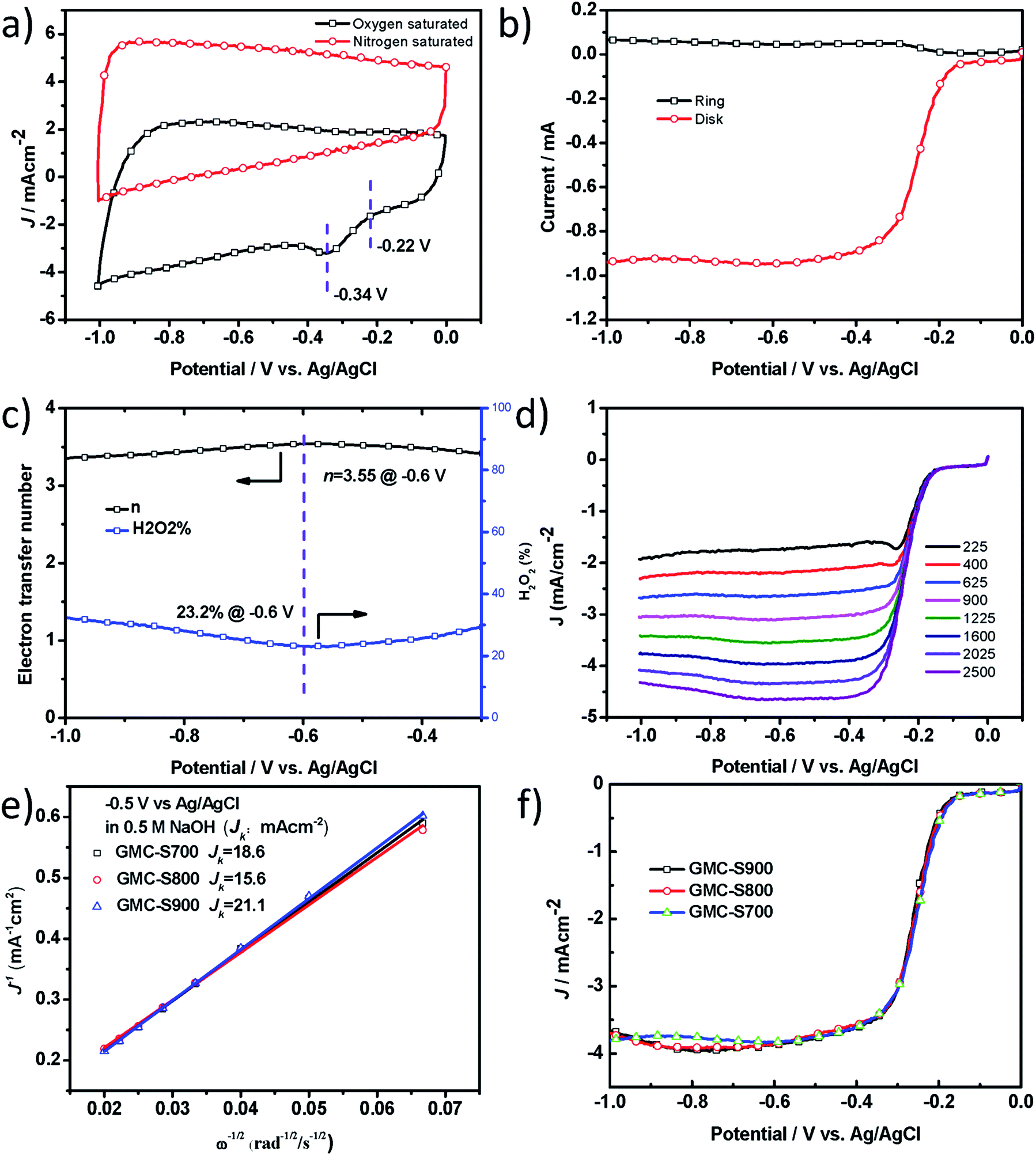
One of the most important applications of heteroatom-doped porous carbons is their use as metal-free catalysts for clean energy production, such as fuel cells or PhotoFuelCells. To accomplish this, we first studied their catalytic performance for oxygen reduction reaction (ORR) in 0.5 M NaOH. Heteroatom-doped, especially, S-doped carbon materials29 have been demonstrated to exhibit promising electrocatalytic performance for ORR in 0.1 M KOH,29,30 but rarely in NaOH solution which is one very widely used electrolyte for PhotoFuelCell. In this work, the electrocatalytic activities of S-doped 2D porous carbons towards the ORR were examined in alkaline conditions (0.5 M NaOH). The ORR catalytic activity of GMC-S was first evaluated by cyclic voltammetry (CV, Fig. 5a and S5†). GMC-S700 showed an ORR onset potential of −0.22 V and a peak potential of −0.34 V versus Ag/AgCl, which were similar to GMC-S800 and GMC-S900 (Fig. S6 and S7†). The electron transfer number (n) and peroxide percentage for GMC-S700 can be calculated as ∼3.55 and 23.2% at −0.60 V respectively from the rotating ring-disk electrode (RRDE) curve (Fig. 5b and c, eqn (4) and (5)). The Koutecky–Levich plots (K–L plots, Fig. 5e) with a good linear relationship for the GMC-S electrode were calculated from linear sweep voltammetry (LSV) curves (Fig. 5d) at various rotation rates. Notably, the mean transferred electron number per O2 molecule involved in the ORR, which can be determined from the K–L equation (eqn (1)), was approximately 4.0 between −0.40 V and −0.90 V for all three GMC-S-X. These results highlight that the ORR proceeds via a major four-electron transfer pathway. This finding was verified by studying the operation of PhotoFuelCells as will be seen below. The kinetic-limiting current density reached 18.6, 15.6 and 21.1 mA cm−2 at −0.5 V for GMC-S700, GMC-S800 and GMC-S900 respectively (Table S1†). The diffusion-limiting current densities (JDL) of −3.9 ± 0.1 mA cm−2 for GMCs catalysts, the mainly four-electron transfer pathway, together with the very low HWP potential (−0.25 V), make GMCs promising as metal-free electrocatalysts for ORR.
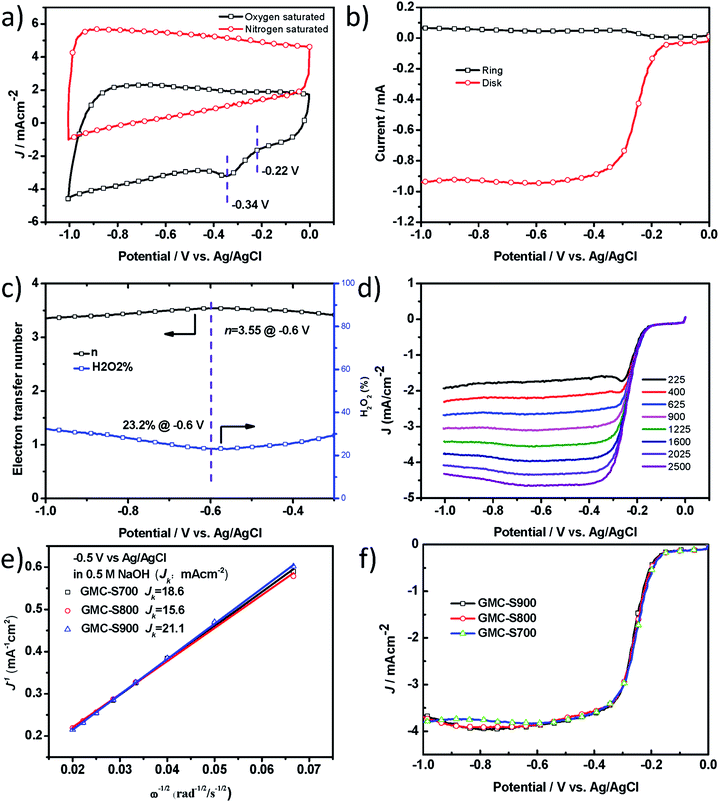 |
| | Fig. 5 Graphene-based 2D porous carbons (GMC-SX, X = 700, 800 and 900) and their ORR performances in 0.5 M NaOH. (a) CV curves of GMC-S700 in N2-saturated and O2-saturated 0.5 M NaOH at a scan rate of 50 mV s−1; (b) RRDE curve of GMC-S700 at 1600 rpm; (c) calculated electron transfer number and percentage of peroxide; (d) RDE curves of GMC-S700 in O2-saturated 0.5 M NaOH with different speeds at a scan rate of 5 mV s−1; (e) the comparison of electrochemical activity given as the kinetic-limiting current density (Jk) at −0.5 V (versus Ag/AgCl); (f) RDE curves of GMC-S700/800/900 in O2-saturated 0.5 M NaOH. | |
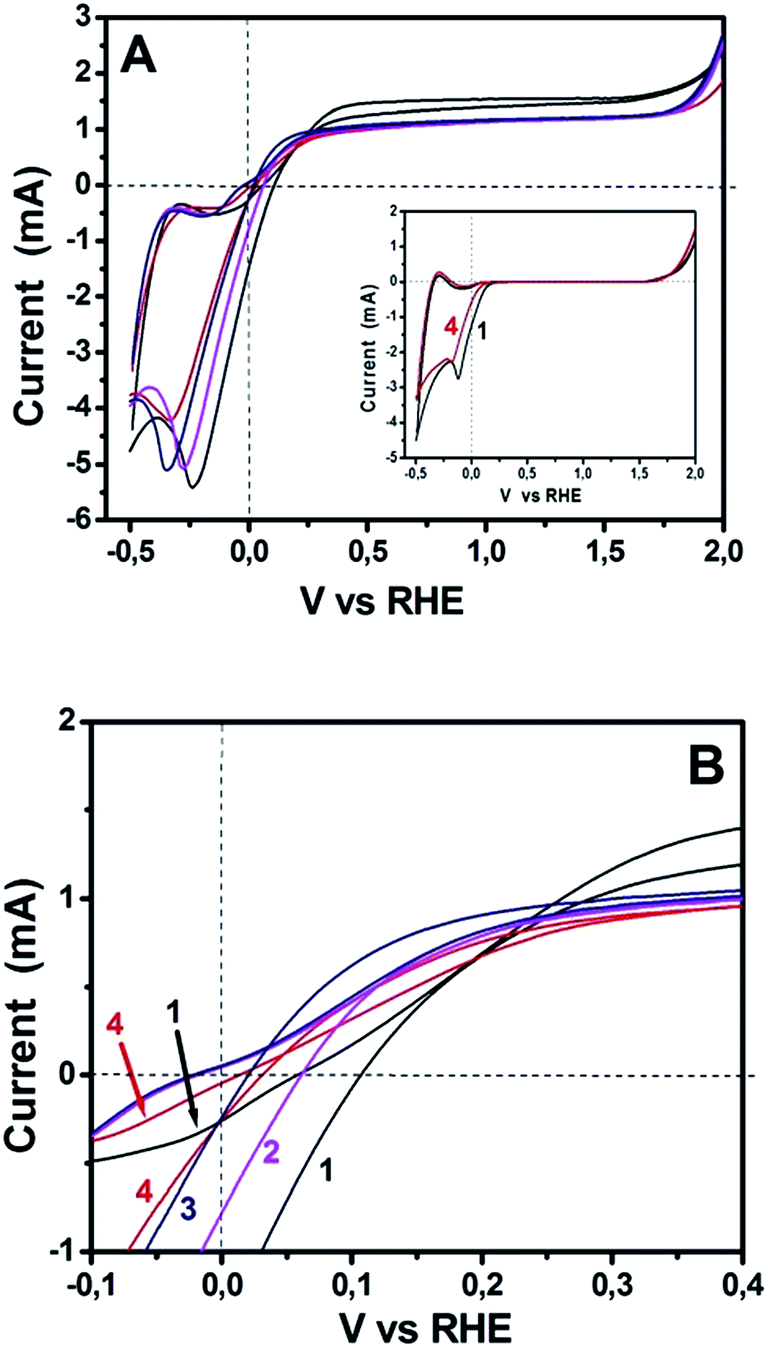
The above data demonstrate excellent functionality of sulfur-doped porous carbon nanosheets to act as oxygen reduction electrocatalysts. Since the ultimate goal of this work was to test their applicability to PhotoFuelCells, we have constructed such devices and studied their behavior. PhotoFuelCells have been constructed as detailed in the Experimental section and illustrated in Fig. 1. Photoanodes were made of np-TiO2 and were sensitized, when necessary, by CdS quantum dots deposited by standard procedures.8,9,18 As cathode (counter) electrode we used either porous carbon nanosheets on carbon cloth, GMC-S/CC, or platinum and carbon black on carbon cloth, Pt/CC, for comparison. In order to appreciate the electrochemical behavior of GMP-S/CC cathodes in relation with the standard Pt/CC electrode, cyclic voltammograms have been recorded and presented in Fig. 6. The general aspect of the curves of Fig. 6A reveals similar characteristics for both standard and porous carbon nanosheet based cathodes. All curves cross the horizontal axis at current zero for both anodic and cathodic scan in a narrow potential range between 0 and 0.1 V. These crossings mark the onset for anodic photocurrent. Interestingly, as seen in Fig. 6B, showing curves at a scale that allows to separate them, onset for anodic photocurrent appeared at higher potential in the case of Pt/CC than for GMP-S/CC. This means that the former necessitates application of a small forward bias while the porous carbon nanosheet-based cells run without any necessity for bias. Even though, these differences are small and without practical importance, they do mark a definite advantage by the employment of the presently introduced electrocatalysts. The insert of Fig. 6A shows two selected voltammograms recorded in the dark. Photocurrent was zero as expected. The rest of the characteristics of the curves in Fig. 6A (including the insert) are those expected for the studied system. Water electrolytic oxidation onset appears between 1.5 to 2.0 Volts and electrolytic reduction around 0 Volts. These general characteristics are common for both Pt/CC and GMP-S/CC. Photocatalytic water oxidation, i.e. photocurrent onset, is characteristic of np-TiO2 and differs little from one electrocatalyst to the other, as already mentioned above. The anodic and cathodic peaks appearing in all curves at about −0.4 and −0.2 V, respectively, are due to adsorption–desorption of cationic species on the mesoporous titania film and depend on the nature of the cation and the thickness of the film.31 All the above data indicate that the presently studied porous carbon electrocatalysts can efficiently substitute the standard Pt/CC electrocatalyst. This conclusion, which is justified by the data of Fig. 6, has been further elaborated by modifying both electrolyte and photoanode.
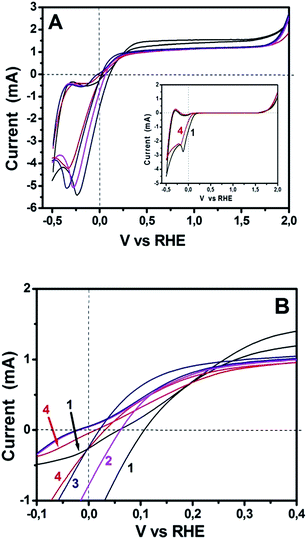 |
| | Fig. 6 Cyclic voltammograms recorded with a np-TiO2 photoanode under illumination and various counter electrodes using a hydrogen electrode as reference. (A) shows curves in the whole recorded range and (B) shows a detail where curves can be distinguished. Curve (1) corresponds to Pt/CC and the rest to GMC-S/CC: (2) GMC-S800; (3) GMC-S900 and (4) GMC-S700. Aqueous electrolyte contained only 0.5 M NaOH. Insert in (A) shows corresponding data recorded in the dark. | |
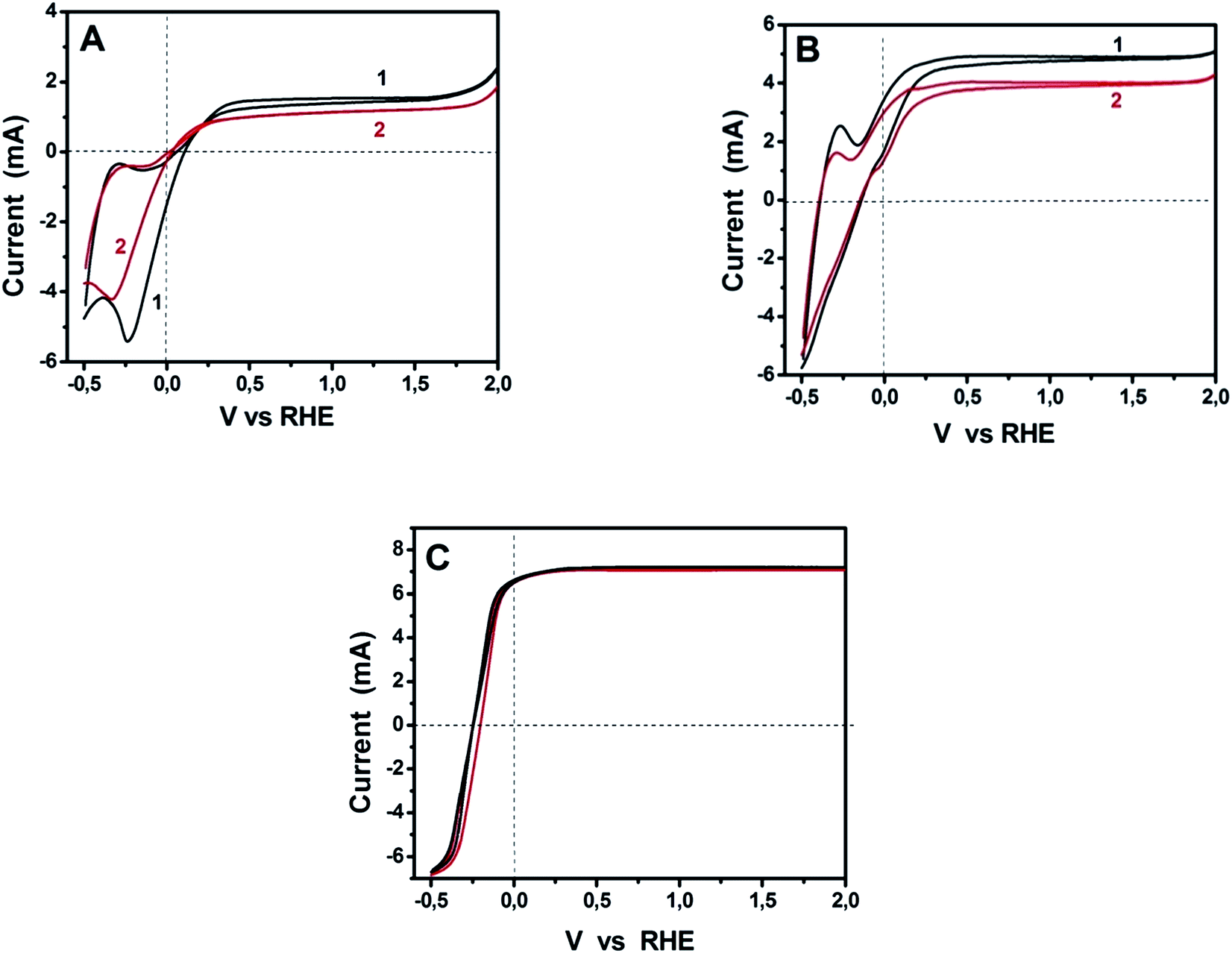
The above data are applicable to purely water-splitting considerations. We proceeded by further studies in the presence of a fuel and in the presence of a sensitizer. Due to the comparable ORR performance for GMC-S700, GMC-S800 and GMC-S900 in 0.5 M NaOH and the relatively high sulfur content for GMC-S700, we have chosen GMC-S700 as representative of the studied materials. Fig. 7 shows cyclic voltammograms recorded with Pt/CC and GMC-S700/CC for three different situations: bare titania photoanode and pure alkaline electrolyte; bare titania photoanode and ethanol added in the electrolyte; and sensitized photoanode with ethanol-containing electrolyte. Fig. 7A is an extract of Fig. 6A. It is seen that the curve corresponding to GMP-S/CC demonstrated a slightly slower response to water reduction than Pt/CC and its maximum photocurrent was smaller than that of the standard platinum containing electrode. The smaller photocurrent is not an absolute means of comparison since the quantities of both electrocatalysts are not optimized. Interestingly, when ethanol was added to the electrolyte, the photocurrent increased in both electrocatalyst cases and the difference between them equally increased but the difference in the onset of water reduction was eliminated. The onset of anodic photocurrent was the same for both electrodes while both photocurrent and reduction onset were shifted to lower potential. This shift is due to the presence of ethanol, which consumes holes, liberates more electrons and thus enhances the electronegativity of the titania conduction band. Finally, in the presence of a sensitizer (Fig. 7C), anodic photocurrent increased and all differences between the cells with the two different electrocatalysts were eliminated. The same holds true for the hysteresis between the anodic and cathodic plot, which now completely coincide. These results have promoted the presently used materials to being ideal electrocatalysts and the absolute alternative to platinum, at least for PhotoFuelCells. Moreover, it is worth noting that in both cases of electrocatalysts the presence of ethanol brought an approximate doubling of the observed current. This phenomenon is known as the “Current Doubling” effect2,32 and it is believed to take place by the formation of unstable radicals and injection of additional electrons into the conduction band of titania. This phenomenon does not depend on the counter electrode and it is thus observed with both electrocatalyst types.
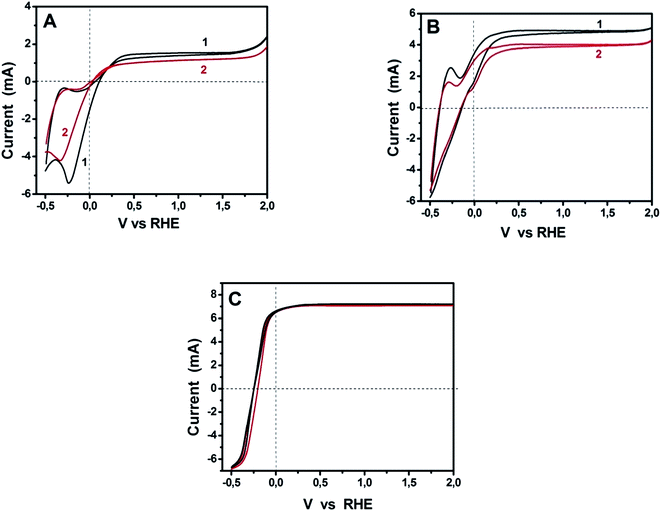 |
| | Fig. 7 Cyclic voltammograms recorded with Pt/CC (1) and GMC-S700/CC (2) counter electrodes in three different cases: (A) bare titania photoanode and pure alkaline electrolyte; (B) bare titania photoanode and ethanol added in the electrolyte; and (C) CdS sensitized np-TiO2 photoanode with ethanol-containing electrolyte. The concentration of NaOH was 0.5 M and that of ethanol 5 vol%. | |
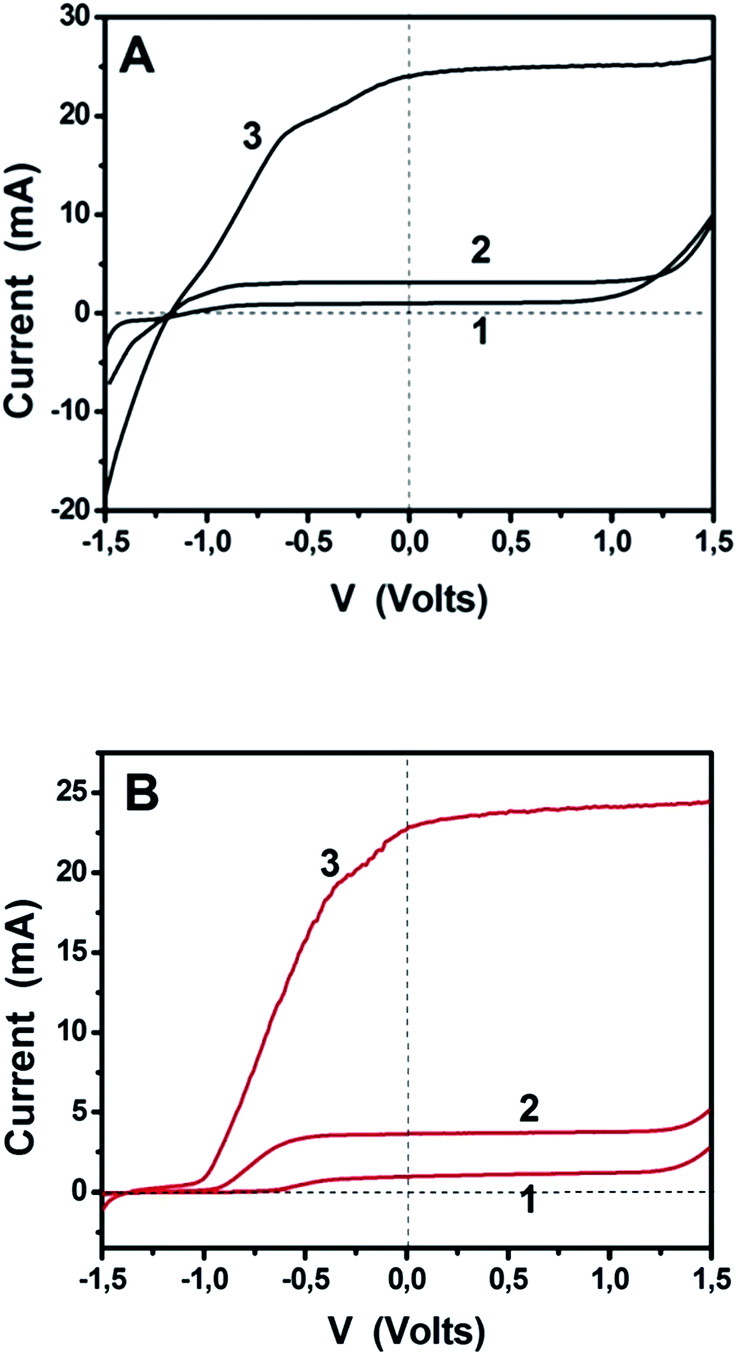
The above results encourage the utilization of doped-porous carbon nanosheets as platinum-free alternatives for electrocatalysis. The data recorded so far, refer to biased photoelectrochemical cells operating in a 3-electrode configuration. In order to further demonstrate the applicability of these materials, we have studied unbiased cells functioning under simulated solar radiation in a 2-electrode configuration, similar to that depicted in Fig. 1. We have recorded current–voltage data for the three different situations described in the previous paragraph, i.e., a cell working with bare titania, the same cell with added fuel (ethanol) and a cell bearing a sensitized photoanode. The data of Fig. 8 and Table 2 have been recorded in a 2-electrode configuration without any reference electrode. The corresponding solar cell is a pure PhotoFuelCell that can be used to produce electricity by consuming a fuel. In the absence of organic fuel, water itself plays the role of fuel. In order to have a point of reference, the data of Fig. 8 have been recorded by using first Pt/CC and then GMC-S700 as counter electrode. Fig. 8A demonstrates the typical behavior of a PhotoFuelCell, which constitutes an alternative means to transform solar energy into electricity.2,5–9,19 The short-circuit current Isc produced in the absence of fuel and by using non-sensitized titania was about 1 mA and increased in the presence of ethanol. When CdS-sensitized photoanode was employed, Isc jumped to 25 mA. These values are very satisfactory and are very promising for upscaling and commercial applications. The open-circuit voltage Voc (voltage at I = 0) did not change much from one situation to the other and this is expected since Voc is defined by the potential difference between the titania conduction band (−0.3 V vs. SHE) and the redox level of the reduction processes taking place at the cathode. In the presence of oxygen, the following reduction process is expected to take place:2
| | | O2 + 4H+ + 4e− → 2H2O (+1.23 V vs. SHE) | (6) |
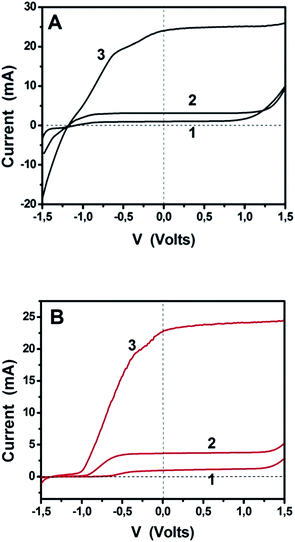 |
| | Fig. 8
IV curves recorded in 2-electrode configuration (without reference electrode) using free-standing PhotoFuelCells activated by illumination with simulated solar radiation. Curves (1) correspond to a photoanode carrying bare titania and to an aqueous alkaline electrolyte. Curves (2) were recorded by adding 5 vol% ethanol and using the same photoanode, while curves (3) were recorded with the same ethanol-containing electrolyte but a CdS-sensitized photoanode. (A) corresponds to a Pt/CC and (B) to a GMC-S700/CC counter electrode. The electrolyte always contained 0.5 M NaOH. The active size of both photoanode and counter electrodes was 10 cm2 (3 cm × 3.3 cm). | |
Table 2 Data extracted from Fig. 8
| Cell status |
Pt/CC cathode |
GMC-S700/CC cathode |
|
I
sc (mA) |
V
oc (V) |
I
sc (mA) |
V
oc (V) |
|
Bare titania photoanode and alkaline electrolyte without fuel.
Bare titania photoanode and alkaline electrolyte containing ethanol.
CdS sensitized titania and alkaline electrolyte containing ethanol.
|
| Case 1a |
1.0 |
1.0 |
1.0 |
0.7 |
| Case 2b |
3.1 |
1.2 |
3.6 |
0.9 |
| Case 3c |
24.0 |
1.2 |
22.7 |
1.0 |
However, at alkaline pH as in the present case, both states move to more negative potentials according to the relation ΔV = −0.059 × pH. Thus for pH 13, the titania conduction band is at −0.3 − 0.77 = −1.07 V and the reduction reaction is modified to:
| | | O2 + 2H2O + 4e− → 4OH− (+0.46 V for pH 13) | (7) |
The expected potential difference can be as high as +0.46 − (−1.07) = 1.53 V. As seen in Table 2, Voc was actually 1.0 V and increased to 1.2 V in the presence of ethanol. Of course, the presence of the fuel results to the consumption of holes and liberation of more electrons, which increase the electronegativity of the conduction band and thus result in higher voltage. The presence of the sensitizer did not further increase voltage, since the latter is defined by the conduction band of the majority species, i.e. of titania, where photogenerated electrons are injected and end up. The PhotoFuelCell then not only produces substantial current but it also generates high voltage making it a highly promising solar conversion device. When the Pt/CC cathode was substituted by GMC-S700/CC the current produced was equivalent with that generated by the Pt/CC based cell. On the contrary, Voc was in all cases approximately 0.3 V lower in the case of GMC-S700/CC than in the case of Pt/CC. This difference was however lower in the case of GMC-S800/CC and GMC-S900/CC, possibly because of different electrocatalyst potentials in these cases. In all cases, the results support the oxygen reduction model of reactions (6) and (7), which is a 4-electron reduction process justifying the above findings. The curves of Fig. 8 were recorded as linear plots by scanning voltage from −1.5 up to +1.5 V. It is then seen that within a relatively large range of negative bias, differing from one curve to the other, the current produced by the cell stayed close to zero. This happened in all three cases, i.e. curves 1–3. GMC-S/CC electrodes then seem to resist the flow of reverse current and to encourage only the flow of forward anodic current. Only for very strong negative bias, i.e. >1.5 V (not shown) did those electrodes allow reverse current to flow. This behavior was not observed by cyclic voltammetry (Fig. 6 and 7) and was not observed with the Pt/CC or other electrodes (not shown). We believe that it is related to the capacity of these electrodes to absorb ions that temporarily affect their behavior. In the course of studying them, we observed that the appearance of the curves in the range of −1.5 to −0.5 V may be modified by repeated linear plots. Such a result supports the model of ion adsorption.
4. Conclusions
We have demonstrated that porous sulfur-doped carbon nanosheets can act as efficient electrocatalysts for oxygen reduction reaction. PhotoFuelCells employing carbon cloth electrodes carrying these porous carbon nanosheets produced comparable currents with the standard platinum–carbon black electrocatalysts traditionally employed in fuel cells but they generated a little smaller open-circuit voltage. These results render the presently developed sulfur-doped carbon nanosheets as a new type of high performance active materials for PhotoFuelCells, possibly, applicable as electrocatalysts also to other similar systems.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Conflict of interest
The authors declare no competing financial interest.
Acknowledgements
This work was supported by the Bilateral R&T cooperation program between Germany and Greece, project INSOLCELL. X. Z. and X. F. thank the financial support from 973 Program of China (2013CBA01602), ERC Grant on 2DMATER, and Natural Science Foundation of China (51403126).
References
- M. Kaneko, J. Nemoto, H. Ueno, N. Gokan, K. Ohnuki, M. Horikawa, R. Saito and T. Shibata, Electrochem. Commun., 2006, 8, 336–340 CrossRef CAS.
- P. Lianos, J. Hazard. Mater., 2011, 185, 575–590 CrossRef CAS PubMed.
- A. E. Becquerel, C. R. Hebd. Seances Acad. Sci., 1839, 9, 561–567 Search PubMed.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- Y. Guo, J. He, S. Wu, T. Wang, G. Li, Y. Hu, H. Xue, X. Sun, J. Tang and M. Liu, J. Power Sources, 2012, 208, 58–66 CrossRef CAS.
- S. Wu, J. He, J. Zhou, T. Wang, Y. Guo, J. Zhao and X. Ding, J. Mater. Chem., 2011, 21, 2852–2854 RSC.
- T. Wang, J. Tang, S. Wu, X. Fan and J. He, J. Power Sources, 2014, 248, 510–516 CrossRef CAS.
- M. Antoniadou, D. I. Kondarides, D. D. Dionysiou and P. Lianos, J. Phys. Chem. C, 2012, 116, 16901–16909 CAS.
- S. Sfaelou, L. Sygellou, V. Dracopoulos, A. Travlos and P. Lianos, J. Phys. Chem. C, 2014, 118, 22873–22880 CAS.
- S. Sfaelou, M. Antoniadou, G. Trakakis, V. Dracopoulos, D. Tasis, J. Parthenios, C. Galiotis, K. Papagelis and P. Lianos, Electrochim. Acta, 2012, 80, 399–404 CrossRef CAS.
- N. Balis, V. Dracopoulos, M. Antoniadou and P. Lianos, Electrochim. Acta, 2012, 70, 338–343 CrossRef CAS.
- J. Tang, J. Liu, C. Li, Y. Li, M. O. Tade, S. Dai and Y. Yamauchi, Angew. Chem., Int. Ed., 2015, 54, 588–593 CAS.
- J. Tang, T. Wang, X. Pan, X. Sun, X. Fan, Y. Guo, H. Xue and J. He, J. Phys. Chem. C, 2013, 117, 16896–16906 CAS.
- X. Lu, S. Xie, H. Yang, Y. Tong and H. Ji, Chem. Soc. Rev., 2014, 43, 7581–7593 RSC.
- X. Zhuang, F. Zhang, D. Wu, N. Forler, H. Liang, M. Wagner, D. Gehrig, M. R. Hansen, F. Laquai and X. Feng, Angew. Chem., Int. Ed., 2013, 52, 9668–9672 CrossRef CAS PubMed.
- Y. Liang, H. Wang, P. Diao, W. Chang, G. Hong, Y. Li, M. Gong, L. Xie, J. Zhou, J. Wang, T. Z. Regier, F. Wei and H. Dai, J. Am. Chem. Soc., 2012, 134, 15849–15857 CrossRef CAS PubMed.
-
W. Xing, G. Yin, J. Zhang, Rotating Electrode Methods and Oxygen Reduction Electrocatalysts, Elsevier, 2013 Search PubMed.
- Y. F. Nicolau, Appl. Surf. Sci., 1985, 22–23(part 2), 1061–1074 CrossRef CAS.
- M. Antoniadou and P. Lianos, Appl. Catal., B, 2010, 99, 307–313 CrossRef CAS.
- S. Stankovich, D. A. Dikin, G. H. B. Dommett, K. M. Kohlhaas, E. J. Zimney, E. A. Stach, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Nature, 2006, 442, 282–286 CrossRef CAS PubMed.
- S. Yang, X. Feng, L. Wang, K. Tang, J. Maier and K. Müllen, Angew. Chem., Int. Ed., 2010, 49, 4795–4799 CrossRef CAS PubMed.
- R. Chinchilla and C. Nájera, Chem. Rev., 2007, 107, 874–922 CrossRef CAS PubMed.
- N. Kobayashi and M. Kijima, J. Mater. Chem., 2007, 17, 4289–4296 RSC.
- Y. Zhao, K. Watanabe and K. Hashimoto, J. Am. Chem. Soc., 2012, 134, 19528–19531 CrossRef CAS PubMed.
- M. Zhong, E. K. Kim, J. P. McGann, S.-E. Chun, J. F. Whitacre, M. Jaroniec, K. Matyjaszewski and T. Kowalewski, J. Am. Chem. Soc., 2012, 134, 14846–14857 CrossRef CAS PubMed.
- N. Kang, J. H. Park, J. Choi, J. Jin, J. Chun, I. G. Jung, J. Jeong, J.-G. Park, S. M. Lee, H. J. Kim and S. U. Son, Angew. Chem., Int. Ed., 2012, 51, 6626–6630 CrossRef CAS PubMed.
- J. P. Paraknowitsch, B. Wienert, Y. Zhang and A. Thomas, Chem.–Eur. J., 2012, 18, 15416–15423 CrossRef CAS PubMed.
- D. Portehault, V. Maneeratana, C. Candolfi, N. Deschler, I. Veremchuk, Y. Grin, C. Sanchez and M. Antonietti, ACS Nano, 2011, 5, 9052–9061 CrossRef CAS PubMed.
- J. Liang, Y. Jiao, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2012, 51, 11496–11500 CrossRef CAS PubMed.
- S. B. Yang, L. J. Zhi, K. Tang, X. L. Feng, J. Maier and K. Mullen, Adv. Funct. Mater., 2012, 22, 3634–3640 CrossRef CAS.
- L.-C. Pop, S. S. Faelou and P. Lianos, Electrochim. Acta, 2015, 156, 223–227 CrossRef CAS.
- A. Fujishima, T. Kato, E. Maekawa and K. Honda, Bull. Chem. Soc. Jpn., 1981, 54, 1671–1674 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Contains complementary experimental data. See DOI: 10.1039/c5ra02027a |
| ‡ These authors contributed equally. |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence