
Methodology for understanding interactions between electrolyte additives and cathodes: a case of the tris(2,2,2-trifluoroethyl)phosphite additive†
Ritu
Sahore
 a,
Adam
Tornheim
a,
Cameron
Peebles
a,
Juan
Garcia
b,
Fulya
Dogan
a,
Daniel C.
O'Hanlon
a,
Chen
Liao
bc,
Hakim
Iddir
b,
Zhengcheng
Zhang
a,
Javier
Bareño
a and
Ira
Bloom
*a
a,
Adam
Tornheim
a,
Cameron
Peebles
a,
Juan
Garcia
b,
Fulya
Dogan
a,
Daniel C.
O'Hanlon
a,
Chen
Liao
bc,
Hakim
Iddir
b,
Zhengcheng
Zhang
a,
Javier
Bareño
a and
Ira
Bloom
*a
aChemical Sciences and Engineering Division, Argonne National Laboratory, Lemont, Illinois 60439, USA. E-mail: ira.bloom@anl.gov
bMaterials Science Division, Argonne National Laboratory, Lemont, Illinois 60439, USA
cJoint Center for Energy Storage Research (JCESR), Argonne National Laboratory, Lemont, Illinois 60439, USA
First published on 30th November 2017
Abstract
Use of electrolyte additives is a promising route to address surface destabilization issues of lithium transition metal (TM)-oxide cathodes (for example, lithium nickel-manganese-cobalt oxides (NMCs)) that occur as they are charged to high voltages (>4.3 V vs. Li/Li+). Despite the successful discovery of several additives, their working mechanisms are often vaguely understood. In this work, we provide a methodology to comprehensively understand additive/cathode interactions in lithium-ion batteries. A case of the tris(2,2,2-trifluoroethyl)phosphite (TTFP) additive is presented where its decomposition behavior was investigated at 4.6 V vs. Li/Li+ in a Li4Ti5O12 (LTO)/Li1.03(Ni0.5Mn0.3Co0.2)0.97O2 (NMC532) cell. Overall, we found that while some of the additive does modify the surface film on the cathode and binds at the surface, it does not passivate the cathode surface towards electrolyte oxidation. Rather, the majority of the TTFP forms stable, free tris(2,2,2-trifluoroethyl)phosphate (TTFPa) molecules by removing O atoms from the charged NMC cathode surface, some of which then further react with the electrolyte solvents and stay in solution. Finally, we propose a stable configuration in which TTFP is bound to the cathode surface via a P–O–TM bond, with one of the –CH2CF3 side groups removed, leading to the formation of BTFPa (bis(2,2,2-trifluoroethyl)phosphate). We anticipate that these techniques and findings could be extended to other additives as well, especially phosphite-based additives, allowing the effective design of future additives.
1. Introduction
There is a need to improve the energy density of current lithium-ion batteries (LIBs). Three component lithium-containing transition metal (TM) oxides (LiNixMnyCozO2 (NMC), 0 < x, y, z < 1, x + y + z = 1) are superior to their single metal counterparts (e.g., LiMO2, M = Ni, Co, or Mn) in that regard.1–3 However, the full energy density of these NMCs has not been utilized, as they are typically charged to a cut-off of 4.2–4.3 V vs. Li/Li+ because of the several electrode/electrolyte stability issues that occur if they are charged to high voltages (V > 4.3 V).4–6 The typically used carbonate-based electrolytes oxidatively decompose at the cathode–electrolyte interface under high-voltage conditions, which has been correlated with faster capacity fade of the cells and greater rise in the impedance of the cathodes.7–9 TM dissolution, oxygen loss, and surface reconstruction to the rock-salt phase with poor lithium-ion conductivity have been the most commonly observed results of high-voltage operation that are commonly linked with the observed impedance rise and capacity fade.10–12Attempts have been made to stabilize the cathode–electrolyte interface via use of either coatings13–15 or electrolyte additives.16 Electrolyte additives are molecules added in small quantities to the electrolyte to add some functional property/properties without changing its bulk properties. The additives for cathodes typically have oxidation potentials lower than the electrolyte solvents, so that during the initial few charge steps, they can oxidatively decompose to form a layer on the cathode surface. This layer ideally is stable, Li-ion conducting, and passivating, i.e., it prevents further electrolyte oxidation, therefore enabling longer battery life. Some examples of additives that improved the cycling performance of high-voltage cathodes are lithium difluoro(oxalato)borate (LiDFOB), prop-1-ene-1,3-sultone (PES), tris(trimethylsilyl)borate (TMSB), lithium bis(oxalato)borate (LiBOB), and tri(hexafluoro-iso-propyl)phosphate (HFiP).17–21 The improvement in capacity retention and/or reduction in TM dissolution and impedance rise is usually attributed to the formation of a protective film on the cathode particle surface, based on X-ray photoelectron spectroscopy (XPS) and scanning electron microscopy (SEM) results showing signs of some deposits containing additive derivatives on the surface. However, the improvement of cycling performance and presence of such deposits on the cathode surface may be due to a different effect and the films may not have the passivating properties, as is usually suggested. Hence, a better understanding of the working mechanism of these additives is needed, not just at the electrochemistry level, but at the reaction mechanism level as well.22 It is important to note here that rather than single additives, usually binary, ternary, or even higher number of additives in an electrolyte have been shown to deliver the best electrochemical performance due to synergistic effects between the additives.23 Now, if the working mechanisms of even single additives are only vaguely understood, the ability to design and predict better performing additive combinations without simply experimentally trying different combinations, is almost impossible.
Therefore, in this work we take tris(2,2,2-trifluoroethyl)phosphite (TTFP) as a model additive of study and provide a methodology to achieve mechanistic understanding of this additive's interaction with the cathode surface at high voltage. Since no single technique can provide a complete picture, we combined electrochemistry, post-mortem analysis and theoretical calculations to comprehensively understand the reactions of the tris(2,2,2-trifluoroethyl)phosphite (TTFP) additive at high voltage (4.6 V vs. Li+/Li), by analyzing the additive decomposition products both at the electrode surface as well as in the electrolyte. First, we use a controlled electrochemical aging protocol (see the Experimental section) to specifically test whether TTFP (as a model additive) “oxidation” under high voltage leads to formation of a passivating layer, which is typically suggested in the literature as the mechanism. This is followed by chemical analysis of the aged electrolyte using analytical techniques such as nuclear magnetic resonance (NMR) spectroscopy, high performance liquid chromatography coupled with electrospray ionization-mass spectrometry (HPLC/ESI-MS), and inductively coupled plasma mass spectrometry (ICP-MS). The electrolyte and additive decomposition products can be exotic molecules. Often they are not commercially available, making comparisons to known species difficult. Therefore, here we demonstrate “isotope-peak analysis” of the HPLC/ESI-MS data for species identification. The chemical composition of the aged cathode surface film was analyzed using XPS. Finally, density functional theory (DFT) calculations were used to augment some of our experimental observations.
TTFP has been shown to improve the cycling performance of Li-rich NMC and Li1.03(Ni0.5Mn0.3Co0.2)0.97O2 (NMC532) cathodes when cycled in a full cell with a graphite anode.24,25 The work with Li-rich NMC attributed the cycle stability to the oxygen scavenging ability of TTFP, thereby retarding electrolyte decomposition due to active oxygen. The work with the NMC532 cathode attributed the improved cyclability to either inhibition of “catalytic-centers” on the cathode surface or to the formation of a protective coating consisting of TTFP decomposition products, both of which prevented further oxidation of the electrolyte. While determining the rationale of performance improvement in the literature is not the goal of the present work, in this work however, we show that TTFP does not passivate the cathode surface toward electrolyte oxidation at high voltage, by conducting potentiostatic hold tests of Li4Ti5O12 (LTO)/NMC532 full cells at 4.6 V vs. Li/Li+. We found that the oxidation current at the end of the hold increased as the initial TTFP concentration in the electrolyte was increased, along with increased charge transfer impedance and TM dissolution, both of which are consistent with the formation of a non-passivating surface film during the hold. XPS of aged cathodes confirmed that TTFP modified the cathode-electrolyte interface. However, the concentration of TTFP did not greatly affect either the chemical composition or coverage of this layer. Based on XPS and DFT calculations, we proposed a mechanism for the attachment of TTFP to the surface, where removal of one of the trifluoroethyl (–CH2CF3) groups results in a much more stable configuration, BTFPa (bis(2,2,2-trifluoroethyl)phosphate), bound to the surface through P–O–TM bonds. However, NMR and HPLC showed that the majority of the TTFP additive oxidized to TTFPa (tris(2,2,2-trifluoroethyl)phosphate) during aging. Apart from TTFPa, several TTFPa-derived molecules were detected in the electrolyte via HPLC/ESI-MS that were found to be the reaction products of TTFPa and electrolyte solvents. DFT calculations indicated that TTFP can react with a terminal oxygen atom from the cathode surface, resulting in a TTFPa molecule, where the change in total energy due to the TTFPa formation was −3.1 eV, indicating a strong thermodynamic driving force for the reaction. Although the additive concentration was not optimized for the cathode we used, we believe the methodology we employ and the mechanisms we provide in this manuscript would still be valid irrespective of TTFP's concentration in the electrolyte.
2. Experimental
2.1 Cell assembly and electrochemical testing
The composition of the electrodes used in this work is given in Table 1. Both electrodes were dried in a vacuum oven at ∼70 °C for at least 4 h before being transferred into an oxygen- and moisture-free glovebox (MBraun). The N/P ratio (areal capacity ratio of negative to positive electrodes) for the electrode pair was approximately three. The baseline electrolyte consisted of 1.2 M LiPF6 in EC/EMC (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7 by wt; Tomiyama); various amounts of TTFP were added (0 to 10 wt%). 56 μl of electrolyte was used in each cell, corresponding to roughly three times the total pore volume of the cathode, anode, and separator. A minimum of three coin cells were assembled for each electrolyte mixture.
:7 by wt; Tomiyama); various amounts of TTFP were added (0 to 10 wt%). 56 μl of electrolyte was used in each cell, corresponding to roughly three times the total pore volume of the cathode, anode, and separator. A minimum of three coin cells were assembled for each electrolyte mixture.
| Cathode | Anode |
|---|---|
| 90 wt% Li1.03(Ni0.5Mn0.3Co0.2)0.97O2 (Toda) | 87 wt% Li4Ti5O12 (NEI) |
| 5 wt% PVDF (Solvay) binder | 8 wt% Kureha 9300 PVDF |
| 5 wt% Timcal C45 conductive additive | 5 wt% Timcal C45 conductive additive |
| Total material loading: 4.08 mg cm−2 | Total material loading: 26.07 mg cm−2 |
| Coating thickness: 24 μm | Coating thickness: 145 μm |
| Al foil thickness: 20 μm | Al foil thickness: 20 μm |
All cycling was performed at room temperature. After assembly, the coin cells were allowed to rest at open circuit to allow for adequate electrolyte wetting. After the rest, the cells were subjected to the electrochemical profile shown in Fig. 1a using a MACCOR Series 4000 Battery Tester. First, the cells were cycled between 2.85 V and 1.45 V (cathode voltage varying between 4.4 V and 3.0 V vs. Li/Li+), once at approximately the C/8 rate followed by ten cycles at the C/3 rate. Second, the cells were charged at the C/8 rate to 3.05 V (Vcath = 4.6 V vs. Li/Li+) and held at that voltage for 60 h, with the current recorded every 5 min. At the end of the potentiostatic hold, the cells were discharged at the C/8 rate to 1.45 V and cycled again between 2.85 V and 1.45 V, once at approximately the C/8 rate and ten additional cycles at the C/3 rate. Finally, the cells were charged at the C/8 rate to 2.75 V (Vcath = 4.3 V vs. Li/Li+) and held at that potential for 20 h. After this protocol, the cells were characterized using electrochemical impedance spectroscopy (EIS) testing using a Solartron Analytical 1400 Cell Test System equipped with a 1455 A frequency response analyzer at open circuit between 1 MHz and 0.01 Hz (1 mHz for 5 wt% and 10 wt% TTFP) with a 10 mV perturbation.
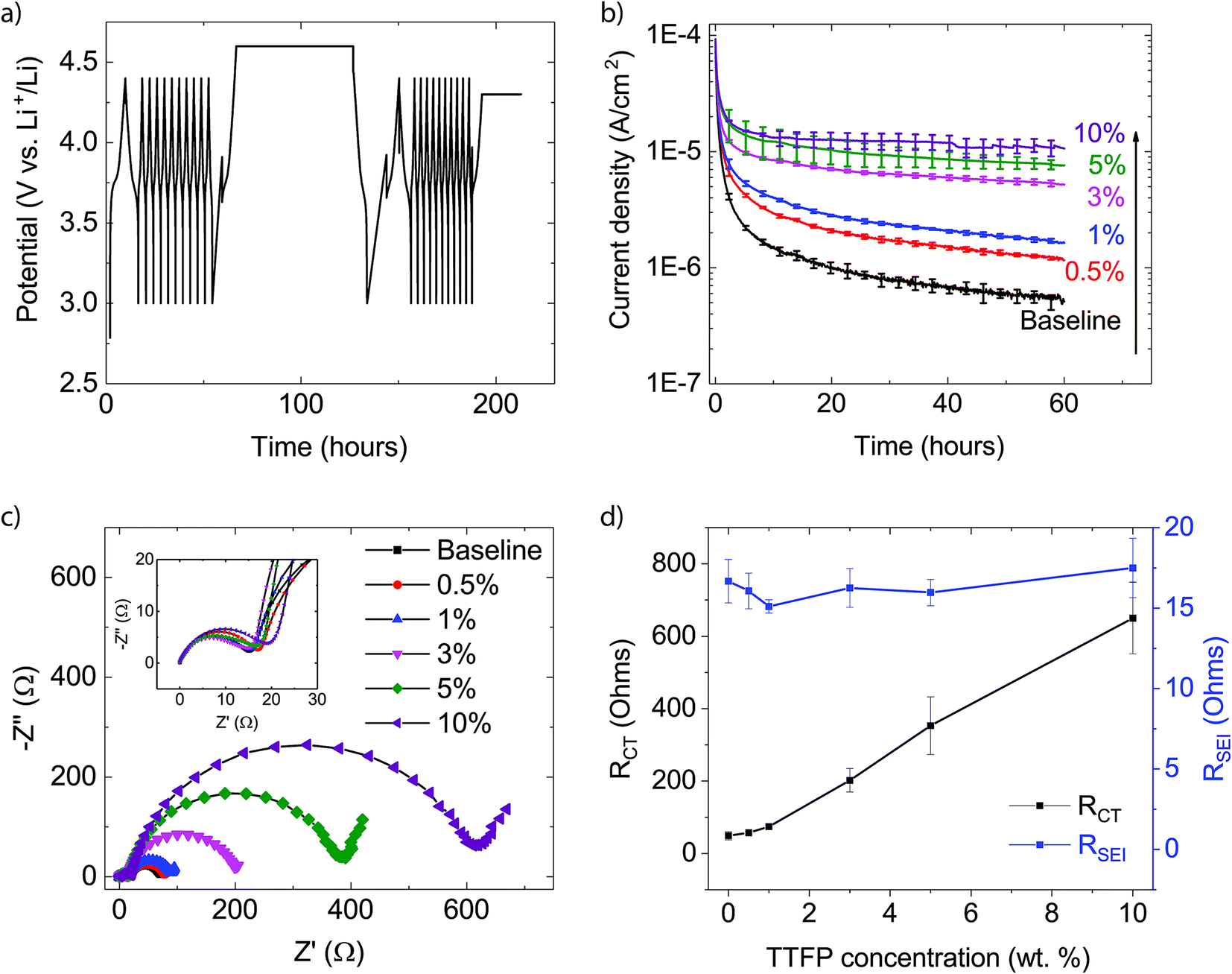 | ||
| Fig. 1 (a) Electrochemical protocol used to evaluate the oxidation reaction rate of the electrolytes at the cathode surface charged to 4.6 V vs. Li/Li+ in a LTO/NMC532 full cell. (b) Leakage current density vs. time profile for electrolytes with increasing TTFP concentration, obtained during the 4.6 V hold in the middle of the profile shown in (a). (c) EIS of the cells at the end of the protocol presented in (a). The inset in the figure provides a magnified view of the high-frequency range of the spectra. (d) Charge transfer (RCT) and SEI (RSEI) resistances extracted from the EIS spectra shown in (c). | ||
2.2 Sample harvesting for characterization
All cells were disassembled using a de-crimper in an argon-filled glovebox. The electrodes of one of the three cells for each TTFP concentration were removed and used for XPS analysis. The cathode was rinsed in 1.0 ml of dimethyl carbonate (DMC; Sigma), dried, and mounted on a sample holder for XPS examination (see below). The anode was not rinsed, but simply allowed to dry and mechanically scraped from the current collector. The recovered anode powder was removed from the glovebox and submitted to the Argonne Analytical Chemistry Laboratory for elemental analysis by ICP-MS.The electrode stack (electrodes, separator, and spacers) was removed from the other two cells for each electrolyte composition and soaked in ∼2.0 ml of DMC and sonicated at room temperature for approximately 1 min to extract all of the soluble components of the cycled electrolyte. The recovered solution was centrifuged to remove loose particles. To this, a dilute aqueous solution of Na2CO3 was added to neutralize any HF present. 5 ml of dichloromethane was added to the solution, resulting in an immediate phase separation. After 4 h, the bottom, salt-free organic layer was recovered with a pipette and the dichloromethane was left to evaporate overnight. The residue was dissolved in a mixture of 0.6 ml water and 0.4 ml acetonitrile for characterization using HPLC/ESI-MS.
A fourth cell was made with an electrolyte containing 10 wt% TTFP and subjected to the electrochemical protocol discussed above. This cell was de-crimped, and its electrode stack was removed and gently stirred in 1.0 ml of DMC to extract its electrolyte, as described above. The DMC extract was then put into a glass capillary insert, and the glass capillary was placed inside an NMR tube containing deuterated benzene for solution NMR analysis.
2.3 XPS
XPS analysis was performed with a PHI 5000 VersaProbe II System (Physical Electronics), with a base pressure of ∼2 × 10−9 Torr. The spectra were obtained using an Al Kα radiation (hν = 1486.6 eV) beam (100 μm, 25 W), Ar+ and electron beam sample neutralization, in Fixed Analyzer Transmission mode. Peak fitting was performed using Shirley background correction and the Gaussian–Lorentzian curve synthesis available in CasaXPS software. XPS spectra of the cathodes were aligned to the carbon black component in the C1s spectra at 284.7 eV. Manufacturer-reported relative sensitivity factors (RSFs) were used to normalize the spectra for plotting, such that the area under a region corresponds to the atomic percent of that element in the sample. This normalization allows direct visual comparison of concentration across samples in the series.2.4 HPLC/ESI-MS
An Agilent Technologies 1260 Infinity Liquid Chromatograph equipped with an Agilent 6120 Quadrupole Electrospray-Ionization Mass Spectrometer Detector was used. A 50 μl aliquot was used from each sample. Component separation was achieved using a ZORBAX ODS® column (5 μm; 4.6 × 250 mm) at 25 °C using a flow rate of 1 ml min−1. The mobile phase consisted of 60% water (HPLC grade, Sigma) and 40% acetonitrile (HPLC grade, Sigma), containing 0.1 vol% formic acid (HPLC grade, Sigma). The detector capillary voltage was set at 3000 V; the drying gas (N2) flow rate was 12 l min−1; the nebulizer pressure was 35 psig; the drying gas temperature was 350 °C; and the fragmentor voltage was 70 V.An estimate of the number of carbons in a pseudomolecular ion detected via ESI-MS was calculated using eqn (1). Here, the isotopic ratio of intensities in the mass spectrum is related by the Poisson distribution to the total number of carbon atoms in the pseudomolecular ion,
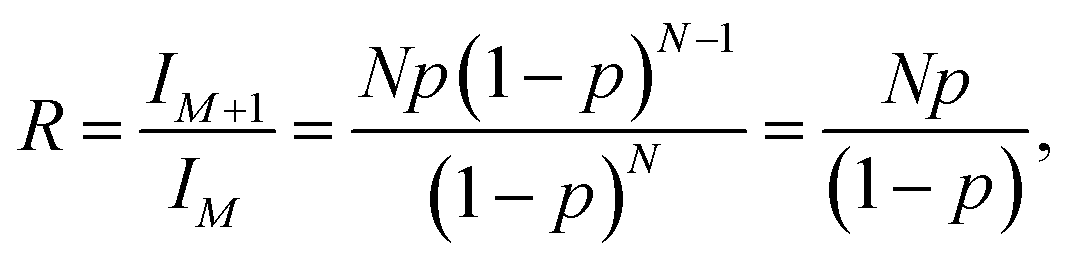 | (1) |
2.5 NMR spectroscopy
NMR spectra of the cycled electrolyte solution were obtained using a Bruker Avance™ III HD 300 MHz spectrometer. The DMC extract was analyzed by 31P NMR spectroscopy using a plastic insert which would prevent any interaction between the deuterated NMR solvent (benzene-d6) and the electrolyte sample. The 31P nucleus was internally referenced to LiPF6 (−146.0 ppm) present in the electrolyte. A 30° angle single-pulse experiment with 10 μs pulse width and 10 s pulse delay was used.2.6 DFT methods
Spin-polarized DFT calculations were carried out as implemented in the Vienna Ab Initio Simulation Package (VASP).26,27 The exchange-correlation potentials are treated by the generalized gradient approximation (GGA) parametrized by Perdew, Burke, and Ernzerhof (PBE).28 The interaction between valence electrons and ion cores is described by the projected augmented wave (PAW) method.29 The GGA + U Dudarev's approach is used for applying the on-site correlation effects among 3d electrons of the TMs. The effective on-site Coulomb (U) and exchange (J) parameter difference (U − J) is set to 5.96, 5.00, and 4.84 eV for Ni, Co, and Mn, respectively.30 The wave functions were expanded in the plane wave basis up to a kinetic energy of 500 eV. All surface calculations were performed using a periodically repeating slab separated by vacuum layers along the surface normal. A vacuum thickness of 10 Å was adopted to remove interaction between the slab layers. The lattice parameter of the supercell was fixed at its bulk value. All the ions were allowed to relax until the total energy differences were no more than 0.003 eV. Additionally, calculations of reaction in solution were performed with the Gaussian 09 implementation of DFT at the B3LYP/6-31+G(d,p) level of theory.31 Frequency calculations were performed for all structures in order to determine the nature of the stationary points. The ultrafine integration grid was employed in all calculations. Bulk solvent effects were accounted for by using a conductor polarizable continuum model (CPCM).2.7 ICP-MS
To perform ICP-MS analysis of the scraped-off anode powders, samples were first weighed in Pt crucibles and heated at 700 °C overnight to burn off carbon. In the same crucibles, 2 ml of concentrated HNO3 and 0.2 ml of concentrated HF (Optima grade) were added and the mixture was heated at 220 °C on a hot plate for 1 h to dissolve the ignition residue. The solutions were quantitatively transferred to 50 ml plastic centrifuge tubes and brought to 25 ml with de-ionized (DI) water before analysis by ICP-MS to determine Ni, Co, and Mn. ICP-MS measurements were carried out with a Perkin Elmer/Sciex ELAN® DRC-II instrument. Elemental concentrations were calculated from the measured concentrations, the volume of the solution, and the mass of the sample taken.3. Results
3.1 Electrochemical characterization
The electrochemical protocol used in this work consisted of three steps: cycling, a potentiostatic hold, and further cycling (Fig. 1a). This allowed for evaluation of the system through cycling, followed by an abusive high voltage hold test to determine oxidative decomposition rates, and then further cycling to evaluate the performance impact of the potentiostatic hold. The N/P ratio for the cells used in the experiments was approximately three, which allowed for sufficient surplus capacity in the anode for anode-lithiation reactions to compensate the oxidative decomposition reactions at the cathode. Furthermore, the high potential of the LTO anode (1.55 V vs. Li/Li+) minimized electrolyte reduction reactions at the anode. Therefore, the cell current measured during the potentiostatic hold could be attributed to electrolyte oxidation at the cathode. For further details about this approach, the reader is referred to studies by Tornheim et al.32,33Fig. 1b shows the average current density measured as a function of time during the potentiostatic hold step. A minimum of three cells were tested for each condition. Error bars (±σ) are included at every 2.5 h. In contrast to the expectation for a passivating layer-forming additive, the addition of TTFP to the electrolyte not only failed to eliminate the electrolyte oxidation current, but rather the current increased monotonically with TTFP concentration in the electrolyte. This indicates that TTFP does undergo an oxidation reaction at the surface. The anodic instability of TTFP under a high voltage on a Pt electrode has also been reported by M. He et al.25 If the cathode surface were passivated by TTFP, the final oxidation current should be lower than that of the baseline. The higher currents compared to baseline throughout the potentiostatic hold indicate that each TTFP-containing electrolyte has not passivated the cathode over the course of the potentiostatic hold (Fig. 1b).
Fig. 1c shows the EIS spectra of a representative cell recorded at the end of the electrochemical profile described above. The inset in the figure magnifies the high-frequency range of the spectra. Each spectrum contains two depressed semicircles, followed by a Warburg tail at low frequencies. The smaller, high-frequency semicircle is dominated by contributions from the solid-electrolyte interface (SEI) layer, while the bigger, mid-frequency semicircle is dominated by the charge-transfer process.34 The resistance of the SEI layer, RSEI, was determined from the distance along the real impedance axis between the zero crossing and the first minimum of the imaginary impedance; i.e., the diameter of the high-frequency semicircle. The charge transfer resistance, RCT, was similarly estimated as the diameter of the mid-frequency semicircle, determined from the difference of the real parts of the impedance between the first and second minima of the spectrum. These values are plotted as a function of TTFP concentration in Fig. 1d. It is clear from Fig. 1c and d that, while RSEI is relatively independent of TTFP concentration, RCT increases markedly for TTFP concentrations higher than 1%.
Fig. 2a shows the amount of Ni, Co, and Mn found on the anodes (ppm, by weight) as a function of TTFP concentration in the electrolyte. The TM concentration was found to be relatively unchanged until the concentration of TTFP in the electrolyte was ∼1 wt%. After that, the increase in TM content in the anode was clearly dominated by an enormous increase in Ni content, although both Mn and Co content also increased. The dominance of Ni in the anode at higher TTFP concentrations is consistent with previous reports on NMC532 materials at high voltage.11,12,35
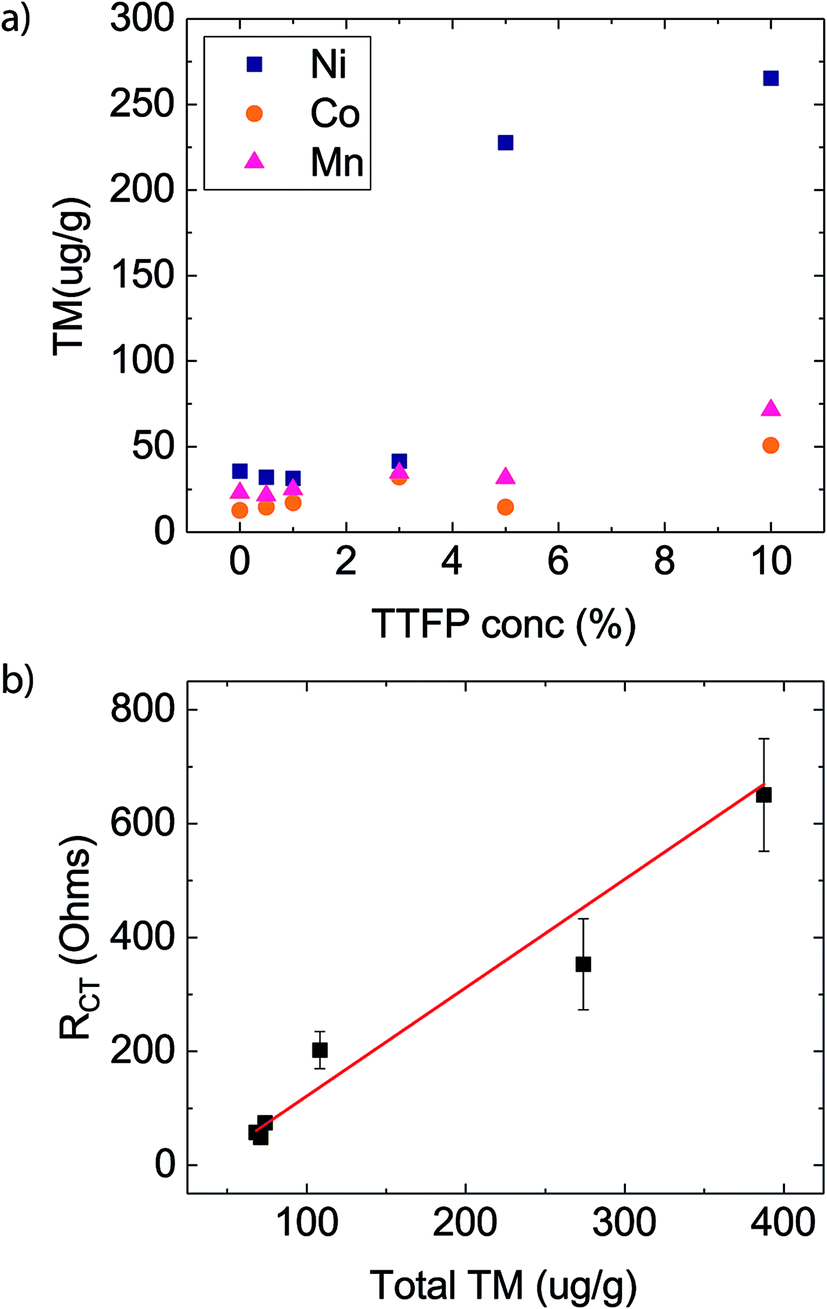 | ||
| Fig. 2 (a) Concentration of transition metals (TM) measured via ICP-MS as a function of TTFP concentration in the electrolyte, on anodes of cells subjected to the electrochemical protocol presented in Fig. 1a. (b) Charge transfer resistance, taken from Fig. 1d, as a function of total TM concentration in the anode. The solid curve represents a linear fit of the results (slope = 1.9 ± 0.35; y-axis intercept = −69.3 ± 25.6; correlation coefficient = 0.85). | ||
Fig. 2b shows the plot of RCTvs. total TM concentration. Least-squares regression using these data shows that the value of the regression coefficient, r2, was high, ∼0.85. The strong linear relationship indicates that RCT was directly proportional to the total TM concentration in the anode.
3.2 XPS
Fig. 3a–d show the C1s, O1s, F1s, and P2p XPS spectra, respectively, of the top ∼10 nm of material on the surface. The TTFP content of the corresponding cells is indicated by the color code shown in the legend. Pristine NMC532 refers to a cathode that was never exposed to the electrolyte, and is included for comparison.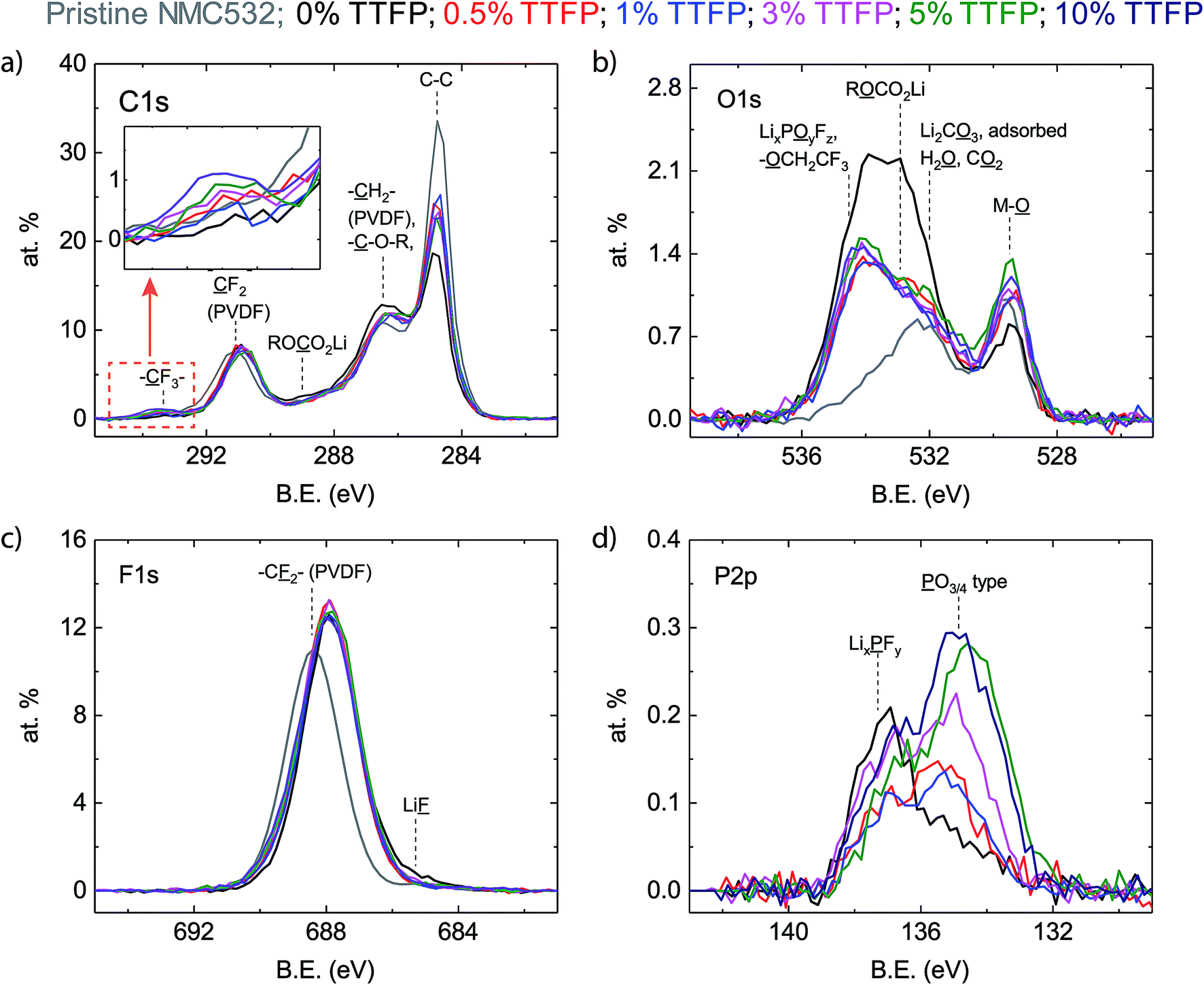 | ||
| Fig. 3 XPS characterization of cathodes harvested from cells subjected to the electrochemical protocol shown in Fig. 1a. The spectra are color coded, as indicated in the legend, according to the TTFP content in the electrolyte. (a) C1s region. (b) O1s region. (c) F1s region. (d) P2p region. The inset in (a) shows the high binding energy region of the C1s spectra. | ||
The C1s spectrum (Fig. 3a) of the pristine cathode sample (grey) consists of two main peaks, at 284.7 eV and 291.1 eV, with the 284.7 eV peak displaying a pronounced high binding energy (BE) shoulder at around 286.5 eV. The peak at 284.7 eV corresponds to the carbon black additive, while the peak at 291.1 eV and the shoulder at 286.5 eV can be attributed to CF2 and CH2 moieties in the PVDF binder of the cathode laminate.36 The peaks at 284.7 eV (C–C) and 291.1 eV (CF2) are still present in the harvested electrode samples, regardless of the TTFP concentration in the electrolyte. The intensity of the CF2 peak is almost the same as that in the pristine electrode and seems independent of TTFP concentration. The shoulder at 286.5 eV is more intense than that in the pristine electrode for all samples. While its intensity does not seem to change with TTFP concentration, it is slightly higher in the sample in which the electrolyte did not contain TTFP, which is consistent with an increased concentration of C–O environments from ether-containing species, which have been shown to form in carbonate-containing electrolytes. In contrast, the peak at 284.7 eV is consistently smaller than in the pristine electrode. While its intensity does not seem to change with TTFP concentration, it is even smaller in the sample in which the electrolyte did not contain TTFP. Finally, there is a small peak at ∼293.2 eV (see the inset in Fig. 3a) consistent with incorporation of CF3 groups into the formed surface film (Fig. S1a†). This peak is absent from the pristine and baseline electrolyte cells, and its intensity seems, overall, to increase with TTFP concentration.
The O1s spectrum of the pristine cathode sample (Fig. 3b) consists of two main peaks, at 529.7 eV and 532.2 eV. The peak at 529.7 eV corresponded to oxygen atoms in a metal oxide environment MOx and can be attributed to the lattice oxygen in the NMC particles in the electrode.37 Since the pristine electrode sample was stored in air before examination, the broad peak at 532.2 eV, which also displays a long tail toward high binding energy, can be attributed to a series of surface contaminants, such as carbonates (Li2CO3 at 531.8 eV)38 and physisorbed CO2 (534–535 eV)39 or water (533.2 eV).40 The O1s spectra from the cycled cathode samples also consist of two main peaks. The NMC MOx lattice oxygen peak is still visible at ∼529.6 eV. The peak at higher binding energy has broadened, shifted to ∼533–534 eV, and represents a greater contribution to the atomic concentration than in the pristine sample. This broad peak can be described by three components, centered at ∼534.6 eV, ∼533.2 eV, and ∼531.6 eV. The ∼534.6 eV component is consistent with oxygen in lithium fluorophosphates (LixPOyFz, 534.5 eV)41 and in –O–CH2–CF3 (533.7 eV, Fig. S1b†). The ∼533.2 eV component is consistent with lithium alkyl carbonates (O with an asterisk in RO*CO2Li).42 The lower binding energy peak at ∼531.6 eV is consistent with lithium carbonate (531.8 eV) and phosphate (531.5 eV).43 Other organic O moieties are also likely to be contributing intensity to the high energy O1s peak, such as ethers, alcohols, and esters.42 Unfortunately, the data cannot be deconvoluted with sufficient resolution to identify all these components.
The impact of TTFP in the electrolyte on the O1s spectra of the cycled electrodes is much clearer than in the C1s spectra. The changes in the O1s environment depend more on whether or not the electrolyte contained TTFP than on its concentration. The intensity of the MOx peak at low binding energy is higher for electrolytes containing TTFP than in the additive-free electrolyte case. This suggests that TTFP-containing electrolytes lead to formation of a thinner layer on the oxide particles compared to the additive-free electrolyte. On the other hand, the intensity of the broad, organic oxygen peak at ∼533–534 eV is lower in the samples containing TTFP than in the 0% TTFP electrolyte sample. The intensity reduction is more prominent in the low binding energy components on the peak, resulting also in a change of peak shape, which is now skewed toward high binding energy. This change of shape is consistent with the addition of fluorinated, oxygen-containing moieties to the surface film, such as –O–CH2–CF3.
The F1s spectrum of the baseline cathode (Fig. 3c) consists of a single broad peak, centered at ∼688.4 eV, dominated by CF2 moieties in the PVDF binder. All cycled samples displayed an F1s spectrum consisting also of a single broad peak, shifted to around ∼688.1 eV. These peaks are more intense and slightly broader than in the pristine electrode case and are insensitive to TTFP concentration in the electrolyte. These peaks could not be uniquely resolved into components; however, the baseline electrolyte peak displays a clear tail on the low binding energy side of the peak, which is consistent with LiF at ∼685.5 eV.42 The amount of LiF deposition is small, consistent with our previous experience of looking at cathodes cycled with the baseline electrolyte.44
The P2p spectra (Fig. 3d) show the largest dependence of TTFP concentration in the electrolyte. The P2p spectrum of the pristine cathode is not included as there is no source of P in it. The spectra from the cycled electrode samples consist of two main peaks, centered at ∼137.4 eV and ∼135.3 eV. The P2p spectrum of the baseline electrolyte sample is dominated by the peak at ∼137.4 eV, which can be attributed to LiPF6 and derived LixPFy species.41 Conversely, the P2p spectra of the TTFP-containing samples are dominated by the ∼135.3 eV peak, which can be attributed to phosphate or phosphite species PO3–4 (Fig. S1d†).41 Overall, the concentration of these species increases with TTFP content in the electrolyte.
Overall, the biggest changes to the XPS spectra described above depend mostly on whether or not the electrolyte contains TTFP, and not as much on the actual amount of TTFP in the electrolyte. However, the CF3-like C1s peak around 293.2 eV and the PO3–4 P2p peak around 135.3 eV do seem to increase at higher TTFP concentrations. Fig. 4 shows a plot of the areas under these two peaks, with PO3–4 concentration on the x-axis, and CF3 concentration on the y-axis. The amounts of these environments seem to be moderately correlated (r2 = 0.6). The slope of the least-squares line was 2.1 ± 0.75. Despite this moderate correlation coefficient, the correlation between these two values is a strong indicator that (1) TTFP is reacting and incorporating into the cathode surface film; and (2) on average, for every phosphorus atom incorporated through this TTFP reaction(s), two CF3 groups are also incorporated. It is worth noting that if TTFP incorporation into the surface proceeded by a simple attachment of TTFP molecules to the surface, three CF3 groups would be expected per phosphorus atom. Therefore, the XPS data presented above are indicative of, on average, the loss of one trifluoroethyl group per TTFP molecule incorporated into the cathode surface film.
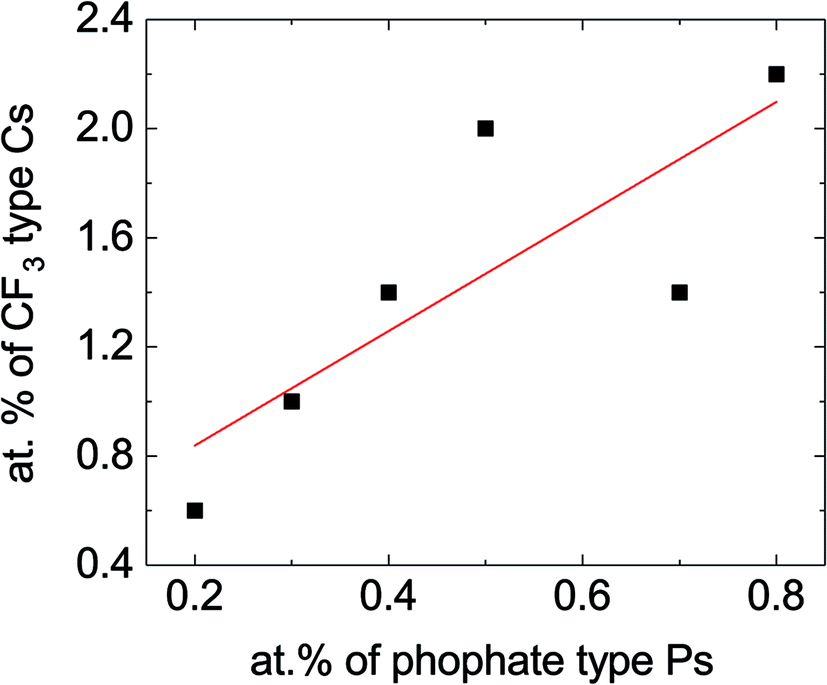 | ||
| Fig. 4 Concentration of –CF3 groups vs. concentration of phosphate/phosphite based on the data shown in the inset of Fig. 3a. The slope of the line was 2.10 ± 0.75, with a y-axis intercept of 0.41 ± 0.40 and correlation coefficient r2 = 0.6. There are several factors that could contribute to lower this correlation coefficient, including the difficulty in estimating the areas under these peaks due to a sloping background, in particular for the CF3 peak, and contributions from other atomic environments in the same binding energy range. | ||
3.3 HPLC/ESI-MS
The XPS analysis detailed above provides information on the films formed on the cathode surface during the electrochemical aging protocol. Since a growing surface layer with increased additive concentration was not evident from the XPS analysis, it was likely to find that the additive or its decomposition products were dissolved in the electrolyte. To test this assertion, electrolyte samples were extracted from the aged cells and evaluated with HPLC/ESI-MS. Since the presence of LiPF6 generates HF, which is detrimental to the LC column as well as MS detector, a procedure similar to that reported by Petibon et al. was adopted to remove the LiPF6 salt and HF (details are provided in the Experimental section).45 Since the process involves exposure of the sample to air and water, it is expected that the decomposition products in the aged electrolytes would react with water and oxygen (for example, hydrolysis or oxidation) and change their original structures depending upon their reaction kinetics. It is, however, not expected that the TTFP would react with electrolyte solvents as aggressively during the salt-removal procedure, as it would under highly oxidative conditions during the potentiostatic holds. To confirm that, as well as to set up a baseline of the artifacts of this salt-removal procedure, we compared the HPLC/ESI-MS signals of aged electrolytes with an uncycled baseline electrolyte (0% TTFP) and a 10% TTFP-containing electrolyte that underwent the same salt-removal procedure.Fig. 5a shows the total-ion chromatograms (TICs) of electrolyte extracts after salt removal. The chromatograms of the uncycled electrolytes are available in the ESI (Fig. S2†). Several new peaks are observed in the TTFP-containing electrolytes compared with the additive-free electrolyte or uncycled electrolytes. These peaks indicate the presence of additional decomposition species in those electrolytes. Mass spectra at any time of a chromatogram give the masses (m/z) of pseudomolecular ions ([M + X]+, X = H, Na, or NH4) detected at that time. The most prominent peak at ∼3.0 min in all chromatograms corresponds to EC, as was confirmed by running pure EC under the same conditions (Fig. S3†). Moreover, the m/z of the ion eluting at that time is 89, agreeing with singly protonated EC (M.W. = 88 u). The other large peak at ∼33.5 min comes from the ion corresponding to oxidized TTFP, i.e., TTFPa. This was also confirmed by running pure TTFPa under the same conditions (Fig. S3†). Moreover, the m/z of the ion eluting at that time is 345, agreeing with singly protonated TTFPa (M.W. = 344 u). Since the area under the EC peak is somewhat the same in all samples, it means that roughly the same amount of cycled electrolyte was extracted from each cell, assuming the amount of EC consumed during aging is very little. Analysis of changes in concentrations of other species as a function of the initial TTFP concentration was done by normalizing their peak areas to EC's peak area.
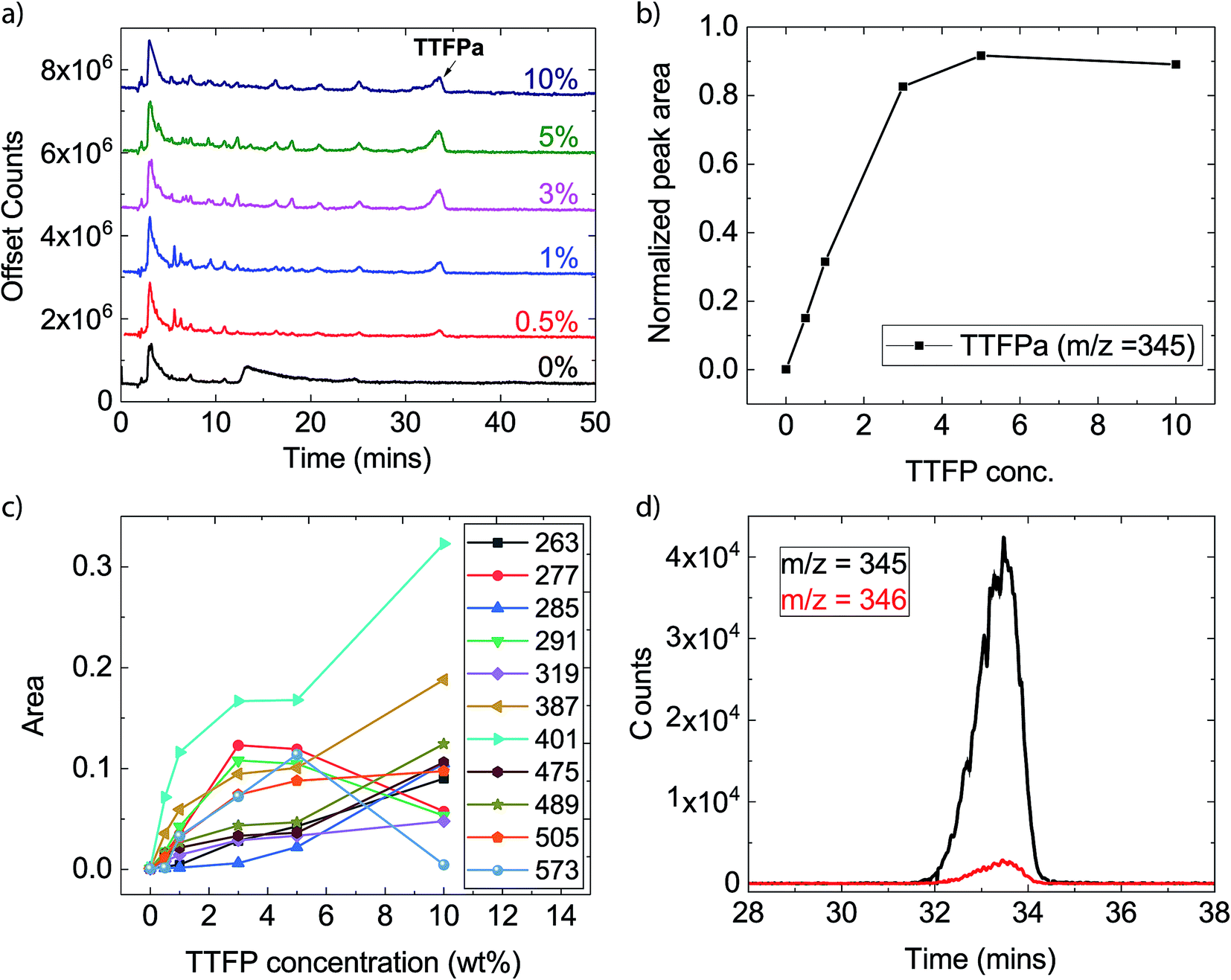 | ||
| Fig. 5 HPLC-MS of sample electrolytes. (a) Total-ion chromatograms of the different TTFP concentrations explored (indicated in the figure). (b) Signal intensity, relative to EC, of TTFPa signal as a function of initial TTFP concentration. (c) Signal intensities, relative to EC, of other characteristic ions producing the strongest signals as a function of the initial TTFP concentration. (d) An example of time traces of an ion (m/z = 345) and its isotopic peak; the ratio of these peak areas was used to calculate the number of carbon atoms in the molecule. | ||
Fig. 5b shows the change in area under the TTFPa peak normalized to the EC peak. It increases with TTFP concentration up to 3%, after which it plateaus until 10%. However, with this technique it is difficult to tell what fraction of the signal comes from TTFPa that formed due to oxidation of TTFP during aging in the cell. This is because some of that signal could have originated from unreacted TTFP, which could have oxidized to form TTFPa during the salt-removal process (Fig. S2 and S3†). However, we were able to confirm via NMR spectroscopy that the majority of TTFPa formation occurred during aging, as discussed in the next section.
In addition to EC and TTFPa, several other ions with m/z of ∼200–500 were detected which correspond to the remainder of the peaks in the chromatograms of TTFP-containing electrolytes (Table 2). Time traces (counts of an ion as a function of time) of all the ions for each TTFP concentration are available in the ESI (Fig. S4†). Interestingly, the majority of these ions have zero signal in the TTFP-free electrolyte, which strongly suggests that these species are TTFP derivatives. Fig. 5c plots the normalized areas of these ion peaks versus the initial TTFP concentration. The majority of the ions showed a continual increase in signal with TTFP concentration while others had maxima at an intermediate concentration. Fig. S5† plots the normalized areas of signal peaks of ions that were present in the TTFP-free electrolyte. Different types of evolution trends as a function of TTFP concentrations could be observed. While the concentration of some of these ions increased continuously with the initial TTFP concentration, others either showed no change in concentration or some other more complicated trend. Further investigations of these ion concentration trends, along with the knowledge of their corresponding molecular structures, could provide even deeper insights into the decomposition reactions going on in the cells, but that is beyond the scope of this paper. The next section describes our method for estimating the molecular structures of the observed ions.
| m/z observed | M.W. of the molecule | Estimated number of carbons in the molecule | Adduct in the pseudomolecular ion |
|---|---|---|---|
| 89 | 88 | 3 | H+ |
| 158 | 140 | 9–10 | NH4+ |
| 205 | 182 | 5–6 | Na+ |
| 229 | 206 | 7–8 | Na+ |
| 253 | 252 | 6 | H+ |
| 263 | 262 | 4 | H+ |
| 267 | 244 | 7 | Na+ |
| 277 | 276 | 5 | H+ |
| 285 | 262 | 7 | Na+ |
| 291 | 290 | 5–6 | H+ |
| 319 | 296 | 7 | Na+ |
| 345 | 344 | 6 | H+ |
| 387 | 364 | 8 | Na+ |
| 401 | 378 | 9 | Na+ |
| 475 | 452 | 11 | Na+ |
| 489 | 466 | 11–12 | Na+ |
| 505 | 482 | 8–9 | Na+ |
| 573 | 550 | 9–10 | Na+ |
In order to be able to estimate the structures of these ions, we started by calculating the number of carbons in each ion by performing isotope peak analysis. Out of all the elements that could be present in these ions (C, O, F, P, H), only C and H have a naturally occurring stable M + 1 isotope (13C and 2H) with abundances of 1.1% and 0.02%, respectively. Since the abundance of 2H is much lower compared to 13C, majority of the M + 1 intensity in the mass spectrum of an ion can be safely assumed to be coming from the 13C isotope. Fig. 5d shows, as an example, time traces of protonated TTFPa (m/z = 345) and its isotope peak (m/z = 346). The ratio of the areas of those peaks (I346/I345) comes out to be 0.066, which corresponds to 5.9 (∼6) carbons according to eqn (1) (Experimental section), which agrees with the fact that TTFPa contains 6 carbons. Therefore, using this method, the number of carbons in each of the detected ions was calculated (listed in Table 2). Finally, based on the molecular weight of the molecule (estimated from m/z of the ion, as described in the ESI†) and the number of carbons in it, the molecular structures of all the ions that were likely TTFP derivatives (i.e., zero intensity in the TTFP-free electrolyte sample) were proposed, as shown in Chart 1. The proposed molecular structures are consistent with the observed molecular masses and number of carbon atoms, as well as with other structures and formation mechanisms proposed in the literature.46–48 While these structural assignments are not unique, they provide clear evidence for oxidation of TTFP and derivatization of the resulting TTFPa. All these species were found to be derivatives of the TTFPa molecule formed by its further reaction with the other electrolyte components: EC, EMC, and LiPF6.
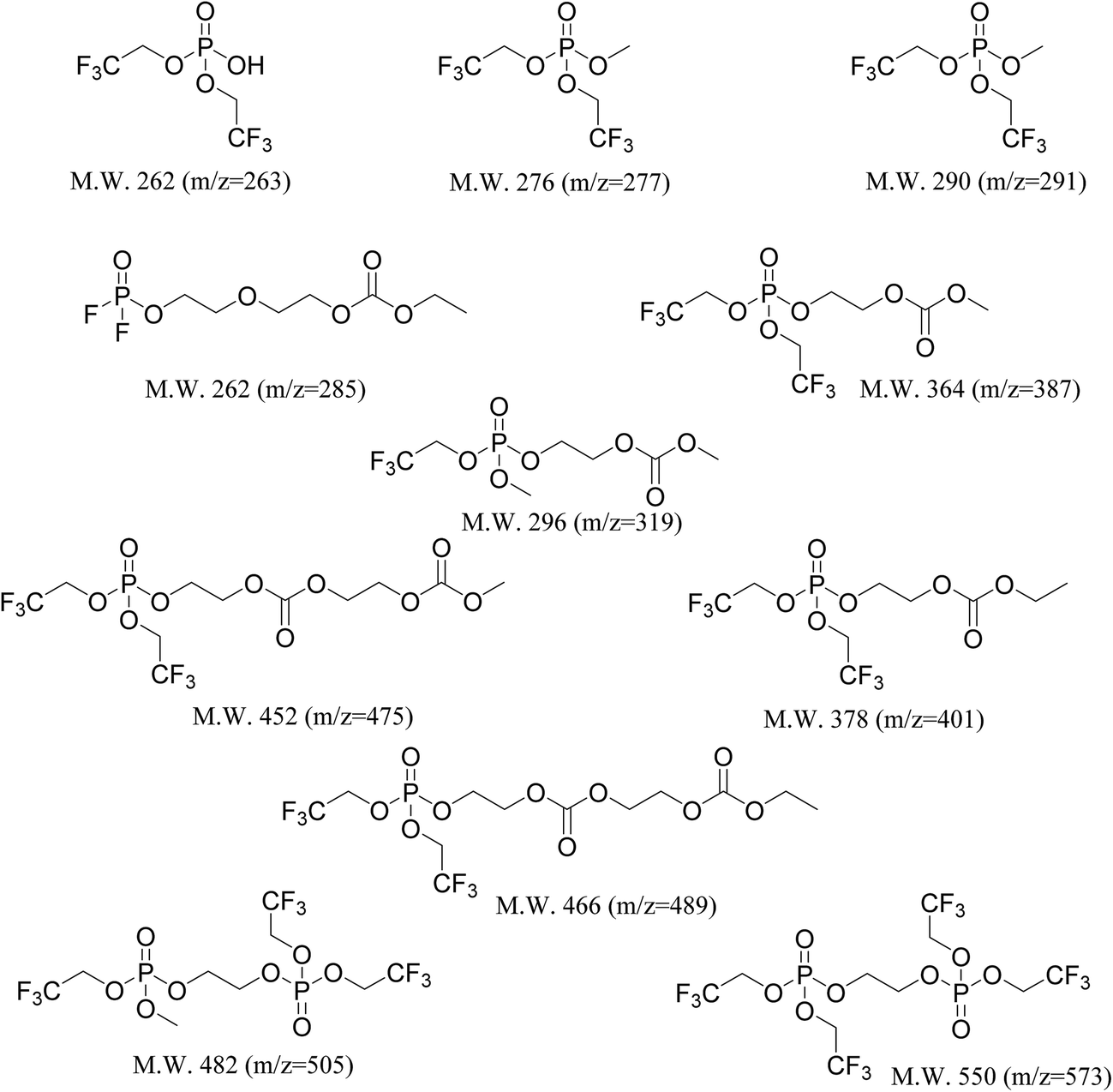 | ||
| Chart 1 Molecular structures of the decomposition species that were present only in the TTFP-containing aged electrolytes, detected via HPLC/ESI-MS. Structural assignments were made based on the molecular weight of the molecule and the number of carbons in the molecule as listed in Table 2. | ||
Overall, from these results it could be concluded that TTFP oxidizes to TTFPa during aging, which then further reacts with the electrolyte, and these decomposition products stay dissolved in the electrolyte.
3.4 NMR
In order to verify that the TTFPa detected by HPLC is formed in the cells during the voltage holds, rather than being a sampling artifact, we conducted NMR analysis of the 10% TTFP electrolyte, harvested after the electrochemical protocol shown in Fig. 1a, as well as of two pristine reference solutions of 10 wt% TTFP or TTFPa in the baseline electrolyte. Here, the electrolyte sampling protocol, described in the Experimental section, did not involve hydrolysis or exposure to air.Fig. 6a shows a 31P solution NMR spectra of the aforementioned samples. All spectra show a large septet at −146.0 ppm (JP–F 708 Hz) belonging to PF6−. In addition to this, the TTFP and TTFPa references show a singlet each, located at 139.0 ppm and at −6.0 ppm, respectively.
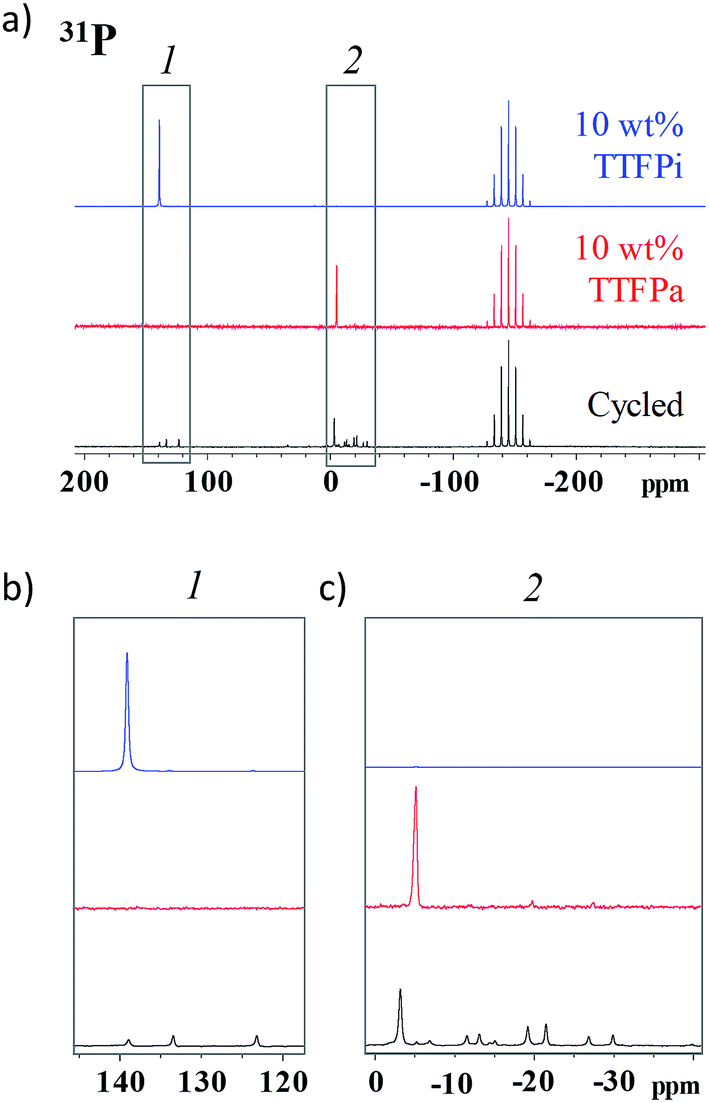 | ||
| Fig. 6 (a) 31P spectra of 10 wt% TTFP and 10 wt% TTFPa reference solutions and cycled electrolyte. The spectra in (b) and (c) are enlargements of selected regions in (a). | ||
Fig. 6b and c show magnified views of the spectral regions around these shifts for the three samples considered. The harvested electrolyte sample shows three singlets in the region between 120 ppm and 140 ppm, corresponding to different phosphites, and several peaks in the region between −40 ppm and 10 ppm, corresponding to different phosphates. Of all peaks observed in the harvested electrolyte sample, a small singlet at 140 ppm and a tall singlet at −4.7 ppm are the closest ones to the characteristic singlets in the TTFP and TTFPa standards, respectively, and can be attributed to the remaining TTFP and formed TTFPa in the harvested electrolyte, with the slight change in NMR shift from the reference sample potentially being due to small solvation changes in the harvested electrolyte. A comparison of the integrated areas of these singlets provides an estimate of the TTFP/TTFPa ratio in the harvested electrolyte as ≲1:5.
In fact, some shift is observed in the 19F spectra as well (Fig. S6†). The 19F NMR spectrum of TTFPa shows a triplet at −74.2 ppm (8.2 Hz), while the TTFP spectrum shows a sextet at −74.0 ppm (8.5 Hz). The near identical proximity of the TTFPa peaks between the known and unknown solutions, as well as matching of the J coupling constants between the known TTFPa and unknown TTFPa peaks (both 8.2 Hz), provides further evidence to support the oxidation of TTFP to TTFPa. Another reason for the likelihood of that peak to be from TTFPa would be the fact that this peak's intensity is much higher compared to other peaks in the spectrum of the aged electrolyte. This is consistent with our HPLC results where the signal intensity from TTFPa was observed to be around three times the signal of the next most intense species (apart from EC).
Several other smaller intensity peaks could be observed in the 31P and 19F spectra of the 10% TTFP containing aged electrolyte which are absent in the 0% TTFP aged electrolyte (Fig. S7†). Hence, those peaks could be attributed to the presence of TTFP derived decomposition species which were also detected via HPLC/ESI-MS. It was not possible to confirm the structure of those molecules via NMR due to the lack of availability of those pure molecules commercially.
3.5 DFT calculations
Theoretical calculations were performed to understand the energetics of possible interactions between TTFP and the NMC surface. Here, we used ab initio molecular dynamics to explore possible adsorption configurations of TTFP. Subsequently, we optimized the geometry of the system in order to find the lowest energy configuration. We found a weak interaction for all possible configurations of TTFP on the fully lithiated NMC surface. The –CF3 group did not bind to any Li, TM, or oxygen atom. Fig. 7a shows a configuration of the adsorption where the –CF3 group weakly interacts with surface lithium atoms. For configurations where TTFP was parallel to the surface, the phosphite group in TTFP did not interact with the surface, either. Therefore, the oxidation of TTFP to TTFPa is very unlikely on the pristine, fully lithiated (fully discharged) cathode surface.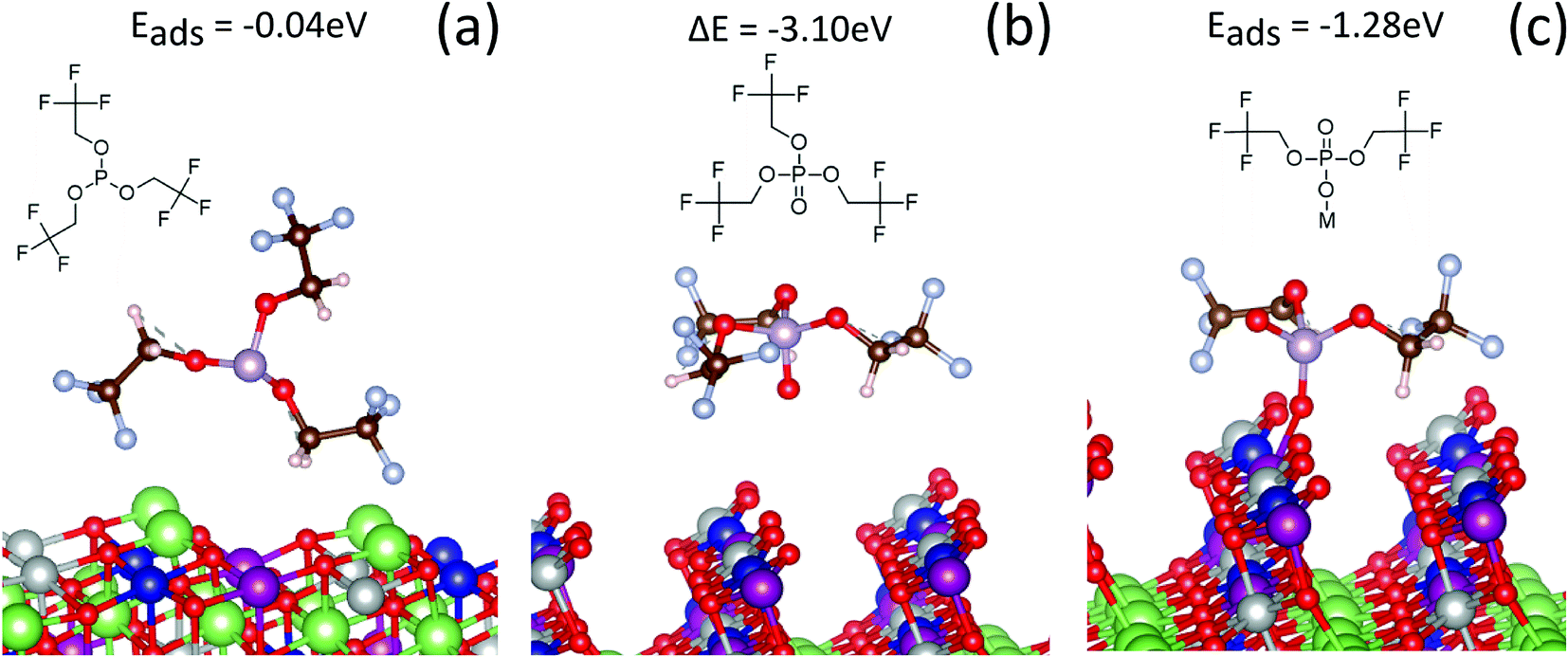 | ||
| Fig. 7 Adsorption configurations of TTFP and its derivatives on the NMC (012) surface. (a) Interaction of TTFP on fully lithiated NMC. (b) TTFPa formed and detached from the delithiated surface. (c) A TTFP derivative with two –CH2CF3 groups absorbed on one of the bridging oxygens of the delithiated surface. The green spheres represent Li; red spheres, oxygen; white spheres, hydrogen; light blue spheres, F; blue spheres, Co; purple spheres, Mn; gray spheres, Ni; and the pink spheres represent P. | ||
In contrast, a strong interaction of the TTFP molecule with the surface was observed when 80% of the lithium was removed from the cathode. The surface oxygen interacted with the phosphorus atom, promoting one electron from the lone pair in the sp3-hybridized molecular orbital to an unoccupied d orbital of phosphorus, forming a π-bond with the oxygen atom. This configuration produced a stable, free phosphate molecule by removing an oxygen atom from the NMC surface (Fig. 7b). An oxygen vacancy was also produced during this process, which left two unpaired electrons in the lattice and, hence, reduced the surface TMs (e.g., Ni). The change in total energy due to the reaction was −3.1 eV, indicating a strong thermodynamic driving force and the formation of a P–O bond with covalent character. This value was very close to our calculations of the formation of TTFPa from TTFP in solution (−3.5 eV), indicating that the oxygen vacancy formation energy at this state of charge is very small.49 However, there was no indication that the TTFPa thus created was bound to the surface.
However, if one of the –CH2CF3 groups is removed from the molecule (Fig. 7c), the absorption energy of the resulting BTFPa (bis(2,2,2-trifluoroethyl)phosphate) is significant, 1.28 eV. In this case, the phosphate binds to the surface oxygen, creating a P–O bond that, although weaker than that created in TTFPa, is of comparable strength to M–O bonds at the NMC surface. This stable configuration has only two –CF3 groups per phosphorus atom, in agreement with XPS measurements described above in which a slope of 2.10 ± 0.75 was obtained when the at% of CF3 type C was plotted against the at% of phosphate/phosphite type P (Fig. 4).
4. Discussion
One of the goals of this study was to test whether TTFP oxidation leads to the formation of a passivating layer on the cathode. The data presented above indicate that this is not the case. First, the current density at the end of the 60 h, 4.6 Vcathvs. Li/Li+ hold (Fig. 1b) does not decrease upon the addition of TTFP to the electrolyte. In contrast, this current increases with TTFP concentration. Since a minimal reduction is expected at the high potential of the LTO anode of either TTFP or the carbonate solvents, the excess current can be attributed to oxidation of TTFP at the cathode surface, in parallel to electrolyte solvent decomposition. TTFP oxidation at the cathode surface is also borne by our DFT results (Fig. 7a and b), which further indicate that the oxidation occurs at a charged (delithiated cathode) state.On the other hand, XPS analysis (Fig. 3) clearly indicates formation of a cathode surface film. The composition of this film is relatively independent of TTFP concentration, but different than that formed in the absence of TTFP. In particular, stronger signals from the underlying metal oxide and carbon black suggest that TTFP induces the formation of a film that is thinner or that covers the underlying NMC, binder, and carbon black more sparsely than without TTFP. One possible reason for the near independence of the surface film on TTFP concentration is that its formation is limited by the number of active sites available on the surface for TTFP to react and bind. In this scenario, further availability of TTFP is not going to change the nature of the film once all the surface sites are occupied.
However, RCT increases with TTFP concentration (Fig. 1d), despite the near independence of the cathode surface film on TTFP concentration. This RCT increase is strongly correlated with TM dissolution from the cathode (Fig. 2b). One possibility for the increase in RCT is due to TM dissolution and deposition on the anode, which has been shown to modify the SEI layer to possibly create a high impedance layer.50 Another possibility is that the RCT increase is predominantly from the cathode. One possible mechanism for this is suggested by the DFT observation (Fig. 7b) that oxidation of TTFP to TTFPa at the delithiated cathode surface is more likely to result in the creation of an oxygen vacancy than in the binding of the TTFPa molecule to the surface. TTFP oxidation provides a facile mechanism for oxygen loss, which along with TM dissolution can trigger changes of crystal structure in the near-surface region of NMC particles which, in turn, could be the cause of the increased RCT.11,51–53
This scenario is not incompatible with the composition of the cathode surface film being nearly independent of TTFP concentration. The constant composition observed by XPS would be a result of a dynamic, rather than static, equilibrium. This hypothesis is supported by the observation (Fig. 5b) that TTFPa content in the electrolyte increases with initial TTFP concentration (until 3%) while the composition of the cathode surface film stays constant. The combination of oxygen loss to TTFPa and TM dissolution could also result in a roughening of the cathode surface, leading to an increase in surface area, which could create more active sites and explain a weak positive correlation between the amount of POx and CF3 in the cathode surface and the concentration of TTFP in the electrolyte. A similar roughening process has been documented during chemical vapor deposition growth of doped semiconductors.54
TTFP oxidation at the delithiated cathode surface also helps to explain the observation that TM dissolution is dominated by Ni at TTFP concentrations ≳3%. When TTFP removes an oxygen atom from the surface as it oxidizes to TTFPa at the cathode surface, two electrons from the oxygen anion are transferred to the cathode. Crystal field considerations and preliminary DFT calculations show that when this process occurs in the discharged (fully lithiated) state, the electron density is increased preferentially at a MnIV atom. However, when the oxidation occurs in the charged (delithiated) state, the electron density increase is localized to an already oxidized NiIV atom. Therefore, TTFP oxidation-mediated TM reduction during our long voltage hold in the charged state can be expected to result in preferential dissolution of the preferentially reduced Ni. As the TTFP concentration increases and its oxidation becomes more dominant, this effect can be expected to become more prominent, as observed. Additionally, the increased Ni concentration observed at the anode at high TTFP concentrations could also be due to TTFP's affinity for NiII as a ligand. Carbonate solvents are weak electron donors and do not strongly stabilize solubilized cations.55 In contrast, TTFP is a strong π-acceptor, and binds much more strongly to TM cations.56 Since the rate of overall TM dissolution at the cathode is expected to depend on the energetics of the solubilized species, the addition of strongly binding phosphite ligands to the electrolyte will increase the rate of TM dissolution. Phosphites in general are classified as soft ligands and have a preferential affinity for softer metal species, such as NiII, over harder metal species, such as MnII, explaining the observation that TM dissolution is dominated by Ni at higher TTFP concentrations.57
We note that although XPS cannot easily distinguish between POx environments in phosphites and phosphates, our data (Fig. 4) indicate that there are only two CF3 groups per POx in the cathode surface film. This is consistent with the DFT calculation (Fig. 7c) indicating that the loss of one of the –CH2CF3 groups from a TTFPa molecule dramatically increases the binding energy of the resulting BTFPa molecule with respect to TTFPa. It is not clear at this time whether this loss occurs during TTFP oxidation, before the TTFPa molecule leaves the NMC surface, or through interaction of a phosphite or phosphate molecule containing two trifluoroethoxy groups with an oxygen vacancy or atom on the NMC surface. However, our HPLC/ESI-MS analysis (Chart 1) indicates the presence of several TTFPa derivatives missing at least one trifluoroethoxy. NMR analysis of the 10% TTFP extracted electrolyte (Fig. 6) did find indications of remaining TTFP with an estimated TTFP/TTFPa ratio of 1:5. This is surprising, since, as determined by HPLC (Fig. 5b), the TTFPa concentration in the extracted electrolyte saturates somewhat between 3% and 5% TTFP. The TTFPa concentration in the 5% TTFP electrolyte cannot exceed 5%, i.e., if all TTFP were oxidized and none of the produced TTFPa were consumed by further reactions. When increasing the TTFP concentration to 10%, if TTFP oxidation to TTFPa were the only TTFP consumption mechanism, we would expect at least a 1:1 TTFP/TTFPa ratio due to the additional unreacted TTFP. Therefore, this is a clear indication of other TTFP-consuming reactions, i.e., continuing TTFP oxidation to TTFPa, followed by increased consumption of TTFPa by further reactions, or both. This agrees with our observation via HPLC that, while the amount of TTFPa generated saturates above 3% TTFP concentration, the amounts of the majority of the TTFPa derivatives detected increase continuously. Hence, the consumption of some of the formed TTFPa to its derivatives could be the reason for the observed plateau of TTFPa content.
Finally, we note that the failure of the TTFP additive to passivate the cathode surface against electrolyte oxidation reported here does not invalidate previous reports of its ability to enhance performance. In particular, none of our observations disproves the previously reported increased capacity retention in NMC/graphite systems, but only its attribution to passivation of the cathode surface. Since, we did show oxidation of TTFP through electrochemistry, simulations, and in the products detected by HPLC, this oxidation would manifest as extra discharge capacity and possibly lead to the increased capacity retention.58 Besides, it is extremely likely that the interactions of additive decomposition products would be very different on a lithiated graphite surface versus lithiated LTO surface. However, unless those species after reducing at the graphite migrate to the cathode and passivate the cathode's surface, we believe the reaction outlined here where TTFP grabs O from the charged cathode surface to form TTFPa, leading to impedance rise and TM dissolution, would keep on happening and ultimately degrade the cathode.
Another possible competing reaction to form TTFPa from TTFP could be through the reaction with trace water in the cell. While the thermodynamic calculations (ESI†) show that the reaction is possible (negative enthalpy and free energy of reaction), that reaction pathway is unlikely as it would likely have to overcome a big energy barrier to break a water molecule. It is more probable that TTFP takes the oxygen from other sources (other organic molecules in the electrolyte or from the cathode surface). The energy barrier of oxygen atoms (at cathode surface) at high state-of-charge to react with TTFP is expected to be very low compared to breaking a water molecule. Carrying out those calculations would be quite extensive and we believe is beyond the scope of this work. Besides, rather than the oxidation reaction of TTFP with water, its hydrolysis reaction is more likely.59,60 Additionally, hydrolysis of LiPF6 is a well understood and experimentally proven mechanism.61 Since LiPF6 is present in much higher concentration in the electrolyte compared to TTFP, we believe the former is, statistically speaking, more likely to react with water compared to TTFP.
5. Conclusions
The degradation mechanism of the TTFP additive under high-voltage conditions was investigated by using a combination of electrochemical aging, post-mortem chemical analysis with XPS, HPLC/ESI-MS, NMR, ICP-MS, and DFT calculations. Potentiostatic holds at 4.6 V were performed on electrolytes with increasing TTFP concentrations (0–10 wt%) to measure the extent of electrolyte oxidation. Rise in the leakage currents during the hold as the initial TTFP concentration was raised, along with higher charge transfer resistance and TM dissolution after aging, confirms that TTFP does not passivate the NMC532 cathode surface toward electrolyte oxidation. XPS analysis shows that TTFP does attach to the surface, but the coverage or composition of this film does not change significantly with TTFP concentration. Based on XPS analysis and DFT calculations, we proposed a mechanism for TTFP's attachment to the surface whereby a P–O–TM bond forms with the surface lattice oxygen via cleavage of one of the –CH2CF3 side groups. HPLC and NMR analysis confirm oxidation of TTFP to TTFPa during aging as well as further reaction of TTFPa with the electrolyte solvents or their decomposition products. This was corroborated by DFT calculations wherein the change in total energy due to the TTFPa formation was −3.1 eV, indicating a strong thermodynamic driving force for the reaction. Overall, these results point toward the presence of other mechanisms at play whenever improved cycling performance of this additive is reported in a full cell. The unique combination of different techniques employed in this work helped generate a deeper understanding of the decomposition behavior of the TTFP additive under high voltage, which could be easily translated to other additives as well.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Analytical Chemistry Laboratory at Argonne National Laboratory for performing the ICP-MS measurements (elemental analysis). The authors gratefully acknowledge support from the U. S. Department of Energy (DOE), Office of Energy Efficiency and Renewable Energy, Vehicle Technologies Office. Argonne National Laboratory is operated for DOE Office of Science by UChicago Argonne, LLC, under contract number DE-AC02-06CH11357.References
- M. S. Whittingham, Chem. Rev., 2004, 104, 4271–4302 CrossRef CAS PubMed
.
- K. Kang, Y. S. Meng, J. Bréger, C. P. Grey and G. Ceder, Science, 2006, 311, 977–980 CrossRef CAS PubMed
- A. Kraytsberg and Y. Ein-Eli, Adv. Energy Mater., 2012, 2, 922–939 CrossRef CAS
- H. Zheng, Q. Sun, G. Liu, X. Song and V. S. Battaglia, J. Power Sources, 2012, 207, 134–140 CrossRef CAS
- L. Yang, B. Ravdel and B. L. Lucht, Electrochem. Solid-State Lett., 2010, 13, A95–A97 CrossRef CAS
- V. Etacheri, R. Marom, R. Elazari, G. Salitra and D. Aurbach, Energy Environ. Sci., 2011, 4, 3243–3262 CAS
- K. Xu, Chem. Rev., 2004, 104, 4303–4418 CrossRef CAS PubMed
- P. Arora, R. E. White and M. Doyle, J. Electrochem. Soc., 1998, 145, 3647–3667 CrossRef CAS
- D. Zhang, B. S. Haran, A. Durairajan, R. E. White, Y. Podrazhansky and B. N. Popov, J. Power Sources, 2000, 91, 122–129 CrossRef CAS
- F. Lin, I. M. Markus, D. Nordlund, T.-C. Weng, M. D. Asta, H. L. Xin and M. M. Doeff, Nat. Commun., 2014, 5, 3529 Search PubMed
- S.-K. Jung, H. Gwon, J. Hong, K.-Y. Park, D.-H. Seo, H. Kim, J. Hyun, W. Yang and K. Kang, Adv. Energy Mater., 2014, 4, 1300787 CrossRef
- J. A. Gilbert, I. A. Shkrob and D. P. Abraham, J. Electrochem. Soc., 2017, 164, A389–A399 CrossRef CAS
- B. C. Park, H. B. Kim, S. T. Myung, K. Amine, I. Belharouak, S. M. Lee and Y. K. Sun, J. Power Sources, 2008, 178, 826–831 CrossRef CAS
- Y. Bai, X. Wang, S. Yang, X. Zhang, X. Yang, H. Shu and Q. Wu, J. Alloys Compd., 2012, 541, 125–131 CrossRef CAS
- W. Liu, M. Wang, X. l. Gao, W. Zhang, J. Chen, H. Zhou and X. Zhang, J. Alloys Compd., 2012, 543, 181–188 CrossRef CAS
- A. M. Haregewoin, A. S. Wotango and B.-J. Hwang, Energy Environ. Sci., 2016, 9, 1955–1988 CAS
- M. Hu, J. Wei, L. Xing and Z. Zhou, J. Appl. Electrochem., 2012, 42, 291–296 CrossRef CAS
- B. Li, Y. Wang, H. Rong, Y. Wang, J. Liu, L. Xing, M. Xu and W. Li, J. Mater. Chem. A, 2013, 1, 12954–12961 CAS
- J. Li, L. Xing, R. Zhang, M. Chen, Z. Wang, M. Xu and W. Li, J. Power Sources, 2015, 285, 360–366 CrossRef CAS
- S. Dalavi, M. Xu, B. Knight and B. L. Lucht, Electrochem. Solid-State Lett., 2011, 15, A28–A31 CrossRef
- S. Tan, Z. Zhang, Y. Li, Y. Li, J. Zheng, Z. Zhou and Y. Yang, J. Electrochem. Soc., 2013, 160, A285–A292 CrossRef CAS
- S. Nowak and M. Winter, J. Electrochem. Soc., 2015, 162, A2500–A2508 CrossRef CAS
- J. C. Burns, A. Kassam, N. N. Sinha, L. E. Downie, L. Solnickova, B. M. Way and J. R. Dahn, J. Electrochem. Soc., 2013, 160, A1451–A1456 CrossRef CAS
- J. Pires, A. Castets, L. Timperman, J. Santos-Peña, E. Dumont, S. Levasseur, C. Tessier, R. Dedryvère and M. Anouti, J. Power Sources, 2015, 296, 413–425 CrossRef CAS
- M. He, C.-C. Su, C. Peebles, Z. Feng, J. G. Connell, C. Liao, Y. Wang, I. A. Shkrob and Z. Zhang, ACS Appl. Mater. Interfaces, 2016, 8, 11450–11458 CAS
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef
- H. Iddir and R. Benedek, Chem. Mater., 2014, 26, 2407–2413 CrossRef CAS
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Gaussian, Inc., Wallingford, CT, USA, 2009 Search PubMed
- A. Tornheim, S. E. Trask and Z. Zhang, J. Electrochem. Soc., 2016, 163, A1717–A1722 CrossRef CAS
- A. Tornheim, M. He, C.-C. Su and Z. Zhang, J. Electrochem. Soc., 2017, 164, A6366–A6372 CrossRef CAS
- D. P. Abraham, E. M. Reynolds, E. Sammann, A. N. Jansen and D. W. Dees, Electrochim. Acta, 2005, 51, 502–510 CrossRef CAS
- W. Choi and A. Manthiram, J. Electrochem. Soc., 2006, 153, A1760–A1764 CrossRef CAS
- M. C. Militello and S. W. Gaarenstroom, Surf. Sci. Spectra, 1999, 6, 141–145 CrossRef CAS
- Y. Su, S. Cui, Z. Zhuo, W. Yang, X. Wang and F. Pan, ACS Appl. Mater. Interfaces, 2015, 7, 25105–25112 CAS
- R. Dedryvère, L. Gireaud, S. Grugeon, S. Laruelle, J. M. Tarascon and D. Gonbeau, J. Phys. Chem. B, 2005, 109, 15868–15875 CrossRef PubMed
- K. Takeuchi, S. Yamamoto, Y. Hamamoto, Y. Shiozawa, K. Tashima, H. Fukidome, T. Koitaya, K. Mukai, S. Yoshimoto, M. Suemitsu, Y. Morikawa, J. Yoshinobu and I. Matsuda, J. Phys. Chem. C, 2017, 121, 2807–2814 CAS
- M. Abu Haija, S. Guimond, A. Uhl, H. Kuhlenbeck and H. J. Freund, Surf. Sci., 2006, 600, 1040–1047 CrossRef CAS
- W. Li and B. L. Lucht, J. Electrochem. Soc., 2006, 153, A1617–A1625 CrossRef CAS
- P. Verma, P. Maire and P. Novák, Electrochim. Acta, 2010, 55, 6332–6341 CrossRef CAS
- W. E. Morgan, J. R. Van Wazer and W. J. Stec, J. Am. Chem. Soc., 1973, 95, 751–755 CrossRef CAS
- C. Peebles, R. Sahore, J. A. Gilbert, J. C. Garcia, A. Tornheim, J. Bareño, H. Iddir, C. Liao and D. P. Abraham, J. Electrochem. Soc., 2017, 164, A1579–A1586 CrossRef CAS
- R. Petibon, L. Rotermund, K. J. Nelson, A. S. Gozdz, J. Xia and J. R. Dahn, J. Electrochem. Soc., 2014, 161, A1167–A1172 CrossRef CAS
- M. Tochihara, H. Nara, D. Mukoyama, T. Yokoshima, T. Momma and T. Osaka, J. Electrochem. Soc., 2015, 162, A2008–A2015 CrossRef CAS
- S. Takeda, W. Morimura, Y.-H. Liu, T. Sakai and Y. Saito, Rapid Commun. Mass Spectrom., 2016, 30, 1754–1762 CrossRef CAS PubMed
- V. Kraft, W. Weber, B. Streipert, R. Wagner, C. Schultz, M. Winter and S. Nowak, RSC Adv., 2016, 6, 8–17 RSC
- J. C. Garcia, J. Bareño, J. Yan, G. Chen, A. Hauser, J. R. Croy and H. Iddir, J. Phys. Chem. C, 2017, 121, 8290–8299 CAS
- D. P. Abraham, T. Spila, M. M. Furczon and E. Sammann, Electrochem. Solid-State Lett., 2008, 11, A226–A228 CrossRef CAS
- D. Mohanty, K. Dahlberg, D. M. King, L. A. David, A. S. Sefat, D. L. Wood, C. Daniel, S. Dhar, V. Mahajan, M. Lee and F. Albano, Sci. Rep., 2016, 6, 26532 CrossRef CAS PubMed
- J. Vetter, P. Novák, M. R. Wagner, C. Veit, K. C. Möller, J. O. Besenhard, M. Winter, M. Wohlfahrt-Mehrens, C. Vogler and A. Hammouche, J. Power Sources, 2005, 147, 269–281 CrossRef CAS
- D. P. Abraham, R. D. Twesten, M. Balasubramanian, I. Petrov, J. McBreen and K. Amine, Electrochem. Commun., 2002, 4, 620–625 CrossRef CAS
- B. Cho, J. Bareño, Y. L. Foo, S. Hong, T. Spila, I. Petrov and J. E. Greene, J. Appl. Phys., 2008, 103, 123530 CrossRef
- M. A. Jones and W. R. Gilkerson, J. Solution Chem., 1979, 8, 871–880 CrossRef CAS
- C. A. Tolman, Chem. Rev., 1977, 77, 313–348 CrossRef CAS
- J. A. Davies and F. R. Hartley, Chem. Rev., 1981, 81, 79–90 CrossRef CAS
- A. J. Smith, J. C. Burns, D. Xiong and J. R. Dahn, J. Electrochem. Soc., 2011, 158, A1136–A1142 CrossRef CAS
- F. H. Westheimer, S. Huang and F. Covitz, J. Am. Chem. Soc., 1988, 110, 181–185 CrossRef CAS
- http://www.inchem.org/documents/sids/sids/122521.pdf .
- L. Terborg, S. Weber, F. Blaske, S. Passerini, M. Winter, U. Karst and S. Nowak, J. Power Sources, 2013, 242, 832–837 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ta08289d |
| This journal is © The Royal Society of Chemistry 2018 |