
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInfluence of H-bond competitors on the solvent-dependent structures of an octaurea-calix[4]tube†‡
Marco Milone a,
Ilenia Pisagattia,
Giuseppe Gattusoa,
Anna Notti*a,
Melchiorre F. Parisi*a,
Giovanna Brancatelli§
b,
Neal Hickeyb and
Silvano Geremia*b
a,
Ilenia Pisagattia,
Giuseppe Gattusoa,
Anna Notti*a,
Melchiorre F. Parisi*a,
Giovanna Brancatelli§
b,
Neal Hickeyb and
Silvano Geremia*b
aDipartimento di Scienze Chimiche, Biologiche, Farmaceutiche ed Ambientali, Università degli Studi di Messina, Viale F. Stagno d’Alcontres 31, Messina 98166, Italy. E-mail: anotti@unime.it; mparisi@unime.it
bCentro di Eccellenza in Biocristallografia, Dipartimento di Scienze Chimiche e Farmaceutiche, Università di Trieste, Via L. Giorgieri 1, Trieste I-34127, Italy. E-mail: sgeremia@units.it
First published on 2nd February 2024
Abstract
A novel octaurea-calix[4]tube (UC4T) has been synthesized in three steps from the original Beer's p-tert-butylcalix[4]tube ionophore. In a polar solvent (DMSO-d6), UC4T rapidly interconverts between two identical conformations with C2v symmetry for the two calix[4]arene subunits. However, in a less polar solvent mixture (CDCl3/DMSO-d6, 98![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2), UC4T adopts a highly distorted asymmetric structure, which hinders the formation of typical tetraurea calix[4]arene capsular assemblies. The complexation of potassium (or barium) cations inside the dioxyethylene ionophoric binding site of UC4T triggers a C2v to C4v symmetry rearrangement of the two calix[4]arene subunits. This rearrangement leads to the formation of a transient capsular dimeric species observed in solution upon the addition of KI or BaCl2·2H2O to a solution (CDCl3/DMSO-d6, 98:2) of the macrocycle. X-ray studies confirm UC4T's ability to adopt different asymmetric conformations, depending on its interactions with solvent molecules. Two distinct crystal forms (α and β) of UC4T have been obtained, each displaying divergent calix[4]arene subunits with pinched-cone conformations. These conformations exhibit distinctive head-to-tail (α) or head-to-head/tail-to-tail (β) orientations of the ureido groups, which are involved in hydrogen bonding with solvent molecules. Notably, the pseudo-capsular 1D supramolecular polymeric arrays observed in the β form of UC4T assemble to create large parallel solvent channels.
:2), UC4T adopts a highly distorted asymmetric structure, which hinders the formation of typical tetraurea calix[4]arene capsular assemblies. The complexation of potassium (or barium) cations inside the dioxyethylene ionophoric binding site of UC4T triggers a C2v to C4v symmetry rearrangement of the two calix[4]arene subunits. This rearrangement leads to the formation of a transient capsular dimeric species observed in solution upon the addition of KI or BaCl2·2H2O to a solution (CDCl3/DMSO-d6, 98:2) of the macrocycle. X-ray studies confirm UC4T's ability to adopt different asymmetric conformations, depending on its interactions with solvent molecules. Two distinct crystal forms (α and β) of UC4T have been obtained, each displaying divergent calix[4]arene subunits with pinched-cone conformations. These conformations exhibit distinctive head-to-tail (α) or head-to-head/tail-to-tail (β) orientations of the ureido groups, which are involved in hydrogen bonding with solvent molecules. Notably, the pseudo-capsular 1D supramolecular polymeric arrays observed in the β form of UC4T assemble to create large parallel solvent channels.
Introduction
Hydrogen-bonded dimers of tetraurea-calix[4]arenes and calix[4]tube-based ionophores emerged as a significant subject in the field of calixarene chemistry at the turn of the century. Building upon the pioneering work of Bohmer1 and Rebek,2 urea-substituted calix[4]arenes gained substantial attention due to their remarkable ability to spontaneously self-assemble. In both solution and the solid state, they form homo- and hetero-dimeric (poly)capsules held together by a cyclic array of 16 hydrogen bonds, akin to a zipper, between their ureido groups.3–5 During the same period, Beer and his colleagues, captivated by the selectivity of calix[4]tube for potassium ions,6 honed the structural features of this tubular receptor.7 Their exploration eventually led them to investigate less rigid calix[4]semitubes8 and ditopic calix[4]semitube urea derivatives.9 Subsequent studies delved into the structural modification of calix[4]tube receptors, including the uptake of rubidium ions by calix[4]-thiacalix[4]tubes10 and the fine tuning of conformational equilibria in functionalized calix[4]tubes. These modulations were achieved through intramolecular hydrogen bonds or weak electrostatic repulsion interactions.11 Notably, these investigations were aimed primarily at influencing the conformational behavior of the molecules, rather than utilizing them as building blocks for supramolecular arrays. On the other hand, the conformational behavior of calix[4]tubes,11,12 bis(calix[4]arenes)13,14 and, more generally, calixarenes,15 is a complex phenomenon highly influenced by various factors, including the nature of the solvent in which they are dissolved and the coordination of ions present in the surrounding environment. For example, metal complexation rigidifies the cone and preorganizes the molecule for host–guest interaction and self-assembly.16–18In our quest to fully explore the supramolecular potential of this class of compounds, we recently synthesized an octaamino-calix[4]tube.19 Our work confirmed the crucial role played by the encapsulation of potassium ion within the cryptand-like binding site of this calix[4]tube derivative in the noncovalent synthesis of discrete porphyrin-calixarene nanostructures.19 Inspired by the self-assembly motif3,20–24 of tetraurea-calix[4]arenes, and in alignment with our focus on designing linear supramolecular polymers,25–31 we decided to shift our attention towards investigating the supramolecular properties of urea-containing calix[4]tubes.
In this contribution, we present the synthesis and structural characteristics of an octatolylurea-calix[4]tube, denoted as UC4T. Additionally, we explore its conformational behavior and self-assembly tendencies in both solution and the solid state.
Results and discussion
Octatolylurea-calix[4]tube, UC4T, was synthesized with a yield of 60% by reacting octaamino-calix[4]tube, AC4T,19 with p-tolylisocyanate (24 equiv.) in anhydrous dimethylsulfoxide at room temperature (Scheme 1). | ||
| Scheme 1 Reagents and conditions: (i) p-tolylisocyanate, dry DMSO, r.t., 3 h. | ||
UC4T, featuring two divergent calix[4]arene cavities equipped with hydrogen-bond donor/acceptor ureido functionalities, is an intriguing building block for the formation of linear supramolecular (poly)capsular assemblies. However, for a hydrogen-bond-driven dimerization of UC4T to occur, at least one calix[4]arene subunit of each calix[4]tube molecule must adopt a regular cone conformation (C4v symmetry of the macroring).6,32 This conformational requirement allows for the formation of an intermolecular array of interdigitated H-bonds between ureido groups.3,32,33 Polycapsular assemblies necessitate even more stringent conformational prerequisites, as both cavities must be preorganized. Additionally, careful consideration of the choice of solvent is required, as the use of competitive hydrogen-bond acceptor solvents may impede intermolecular self-assembly. Due to the unique quadruple-linkage with short ethylene-spacers, the calixarene subunits of the UC4T parent calix[4]tube derivatives6,7,11,19,34,35 display a pinched-cone C2v symmetry in solution and in the solid state. This is a result of the alternation between synclinal (known as gauche) and antiperiplanar (known as anti) conformations of the four linkers. In light of these considerations, we initially examined whether UC4T possesses the necessary conformational flexibility in solution for the two calix[4]arene subunits to adopt a regular cone conformation. Unfortunately, UC4T was found to be insoluble in several solvents typically used to promote the formation of hydrogen-bonded dimers between wide-rim ureido functionalized calix[4]arene cavities, such as benzene, toluene, chloroform, and tetrachloroethane.2,36 However, it readily dissolved in polar DMSO. By analogy with the parent AC4T,19 Beer's p-tert-butylcalix[4]tube,6 and other calix[4]tube derivatives,7,11,14,34,35 in DMSO-d6 solution, the two cavities of the octaurea-calix[4]tube UC4T appear to adopt a time-averaged pinched-cone conformation with C2v symmetry. This is evident from the 1H NMR spectrum, which exhibits, four resonances for the two sets of non-equivalent NHs, two sharp singlets for the aromatic hydrogen atoms (ArH), two distinct sets of different tolyl residues (TolH and CH3), two singlets for the OCH2s and a single AX system for the bridging methylenes of the calixarene scaffold (Fig. 1).
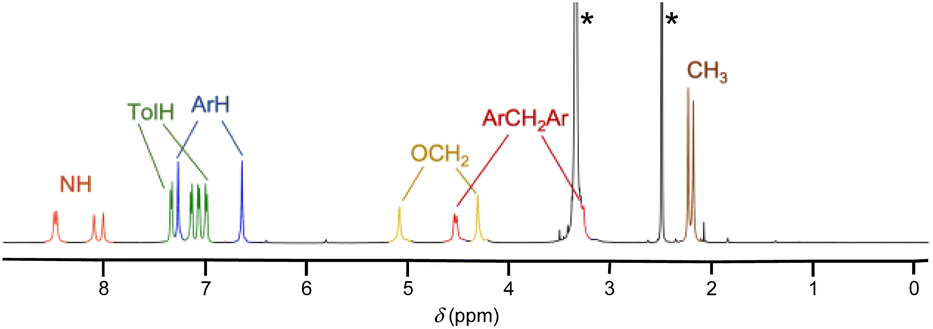 | ||
| Fig. 1 1H NMR spectrum of UC4T (500 MHz, DMSO-d6, 298 K). The asterisks indicate the residual peaks of the solvents. | ||
The interconversion between two equivalent UC4T conformations in solution was confirmed by further NMR analysis of the 2D ROESY spectrum of the same solution (Fig. S1, in the ESI†). This spectrum, revealed the presence of chemical-exchanging cross-peaks, for the ArH, NH and OCH2 resonances, having the same phase as the corresponding diagonal peaks.
With the exception of a few examples of capsules derived from rigid cone-shaped calix[4]arenes37 and mechanically entangled capsular dimers,38 DMSO, owing to its strong propensity to form hydrogen bonds with ureido moieties, is generally not a suitable solvent for the formation of capsular assemblies of tetraurea-calix[4]arenes. After several attempts, UC4T was finally solubilized in a CDCl3/DMSO-d6 (98/2, v/v) solvent mixture. Surprisingly, the 1H NMR spectrum in this solvent system displayed an unexpected complexity (Fig. 2, trace a). As we added increasing amounts of DMSO-d6 to the original solution (up to a final concentration of 8%), a new set of resonances initially coexisted with the first one and then became predominant (Fig. 2, traces b and c). The 1H NMR spectrum in the presence of 8% DMSO-d6 (Fig. 2, trace d) clearly resembles the spectrum of UC4T in neat DMSO-d6 (Fig. 2, trace e), indicating that a higher concentration of a polar solvent disrupts intra- or intermolecular hydrogen bonds, promoting the conversion into a C2v symmetric pinched-cone structure.
 | ||
| Fig. 2 1H NMR spectra (500 MHz, 298 K) of [UC4T] = 1 mM in: (a) CDCl3/DMSO-d6, 98:2; (b) CDCl3/DMSO-d6, 96:4; (c) CDCl3/DMSO-d6, 94:6; (d) CDCl3/DMSO-d6, 92:8 and (e) DMSO-d6. The asterisks indicate the residual peaks of the solvents. | ||
A preliminary study using Diffusion-Ordered NMR SpectroscopY (DOSY) was conducted on the 94:6 CDCl3/DMSO-d6 sample with the aim of distinguishing assemblies of different sizes. This study ruled out the formation of oligo- or polycapsular assemblies in solution due to the very close diffusion coefficients determined for the two species present in solution (Fig. S2†). Extensive 2D NMR analysis of the 98:2 CDCl3/DMSO-d6 sample, including COSY and HSQC (Fig. S3 and S4†), revealed several features. There were eight resonances assigned to the phenolic aromatic hydrogens, displaying meta-coupling cross-peaks. Additionally, four non-equivalent tolyl residues, eight distinct resonances for the NHs and four AX systems for the axial and equatorial hydrogen atoms of the methylene groups bridging the aromatic rings were identified. In contrast to the DMSO-d6 sample, the ROESY spectrum recorded in the 98:2 CDCl3/DMSO-d6 mixture exhibited only positive through-space dipolar couplings (Fig. 3). This observation indicated that all the resonances originated from a “frozen” conformer or conformers. Specifically, cross-peaks were observed between the hydrogen atoms of the bridging methylene groups and those situated on the two flanking aryl rings (i.e., A-eeq-b, B-feq-c, C-geq-d and D-heq-a) and between hydrogen atoms located on the adjacent aryl rings (i.e., A-b, B-c, C-d and D-a). These findings suggested that all these resonances belong to a single calix[4]arene asymmetric subunit of UC4T (Fig. 3). Consequently, it can be inferred that the two calixarene cavities exhibit equivalent resonances and are therefore distorted in a similar manner.
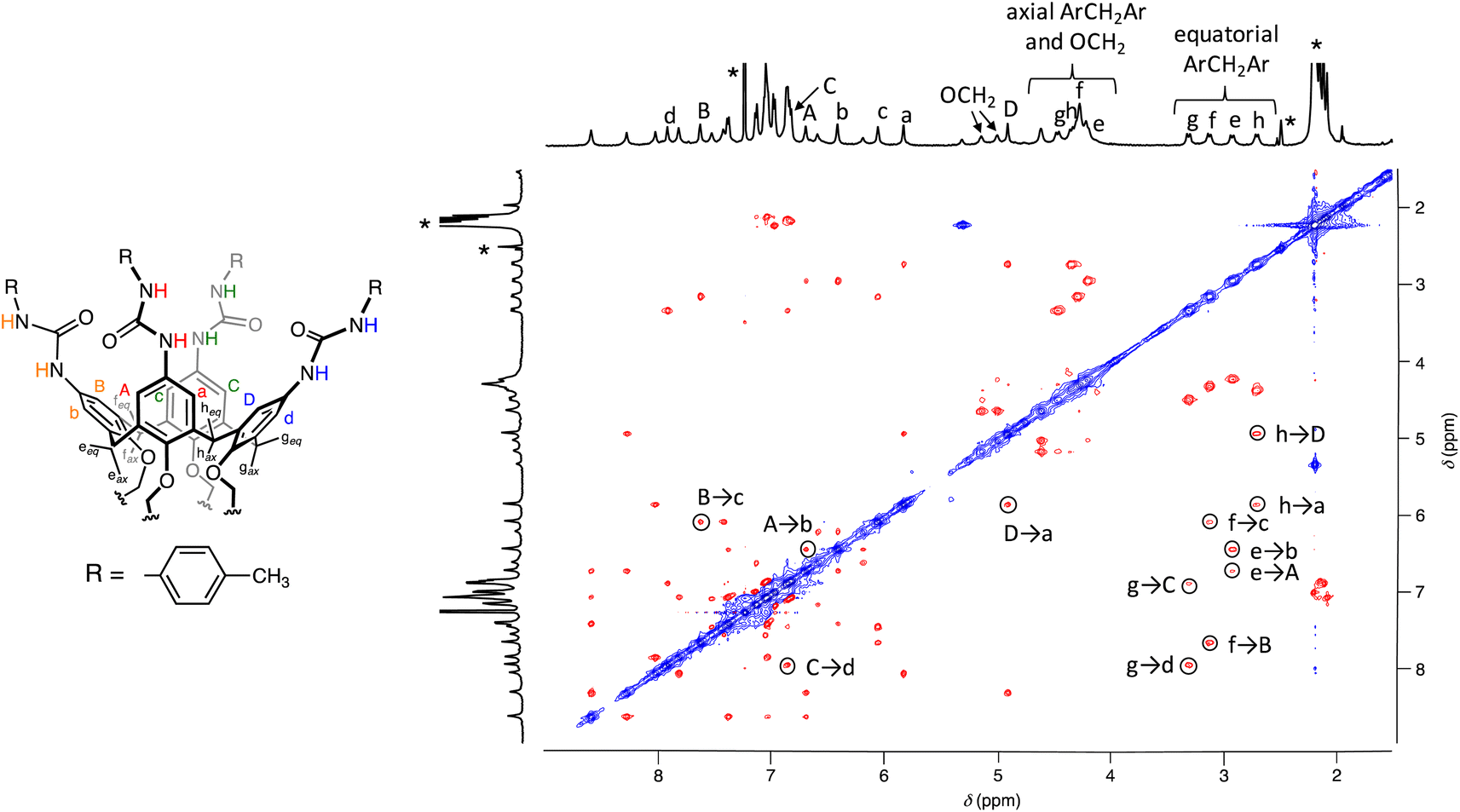 | ||
| Fig. 3 ROESY NMR spectrum (500 MHz, CDCl3/DMSO-d6 98:2, 298 K) of [UC4T] = 1 mM. The asterisks indicate the residual peaks of the solvents. The labels of the assigned NMR signals are shown in the schematic drawing of UC4T on the left. | ||
2D NOESY studies on calix[4]tube derivatives have previously proved to be very useful for the structural elucidation of conformers present in solution, as they are particularly suitable for the quantitative determination of intramolecular atomic distances.11 In our case, the 2D NOESY spectrum of UC4T (Fig. S5†) revealed clear cross-peaks between the aromatic hydrogen atoms a, b, and c and three distinct pairs of urea NH resonances. These contacts are either less pronounced or entirely absent for the corresponding meta-coupled hydrogens A, B, and C. This observation implies that these ureido moieties are fixed in a sort of head-to-tail arrangement, with urea groups oriented in the same direction. Intriguingly, the fourth pair of ureido NHs exhibited NOE contacts with the hydrogen atoms of different phenolic rings, indicating that these NHs are oriented towards the inside of the calixarene cavity. It has been reported that H-bonding or electrostatic interactions can influence the conformational distribution of calix[4]tube derivatives. For instance, in CDCl3, a calix[4]tube distally substituted with adamantyl-ureido moieties exhibits an equal population of two distinct pinched-cone conformers. However, the subsequent addition of CF3CO2D induces a substantial conformational rearrangement, resulting in a 9:1 ratio in favor of one of the conformers.11 Our findings regarding the conformational equilibrium of UC4T in various CDCl3/DMSO-d6 mixtures emphasize the significant role played by hydrogen-bonded solvent molecules in the conformational equilibrium of this calix[4]tube derivative. NMR data suggest that at lower DMSO concentrations, UC4T adopts a “frozen” asymmetric conformation, likely due to H-bond interactions (vide infra). The conformational stability of UC4T in a 98:2 CDCl3/DMSO-d6 solvent mixture was further investigated through a series of VT-NMR experiments conducted in the temperature range of −30 to +50 °C (Fig. S6†). These experiments showed minimal overall spectral changes, while the asymmetric spectral pattern remained consistent across the investigated temperature range. The addition of increasing amounts of the polar H-bond acceptor solvent, DMSO-d6, eventually shifted the equilibrium toward more flexible equivalent C2v–C2v interconverting conformations.
To the best of our knowledge, this represents the first example of a symmetrically-substituted calix[4]tube derivative displaying a stable solvent-dependent asymmetric conformation in solution. Speculating about the formation of specific intra- (ureido–ureido) or intermolecular (ureido-solvent) H-bond interactions that might favor the observed asymmetric conformation in solution is challenging. This is because many of these intra- and intermolecular interactions can coexist due to the simultaneous presence of hydrogen-bonding donor/acceptor ureido groups and solvent molecules (e.g., H2O¶ and DMSO-d6). Based on the aforementioned findings, it was concluded that the formation of the desired (poly)capsular assembly of UC4T does not occur in a 98:2 CDCl3/DMSO-d6 solvent mixture, likely due to conformational constrains.
Considering the importance of cation encapsulation within the cryptand-like dioxyethylene cage for an effective conformational rearrangement (from pinched-cone to regular cone) of a calix[4]tube derivative to occur,34 and the symmetry requirement for promoting the capsular assembly of urea-calix[4]arene derivatives,32,33 KI or BaCl2·2H2O was added to the 98:2 CDCl3/DMSO-d6 solutions of UC4T. Subsequently, 1H NMR spectra were regularly recorded over a two-week period. Over time, the OCH2 resonances gradually merged into a single broad signal at δ 4.30 ppm providing evidence of cation encapsulation inside the dioxyethylene cage of UC4T and, as a result, of the necessary conformational rearrangement of the two calix[4]arene subunits into preorganized cone-shaped cavities. The formation of hydrogen-bond sealed structures of UC4T, interacting through ureido moieties, is further supported by the downfield shift of the NH resonances to δ 9.6 ppm (Fig. S7†).36 Although both samples (UC4T@K+ and UC4T@Ba2+) produced an increasing amount of a powdery precipitate as time went on, the presence of two equally intense AX systems for the bridging methylenes (ArCH2Ar) detected after a few days from cation addition, suggests that a small amount of dimeric calixtube structure, with two nonequivalent calix[4]arene subunits, is initially formed in solution. Most likely, the linear growth of these dimeric structures leads to larger oligomers that tend to precipitate as they become insoluble in the 98:2 CDCl3/DMSO-d6 solvent mixture.
In our efforts to obtain solid-state structures of (poly)capsular assemblies involving UC4T in the presence of K+ or Ba2+ cations, we observed that upon evaporation of 98:2 CHCl3/DMSO solutions containing either KI or BaCl2·2H2O two distinct crystal forms were obtained, designated as α (obtained with a K+/UC4T molar ratio of 1.2) and β (obtained with a Ba2+/UC4T molar ratio of 1.2). Notably, in both α and β forms, the X-ray structures did not reveal any metal ions coordinated within the ionophoric binding site of UC4T. Furthermore, the electron density maps did not support the presence of any localized heavy ion in either structure. Both crystal forms featured UC4T molecules positioned on crystallographic inversion centers (Fig. 4).
 | ||
| Fig. 4 Stick representation of the α and β forms of UC4T, with atomic species depicted in CPK colors. Dashed lines represent H-bonds. UC4T molecules are situated on crystallographic inversion centers in both crystal forms. Only one position of each disordered group is displayed for clarity (details in the ESI†). | ||
The divergent calix[4]arene subunits within the centrosymmetric UC4T molecule adopt a C2v pinched-cone conformation in both crystal forms, meaning that the two symmetry related calixarene subunits are pinched on the same sides (Fig. 5). Specifically, two opposite aryl rings exhibit outward inclinations (136 and 142° in α; 139 and 141° in β) while the other two are nearly orthogonal (88 and 86° in α; 84 and 87° in β) with respect to the corresponding mean plane of the methylene bridging groups of the calixarene subunit.||
 | ||
| Fig. 5 Orthogonal views of the α and β forms of UC4T, with atomic species represented in CPK colors. Both crystal forms feature pinched-cone conformations in the two divergent calix[4]arene subunits. For clarity, only one position of each disordered group is displayed (details in the ESI†). | ||
This pinched-cone conformation significantly affects the relative positions of the four ethylene linkers located at the narrow rim of the calix[4]arene subunits. Opposite pairs of oxygen atoms of the same calixarene subunits are pushed further apart on the pinched sides and brought closer together on the other sides (O⋯O distances of 5.43 and 3.27 Å for α and 5.39 and 3.26 Å for β). Consequently, the pairs of O atoms connected by the ethylene linkers on the pinched sides are closer together (2.71 and 2.74 Å for α and β, respectively), while they are further apart on the other sides (3.61 and 3.63 Å for α and β, respectively). These geometric features align with the observed conformation of the ethylene linkers (Fig. 5). In the α form, the torsion angles of the C–O–C–C–O–C chains are 171° (ap, antiperiplanar), 20° (sp, synperiplanar) and 171° (ap) for the less extended chain, while for the two disordered positions of the more extended chain, they are 104°/95° (+ac, anticlinal+), 155°/160° (ap) and 112°/123° (+ac). In the β form, the corresponding chain conformations are 163° (ap), 36° (+sc, synclinal+), and 162° (ap) for the less extended chain and 98° (+ac), 154° (ap) and 116° (+ac) for the more extended chain. Due to the inversion center, symmetry related chains exhibit the same torsion angles with opposite signs.
The orientation of the ureido groups differ significantly in the two crystal forms. In the α form, neighboring ureido groups all exhibit head-to-tail orientations, where the carbonyl oxygen atom of one ureido group points toward the NHs of an adjacent group in either a clockwise or anticlockwise sense of rotation. Conversely, in the β form, they are less symmetrically arranged, displaying a general head-to-head (carbonyl toward carbonyl) and tail-to-tail (NHs toward NHs) orientation (Fig. 6).
 | ||
| Fig. 6 Top views depicting the orientation of the ureido groups in the α and β forms of UC4T. Atomic species are represented in CPK colors. For clarity, only the macrocyclic skeleton and the ureido groups of a single calix[4]arene subunit are presented. | ||
In the α form, in conjunction with the crystallographic inversion center, a pseudo twofold axis passing through the midpoint of the pinched calix[4]arene subunits results in a pseudo C2h point symmetry of the molecule (Fig. 5). It is worth noting that ethylene linkers with an ap/sp/ap chain conformation are compatible with a horizontal symmetry plane, while linkers with a more extended +ac/ap/+ac chain conformation are not compatible with C2h symmetry. Intriguingly, in the α form these more extended chains exhibit a statistical disorder, which could be explained by the overlap of two equivalent molecules related by the pseudo horizontal plane. In contrast, in the β form, the complete absence of the twofold axis due to the ureido group orientations reduces the point symmetry of the molecule to Ci. Further details regarding the orientation and deformation of the tolylureido arms are available in the ESI.†
The two crystallographic structures of UC4T clearly highlight the crucial role played by hydrogen bonding with DMSO molecules in determining the solid-state conformational arrangement of UC4T. In the α form all NH groups participate in H-bonds with co-crystallized DMSO solvent molecules. Specifically, the two NH groups of the A arm act as H-bond donors to two O-atom acceptors in distinct DMSO molecules, while the other three ureido groups engage in bifurcated H-bonds, involving two donors and one acceptor, with single DMSO molecules (Fig. 7). Conversely, the carbonyl groups of the ureido moieties are not involved in any H-bond interactions.
 | ||
| Fig. 7 H-bonds identified in the α and β forms of UC4T. Atomic species are represented in CPK colors. For clarity, only the crystallographically independent unit of UC4T and one position of each disordered group are displayed. H-bonds are illustrated with dashed lines, with cyan indicating H-bonds between UC4T and solvent molecules, and red indicating H-bonds between UC4T molecules related by crystallographic symmetry. | ||
In the β form, only the tolylureido arms of the pinched phenolic rings (B and D, Fig. 7) are involved in bifurcated H-bonds, with two donors and one acceptor, with co-crystallized DMSO solvent molecules. The two NH groups of the A arm (Fig. 7) act as H-bond donors in a bifurcated interaction with a co-crystalized water molecule, which had been stoichiometrically added to the crystallization solution as BaCl2·2H2O salt. The NH groups of the opposite tolylureido arm (C), which exhibits a two-position disorder of the p-tolyl end-group, form bifurcated H-bonds with the ureido carbonyl group of a pinched tolylureido arm (B) of a symmetry related UC4T molecule (Fig. 7). Consequently, this type of bifurcated intermolecular H-bond, involving two pairs of ureido groups related by an inversion center per molecule (one donor and one acceptor of H-bonds), results in a closely packed 1D array of UC4T molecules interconnected by a complementary H-bond network along the c axis of the unit cell (Fig. 8). Additional packing details are outlined in the ESI.†
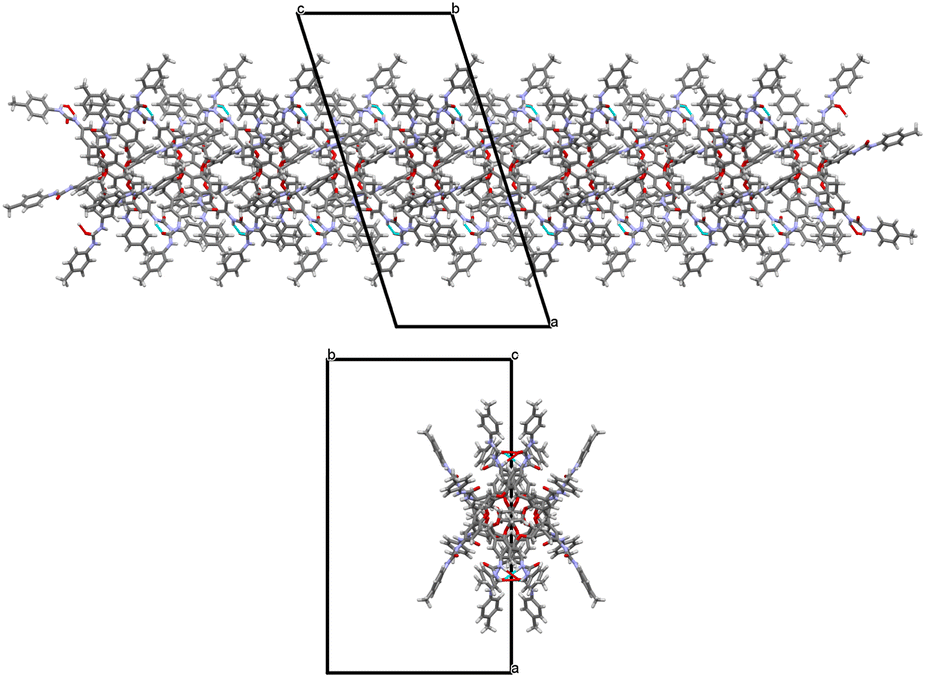 | ||
| Fig. 8 Side and top views of the closely packed 1D array of UC4T molecules interconnected by a complementary H-bond network (shown as cyan dashed lines) observed along the c axis of the monoclinic unit cell in the β form. | ||
These 1D arrays assemble into a C centered monoclinic lattice, creating large parallel solvent channels along the c axis of the monoclinic unit cell, which are partially occupied by highly disordered solvent molecules, as observed in the crystal packing of the β form (Fig. S11†). In contrast, the α form exhibits a more compact packing with small cavities filled by DMSO molecules (Fig. S12†).
Experimental
Procedure for synthesis of UC4T
p-Tolylisocyanate (617 mg; 4.63 mmol) was added to a solution of AC4T (ref. 19) (207 mg; 0.193 mmol) in anhydrous DMSO (30 mL). The reaction mixture was stirred for 3 h at r.t. under an argon atmosphere. The suspension formed upon addition of acetone (20 mL) was heated under reflux for 1 h and the precipitate formed finally collected by suction filtration, to provide UC4T as a white powder (246 mg; 60% yield). 1H NMR (DMSO-d6) δ 8.48 (s, 4H, NH), 8.46 (s, 4H, NH), 8.09 (s, 4H, NH), 8.00 (s, 4H, NH), 7.34 (d, J = 8.1 Hz, 8H, TolH), 7.26 (s, 8H, ArH), 7.14 (d, J = 8.1 Hz, 8H, TolH), 7.07 (d, J = 8.1 Hz, 8H, TolH), 6.99 (d, J = 8.1 Hz, 8H, TolH), 6.63 (s, 8H, ArH), 5.08 (s, 8H, OCH2), 4.31 (s, 8H, OCH2), 4.53 and 3.29 (AX, J = 12.2 Hz, 16H, ArCH2Ar), 2.23 and 2.18 (s × 2, 24H, CH3) ppm; 13C NMR (DMSO-d6) δ 153.3, 152.7, 152.3, 150.5, 137.3, 137.1, 135.4, 134.0, 133.3, 132.7, 130.4, 129.1, 118.9, 118.3, 118.0, 72.9, 31.8 and 20.3 ppm.Materials and methods
General
All reagents and solvents were commercially available and used without further purification. 1H and 13C NMR spectra were recorded at 500 and 125 MHz, respectively, on a Varian Vnmrs-500 spectrometer in DMSO-d6 or CDCl3/DMSO-d6 mixtures and chemical shifts were reported on the delta scale in ppm relative to residual DMSO-d5 (δ = 2.49 and 39.5 for 1H and 13C, respectively). DOSY experiments were recorded in CDCl3/DMSO-d6, 94:6 at 25 ± 0.1 °C, using a gradient stimulated echo with spin-lock and a convection compensation pulse sequence.
Crystallography
Single-crystal structure determination of α and β forms of UC4T
Single crystals suitable for X-ray investigation were obtained by evaporation of DMSO/chloroform solutions obtained by dissolving 4 mg of UC4T and an inorganic salt of potassium or barium in 20 μl of anhydrous DMSO, to which 980 μl of chloroform was successively added. Two different crystal forms (α and β) were obtained from two different crystallization trials, one in which the DMSO solution contained KI (K+/UC4T molar ratio of 1.2) and one in which it contained BaCl2·2H2O (Ba2+/UC4T molar ratio of 1.2). Data collection was carried out at the XRD1 beamline of the Elettra synchrotron (Trieste, Italy), employing the rotating-crystal method with a Dectris Pilatus 2M area detector. Single crystals were dipped in paratone cryoprotectant, mounted on a loop and flash-frozen under a nitrogen stream at 100 K. Diffraction data were indexed and integrated using the XDS package,39 while scaling was carried out with XSCALE.40 Structures were solved using the SHELXT41 program and refinement was performed with SHELXL-18/3,42 operating through the WinGX GUI,43 by full-matrix least-squares (FMLS) methods on F2. Non-hydrogen atoms were anisotropically refined with exclusion of atoms with a low occupancy factor, which were refined isotropically. Hydrogen atoms were added at the calculated positions and refined using the riding model. Due to the low reflection/parameters ratio, the β form was refined using the SIMU and ISOR cards together with several geometrical constraints (aromatic rings) and restraints (DMSO solvent molecules). Crystallographic data and refinement details are reported in Table S1.†Conclusions
The synthesis of the first calix[4]tube derivative bearing eight ureido substituents (UC4T) has been successfully achieved. Both solution and solid-state studies have shed light on the pivotal role of hydrogen-bonded solvent molecules in shaping the conformational arrangement of this derivative. While drawing a direct parallel between solution and solid-state structures can be problematic, single crystal X-ray studies have provided confirmation of UC4T's capability to adopt distinct conformations contingent upon its interaction with polar solvent molecules. It is reasonable to assume that in a solvent mixture containing a small fraction of polar molecules, such as a 98/2 CDCl3/DMSO-d6 ratio, DMSO molecules act as stabilizers and, in doing so, force UC4T to adopt a non-symmetric conformation. This, in turn, hinders the formation of (poly)capsular assemblies. The complexation of suitably sized metal ions (K+ and Ba2+) within the dioxyethylene cage gradually led to the adoption of more regular cone-shaped calix[4]arene subunits in solution, subsequently resulting in the formation of transient H-bonded dimers.In the two distinct crystal forms (α and β) obtained from separate crystallization trials in the presence of K+ and Ba2+, the centrosymmetric UC4T molecule exhibit divergent calix[4]arene subunits adopting a C2v pinched-cone conformation. Intriguingly, different orientations of the ureido groups were observed in the solid state. In the α form, there are head-to-tail orientations, while in the β form, head-to-head and tail-to-tail orientations are present. Furthermore, in the β form, a closely packed 1D array of UC4T molecules are interconnected by a complementary H-bond network, running along the c axis of the unit cell, which generates large parallel solvent channels partially occupied by DMSO molecules.
Author contributions
A. N., M. F. P. and S. G. conceived and supervised this project. A. N., M. M. and I. P. synthesized the compound and studied its properties by NMR spectroscopy. G. B., N. H. and S. G. performed crystallization, X-ray data collection and refinement. G. G. gave support for data interpretation and in revising the manuscript. All authors participated in the discussion of the results and contributed to the preparation of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge for financial support MIUR through the PRIN project 20179BJNA2 and Università di Messina through the APC initiative.Notes and references
- O. Mogck, E. F. Paulus, V. Böhmer, I. Thondorf and W. Vogt, Hydrogen-bonded dimers of tetraurea calix[4]arenes: unambiguous proof by single crystal X-ray analysis, Chem. Commun., 1996, 2533–2534 RSC.
- K. D. Shimizu and J. Rebek Jr, Synthesis and assembly of self-complementary calix[4]arenes, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 12403–12407 CrossRef CAS PubMed.
- J. Rebek Jr, Host–guest chemistry of calixarene capsules, Chem. Commun., 2000, 637–643 RSC.
- R. K. Castellano and J. Rebek Jr, Formation of discrete, functional assemblies and informational polymers through the hydrogen-bonding preferences of calixarene aryl and sulfonyl tetraureas, J. Am. Chem. Soc., 1998, 120, 3657–3663 CrossRef CAS.
- R. K. Castellano, D. M. Rudkevich and J. Rebek Jr, Polycaps: reversibly formed polymeric capsules, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 7132–7137 CrossRef CAS PubMed.
- P. Schmitt, P. D. Beer, M. G. B. Drew and P. D. Sheen, Calix[4]tube: a tubular receptor with remarkable potassium ion selectivity, Angew. Chem., Int. Ed. Engl., 1997, 36, 1840–1842 CrossRef CAS.
- S. E. Matthews, V. Felix, M. G. B. Drew and P. D. Beer, Halo-derivatised calix[4]tubes, Org. Biomol. Chem., 2003, 1, 1232–1239 RSC.
- P. R. A. Webber, A. Cowley, M. G. B. Drew and P. D. Beer, Potassium selective calix[4]semitubes, Chem.–Eur. J., 2003, 9, 2439–2446 CrossRef CAS PubMed.
- P. R. A. Webber and P. D. Beer, Ion-pair recognition by a ditopic calix[4]semitube receptor, Dalt. Trans., 2003, 2249–2252 RSC.
- E. Khomich, M. Kashapov, I. Vatsouro, E. Shokova and V. Kovalev, Substituent control of potassium and rubidium uptake by asymmetric calix[4]-thiacalix[4]tubes, Org. Biomol. Chem., 2006, 4, 1555–1560 RSC.
- K. Puchnin, D. Cheshkov, P. Zaikin, I. Vatsouro and V. Kovalev, Tuning conformations of calix[4]tubes by weak intramolecular interactions, New J. Chem., 2013, 37, 416–424 RSC.
- J. Budka, P. Lhoták, I. Stibor, J. Sýkora and I. Císařová, Solid state calix[4]arene tubular assemblies based on cation–π interactions, Supramol. Chem., 2003, 15, 353–357 CrossRef CAS.
- I. Lentin, A. Gorbunov, S. Bezzubov, V. Nosova, D. Cheshkov, V. Kovalev and I. Vatsouro, Shrinkable/stretchable bis(calix[4]arenes) comprising photoreactive azobenzene or stilbene linkers, Org. Chem. Front., 2023, 10, 1470–1484 RSC.
- J. Budka, P. Lhoták, I. Stibor, V. Michlová, J. Sykora and I. Cisarová, A biscalix[4]arene-based ditopic hard/soft receptor for K+/Ag+ complexation, Tetrahedron Lett., 2002, 43, 2857–2861 CrossRef CAS.
- J.-D. van Loon, W. Verboom and D. N. Reinhoudt, Selective functionalization and conformational properties of calix[4]arenes, a review, Org. Prep. Proced. Int., 1992, 24, 437–462 CrossRef CAS.
- N. Hickey, V. Iuliano, C. Talotta, M. De Rosa, A. Soriente, C. Gaeta, P. Neri and S. Geremia, Solvent and guest-driven supramolecular organic frameworks based on a calix[4]arene-tetrol: channels vs. molecular cavities, Cryst. Growth Des., 2021, 21, 6357–6363 CrossRef CAS.
- M. Giuliani, I. Morbioli, F. Sansone and A. Casnati, Moulding calixarenes for biomacromolecule targeting, Chem. Commun., 2015, 51, 14140–14159 RSC.
- D. Coquière, A. de la Lande, S. Martí, O. Parisel, T. Prangé and O. Reinaud, Molecular recognition and self-assembly special feature: multipoint molecular recognition within a calix[6]arene funnel complex, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 10449–10454 CrossRef PubMed.
- M. Gaeta, E. Rodolico, M. E. Fragalà, A. Pappalardo, I. Pisagatti, G. Gattuso, A. Notti, M. F. Parisi, R. Purrello and A. D'Urso, Self-assembly of discrete porphyrin/calix[4]tube complexes promoted by potassium ion encapsulation, Molecules, 2021, 26, 704 CrossRef CAS PubMed.
- Y. L. Cho, D. M. Rudkevich, A. Shivanyuk, K. Rissanen and J. Rebek Jr, Hydrogen-bonding effects in calix[4]arene capsules, Chem.–Eur. J., 2000, 6, 3788–3796 CrossRef CAS PubMed.
- L. Wang, M. O. Vysotsky, A. Bogdan, M. Bolte and V. Böhmer, Multiple catenanes derived from calix[4]arenes, Science, 2004, 304, 1312–1314 CrossRef CAS PubMed.
- Y. Rudzevich, V. Rudzevich, C. Moon, G. Brunklaus and V. Böhmer, Self-assembled dendrimers with uniform structure, Org. Biomol. Chem., 2008, 6, 2270–2275 RSC.
- A. Bogdan, M. Bolte and V. Böhmer, Molecules with new topologies derived from hydrogen-bonded dimers of tetraurea calix[4]arenes, Chem.–Eur. J., 2008, 14, 8514–8520 CrossRef CAS PubMed.
- O. Molokanova, G. Podoprygorina, M. Bolte and V. Böhmer, Multiple catenanes based on tetraloop derivatives of calix[4]arenes, Tetrahedron, 2009, 65, 7220–7233 CrossRef CAS.
- A. Notti, I. Pisagatti, F. Nastasi, S. Patanè, M. F. Parisi and G. Gattuso, Stimuli-responsive internally ion-paired supramolecular polymer based on a bis-pillar[5]arene dicarboxylic acid monomer, J. Org. Chem., 2021, 86, 1676–1684 CrossRef CAS PubMed.
- D. Zuccaccia, R. Pinalli, R. De Zorzi, M. Semeraro, A. Credi, C. Zuccaccia, A. Macchioni, S. Geremia and E. Dalcanale, Hierarchical self-assembly and controlled disassembly of a cavitand-based host-guest supramolecular polymer, Polym. Chem., 2021, 12, 389–401 RSC.
- N. Manganaro, I. Pisagatti, A. Notti, M. F. Parisi and G. Gattuso, Self-sorting assembly of a calixarene/crown ether polypseudorotaxane gated by ion-pairing, New J. Chem., 2019, 43, 7936–7940 RSC.
- N. Manganaro, I. Pisagatti, A. Notti, A. Pappalardo, S. Patanè, N. Micali, V. Villari, M. F. Parisi and G. Gattuso, Ring/chain morphology control in overall-neutral, internally ion-paired supramolecular polymers, Chem.–Eur. J., 2018, 24, 1097–1103 CrossRef CAS PubMed.
- C. Capici, Y. Cohen, A. D'Urso, G. Gattuso, A. Notti, A. Pappalardo, S. Pappalardo, M. F. Parisi, R. Purrello, S. Slovak and V. Villari, Anion-assisted supramolecular polymerization: from achiral AB-type monomers to chiral assemblies, Angew. Chem., Int. Ed., 2011, 50, 11956–11961 CrossRef CAS PubMed.
- F. Tancini, R. M. Yebeutchou, L. Pirondini, R. Dezorzi, S. Geremia, O. A. Scherman and E. Dalcanale, Host–guest-driven copolymerization of tetraphosphonate cavitands, Chem.–Eur. J., 2010, 16, 14313–14321 CrossRef CAS PubMed.
- R. M. Yebeutchou, F. Tancini, N. Demitri, S. Geremia, R. Mendichi and E. Dalcanale, Host–guest driven self-assembly of linear and star supramolecular polymers, Angew. Chem., Int. Ed., 2008, 47, 4504–4508 CrossRef CAS PubMed.
- G. Podoprygorina, M. Bolte and V. Böhmer, Tetra-urea calix[4]arenes 1,3-bridged at the narrow rim, Org. Biomol. Chem., 2009, 7, 1592–1598 RSC.
- I. Thondorf, F. Broda, K. Rissanen, M. Vysotsky and V. Böhmer, Dimeric capsules of tetraurea calix[4]arenes. MD simulations and X-ray structure, a comparison, J. Chem. Soc., Perkin Trans. 2, 2002, 1796–1800 RSC.
- S. E. Matthews, P. Schmitt, V. Felix, M. G. B. Drew and P. D. Beer, Calix[4]tubes: a new class of potassium-selective ionophore, J. Am. Chem. Soc., 2002, 124, 1341–1353 CrossRef CAS PubMed.
- K. Puchnin, P. Zaikin, D. Cheshkov, I. Vatsouro and V. Kovalev, Calix[4]tubes: an approach to functionalization, Chem.–Eur. J., 2012, 18, 10954–10968 CrossRef CAS PubMed.
- O. Mogck, V. Böhmer and W. Vogt, Hydrogen bonded homo- and heterodimers of tetra urea derivatives of calix[4]arenes, Tetrahedron, 1996, 52, 8489–8496 CrossRef CAS.
- M. O. Vysotsky, O. Mogck, Y. Rudzevich, A. Shivanyuk, V. Böhmer, M. S. Brody, Y. L. Cho, D. M. Rudkevich and J. Rebek Jr, Enhanced thermodynamic and kinetic stability of calix[4]arene dimers locked in the cone conformation, J. Org. Chem., 2004, 69, 6115–6120 CrossRef CAS PubMed.
- M. O. Vysotsky, I. Thondorf and V. Böhmer, Hydrogen bonded calixarene capsules kinetically stable in DMSO, Chem. Commun., 2001, 1890–1891 RSC.
- W. Kabsch, XDS, Acta Crystallogr. Sect. D: Biol. Crystallogr., 2010, 66, 125–132 CrossRef CAS PubMed.
- W. Kabsch, Integration, scaling, space-group assignment and post-refinement, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 133–144 CrossRef CAS PubMed.
- G. M. Sheldrick, SHELXT – Integrated space-group and crystal-structure determination, Acta Crystallogr. Sect. A: Found. Crystallogr., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Crystal structure refinement with SHELXL, Acta Crystallogr. Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- L. J. Farrugia, WinGX and ORTEP for windows: an update, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, additional 1H NMR spectra and DOSY plots, X-ray figures, and tables of crystal data of α and β forms of UC4T. CCDC 2299675 and 2299676. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ra08764f |
| ‡ In memory of the late dr Volker Böhmer and dr Myroslav O. Vysotsky. |
| § Current address: Ardena Oss, Solid State Research Unit, Kloosterstraat 9, 5349 AB Ross, The Netherlands. |
| ¶ Water molecules are present in variable trace amounts in deuterated solvents. |
| || Angles smaller (or larger) than 90° indicate that the tolylureido moieties at the wide rims are inward (or outward) oriented with respect to the center of the calixarene cavity. |
| This journal is © The Royal Society of Chemistry 2024 |