
Geometric and electronic structure of a peroxomanganese(iii) complex supported by a scorpionate ligand†
Abstract
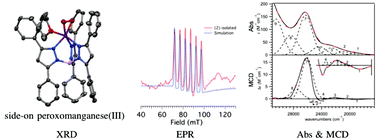
A monomeric MnII complex has been prepared with the facially-coordinating TpPh2 ligand, (TpPh2 = hydrotris(3,5-diphenylpyrazol-1-yl)borate). The X-ray crystal structure shows three coordinating solvent molecules resulting in a six-coordinate complex with Mn–ligand bond lengths that are consistent with a high-spin MnII ion. Treatment of this MnII complex with excess KO2 at room temperature resulted in the formation of a MnIII–O2 complex that is stable for several days at ambient conditions, allowing for the determination of the X-ray crystal structure of this intermediate. The electronic structure of this peroxomanganese(III) adduct was examined by using electronic absorption, electron paramagnetic resonance (EPR), low-temperature magnetic circular dichroism (MCD), and variable-temperature variable-field (VTVH) MCD spectroscopies. Density functional theory (DFT), time-dependent (TD)-DFT, and multireference ab initio CASSCF/NEVPT2 calculations were used to assign the electronic transitions and further investigate the electronic structure of the peroxomanganese(III) species. The lowest ligand-field transition in the electronic absorption spectrum of the MnIII–O2 complex exhibits a blue shift in energy compared to other previously characterized peroxomanganese(III) complexes that results from a large axial bond elongation, reducing the metal–ligand covalency and stabilizing the σ-antibonding Mn dz2 MO that is the donor MO for this transition.
 Please wait while we load your content...
Please wait while we load your content...

