
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Microengineered peripheral nerve-on-a-chip for preclinical physiological testing
Renee M.
Huval
a,
Oliver H.
Miller
b,
J. Lowry
Curley
a,
Yuwei
Fan
a,
Benjamin J.
Hall
bc and
Michael J.
Moore
*ab
aTulane University - Biomedical Engineering, New Orleans, Louisiana, USA. E-mail: mooremj@tulane.edu
bTulane University - Neuroscience Program, New Orleans, Louisiana, USA
cTulane University - Cell & Molecular Biology, New Orleans, Louisiana, USA
First published on 27th March 2015
Abstract
The use of advanced in vitro testing is a powerful tool to develop predictive cellular assays suitable for improving the high attrition rates of novel pharmaceutical compounds. A microscale, organotypic model of nerve tissue with physiological measures that mimic clinical nerve compound action potential (CAP) and nerve fiber density (NFD) tests may be more predictive of clinical outcomes, enabling a more cost-effective approach for selecting promising lead compounds with higher chances of late-stage success. However, the neurological architecture, physiology, and surrounding extracellular matrix are hard to mimic in vitro. Using a dual hydrogel construct and explants from rat embryonic dorsal root ganglia, the present study describes an in vitro method for electrophysiological recording of intra- and extra-cellular recordings using a spatially-controlled, microengineered sensory neural fiber tract. Specifically, these 3D neural cultures exhibit both structural and functional characteristics that closely mimic those of afferent sensory peripheral fibers found in vivo. Our dual hydrogel system spatially confines growth to geometries resembling nerve fiber tracts, allowing for a high density of parallel, fasciculated neural growth. Perhaps more importantly, outputs resembling clinically relevant test criteria, including the measurement of CAP and NFD are possible through our advanced model. Moreover, the 3D hydrogel constructs allow flexibility in incorporated cell type, geometric fabrication, and electrical manipulation, providing a viable assay for systematic culture, perturbation, and testing of biomimetic neural growth for mechanistic studies necessitating physiologically-relevant readouts.
Introduction
Recent innovation in 3D hydrogel culture systems, microscale tissue engineering, and microfluidics have paved the way for the development of biomimetic “organoid-on-a-chip” tissue models for studying physiology and disease, toxicity screening, and drug discovery.1–4 The use of advanced in vitro testing is a powerful tool to develop predictive cellular assays suitable for improving the high attrition rates of novel pharmaceutical compounds.5 Application of such models have been recently reviewed in cardiac,6 liver,7 skeletal muscle8 and neuronal developmental9,10 systems.Inability of current models, at both the pre-clinical in vitro and in vivo levels, to correctly predict the efficacy of novel therapeutics in humans is the leading cause of late stage failure, with unforeseen toxicity a close second.11,12 The increased burden to patients and healthcare costs demonstrate that current pre-clinical methodology is insufficient.11,13 In order to better recapitulate clinical relevancy using benchtop models, quantifiable outcomes must be representative of organ-specific physiological endpoints. It is critical that such model systems move beyond 3D versions of conventional cell based assays to models that truly recapitulate functional aspects of organ physiology that can be evaluated to screen for drug safety and efficacy.14
Such assessment is especially challenging for neural applications, where changes in electrophysiological behaviors may arguably be the most relevant functional outcome. For this reason, 3D tissue models of nervous tissue are lagging those of epithelial, metabolic, and tumor tissues, where soluble analytes serve as appropriate metrics. The potentially rapid application of electrophysiological techniques has been shown possible through multi-electrode array (MEA) technologies for the screening of environmental toxins15,16 as well as disease modeling and therapeutic testing.17,18 This technology is ground-breaking for the study of central nervous system (CNS) applications, being applied to both brain slices and dissociated cultures.19–21 Some tradeoff is required between simple, high-throughput screening and advanced models yielding high-content data. MEA testing improves on throughput compared to intracellular patch-clamp experiments and allows for the measurement of network dynamics. Patch-clamp is still the gold standard for studies requiring precise stimulation of single-cells or recording of sub-threshold events. However, the difficulty in recapitulating the 3D structure and measurement of population-level biological conduction over long distances, which are critical for organotypic models of peripheral nerve tissue, has limited in vitro testing to simple 2D models.22–24 Rather than examining stochastic network formation and electrophysiological data, a more biomimetic assay will better recapitulate clinical methods of investigating peripheral nerve function, including the measurement of compound action potentials (CAP) and nerve fiber density (NFD) using morphometric analysis of skin biopsies.25
A microscale, organotypic model of peripheral nerve tissue with physiological measures that mimic clinical CAP and NFD tests may be more predictive of clinical outcomes, enabling a more cost-effective approach for selecting promising lead compounds with higher chances of late-stage success. Here, we describe the fabrication and physiological assessment of a 3D microengineered system which enables the growth of a uniquely dense, highly parallel neural fiber tract, where polarity in axonal outgrowth can be controlled. Due to the confined nature of the tract, our model allowed stimulation at the axon bundles distally from neuronal somas, providing a unique approach for studying electrophysiological properties within a 3D in vitro system. This arrangement made possible the measuring of both CAPs and intracellular patch clamp recordings with distally-placed stimulation. Subsequent confocal and transmission electron microscopy (TEM) analysis allows for structural analysis. While perhaps not suited for high-throughput drug discovery, the balance of high-content and medium throughput may position this model for later-stage preclinical development. Taken together, our in vitro model system has the novel ability to assess tissue morphometry and population electrophysiology, analogous to clinical histopathology and nerve conduction testing, representing the first step towards an advanced predictive assay of peripheral nerve behavior.
Experimental
Fabrication of 3D dual hydrogel constructs
3D dual hydrogel constructs were micropatterned according to previously published procedures from our lab26,27 using a unique apparatus for dynamic mask projection photolithography as seen in Fig. 1 below. Briefly, an ultraviolet light source with a 320–500 nm filter (OmniCure 1000, EXFO, Quebec Canada) and collimating adapter (EXFO) was projected onto a digital micromirror device, or DMD, (Discovery™ 3000, Texas Instruments, Dallas, TX) providing a dynamic photomask. An intensity of 181 mW cm−2 at the surface of the membrane, as measured with a radiometer, (306 UV Powermeter, Optical Associates, San Jose, CA) and output of 55 seconds from the UV light source allowed patterned crosslinking throughout the depth of the hydrogel solution in a single irradiation. | ||
| Fig. 1 3D dual hydrogel construct with growth-permissive (Puramatrix) and growth-inhibitive (PEG) regions for confining neurite growth in desired 3D geometry (A). Bright field images of: 3D dual hydrogel construct containing DRG explant with neurite outgrowth after 7 DIV (B). | ||
The walls of 6-well, permeable, collagen-coated PTFE cell culture inserts (Transwell®, Corning Inc., Corning, NY) with a pore size of 0.4 μm were treated with Rain-X (SOPUS Products, Houston, TX) to reduce meniscus formation, and 500 μl of photocrosslinkable solution containing 10% w/v poly(ethylene gycol) dimethacrylate (PEG) with an average molecular weight (MW) of 1000 Da (Polysciences, Warrington, PA) and 0.5% w/v Irgacure 2959 photoinitiator (Ciba Specialty Chemicals, Basel, Switzerland) in PBS was added per cell culture insert. After irradiation, uncrosslinked hydrogel solution was rinsed with PBS and the micropatterned PEG construct remained attached to the membrane surface. Using a pipette, 0.15% w/v Puramatrix (BD Biosciences, Bedford, MA) supplemented with 1 μg ml−1 of soluble laminin (Invitrogen, Carlsbad, CA) was carefully added to voids within the PEG construct. Inserts containing dual-hydrogel constructs were placed in 6-well plates and 1.5 ml of growth medium was added to each well to initiate self-assembly of the peptide gel, Puramatrix. The process has been outlined previously in detail.26–30
Incorporation of dorsal root ganglia explants
All animal handling and tissue harvesting procedures were performed under observation of guidelines set by NIH (NIH Publication #85-23 Rev. 1985) and the Institutional Animal Care and Use Committee (IACUC) of Tulane University. Neural explants were incorporated into dual hydrogel constructs as previously described.28 Briefly, 6-well collagen-coated PTFE cell culture inserts were soaked overnight in adhesion media consisting of Neurobasal medium supplemented with penicillin/streptomycin, nerve growth factor (NGF), 10% fetal bovine serum (FBS), and L-glutamine (Gibco-Invitrogen, Carlsbad, CA). Four dorsal root ganglia (DRG) isolated from Long-Evans rat embryonic day 15 pups (Charles River, Wilmington, MA) were placed on a hydrated cell culture insert and incubated in adhesion media for 2 hours at 37 °C and 5% CO2 to adhere. Adhesion media was then replaced by 500 μl of 10% PEG/0.5% Irgacure 2959 in PBS for construct polymerization.The projected photomask pattern for the PEG construct was aligned around an adhered DRG using visible light and an inverted microscope. UV light was used to project the same photomask for 55 seconds, as described in section 3.1, and effectively confined the DRG within a polymerized PEG construct. The time tissue cultures spent outside of the biosafety cabinet was kept to a minimum to help prevent contamination, and uncrosslinked hydrogel solution was rinsed 3 times with PBS containing 1% penicillin/streptomycin (Gibco-Invitrogen, Carlsbad, CA) to remove unpolymerized PEG solution and improve culture sterility. Excess PBS was removed from patterned voids inside PEG, and approximately 2 μl of Puramatrix was carefully pipetted into the remaining space. In one set of experiments, Puramatrix was omitted from the procedure, causing all neurite outgrowth to grow along the 2D surface of the insert membrane. Finally, the insert containing the dual hydrogel constructs, each with a live DRG explant, was immediately placed in 1.5 ml of growth media (Neurobasal medium supplemented with NGF, penicillin/streptomycin, L-glutamine, and B27; Gibco-Invitrogen, Carlsbad, CA) to initiate the self-assembly of the Puramatrix and maintained at 37 °C and 5% CO2, with medium changes every 48 hours. Experiments were initiated after 7 days to permit neurite outgrowth and neuronal maturation.
Immunocytochemistry
Specimens evaluated with immunohistochemistry were fixed in 4% paraformaldehyde (Electron Microscopy Sciences, Hatfield, PA) for 2 hours at 37 °C. Cell nuclei were stained with DAPI nucleic acid stain according to manufacturer's instructions (Molecular Probes, Eugene, OR). Neurites were tagged using mouse monoclonal [2G10] neuron-specific beta III tubulin primary antibody (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :200), followed by fluorescent tagging with Cy3.5-conjugated goat anti-mouse immunoglobulinG (H + L) secondary antibody (1:100; Abcam, Cambridge, MA). Glial cells were stained using rabbit polyclonal S100-specific primary antibody (1:500, Abcam, Cambridge, MA) and Cy2-conjugated goat anti-rabbit immunoglobulinG (H + L) secondary antibody (1:100, Jackson ImmunoResearch Laboratories, Westgrove, PA). Antibody tagging steps were carried out in PBS with 0.1% saponin and 2% bovine serum albumin (Sigma-Aldrich, St. Louis, MO) overnight at 4 °C, followed by 3 10 minute washes in PBS with 0.1% saponin at room temperature. For constructs stained for myelin, neurites were tagged using mouse monoclonal [2G10] neuron-specific beta III tubulin primary antibody (1:200), followed by fluorescent tagging with Cy2-conjugated goat anti-mouse immunoglobulinG (H&L) secondary antibody (1:500; Abcam, Cambridge, MA). Myelin was stained using Fluoromyelin™ Red Fluorescent Myelin Stain (Molecular Probes, Eugene, OR) for 40 minutes according to manufacturer's recommended preparation.
:200), followed by fluorescent tagging with Cy3.5-conjugated goat anti-mouse immunoglobulinG (H + L) secondary antibody (1:100; Abcam, Cambridge, MA). Glial cells were stained using rabbit polyclonal S100-specific primary antibody (1:500, Abcam, Cambridge, MA) and Cy2-conjugated goat anti-rabbit immunoglobulinG (H + L) secondary antibody (1:100, Jackson ImmunoResearch Laboratories, Westgrove, PA). Antibody tagging steps were carried out in PBS with 0.1% saponin and 2% bovine serum albumin (Sigma-Aldrich, St. Louis, MO) overnight at 4 °C, followed by 3 10 minute washes in PBS with 0.1% saponin at room temperature. For constructs stained for myelin, neurites were tagged using mouse monoclonal [2G10] neuron-specific beta III tubulin primary antibody (1:200), followed by fluorescent tagging with Cy2-conjugated goat anti-mouse immunoglobulinG (H&L) secondary antibody (1:500; Abcam, Cambridge, MA). Myelin was stained using Fluoromyelin™ Red Fluorescent Myelin Stain (Molecular Probes, Eugene, OR) for 40 minutes according to manufacturer's recommended preparation.
Fluorescence microscopy and image processing
Bright field and conventional fluorescent images were acquired with a Nikon AZ100 stereo zoom microscope using 1× and 2× objectives (Nikon, Melville, NY), while confocal images were taken using a Leica TCS SP2 laser scanning microscope and 20× objective (Leica Microsystems, Buffalo Grove, IL). Confocal z-stacks were acquired through the maximum depth of visible neurite growth with thicknesses ranging between 55–65 μm imaged over 20 slices, each 512 × 512. Image processing was performed using ImageJ (National Institutes of Health, Bethesda, MA). For color coding depth in confocal z-stacks, the Z Code Stack function with a Rainbow LUT was applied using the MacBiophotonics Plugin package for ImageJ. Projections of z-stacks were taken as maximum intensity projections. V3D-Viewer software (Janelia Farm Research Campus, Howard Hughes Medical Institute, Ashburn, VA) allowed 3D rendering and visualization of the confocal z-stack images.Transmission electron microscopy
Transmission electron microscopy was used to qualitatively assess morphology, spatial distribution, and nanoscale features of neural cultures. After 7 days in vitro (DIV), constructs were fixed in 4% paraformaldehyde for 2 hours at 37 °C, washed 3 times for 10 minutes with PBS, and sectioned to reveal regions of interest. Post-fixation using 1% OsO4 for 1 hour and 2% uranyl acetate for 30 minutes was performed in limited-light settings with 3 10 minute PBS washes in between. The samples were dehydrated with ethanol (50, 70, 95, and 2 × 100%, 30 minutes each) and embedded in 1:1 propylene oxide-Spurr resin for 45 minutes and 100% spur resin overnight (Low Viscosity Embedding Kit, Electron Microscopy Sciences, Hatfield, PA). Polymerization of specimens occurred at 70 °C over 24 hours.
Embedded samples were trimmed and sliced with thicknesses varying from 80 to 100 nm using a Reichert Ultracut S ultratome (Leica Microsystems, Buffalo Grove, IL) and Ultra 45° diamond knife (Diatome, Fort Washington, PA). Slices were placed on Formvar carbon-coated copper grids with 200 mesh and stained with 2% uranyl acetate and 0.1% lead citrate (20 minutes each). Samples were mounted on a single-tilted stage and examined with a FEI Tecnai G2 F30 Twin transmission electron microscope (FEI, Hillsboro, OR) using an accelerator voltage of 200 kV. Images were taken at 3000–20000× magnifications with 4000 × 4000 pixel resolution. All materials and reagents used for sample preparation were obtained from Electron Microscopy Sciences (Hatfield, PA).
Field potential recording
After 7 DIV, dual hydrogel constructs containing live DRG explants were transferred to an interface chamber held at room temperature and perfused with bicarbonate-buffered artificial cerebrospinal fluid (ACSF) made of, in mM, 124 NaCl, 5 KCl, 26 NaHCO3, 1.23 NaH2PO4, 4 MgSO4, 2 CaCl2, and 10 glucose. ACSF was bubbled with 95% O2, 5% CO2 at all times to maintain consistent oxygenation and pH. Constructs were stained for contrast with 1% Bromophenol Blue (Sigma-Aldrich, St. Louis, MO) and visualized using an SMZ 745 stereomicroscope (Nikon, Melville, NY). Thin-walled borosilicate glass pipettes (OD = 1.5, ID = 1.6; Warner Instruments, Hamden, CT) were pulled to resistances between 3 and 7 MΩ using a P-97 Flaming/Brown micropipette puller (Sutter Instrument Co., Novato, CA) and backfilled with ACSF. As shown in Fig. 5A, recording electrodes were placed near cell somata in the vicinity of each ganglion, and constructs were stimulated with a concentric bi-polar electrode (CBARB75, FHC, Bowdoin, ME) at varying distances away from the ganglion along neurite tracts. An Axopatch-1C amplifier (Molecular Devices, Sunnyvale, CA), coupled with an isolated pulse stimulator (Model 2100; A-M Systems, Sequim, WA), PowerLab 26T digitizer (AD Instruments, Colorado Springs, CO), and LabChart software (AD Instruments, Colorado Springs, CO) was used for recording, stimulating, and data acquisition. Recordings were filtered at 5 kHz, displayed on Tektronix oscilloscopes, and analyzed offline using custom written routines in Igor Pro (WaveMetrics, Portland, OR). Standard deviations were calculated when appropriate. The statistical values were calculated using 2-tailed, paired t-tests with a p value <0.05 considered significant. All values are reported with errors as standard error of the mean (SEM).20 μM DNQX (6,7-dinitroquinoxaline-2,3-dione) and 50 μM APV (2R)-amino-5-phosphonopentanoate) were used to identify and block synaptic activity. 0.5 μM tetrodotoxin (TTX) was used to as a complete blockade of Na+ channel activity. All drugs and salts used in experimental solutions were obtained from Tocris (Minneapolis, MN) and Sigma-Aldrich (St. Louis, MO) respectively.
Whole-cell patch clamp recording
After 7 DIV, constructs were transferred to a submersion recording chamber at room temperature and allowed to equilibrate for 20 minutes. Bicarbonate-buffered ACSF solution (containing, in mM, 124 NaCl, 5 KCl, 26 NaHCO3, 1.23 NaH2PO4, 1.5 MgCl2, 2 CaCl2, and 10 glucose) was bubbled with 95% O2, 5% CO2 at all times to maintain consistent oxygenation and pH. For voltage clamp recordings, borosilicate glass pipettes were filled with a cesium-substituted intracellular solution containing, in mM, 120 CsMeSO3, 1 NaCl, 0.1 CaCl2, 2 ATP, 0.3 GTP, 10 HEPES, and 10 EGTA. For current clamp recordings, pipettes were filled with a potassium gluconate based internal solution containing, in mM, 120 Kgluconate, 10 KCl, 10 Hepes, 10 D-sorbitol, 1 MgCl2·6H20, 1 NaCl, 1 CaCl2, 10 EGTA, 2 ATP. Pipette resistances ranged from 4 to 7 MΩ. Series access resistance ranged from 7 to 15 MΩ and was monitored for consistency. For evoked action potential recordings, concentric bipolar stimulating electrodes (CBARC75, FHC, Bowdoin, ME) were placed in the afferent fibers of the DRG, and after attaining a whole-cell patch, action potentials were evoked using minimum stimulation necessary, typically <0.01 mA. Placement of recording and stimulating electrodes is shown in Fig. 7A below.DRGs were visualized with a BX61WI Olympus upright microscope (Olympus, Center Valley, PA) with live differential interference contrast (DIC) imaging. Whole-cell recordings were made with a PC-505B patch clamp amplifier (Warner Instruments, Hamden, CT). Signals were digitized with a PowerLab 26T digitizer and collected with Lab Chart acquisition software (AD Instruments, Colorado Springs, CO). Signals were amplified, sampled at 20 kHz, filtered to 2 kHz, and analyzed using custom written routines in Igor Pro (WaveMetrics, Portland, OR).
Results
Spatial and morphological characteristics of 3D neural cultures
We developed a novel in vitro 3D neural culture designed to approximate the cyto and macro-scale architecture of native afferent peripheral nervous tissue. The 3D neural constructs consist of DRG tissue explants cultured on a permeable cell culture membrane insert that are contained by PEG constructs confining growth to patterned voids filled with Puramatrix. Narrow tracts guiding neurite growth from the ganglion along the x-axis measure ~490 μm in diameter, up to ~400 μm thick, and ~3 mm in length. A 3D dual hydrogel construct containing DRG neurons, glia, and neurite growth is shown after 7 DIV in Fig. 2.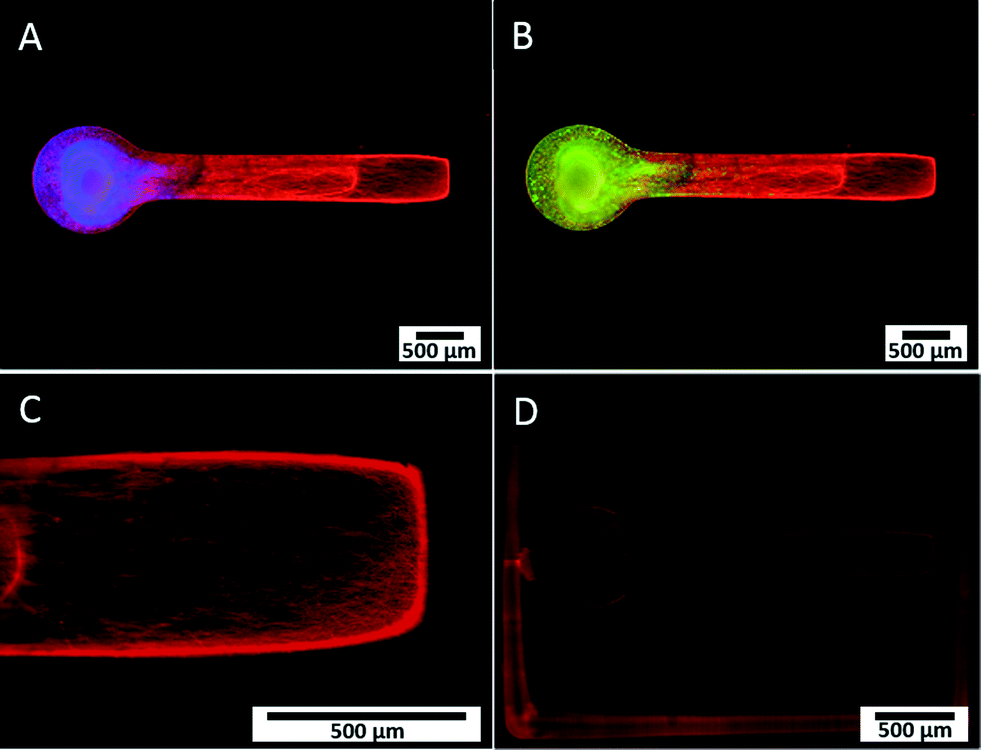 | ||
| Fig. 2 Fluorescence microscopy of DRG neurite growth and cell migration in 3D dual hydrogel constructs after 7 DIV. β-III tubulin-positive neurites (red), DAPI-stained nuclei (blue, A), and S100-positive glial cells (green, B) confined within channel filled with Puramatrix. Neurite growth present near the end of the channel, approximately 3 mm from the ganglion, as measured from the end of the circular region containing ganglion and the start of the straight channel (C). Absence of myelin after Fluoromyelin™ Red Fluorescent Myelin Stain (D). | ||
Similar to previous reports,26,27 the neurites and supportive glial cells were effectively constrained by the geometry of the PEG hydrogel. Simultaneous labeling with anti- β-III tubulin, anti-S100, and DAPI confirmed outgrowth after 7 DIV was consistently robust and all labeled structures were within the Puramatrix portion of the construct [Fig. 2(A, B)]. Presence and migration of supportive cells, including glial cells, spans up to 3/4 of the length of the channel, nearly 1.8 mm away from the ganglion as measured from the start of the straight channel.
Leading neurite growth throughout the depth of the Puramatrix occurred randomly within the channel with a considerable amount of branching and fasciculation at multiple planes of focus [Fig. 2(C)]. Conversely, growth in channels deprived of Puramatrix appears limited and aligned along the fibers of the insert at the membrane surface (data not shown). Consistent with literature suggesting myelin formation occurs only with antioxidant supplementation after 14 DIV,31 3D neural cultures showed no presence of myelin after staining with Fluoromyelin™ Red Fluorescent Myelin Stain (Molecular Probes, Eugene, OR) at 7 DIV [Fig. 2(D)].
Confocal imaging confirmed β-III tubulin-positive neurites occurred in 3 dimensions at the beginning, middle, and end of the channel, as demonstrated previously as well as DAPI-stained nuclei, and S100-positive glial cells throughout the z-stack [Fig. 3(A–C)]. In agreement with reports of preferential growth along interfaces of two materials,32,33 antibody labeling in images suggest denser neurite growth along the edges of the channel [Fig. 2(C) and 3(C)]. Cross-sectional images from transmission electron microscopy (TEM) support the evidence shown by fluorescent microscopy. Slices taken within the ganglion and in the neural tract show high density of parallel, highly fasciculated unmyelinated neurites [Fig. 4(A, B)], presence of Schwann cells [Fig. 4(D)], and the beginning of Schwann cell encapsulation of neurites [Fig. 4(C)].
Population electrophysiological properties of 3D neural fiber tracts
To test the functional properties of the 3D neural culture and determine whether it serves as a physiologically relevant model of afferent peripheral nervous tissue, we conducted intracellular and extracellular electrophysiological experiments after 7 DIV.Using techniques adapted from traditional field potential recordings in acute rodent brain slices, our constructs were studied on an interface chamber permitting the use of a custom rig for extracellular recording. For each experiment, a recording electrode was placed in the ganglion, or somatic region of the construct, and a stimulating electrode in the channel was inserted along the neurite tract [Fig. 5(A)]. Following stimulation, a compound action potential (CAP) propagated in a retrograde manner into the somatic region and was recorded as the resulting extracellular potential change in the ganglion for each construct (n = 19). However, for constructs lacking Puramatrix, and therefore confined to 2D growth, no responses were detected, despite numerous attempts (data not shown). The 3D neural constructs supported field recordings for over an hour and consistently displayed coherent population spikes upon stimulation [Fig. 5(B)]. Similar to compound action potentials recorded from intact nerves, responses consistently exhibited a short latency to onset followed by a single, cohesive event with a graded nature representing the summed effect of each action potential on recruited axons and corresponding cells. The consistent short envelope and delay of onset of the responses are also characteristic of a CAP and suggest a fast event purely driven by action potentials. As with nerve stimulation, more fibers were recruited with higher stimulus intensities yielding stronger responses until maximum excitation occurred (data not shown).
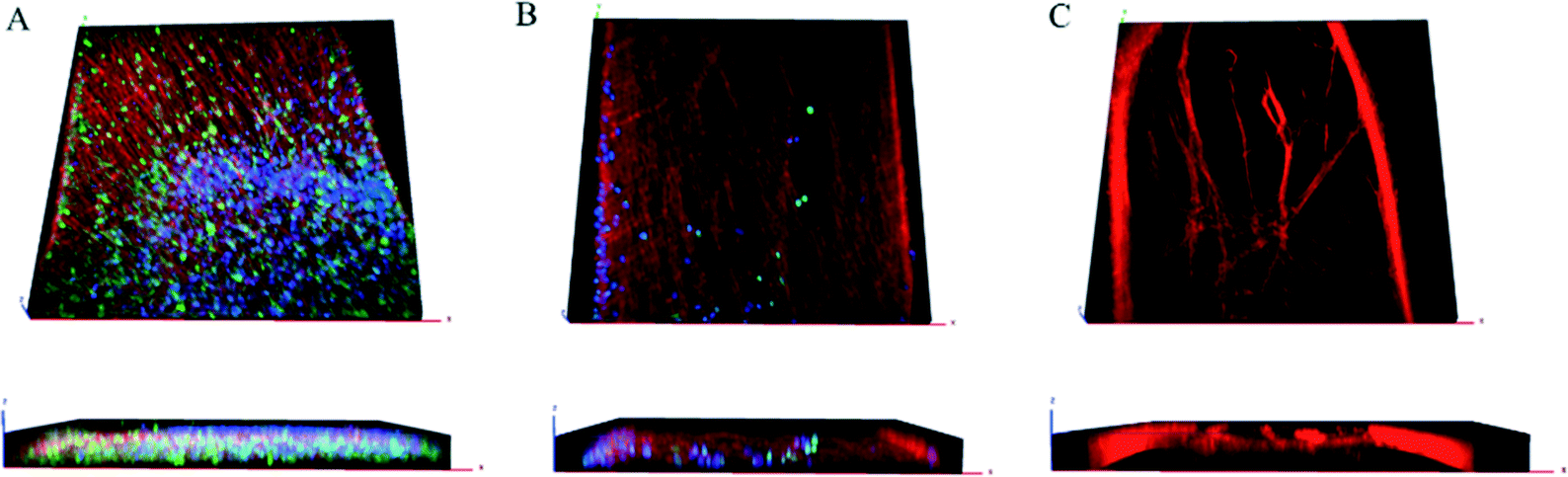 | ||
| Fig. 3 3D rendering of confocal images. β-III tubulin-positive neurites (red), DAPI-stained nuclei (blue), and S100-positive glial cells (green) shown in 3D at beginning (A), middle (B), and end (C) of channel with corresponding cross-sections in the z-plane shown below. | ||
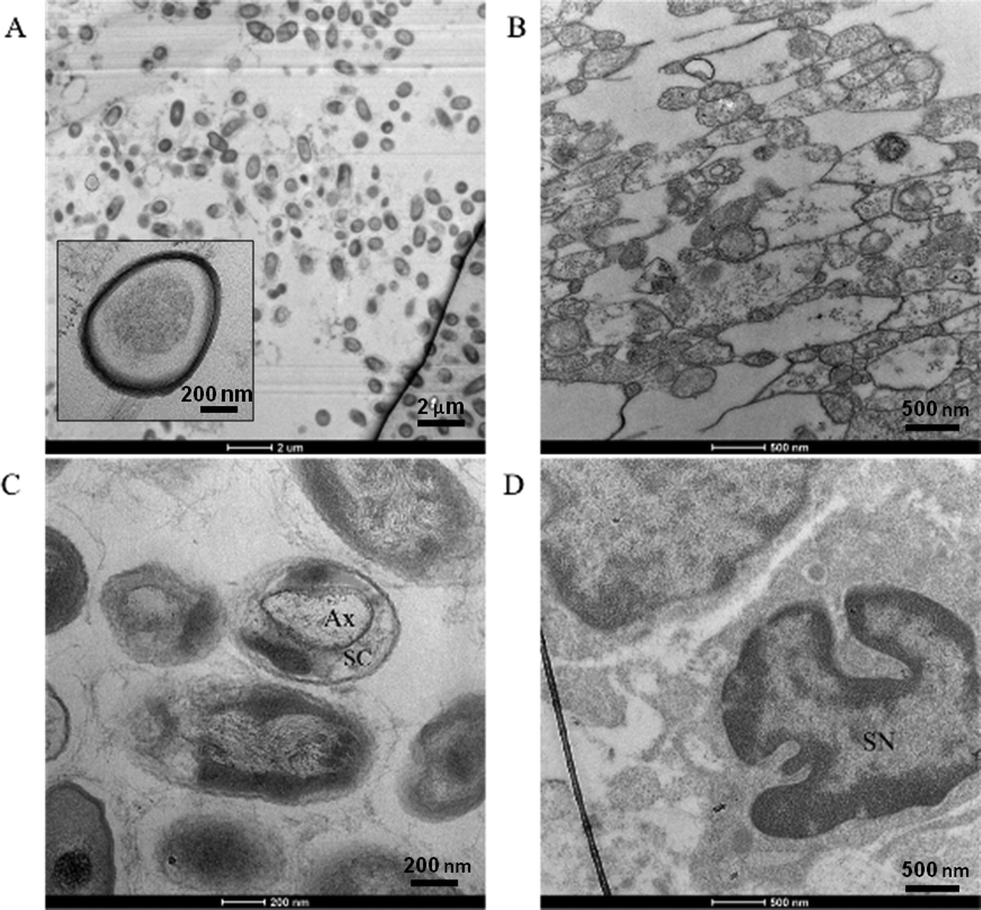 | ||
| Fig. 4 Transmission electron microscopy of neural culture cross-sections. High density of parallel, fasciculated unmyelinated neurites in channel approximately 1.8 mm from ganglion, inset showing zoomed view (A–B). Focus centered on an axon (Ax) encapsulated by a Schwann cell (SC) approximately 1 mm from the ganglion (C). Schwann cell nucleus (SN) found in ganglion (D). All measurements made from the end of the circular region containing ganglion at the start of the straight channel. | ||
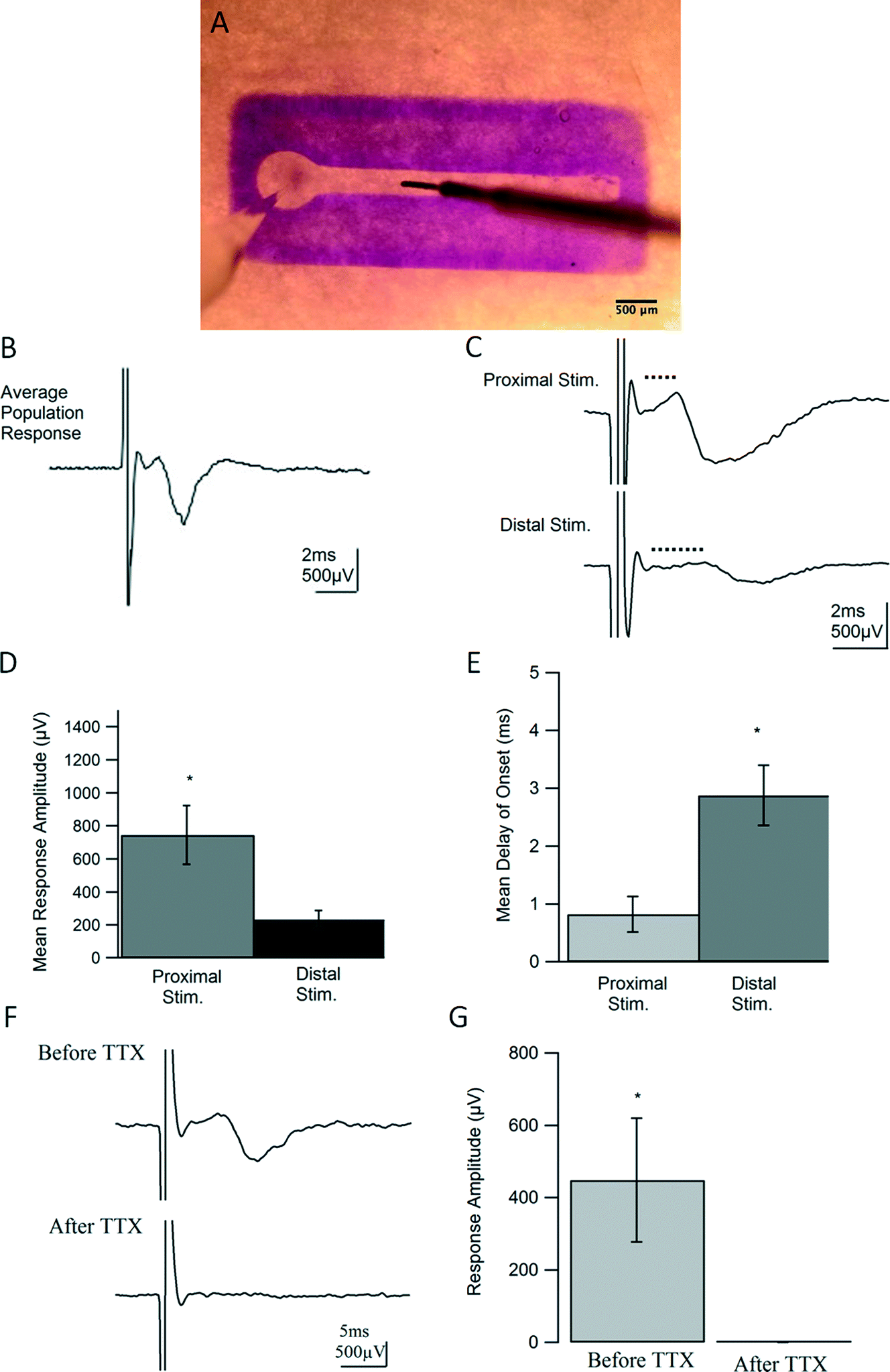 | ||
| Fig. 5 Bromophenol Blue-stained construct with placement of recording (left) and stimulating (right) electrodes placed within ganglion and neural tract in channel, respectively, for field recording (A). Example trace of population response demonstrating successful field potential recordings in 3D neural constructs and waveform properties characteristic of a compound action potential (B, CAP). Field potential evoked in ganglion of 3D neural cultures from proximal (1.5 mm) and distal (2.25 mm) locations, n = 4. Marked by dotted lines, average traces highlight the increase in delay to onset when stimulating distally (C). Distal stimulation caused a significant increase (p = 0.02) in delays to onset with average delay time increasing from 0.82 ms to 2.88 ms, as well as a significant decrease (p = .03) in average response amplitude from 745 μV to 234 μV (D, E). Stimulation distances were measured from the start of the straight channel to the point of stimulation. Delay of onset was measured as the time between the return of the stimulus artifact to baseline to the positive peak of the response. Blockade of Na+ channel activity using 0.5 μM tetrodotoxin (TTX) in 3D neural constructs. Average traces demonstrating abolishment of population response by TTX, n = 3 (F). Response amplitudes were significantly different (p = 0.029), with average amplitudes decreasing from 448.75 μV to 0.04 μV after TTX wash-in (G. Amplitudes were measured from peak-to-peak. | ||
The delay to onset of the response was also increased when the distance between the recording and stimulating electrode was enlarged [Fig. 5(C, E)], confirming the ability of the geometrically-confined neural culture to conduct signals at varying distances along its nerve-like tract. On average, responses only displayed a delay of onset of 0.82 ms when stimulated proximally or within 1.5 mm from the ganglionic region, as measured from the start of the straight channel. However, when the stimulating electrode was moved 2.25 mm from the ganglion, the distal stimulation yielded delays of onset with an average of 2.88 ms that are statistically significant, p < 0.05, from those observed in proximal stimulation [p = 0.02]. As seen in fluorescent microscopy [Fig. 2(C) and 3], 7 days of in vitro growth does not allow for neurites to completely fill the channel, and a decrease in amplitude from 745 μV to 234 μV was exhibited in moving from proximal to distal stimulation [p = .033, Fig. 5(C, D)]. Furthermore, by inhibiting Na+ channel activity, action potentials could no longer be generated upon stimulation. Responses from constructs could be completely abolished within 2 minutes of introducing 0.5 μM TTX, confirming the source and biological nature of the responses. Responses before and after TTX wash-in are statistically significant, p = 0.029, n = 3 [Fig. 5(F, G)].
To investigate whether the responses contained a synaptic component, we introduced glutamate receptor inhibitors DNQX and APV at 20 and 50 μM respectively to block excitatory synaptic transmission. The experiment lasted for 35 minutes with recordings taken every minute and time points referred to as t1–t35 for simple reference. The drug wash-in occurred 5 minutes into the experiment, t6, and wash-out 20 minutes later, or 25 minutes into the experiment at t26. Responses prior to drug wash-in, t1–t5, were compared to responses recorded 10 minutes into the drug wash-in, t16–t20, allowing ample time for drugs to perfuse and take effect. There was no statistically significant difference observed in the response amplitude or duration before and after wash-in of the drugs [Fig. 6(A–C)], suggesting there was no synaptic component of the response to block.
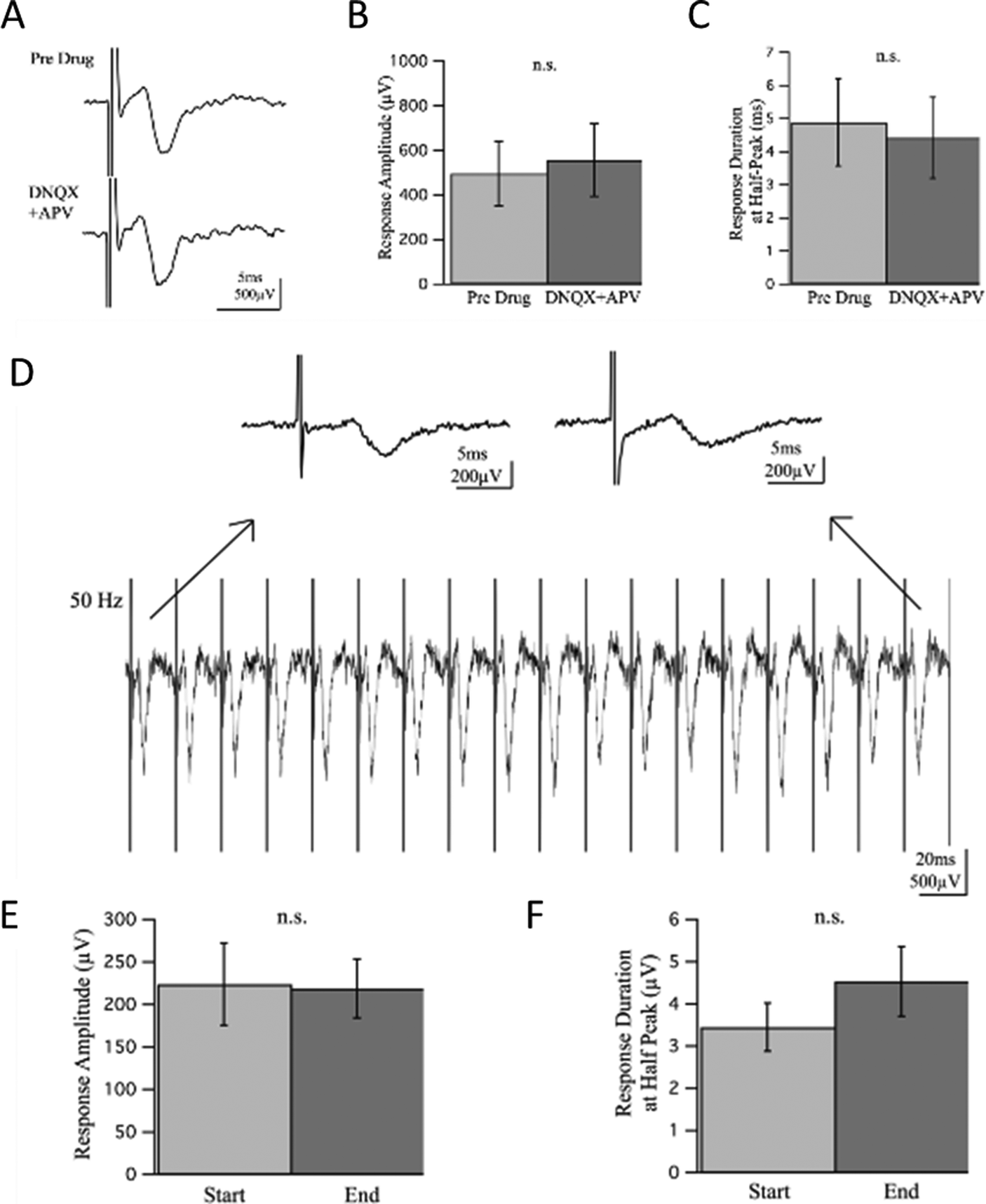 | ||
| Fig. 6 No effect of excitatory glutamate blockers DNQX and APV in 3D neural constructs, n = 4. Average traces of responses before (t1–t5) and after (t16–t20) drug wash-in demonstrate no marked change in response amplitude or duration from drugs (A). Response amplitudes and durations were of no statistical difference after DNQX and APV (B–C). Amplitudes were measured peak-to-peak and durations at half-peak to minimize variance between measurements. Consistent firing of response during high frequency stimulation in 3D neural constructs, n = 3. Example traces demonstrate the consistency of the electrically-evoked population spike during the 50 Hz train, with enlarged traces at the start and end for comparison (D). The amplitudes and duration of responses at the end of the 50 Hz pulse train are not significantly different than those at the start (E–F). Amplitudes were measured from peak-to-peak and durations at half-peak to minimize variance between measurements. | ||
A high frequency train of pulses was also induced to assess characteristics of the response. When 20 pulses at 50 Hz were applied to the cultures, the population spikes maintained a consistent delay of onset, envelope, and amplitude suggesting a strong response capable of repeatedly firing with no depression or facilitation, direct antidromic evocation of action potentials. Response amplitude and duration at half-peak before and after high frequency stimulation were not statistically significant [Fig. 6(D–F)].
Whole-cell electrophysiological properties of neurons in 3D constructs
To verify that the absence of aberrant self-synapsing within the DRG, as well as to observe basal electrical activity within cells in the construct, intracellular recordings were employed. Using this method, we achieved whole-cell patch clamp access inside the 3D neural constructs for over an hour. Modified techniques from whole-cell patch clamping in acute rodent brain slices allowed voltage and current clamp recordings. However, the hydrogel construct in which the tissue was cultured presented challenges. The Puramatrix gel is more adhesive than native brain tissue and the dense DRG explant contains connective tissue. As with field recordings, these features made movement, re-placement, and continued use of the electrodes difficult. Cells within the DRG were densely populated, had less contrast, and were harder to visualize than more sparsely distributed cells in brain slice neuropil that are normally surrounded by features with different diffractive indices. Through repeated visualization in multiple focal planes, positive pressure while navigating through gel, and a tilted electrode approach angle,34 successful whole-cell patch clamp recordings were possible [Fig. 7].Again, a bipolar stimulating electrode was placed in the neurite tract within the channel and recordings were taken from cells in the somatic region of the construct [Fig. 7(A, B)]. Cells supported electrically-evoked antidromic action potentials driven from neurite extensions in the channel [Fig. 7(C)]. Characteristic of responses lacking synaptic input, intracellular responses had fast rise times, averaging 2 ms baseline-to-peak, with distinct, non-graded onsets as shown in [Fig. 7(D)]. There was no rise in potential leading to threshold prior to the onset of the response [Fig. 7(D)] nor were there any smaller, graded events following the response [Fig. 7(C)], yielding no evidence of synaptic input. Moreover, if synaptic activity were contributing to the onset of the response, threshold for the initiation of action potentials would be harder to reach under hyperpolarization. However, the cells were still able to support action potentials when hyperpolarized from resting membrane potential (RMP) to −100 mV, 1.95× less than the RMP on average, and displayed responses no different than when at RMP. Furthermore, spontaneous activity caused by synaptic activation was not observed in baseline recordings under voltage. Finally, recording in current clamp with potassium-gluconate based internal solution revealed no graded voltage fluctuations, but did reveal spontaneous action potentials in a subset of cells [Fig. 7(E, F)].
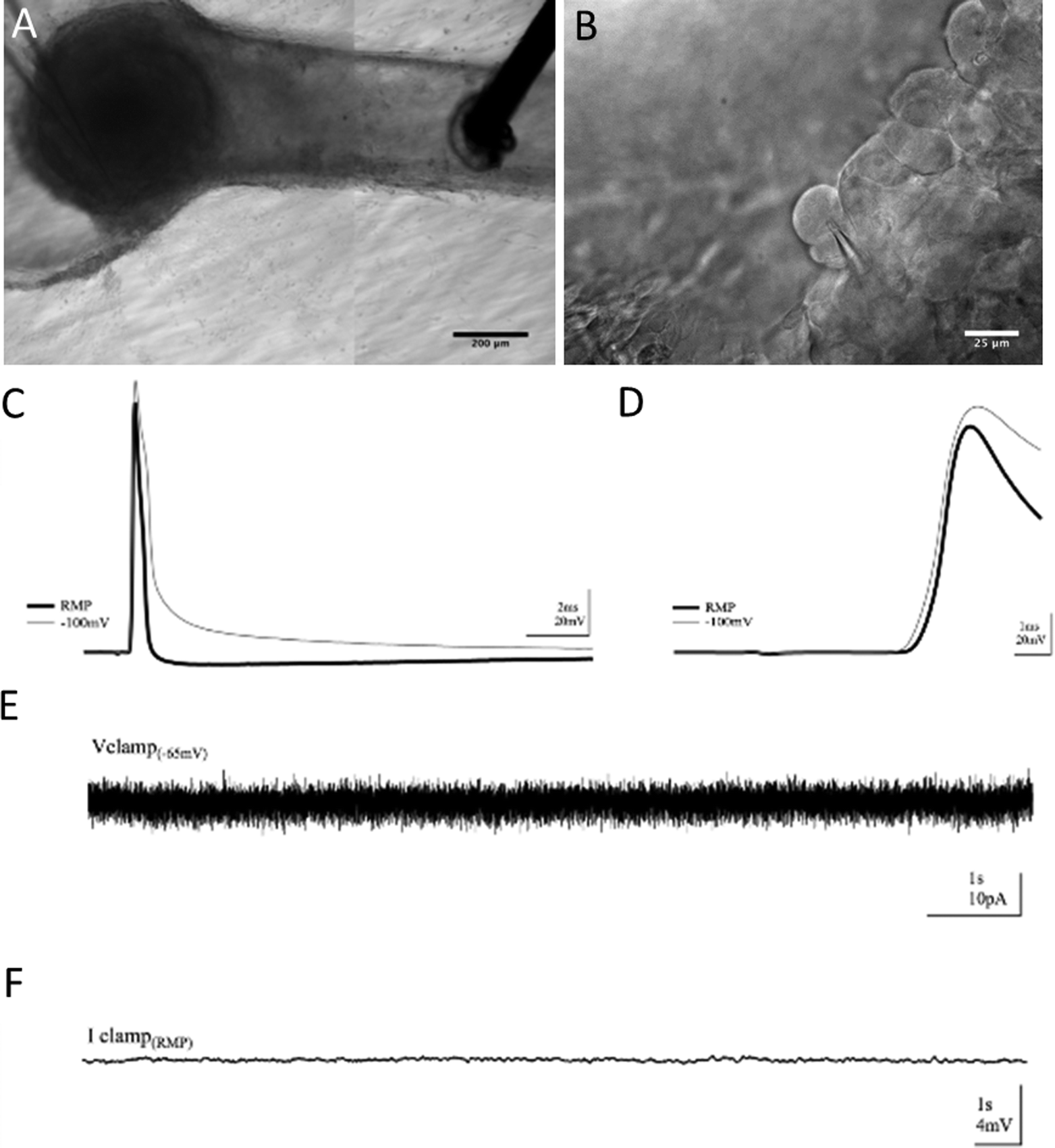 | ||
| Fig. 7 Placement of recording (left) and stimulating (right) electrodes for whole-cell patch clamp (A) and electrode patch of primary sensory neuron (B). Whole-cell patch clamp recordings in 3D neural cultures exhibiting no evidence of synaptic connectivity. n = 3. Example trace displaying an electrically-evoked action potential recorded from a cell in the ganglion (C). Enlarged trace demonstrating quick, non-graded onset of response (D). Voltage clamp trace with no spontaneous currents (E). Current clamp trace exhibiting no spontaneous changes in potential (F). | ||
Discussion
We present a platform for culturing and investigating 3D biomimetic neural models that more closely recapitulates the structural and functional characteristics of native tissue compared to current 2D culture conditions. The need for models to utilize organ specific functional endpoints is becoming increasingly apparent.14 For peripheral nerve applications, electrophysiological studies of compound action potential (CAP) generation and propagation may help provide a link between molecular mechanistic events and changes in the structure-function relationships due to disease pathology, toxic side effects or pharmaceutical interventions. Specifically, our dual hydrogel system spatially confines growth to geometries resembling nerve fiber tracts, allowing for anatomically-relevant neural growth in three dimensions. Perhaps more importantly, outputs resembling clinically relevant test criteria, including the measurement of CAP and nerve fiber density (NFD) are possible through our advanced model.In native tissue, fibers, or axons, of varying density, diameter and degree of myelination conduct sensory information back to the central nervous system at different velocities. Schwann cells support the sensory relay by myelinating axons and providing insulation for swifter conduction. The ability to measure disruption of these structural components of peripheral nerves due to external compounds would have far reaching implications into many applications (e.g. toxicity and diabetic induced neuropathy, pain research and neuroprotection). Although neuronal morphology is a useful indicator of phenotypic maturity, a still more functional sign of neuronal condition is their ability to conduct an action potential.
Normally, electrophysiological experiments are undertaken in either dissociated surface-plated cultures or acute/organotypic slice preparations, all with limitations. Investigation in 2D dissociated cell cultures is limited to the level of single-cell recordings or spontaneous unstructured network activity due to a lack of organized, multi-cellular neuritic architecture, as would be found in slice preparations. Furthermore, 3D cell culture models have demonstrated closer physiological similarity to in vivo conditions for native voltage-gated ion channel functionality, resting membrane potentials, intracellular Ca2+ dynamics, and Na+/H+ exchange over traditional 2D models.20,34–39 Slice preparations have intact neural circuitry and allow both intra and extracellular studies. However, acute brain slices present a complex, simultaneous array of variables lacking the means to control individual factors and are inherently limited in throughput capability. In contrast, our 3D in vitro model with axon growth sufficiently confined to form a dense, polarized arrangement, enabled field recordings of CAPs, as well as whole-cell recordings with distal stimulation, both of which can normally only be done in intact tissue.
Similar to the structure of a human afferent peripheral nerve,40 DRG neurons in our constructs project long, parallel, fasciculated axons confined to a compact tract. Additionally, the 3D growth observed in our construct comprised axons of various diameters in dense, parallel orientation spanning distances up to 3 mm. At 7 DIV we observed no myelination, which is not surprising considering 14 DIV as well as the incorporation of specific antioxidants has been reported as minimum conditions for Schwann envelopment.31 The presence of migratory Schwann cells, as observed in confocal imaging (Fig. 2B and 3A–B), combined with axonal sheathing, as seen in TEM imaging (Fig. 4C), suggests the potential for studies incorporating myelination, or dismyelination, with proper conditions. While the experiments reported herein were restricted to 7 DIV, the constructs have been cultured for up to 5 weeks in vitro, suggesting the feasibility of such studies. As mentioned above, our ability to perform morphometric analysis represents potentially novel methodology to measure one form of clinically relevant endpoints in a benchtop model.
In addition, our 3D hydrogel constructs supported recordings of both single-cell and population-level activity of cultured neurons. The dual hydrogel system successfully adapted whole-cell patch clamp techniques and confined neurite growth to a 3D geometry allowing the measure of synchronous population level events in extracellular field recordings. To our knowledge, the measure of such an endpoint, directly analogous to measurement used in clinical NCD testing, has not previously been demonstrated for purely cellular in vitro studies. In both experimental setups, the DRG primary sensory neurons with a nerve-like tract supported electrically-evoked action potentials, specifically CAP in field recordings, for over an hour. We did attempt to obtain field recordings in 2D cultures constrained by the PEG mold, i.e. lacking Puramatrix. However, after many attempts, we were not able to detect any responses in these cultures. Likely there is simply an exceedingly small margin of error for placement of the electrodes in a manner as to both properly stimulate the bundle of axons and detect sufficient ion flux away from the recording tip in a 2D preparation lacking “tissue” architecture. Electrophysiological results reinforce evidence shown in fluorescent and electron imaging, suggesting the 3D neural culture we present resembles basic functional characteristics of a sensory peripheral nerve.
As demonstrated in Fig. 5, field recordings measured the combined extracellular change in potential caused by signal conduction in all recruited fibers. Electrically-evoked population spikes were graded in nature, comprising the combined effect of action potentials in slow and fast fibers. Spikes were single, cohesive events with swift onsets and short durations characteristic of CAPs or responses comprised purely of action potentials with quick signal conduction in the absence of synaptic input. Voltage-gated Na+ channel blockade with TTX completely and reversibly abolished all compound action potentials in the constructs further indicating the biological nature and driving force of the responses. The 3D neural constructs also supported CAP stimulated from varying distances along the neurite tract or channel, demonstrating the culture's ability to swiftly carry signals from distant stimuli much like an afferent peripheral nerve. Additionally, this ability to support proximal and distal stimulation techniques is an important aspect of future development of our model, as nerve conduction velocity could be easily calculated through careful analysis of stimulation/recording distances.
In native afferent peripheral nerves, there are single axons that span from the dorsal root ganglion to the point of sensation with no need for synapses to relay information until reaching the dorsal horn in the spinal cord. We expected a similar conduction mechanism when electrically stimulating our cultures, as there were no other cell types present with which the DRG primary sensory neurons could synapse. To verify whether neurites were synapsing back onto each other though, experiments designed to detect synaptic activity were performed [Fig. 6]. By inhibiting glutamatergic binding at NMDA and AMPA receptors, excitatory synaptic transmission can be mitigated. Glutamate receptor antagonists DNQX and APV were introduced at typical concentrations of 20 and 50 μM respectively and were allowed ample time to perfuse and take effect during field recordings. If synaptic activity were contributing to responses and thereby blocked by DNQX and APV, a decrease in amplitude and duration of the response would be expected. However, there was no significant change in response amplitude or duration in field potential recordings between pre-drug and drug wash-in conditions. High frequency stimulation was induced to further assess synaptic activity during continuous neural firing. If synaptic input were present, depression or facilitation of the response would occur from either depletion of transmitter-releasing vesicles or an increase in presynaptic Ca2+ ions causing increased transmitter release. A train of 20 pulses at 50 Hz was induced, yet no significant change in response amplitude or duration was observed, indicating no synaptic component was present in the response.
Intracellular recordings supported the notion of purely action potential (AP) driven responses as well. The purpose of our whole-cell recordings was to demonstrate that the 3D preparation is amenable to single-cell evaluation and to provide further evidence that the population responses were indeed compound action potentials. If synaptic input were contributing to electrically-evoked responses, small rises in potential would occur and would be visible under current clamp as graded events either leading to the onset of an AP or following the AP once potential has returned to baseline. Under whole-cell patch clamp recordings of cells in 3D neural cultures, responses displayed spikes void of graded onsets and small increases in potential independent of the AP [Fig. 7]. Cells under whole-cell patch clamp were also hyperpolarized to −100 mV to make threshold more difficult for the cell to reach after stimulation if responses were synaptically-driven. However, APs were also supported under hyperpolarization, confirming an onset not brought on by synaptic input. Furthermore, no spontaneous currents or changes in potential occurred during baseline recordings, supporting the hypothesis of synapse absence and a purely AP-driven response evoked by external stimuli.
The aforementioned results confirm the architecture and physiology of the 3D neural cultures we present. They also demonstrate the ability to integrate electrophysiological experiments and utilize our 3D hydrogel constructs as platforms for culturing and testing biomimetic neural growth. While our investigations have been centered on primary sensory neurons and their simulation of a sensory peripheral nerve, our approach may be utilized to study different neuronal cell types in various geometries in support of a wide range of applications. Further developments will be required in order to ensure a more robust model for drug development, toxicity testing and disease modeling. First, the use of either dissociated primary cells or stem-cell derived neurons, rather than explants, could significantly enhance our throughput. It is also important to point out that any model seeking widespread adoption will require validation with a variety of compounds exhibiting known outcomes.14 The inclusion of a more biomimetic range of growth factors in the culture medium should lead to more robust development of numerous fiber subtypes, as is typical in nerve fiber tracts.41 Similarly, biomolecular cues could be introduced to engineer more advanced spatially controlled models.26,29,30 Towards that end, our past work focused on axonal outgrowth and guidance, and so this model could also be applied to therapeutics targeting nerve damage and/or repair.26–30,42 As mentioned previously, consistent myelination of axons would allow for studies of the pathology of various forms of demyelinating diseases, as well as the development of compounds encouraging remyelination. Additionally, an in depth analysis of the structure-function relationship will allow for more robust analysis of model outcomes.
Conclusions
In conclusion, our easily manufactured, potentially high-throughput 3D hydrogel constructs support robust biomimetic neural cultures that closely approximate native in vivo architecture and physiology. This is the first report of a 3D microengineered neural culture supporting both successful intracellular AP and extracellular CAP recordings. Changes in both signal conduction amplitude and delay were seen based on the location of stimulation. Additionally, we demonstrated the effect of an externally applied drug (TTX) to modulate functional outputs, while blocking synaptic transmission (administration of DNQX and APV) had no consequence. With further development, we propose that such a versatile, 3D in vitro model of peripheral nerve will help bridge the gap between simple cell culture models, which are best for identifying mechanistic pathways, and animal models, best used for investigation of interacting organ systems and simultaneous presentation of many variables.Conflict of interest disclosure
JLC, BJH, and MJM are inventors on a patent that has been filed based on the technology presented in this article. JLC and MJM are co-founders of a start-up company that intends to license this technology.Acknowledgements
The authors thank Dr. Jibao He for assistance with TEM sample preparation and imaging. Funding was provided in part by Tulane University, an NSF CAREER Award to MJM (CBET-1055990), and the DoD (W82XWH-12-1-0246).Notes and references
- D. Huh, Y. S. Torisawa, G. A. Hamilton, H. J. Kim and D. E. Ingber, Lab Chip, 2012, 12, 2156–2164 RSC.
- A. M. Ghaemmaghami, M. J. Hancock, H. Harrington, H. Kaji and A. Khademhosseini, Drug Discovery Today, 2011, 17, 173–181 CrossRef PubMed.
- A. K. Capulli, K. Tian, N. Mehandru, A. Bukhta, S. F. Choudhury, M. Suchyta and K. K. Parker, Lab Chip, 2014, 14, 3181–3186 RSC.
- J. H. Sung, M. B. Esch, J. M. Prot, C. J. Long, A. Smith, J. J. Hickman and M. L. Shuler, Lab Chip, 2013, 13, 1201–1212 RSC.
- L. Kimlin, J. Kassis and V. Virador, Expert Opin. Drug Discovery, 2013, 8, 1455–1466 CrossRef CAS PubMed.
- G. Vunjak Novakovic, T. Eschenhagen and C. Mummery, Cold Spring Harbor Perspect. Med., 2014, 4 Search PubMed.
- S. S. Bale, L. Vernetti, N. Senutovitch, R. Jindal, M. Hegde, A. Gough, W. J. McCarty, A. Bakan, A. Bhushan, T. Y. Shun, I. Golberg, R. Debiasio, B. O. Usta, D. L. Taylor and M. L. Yarmush, Exp. Biol. Med., 2014, 239, 1180–1191 CrossRef PubMed.
- C. S. Cheng, B. N. Davis, L. Madden, N. Bursac and G. A. Truskey, Exp. Biol. Med., 2014, 239, 1203–1214 CrossRef PubMed.
- Z. Hou, J. Zhang, M. P. Schwartz, R. Stewart, C. D. Page, W. L. Murphy and J. A. Thomson, Stem Cell Res. Ther., 2013, 4(Suppl 1), S12 CrossRef PubMed.
- H. T. Hogberg, J. Bressler, K. M. Christian, G. Harris, G. Makri, C. O'Driscoll, D. Pamies, L. Smirnova, Z. Wen and T. Hartung, Stem Cell Res. Ther., 2013, 4(Suppl 1), S4 CrossRef PubMed.
- I. Kola and J. Landis, Nat. Rev. Drug Discovery, 2004, 3, 711–715 CrossRef CAS PubMed.
- D. Schuster, C. Laggner and T. Langer, Curr. Pharm. Des., 2005, 11, 3545–3559 CrossRef CAS.
- J. A. DiMasi, R. W. Hansen and H. G. Grabowski, J. Health Econ., 2003, 22, 151–185 CrossRef.
- A. Astashkina and D. W. Grainger, Adv. Drug Delivery Rev., 2014, 69–70, 1–18 CrossRef CAS PubMed.
- E. R. McConnell, M. A. McClain, J. Ross, W. R. Lefew and T. J. Shafer, NeuroToxicology, 2012, 33, 1048–1057 CrossRef CAS PubMed.
- M. W. de Groot, M. M. Dingemans, K. H. Rus, A. de Groot and R. H. Westerink, Toxicol. Sci., 2013, 137, 428–435 CrossRef PubMed.
- B. J. Wainger, E. Kiskinis, C. Mellin, O. Wiskow, S. S. Han, J. Sandoe, N. P. Perez, L. A. Williams, S. Lee, G. Boulting, J. D. Berry, R. H. Brown, Jr., M. E. Cudkowicz, B. P. Bean, K. Eggan and C. J. Woolf, Cell Rep., 2014, 7, 1–11 CrossRef CAS PubMed.
- C. R. Muratore, H. C. Rice, P. Srikanth, D. G. Callahan, T. Shin, L. N. Benjamin, D. M. Walsh, D. J. Selkoe and T. L. Young-Pearse, Hum. Mol. Genet., 2014, 23, 3523–3536 CrossRef CAS PubMed.
- M. E. Spira and A. Hai, Nat. Nanotechnol., 2013, 8, 83–94 CrossRef CAS PubMed.
- M. Frega, M. Tedesco, P. Massobrio, M. Pesce and S. Martinoia, Sci. Rep., 2014, 4, 5489 CAS.
- V. Benfenati, S. Toffanin, S. Bonetti, G. Turatti, A. Pistone, M. Chiappalone, A. Sagnella, A. Stefani, G. Generali, G. Ruani, D. Saguatti, R. Zamboni and M. Muccini, Nat. Mater., 2013, 12, 672–680 CrossRef CAS PubMed.
- G. Melli and A. Hoke, Expert Opin. Drug Discovery, 2009, 4, 1035–1045 CrossRef CAS PubMed.
- D. Liazoghli, A. D. Roth, P. Thostrup and D. R. Colman, ACS Chem. Neurosci., 2012, 3, 90–95 CrossRef CAS PubMed.
- M. M. Taiana, R. Lombardi, C. Porretta-Serapiglia, E. Ciusani, N. Oggioni, J. Sassone, R. Bianchi and G. Lauria, PLoS One, 2014, 9 Search PubMed.
- A. Z. Burakgazi, W. Messersmith, D. Vaidya, P. Hauer, A. Hoke and M. Polydefkis, Neurology, 2011, 77, 980–986 CrossRef CAS PubMed.
- E. L. Horn-Ranney, J. L. Curley, G. C. Catig, R. M. Huval and M. J. Moore, Biomed. Microdevices, 2013, 15, 49–61 CrossRef CAS PubMed.
- J. L. Curley and M. J. Moore, J. Biomed. Mater. Res., 2011, 99, 532–543 CrossRef PubMed.
- J. L. Curley, S. R. Jennings and M. J. Moore, J. Visualized Exp., 2011, 48, e2636 Search PubMed.
- J. L. Curley, G. C. Catig, E. L. Horn-Ranney and M. J. Moore, Biofabrication, 2014, 6, 035026 CrossRef PubMed.
- E. L. Horn-Ranney, P. Khoshakhlagh, J. W. Kaiga and M. J. Moore, Biomater. Sci., 2014, 2, 1450–1459 RSC.
- J. L. Podratz, E. H. Rodriguez and A. J. Windebank, Glia, 2004, 45, 54–58 CrossRef PubMed.
- R. V. Bellamkonda, Biomaterials, 2006, 27, 3515–3518 CAS.
- V. Lemmon, S. M. Burden, H. R. Payne, G. J. Elmslie and M. L. Hlavin, J. Neurosci., 1992, 12, 818–826 CAS.
- H. R. Irons, D. K. Cullen, N. P. Shapiro, N. A. Lambert, R. H. Lee and M. C. Laplaca, J. Neural Eng., 2008, 5, 333–341 CrossRef PubMed.
- A. Desai, W. S. Kisaalita, C. Keith and Z.-Z. Wu, Biosens. Bioelectron., 2006, 21, 1483–1492 CrossRef CAS PubMed.
- N. Castillon, J. Hinnrasky, J. M. Zahm, H. Kaplan, N. Bonnet, P. Corlieu, J. M. Klossek, K. Taouil, A. Avril-Delplanque, B. Péault and E. Puchelle, Lab. Invest., 2002, 82, 989–998 CrossRef CAS.
- C. Mao and W. S. Kisaalita, Biosens. Bioelectron., 2004, 19, 1075–1088 CrossRef PubMed.
- T. Xu, C. A. Gregory, P. Molnar, X. Cui, S. Jalota, S. B. Bhaduri and T. Boland, Biomaterials, 2006, 27, 3580–3588 CAS.
- Y. Z. Lai, K. Cheng and W. Kisaalita, PLoS One, 2012, 7 Search PubMed.
- Q. H. Hogan, Reg. Anesth. Pain Med., 2010, 35, 306–311 CrossRef PubMed.
- P. Lotfi, K. Garde, A. K. Chouhan, E. Bengali and M. I. Romero-Ortega, Front. Neuroeng., 2011, 4, 11 CAS.
- P. Khoshakhlagh and M. J. Moore, Acta Biomater., 2015, 16, 23–34 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |