
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSurface modified titanium dioxide using transition metals: nickel as a winning transition metal for solar light photocatalysis†
Andraž
Šuligoj
 ab,
Iztok
Arčon
cd,
Matjaž
Mazaj
a,
Goran
Dražić
a,
Denis
Arčon
de,
Pegie
Cool
f,
Urška Lavrenčič
Štangar
bc and
Nataša Novak
Tušar
*ac
ab,
Iztok
Arčon
cd,
Matjaž
Mazaj
a,
Goran
Dražić
a,
Denis
Arčon
de,
Pegie
Cool
f,
Urška Lavrenčič
Štangar
bc and
Nataša Novak
Tušar
*ac
aNational Institute of Chemistry, Hajdrihova 19, SI-1001 Ljubljana, Slovenia. E-mail: natasa.novak.tusar@ki.si
bFaculty of Chemistry and Chemical Technology, University of Ljubljana, Večna Pot 113, 1000 Ljubljana, Slovenia
cUniversity of Nova Gorica, Vipavska 13, SI-1001 Nova Gorica, Slovenia
dJozef Stefan Institute, Jamova 39, SI-1000 Ljubljana, Slovenia
eFaculty of Mathematics and Physics, University pf Ljubljana, Jadranska 19, 1000 Ljubljana, Slovenia
fDepartment of Chemistry, University of Antwerp, Campus Drie Eiken, Universiteitsplein 1, 2610 Wilrijk, Belgium
First published on 9th April 2018
Abstract
Titanium dioxide has been widely used as an antimicrobial agent, UV-filter and catalyst for pollution abatement. Herein, surface modifications with selected transition metals (Me) over colloidal TiO2 nanoparticles and immobilization with a colloidal SiO2 binder as composite films (MeTiO2/SiO2) on a glass carrier were used to enhance solar-light photoactivity. Colloidal TiO2 nanoparticles were modified by loading selected transition metals (Me = Mn, Fe, Co, Ni, Cu, and Zn) in the form of chlorides on their surface. They were present primarily as oxo-nanoclusters and a portion as metal oxides. The structural characteristics of bare TiO2 were preserved up to an optimal metal loading of 0.5 wt%. We have shown in situ that metal-oxo-nanoclusters with a redox potential close to that of O2/O2˙− were able to function as co-catalysts on the TiO2 surface which was excited by solar-light irradiation. The materials were tested for photocatalytic activity by two opposite methods; one detecting O2˙− (reduction, Rz ink test) while the other detecting ˙OH (oxidation, terephthalic acid test). It was shown that the enhancement of the solar-light activity of TiO2 by the deposition of transition metal oxo-nanoclusters on the surface depends strongly on the combination of the reduction potential of such species and appropriate band positions of their oxides. The latter prevented excessive self-recombination of the photogenerated charge carriers by the nanoclusters in Ni and Zn modification, which was probably the case in other metal modifications. Overall, only Ni modification had a positive effect on solar photoactivity in both oxidation and reduction reactions.
1. Introduction
Titanium dioxide (TiO2) is widely used as a photocatalyst for both water and air depollution as well as in self-cleaning surfaces and sensor technology. However, due to its band gap of 3.2 eV it is only able to absorb UV light, i.e. λ ≤ 390 nm, thus limiting its activity under solar light. To overcome this problem, several strategies can be used: (i) doping of the TiO2 bulk to extend the absorption edge or (ii) TiO2 surface modification, to employ surface–interface charge transfer (IFCT). The latter, with the use of transition metals coated on the surface of TiO2, was proposed1 to enhance electron–hole separation, while, in addition, the metal ion clusters located on the surface of TiO2 also serve as co-catalysts being able to be reduced by the electrons from the conduction band of TiO2 and later on to be involved in the production of superoxide anions (O2˙−). The surface of titania commonly exhibits coverage with titanol (Ti–OH) groups which with their electron donor capability offer a good surface for modification with positively charged transition metal ions. Different transition metals have already been applied as surface nanoclusters such as Cu(II),1 Fe(III),2 and Zn(II).3Cu(II)-1,3,4 and Fe(III)-surface2 modified TiO2 have been well described in air cleaning photoreactor set-ups. Increased photocatalytic activity under solar irradiation was ascribed to IFCT from EVB (TiO2) to metal nanoclusters on the surface with the subsequent formation of O2˙− radicals viaeqn (1). These then help the photocatalytic process in addition to hydroxyl radicals (˙OH) and/or holes (h+) themselves.
| O2 + e− → O2˙− | (1) |
| 2O2˙− + 2H2O →→ ˙OH + OH− + O2 | (2) |
Yet, in a liquid system dye degrading Cu2+ surface sites showed a general trend of decreasing the photoactivity of TiO2 due to the thermodynamically favoured transition of e− and h+ to the band edges of CuO which are bracketed by those of TiO2 and hence higher recombination is promoted.5 A positive effect of small additions of copper (0.1 wt%) for phenol degradation was also found6 and was explained by the transfer of e− from CuO to TiO2via Ti–O–Cu bonds, with the addition of particles being smaller and having a higher number of surface OH groups. Thus, it remains unclear why surface modification of TiO2 with some transition metals shows improvement while with others, this is not the case.
Additionally, photoreactivities are commonly measured in air photoreactors towards air-borne alcohols, in oxygen rich atmospheres, which facilitates the above-described mechanism (eqn (1)).3 However, one should note that active oxygen species are significantly less reactive compared to hydroxyl radicals, i.e. their reduction potential at pH 7 is 0.94 V while for ˙OH this value is more than doubled, 2.34 V.7 The contribution of ˙O2− to the activity of catalysts should hence be far less than that of ˙OH. However, ˙OH can be generated from O2˙−, where the additional restraint is the presence of water (eqn (2)). However, more experiments are needed to elucidate the role of O2 presence in the reaction mixture, be it in air or in aqueous environments.
Also, more research should be done in the field of immobilised catalysts in the form of films on carriers such as metal, glass, paper, cellulose fibres, etc. if we are to increase the number of commercial applications of such materials on a large scale.8 Titania catalysts in the form of films with a high surface area have not been used in these kinds of studies, supposedly having too small nanoparticles for enough metal clusters to be formed on them, which represents another gap in the experimental data reported.
Titania in combination with porous silica (TiO2/SiO2) with a large surface area was studied in our previous work.9 Here, we report the fundamental reasons for the improvement of solar light activity to be the suitable band positions of metals (Me) which are deposited on the surface of colloidal TiO2 and immobilised with colloidal SiO2 as films on a glass carrier (MeTiO2/SiO2). Colloidal titania was decorated with selected transition metals spanning from manganese to zinc due to their ability to accept/release electrons and the different positions of their valence (EVB) and conduction bands (ECB). This enabled discussion on the electronic nature of surface nanocluster mechanisms which elucidated the reasons for increased solar-light activity while immobilisation on the soda-lime glass by using colloidal SiO2 enabled comparison of optical properties of the same samples which were then used in reactions. Two different reaction pathways were used to provide a general assessment of the solar-light activity of the catalysts. First, the O2˙− radicals, and consequently the reduction activity, of the modified TiO2 surface were assessed in the test with Resazurin ink, while the direct formation of ˙OH (oxidation) was monitored via oxidation of terephthalic acid.
2. Experimental
Unless stated otherwise all chemicals were used as purchased: tetraethyl orthosilicate (TEOS, 98%) from Acros Organics; acidic TiO2 colloidal suspension CCA 100 AS from Cinkarna company (Slovenia); P-25 TiO2 powder from Evonik Degussa; terephthalic acid (TPA) from Alfa Aesar; hydrochloric acid (37%) and 2-propanol from Carlo Erba; hydroxyethyl cellulose (HEC) from Fluka; ethanol (96%) from Itrij; Levasil 200/30% from Obermeier; 1-propanol from Sigma-Aldrich; and NaOH from VWR. Glycerol (99.5%, Sigma-Aldrich) and hydroxyethyl cellulose (Fluka) were used for the preparation of the Rz ink. Resazurin (dye content ∼92%) was supplied by Sigma-Aldrich. FeCl3·6H2O, NiCl2·6H2O and ZnCl2 were purchased from Sigma-Aldrich, MnCl2·4H2O from Riedel-de Haen, and CuCl2 and CoCl2·6H2O from Merck.2.1 Photocatalyst preparation
The modification of the surface of TiO2 was done according to a modified process, described previously.1 Namely, to the acidic CCA 100 AS colloidal TiO2 suspension (pH = 1.1, 20 mL) an appropriate amount of metal chloride (hydrate) was added in order to obtain a metal loading (m/m) of 0.05, 0.075, 0.1, 0.5, 2, 5, 7.5 and 10% (MeTiO2). Metal chlorides were chosen instead of nitrates, because removal of the latter requires high temperatures (ca. 300–500 °C), while chlorides can be removed by washing at room temperature. High-temperature post-treatment may cause the favourable formation of metal oxides plus the additional growth of anatase particles which reduces the specific surface area.10 The suspension was then heated to 90 °C and stirred at this temperature for 1 h in a round flask in order to coat the surface of TiO2 with metal-oxo-nanoclusters. Importantly, the pH did not rise above 2 during the synthesis, because metal ions are prone to form oxides at higher pH values, which do not enhance the photocatalytic activity remarkably, due to their rigid structure and hence lesser availability to receive and donate electrons. Washing to remove chlorides was done by centrifuging the suspension at 9000 rpm for 2 min. The supernatant was then removed along with Cl− ions. The AgNO3 test was performed and if proven positive, dH2O (2 mL) was added followed by centrifugation and decantation. The process was repeated until the AgNO3 test was negative. Later, EDX analysis was performed on pressed pellets in order to confirm the absence of Cl− on the surface of TiO2. All samples were chlorine-free.Prior to conducting photocatalytic tests, the catalysts (MeTiO2) were immobilised in the form of films on objective glass slides using a colloidal silica binder (MeTiO2/SiO2 composite films). The idea of the so-called glass frit was introduced already in the eighties11 where the crucial condition for good adhesion is the chemical compatibility of surfaces to be bonded, i.e. glass and a SiO2 mesh. Hence, at room temperature, TEOS was added to HCl as a catalyst, diluted with isopropanol (iPrOH) and then left to hydrolyse for 30 min in a molar ratio of TEOS![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :HCl:iPrOH = 0.075:0.005:1. However, in order to preserve the high permeability of the films, colloidal SiO2 (Levasil 200/30%, 25 nm, 0.15 mol) particles were added to the TEOS silica mesh12,13 and left to mix vigorously overnight. This SiO2 mixture was added to the colloidal MeTiO2 suspension (1.5 M) in a Ti:Si molar ratio of 1:1 and mixed vigorously for 5 min. As a reference TiO2 catalyst, the P-25 suspension (1.5 M) was made by suspending 2.4 g P-25 in 10 mL H2O. The composite, made of SiO2 and MeTiO2, was then sonicated in a cold ultrasonic bath for 10 min.
:HCl:iPrOH = 0.075:0.005:1. However, in order to preserve the high permeability of the films, colloidal SiO2 (Levasil 200/30%, 25 nm, 0.15 mol) particles were added to the TEOS silica mesh12,13 and left to mix vigorously overnight. This SiO2 mixture was added to the colloidal MeTiO2 suspension (1.5 M) in a Ti:Si molar ratio of 1:1 and mixed vigorously for 5 min. As a reference TiO2 catalyst, the P-25 suspension (1.5 M) was made by suspending 2.4 g P-25 in 10 mL H2O. The composite, made of SiO2 and MeTiO2, was then sonicated in a cold ultrasonic bath for 10 min.
The glass slides were cleaned with ethanol and deionized water. They were then coated with this catalyst suspension using dip coating at a withdrawal speed of 2 cm min−1. Thermal stabilisation of the films was done at 150 °C for 1 h (ED 23, Binder). The process was then repeated 8–12 times to achieve a surface density of 1 mg cm−2. Thermal treatments at higher temperatures (e.g. 500 °C) were not conducted as this may cause a decrease in the surface area due to sintering, to avoid the detrimental diffusion of sodium ions from the glass support at higher temperatures14 and to minimise the environmental impact.
2.2 Characterization
All physicochemical characterizations were conducted on metal modified TiO2 samples, which were immobilised on a glass carrier using a silica binder (SiO2 50 mol%), which generally preserved structural properties. It should be noted here that silica addition caused the “dilution” of the active TiO2 component, which after this constituted 50 mol%. However, two measurements were conducted on modified TiO2 only, i.e. without the addition of SiO2: (i) isoelectric point measurements to better pinpoint the differences between the samples and (ii) X-ray absorption spectroscopy measurements (XAS) due to the sensitivity of the method with which 0.5 wt% of the investigated metal vs. TiO2 in the sample is just enough to observe the absorption edge jump and to determine the oxidation state of the selected metal (XANES – X-ray Absorption Near Edge Structure).X-Ray diffraction (XRD) patterns were recorded on a PANalytical X'Pert PRO high-resolution diffractometer using CuKα1 radiation (1.5406 Å) in the 2θ range from 5 to 60° (100 s per step of 0.034°) using a fully opened X'Celerator detector.
The elemental composition was determined by the energy-dispersive X-ray (EDX) analysis with an INCA Energy system attached to a Zeiss Supra 3 VP field emission gun (FEG) microscope, operating at 20 kV.
Isoelectric points were determined using a Zetasizer nano ZS instrument (Malvern) with electrophoretic light scattering technology in the pH range of 2–12 adjusted by the addition of either 0.1 M NaOH or 0.1 M HCl and measured using a digital pH meter (Mettler Toledo).
Nitrogen adsorption measurements were done at 77 K on a Tristar 3000 Micromeritics volumetric adsorption analyser. Before the adsorption analysis, the samples were outgassed under vacuum for 2 h at 473 K in the port of the adsorption analyser. The BET specific surface area15 was calculated from the adsorption data in the relative pressure range from 0.05 to 0.25. The total pore volume was estimated on the basis of the amount adsorbed at a relative pressure of 0.97.16 The pore size distributions (PSDs) were calculated from the nitrogen adsorption data using a BJH algorithm based on the ideas of Barrett et al.17 The maxima on the PSD are considered as the primary mesopore diameters of the given samples.
Diffuse reflectance and transmittance UV-VIS spectra were measured using a UV-VIS spectrophotometer equipped with an integrating sphere (LAMBDA 650 UV/Vis with a 150 mm integrating sphere, Perkin Elmer, USA). Indirect band-gap energies were determined by plotting the Kubelka–Munk transformation of the original diffuse reflectance spectra vs. photon energy.18
Transmission electron microscopy (TEM) was performed on a 200 kV field emission gun (FEG) microscope (JEOL JEM 2100). For ARM 200 CF studies a drop of an ethanol diluted nanoparticle solution was placed on a copper grid and dried at room temperature. The specimens were additionally coated with carbon in order to prevent excessive charging and decomposition of the sample under the electron beam (80 kV).
A conventional continuous wave (cw) electron paramagnetic resonance spectrometer operating at a Larmor frequency ESRνL = 9.6 GHz was employed to detect the electron spin resonance. The spectrometer is equipped with a standard Varian E-101 microwave bridge, a Varian rectangular TE102 resonance cavity, and an Oxford Cryogenics continuous-flow helium cryostat. Prior to analysis the samples were annealed at 120 °C overnight.
2.3 Photocatalytic activity
The direct formation of ˙OH (TPA test) was carried out by a method of oxidation of terephthalic acid (TPA) to hydroxyterephthalic acid (HTPA) (Fig. 1) as described elsewhere.19 The tests were done in a UV/VIS photo chamber reactor equipped with 2 LUMILUX L 15W/840 CoolWhite 590 × 38 mm (Osram) solar light lamps. The samples were first preirradiated for 2 h under UVA light. Briefly, a solution of sodium terephthalate was prepared by mixing together TPA (500 mg) and an aqueous solution of NaOH (31 mL, 0.201 M), followed by dilution with ethanol to obtain 50 mL of the TPA solution. Separately, an aqueous solution of HEC (2 wt%, 10 mL) was mixed with ethanol (96%, 30 mL). The final solution was obtained by mixing TPA solution (10 mL) with HEC solution (20 mL). The films of TPA were deposited onto photocatalytic films at 22 °C using a dip-coater with a pulling speed of 10 cm min−1. The films were dried using a hair dryer immediately after the deposition of the film. The kinetics of ˙OH formation was calculated using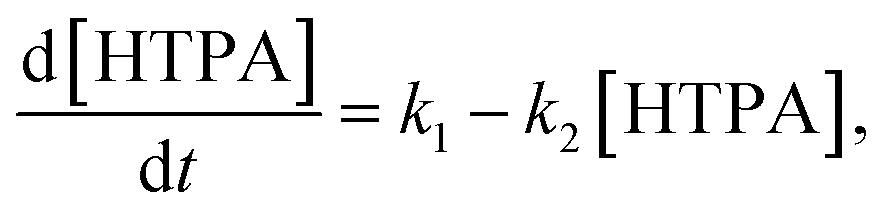 where by solving the differential equation one obtains [HTPA] = k1/k2(1 − e−k2t). Measurements of fluorescence (excitation @320 nm and emission @430 nm) using a microplate reader (Infinite F200, Tecan, Switzerland) were used for quantification.
where by solving the differential equation one obtains [HTPA] = k1/k2(1 − e−k2t). Measurements of fluorescence (excitation @320 nm and emission @430 nm) using a microplate reader (Infinite F200, Tecan, Switzerland) were used for quantification.
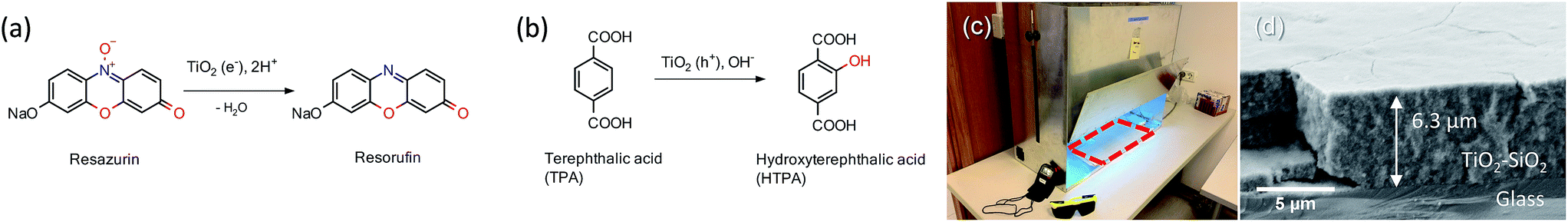 | ||
| Fig. 1 Mechanism of photocatalytically sensitized (a) reduction of Rz ink (blue) to Resorufin (pink) and (b) oxidation of TPA to HTPA (fluorescent). (c) UV/VIS photo chamber reactor which allows UV or visible light illumination – with the red line area under investigation being marked and (d) cross-cut SEM micrograph of the colloidal TiO2 film on glass onto which test molecules from (a) and (b) were loaded. | ||
The second photocatalytic assessment was performed by following the reduction of Rz ink to Resorufin (Fig. 1a).20 The dye is reduced by photogenerated electrons, while holes oxidise the sacrificial electron donor, i.e., glycerol and thus do not interfere with the reaction; hence this test directly follows reduction by e− since oxygen is not directly present in the reaction. In heterogeneous photocatalysis the rate (proportional to kapp) of the process is dependent on the radiant flux (Iθ). θ is a constant with a value of usually 0.5 under high-intensity conditions where recombination dominates or 1.0 under low-intensity conditions where surface reactions dominate.21 An excess of glycerol ensures that electron–hole recombination is not the dominant process, thus ensuring θ → 1. Briefly, to HEC (1.5 wt%, 20 g) glycerol (2 g) was added. Resazurin (20 mg) and Tween 20 (20 mg) were then added and mixed overnight to completely dissolve the dye. The ink (50 μL) was applied on the films, preirradiated for 2 h under UVA light by a doctor blade method. 3 M tape (Scotch®) was taped on the two sides of the coated glass and then the ink was distributed on the films with a glass rod to form a uniform thickness across the whole surface of the layer. The samples were then kept at 70 °C for 10 min to produce an ink film. The films were irradiated with the same lamps in the same chamber configuration under air as in the TPA test. Bleaching of the dye was carried out by DRS in absorption mode, and the spectra were recorded in 10 s irradiation intervals (LAMBDA 650, Perkin Elmer). Three samples were tested independently in order to perform statistical analysis.
3. Results and discussion
3.1 Photocatalytic activity towards oxidation
The photocatalytic formation of ˙OH measured via HTPA formation for TiO2 with a modified surface is shown in Fig. 2. It was shown before22 that reactions that are not specific, e.g. dye degradation, can show even higher improvement of degradation rates for similar modification of the TiO2 surface. The co-catalyst approach is focused on the generation of oxygen radicals on the active sites located on the surface of metal clusters (eqn (1)), while to generate ˙OH radicals further steps are needed (eqn (2)). However, since the lamps in the tests were simulating solar light, they also had a portion of UVA light intensity (Fig. S1†). This part of the spectrum is responsible for the generation of holes with high oxidation power, which oxidise adsorbed hydroxyls or water to form ˙OH which is then detected as fluorescent HTPA. In this way, nanoclusters impede the electron–hole recombination. By using a UV-cutoff filter (Fig. S6†) it was shown that this (h+ oxidation via UV-illumination) is the major pathway for TPA oxidation, which is in agreement with other authors.19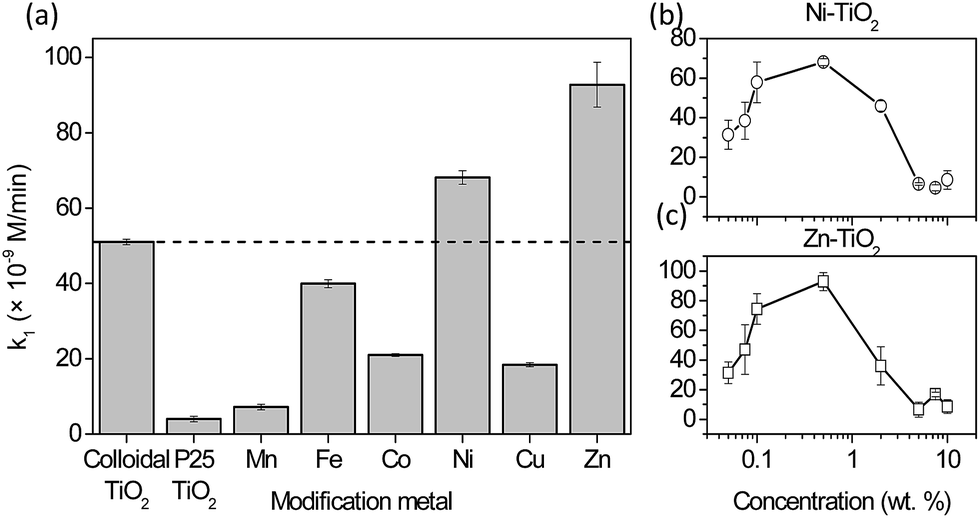 | ||
| Fig. 2 Photocatalytic results of the oxidation of terephthalic acid under visible light irradiation. Activity of the 0.5% modified TiO2 samples with selected transition metals (a) and the dependence of activity on the concentration of metal loading for Ni (b) and Zn (c). For comparison, the activity of P-25 TiO2 immobilized with the same binder is presented. The dashed line represents the activity of unmodified colloidal TiO2. Error bars present the standard error of the mean (N = 3). | ||
Fig. 2a shows that the solar light activity of colloidal TiO2 (marked with a dashed line) was by itself far better than the activity of the P-25 reference material. Interestingly, 0.5% modifications of the titania surface with Mn, Fe, Co and Cu decreased the activity by 86, 21.7, 58.9, and 63.9%, respectively. On the other hand, modifications with Ni and Zn led to the increase of the photocatalytic rate by 33.6 and 82%, respectively.
3.2 Photocatalytic activity towards reduction
In Table 1 the results of the reduction of Rz ink are presented. For this reaction, an optimum metal loading of 0.5 wt% was chosen. The time taken to bleach (ttb) may be estimated by extrapolating the two linear regions (initial degradation and steady-state region) in the absorption at 537 nm vs. time plot and identifying their point of intersection. ttb approximates to ca. 2 × t1/2, which is the time to bleach the dye.23 It must be emphasised that the absorption peak at 608 nm was also monitored which showed similar kinetics. This means that not only is the Rz ink reduced to Resorufin, but the latter is then degraded to further bleaching products, most probably colourless hydroresorufin. The samples which reduced Rz ink the fastest were Mn and Ni modified titania films. On the other hand, Co, Cu and Zn were found to have a negative effect on the solar light activity. In the case of modification with manganese, the results are better but not significantly different than those of bare TiO2, while Ni-modification clearly shows an improvement in times needed for bleaching of Rz ink. Again, P25 TiO2 reduced Rz ink much slower than colloidal TiO2, which is in accordance with TPA oxidation results and our previous reports towards air-borne toluene oxidation.9 These results give an indication that TiO2 modifications with Ni and Mn are also most active towards the degradation of Methylene Blue in the solution; a linear correlation between the initial degradation rates of these two model compounds has been proven by Mills et al.24 This could extend the usefulness of such materials for water treatment as well.| Sample (x–TiO2) | ttb (s) |
|---|---|
| Colloidal TiO2 | 39 ± 2 |
| P25 TiO2 | 141 ± 3 |
| Mn | 34 ± 5 |
| Fe | 46 ± 4.5 |
| Co | 61 ± 3.5 |
| Ni | 30 ± 4 |
| Cu | 53 ± 0.3 |
| Zn | 53 ± 1 |
Overall, Ni was the only modification which had a positive effect on solar photoactivity in both oxidation and reduction reactions.
3.3 Characterization
To rule out that metal ions did not enter the lattice of titanium dioxide, Rietveld refinement of the X-ray diffraction patterns was used to obtain the parameters of the anatase unit cell (Table 2). The results (Fig. 3) showed no major differences among the samples, regardless of the type or weight fraction of the transition metal loaded on the surface of TiO2. Interestingly, metal oxides were not detected even at higher grafting concentrations; probably due to their sizes being too small. All samples showed peaks at 25.3°, 37.9°, 48° and 54° 2θ, corresponding to the anatase structure (ASTM card #00-021-1272). Surface metal-oxo-nanoclusters did not cause any shift of the diffraction peaks; hence, the synthesis temperature (90 °C) did not cause the diffusion of metal ions into the structure of titania. Also, the lattice parameters are very similar among the samples (Table 2) as well as the crystallite sizes. Instead, the metal species are grafted as nanoclusters on the surface of TiO2 nanoparticles.| Sample (x–TiO2) | a = b (Å) | c (Å) | Cell volume (Å3) | d (nm) |
|---|---|---|---|---|
| Colloidal TiO2 | 3.803(5) | 9.51(3) | 137.5(9) | 8(1) |
| Mn | 3.811(2) | 9.550(5) | 138.7(1) | 13(1) |
| Fe | 3.798(10) | 9.51(3) | 137.2(8) | 10.0(9) |
| Ni | 3.800(9) | 9.52(2) | 137.5(8) | 8(1) |
| Cu | 3.806(8) | 9.51(2) | 137.5(8) | 8(1) |
| Co | 3.802(12) | 9.52(3) | 137.7(10) | 9(1) |
| Zn | 3.801(1) | 9.519(4) | 137.5(1) | 9(3) |
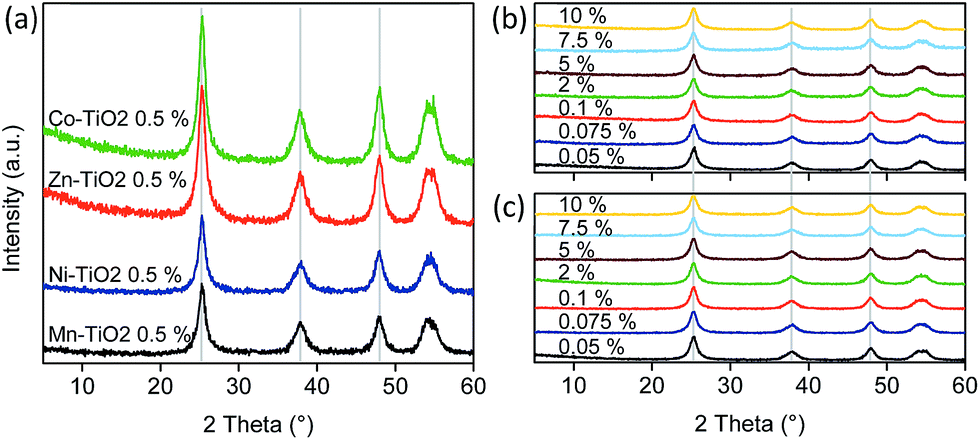 | ||
| Fig. 3 XRD patterns of (A) TiO2 nanoparticles modified with metal-oxo-nanoclusters at 0.5 wt% loading, and patterns of TiO2 particles modified with (B) Ni and (C) Zn nanoclusters. | ||
Modifying the surface of small TiO2 nanoparticles should change the surface area as well as the pore structure and volume due to the occlusion of the micropores. This is clearly seen at higher loadings (higher than 2 wt%) in Fig. 4b and c. The type IV isotherm, which is observed for the parent TiO2 material and typical for mesoporous adsorbents, gradually changes to type II isotherms, which indicates nonporous or macroporous materials.25 Also, at higher loadings, the H2 hysteresis loop is visible which indicates the complex pore structure where network effects are important. Moreover, the gradual decrease in the desorption curve indicates a broad size distribution of the pore necks. From these data, we can assume that grafting nanoclusters onto the surface of TiO2 at high concentrations changed the morphology and pore structure of the samples, where the trend is towards a more plate-like particle morphology. Also, in general, the complex pore structure in low loadings is being partially filled, resulting in simpler pore connectivity.
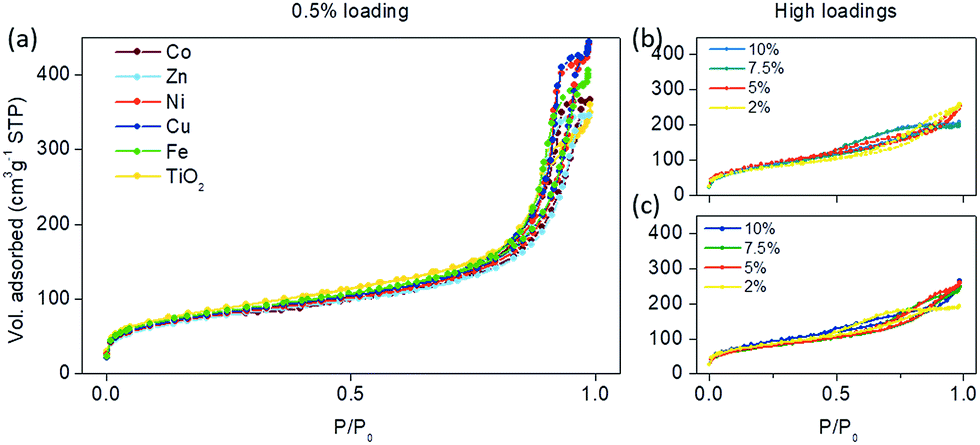 | ||
| Fig. 4 N2-physisorption isotherms of the samples modified with (a) 0.5 wt% of selected transition metals, and higher loadings (2–10%) of (b) Ni and (c) Zn nanoclusters on the surface of TiO2. | ||
However, lower loadings (0.05–0.5 wt%) of metal-oxo-nanoclusters on the surface (Fig. 4a) maintained the H1 adsorption hysteresis, typical for pore channels without obstructions.25 Hence, the pores were uniform and easily accessible either by the adsorption or desorption part of the isotherm; although a decrease in pore size is seen consistently throughout all grafting metals (Table 3). Also, the relatively narrow hysteresis implies that the necks of the pores were not clogged, confirming the accessibility of the pores. This coincides well with the catalytic activities, which showed the largest activities at these concentrations (0.5 wt%) as seen in Fig. 2. The specific surface areas did not differ significantly among different modifications (Table 3), on the basis of which we can conclude that pore distribution and surface area are not the factors influencing the activity.
| Sample (x–TiO2) | S BET (m2 g−1) | V tot (cm3 g−1) | Pore sizea (nm) | Band gapb (eV) |
|---|---|---|---|---|
| a S BET, Vtot and pore sizes determined by N2-sorption isotherms. b Band gaps determined from UV–vis diffuse reflectance spectra. | ||||
| Colloidal TiO2 | 299 | 0.502 | 8.7 | 3.31 |
| Mn | 275 | 0.584 | 3.8 | 3.35 |
| Fe | 283 | 0.628 | 2.9 | 3.34 |
| Co | 267 | 0.567 | 3.7 | 3.33 |
| Ni | 269 | 0.654 | 3.6 | 3.29 |
| Cu | 289 | 0.466 | 3.6 | 3.34 |
| Zn | 264 | 0.535 | 3.1 | 3.29 |
The ATR FT-IR analysis showed that the bare TiO2 surface is more hydroxylated than those which have been modified for all samples (Fig. S3†) as observed by the intensity of the O–H νs vibration at 3350 cm−1. This can be ascribed to the higher number of surface –OH in bare TiO2, while a partial occupancy of these sites by metal-oxo-nanoclusters can be expected in surface modified samples.
Because photocatalysis involves surface reactions, the surface charge of the catalyst was assessed by measuring the zeta (ζ) potential (Fig. S2†). The modification of the surface of titania shifted the isoelectric point to higher values in all samples (Table 4) due to the presence of positively charged ions on the surface of titania. Moreover, all oxides of transition metals used in this study have higher IEPs,26 which contribute to the observations in ζ-potential measurements. This phenomenon was most significant in modifications with copper and zinc, which also possess the highest IEP, i.e. 9.5 and 8.8 for CuO and ZnO, respectively.26 This means that the acid character is extended to higher pH values, which has a consequence of less negative potential at neutral pH. In other words, after modification, the surface became more positive as in bare TiO2. Hence, anionic dyes can be adsorbed on the surface at pH closer to neutral in comparison to bare TiO2.
| Sample (x–TiO2) | Isoelectric point | Potential at pH 7 (mV) |
|---|---|---|
| Colloidal TiO2 | 5.9 | −23 |
| Mn | 6.7 | −3.3 |
| Fe | 6.8 | −2.7 |
| Co | 6.4 | −12.5 |
| Ni | 6.2 | −16 |
| Cu | 6.8 | −4.7 |
| Zn | 6.8 | −3.9 |
This decrease was the most prominent in Ni- and Zn-modified samples, which implies a more uniform coating of the surface by small nanoclusters leading to higher shading of the titanol groups on TiO2.
Photocatalytic activity can be discussed in terms of the relative energy of valence and conduction bands (EVB and ECB, respectively). If we know the values of the band gap (Eg) and absolute electronegativity of the semiconductor (χ), which can be determined by the geometric mean of the electronegativities of constituent atoms,26EVB can be determined as follows:
| EVB = χ − 4.5 + 0.5 Eg | (3) |
where −4.5 eV is the Fermi level of the NHE with respect to the vacuum level. From the DRS measurements (Fig. S5†) the Eg of bare TiO2 was determined to be 3.31 eV and thus EVB equalled −7.46 eV. The band gap values of the modified samples differ only slightly. As a rule, if the band gap depends on the dopant content in the TiO2 matrix, it may be attributed to the dopant introduction in substitutional positions. Here, this is not the case (Table S2, Fig. S5†) and a gradual increment in Ni-concentration shifted the absorption edge to higher wavelengths non-linearly, again proving that the metals were not incorporated into the titania framework.
However, since films appeared semi-transparent (i.e. matt), suggesting partial transmittance of the visible light, light scattering in the visible region is important in terms of maximising light harvesting. Reflection on non-transparent films (which is the case in this research) results from scattered and reflected radiation on macroscopic particles. Thus, the sum of transmittance (T) and reflectance (R) spectra enables the calculation of the true absorbance (A) of TiO2 films at a certain wavelength. It must be emphasised that in this study films which were tested for diffuse reflectance had identical surface properties in both experimental set-ups (DRS and photocatalytic tests). This enables us to directly link optoelectronic properties to their catalytic performances. In most publications this is not the case since light scattering in the sample occurring in DRS measurements is different from that in a photoreactor, especially if the catalyst is in powder i.e. suspension form.27
From Fig. 5 it can be seen that the differences in reflectance spectra of colloidal TiO2 films are quite subtle. This was expected since the absorption of photons directly depends on the number of titania particles, and consequently on the mass of the films on a given surface area, which was uniform among the samples. However, TiO2 modified with zinc and iron had the highest scattering in the 400–450 nm region, followed closely by Co and then Ni, while Mn- and Cu-modified films showed the least scattering in that region. A quick drop in the reflectance value appeared towards larger values because of the small size of the nanoparticles due to the fact that lower wavelengths are scattered more efficiently, i.e. by Mie theory28 scattering depends on the incident light wavelength and size of the particles. True absorption was shifted to longer wavelengths in all modifications, most notably in Ni, Co and Zn samples, although only by e.g. 7 nm for Ni-modification. However, an additional increase of Ni (0.5% → 1%) leads to increased absorption in the 400–450 nm part of the spectrum due to the 3d7 d–d transition in octahedral Ni2+ ions as suggested by EXAFS measurements (see below). Relatively high scattering in the 250–400 nm range and a red-shift in absorption make Zn and Ni the best candidates for photoactive films. This corroborates nicely photocatalytic tests; in the TPA test the highest activity was achieved by Zn- and Ni-modified samples, while in Rz ink reduction Ni modification resulted in the fastest bleaching under solar light.
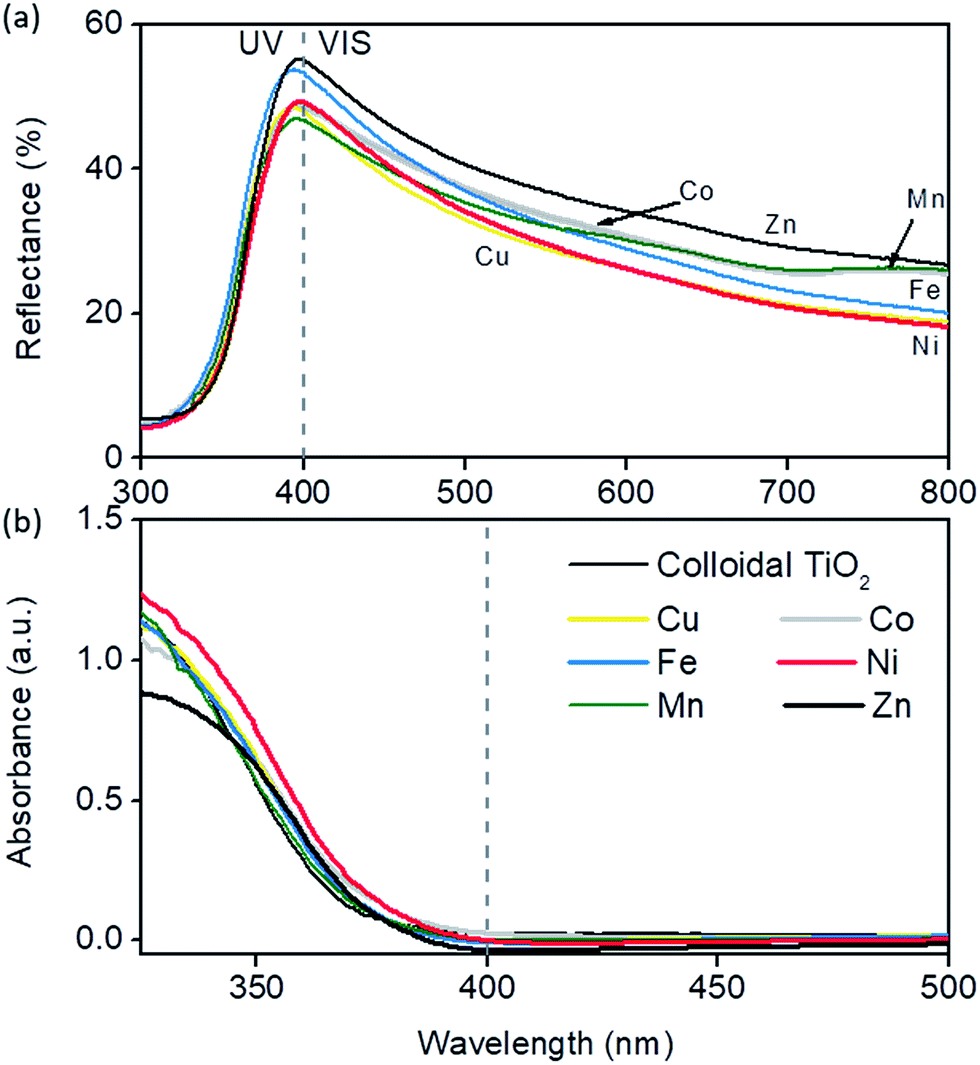 | ||
| Fig. 5 Diffuse reflectance spectra of the TiO2 films prepared from colloidal TiO2 with modification of the surface with 0.5 wt% of selected transition metals. Below, true absorbance in the films obtained by the summation of reflectance and transmittance data. | ||
Transmission electron microscopy of the Ni-modified sample revealed (Fig. 6) the distribution of nanoparticles and Ni-oxo-nanoclusters. A uniform distribution of nanoparticles is seen in Fig. 6A, with an average grain size of 6 nm. A portion of Ti was detected as Ti3+ by EELS analysis of the outside of titania particles (not shown). This is possible due to the presence of reducible Ni clusters which extract oxygen from the surface layer of TiO2 to form NiO clusters thereby reducing titanium to Ti3+. Both claims were also supported by in situ EPR measurements (see below). The distribution of nanoclusters obtained from EDXS mapping and HAADF/STEM is shown in Fig. 6D and F (marked as bright spots), respectively, confirming that Ni is uniformly distributed on the surface of TiO2 particles and that Ni is present in the form of oxo-nanoclusters.
 | ||
| Fig. 6 TEM micrographs of TiO2 modified with 1 wt% Ni. (A) Bright-field micrographs, (B–E) EDXS mapping, (F) HAADF/STEM and (G) electron diffraction SAED. | ||
For the most promising modification (Ni) X-ray absorption investigation was carried out. From the XANES study (Fig. 7a) it can be seen that the Ni-modified TiO2 sample exhibits the same Ni K-edge energy position and a similar edge profile to those of the reference Ni2+ compounds Ni hyaluronate complex29 and NiO,30 where Ni2+ is octahedrally coordinated to 6 oxygens in the first coordination shell, indicating that Ni cations in the Ni-modified TiO2 sample are also in a divalent state, octahedrally coordinated to 6 oxygen atoms in its first coordination sphere.
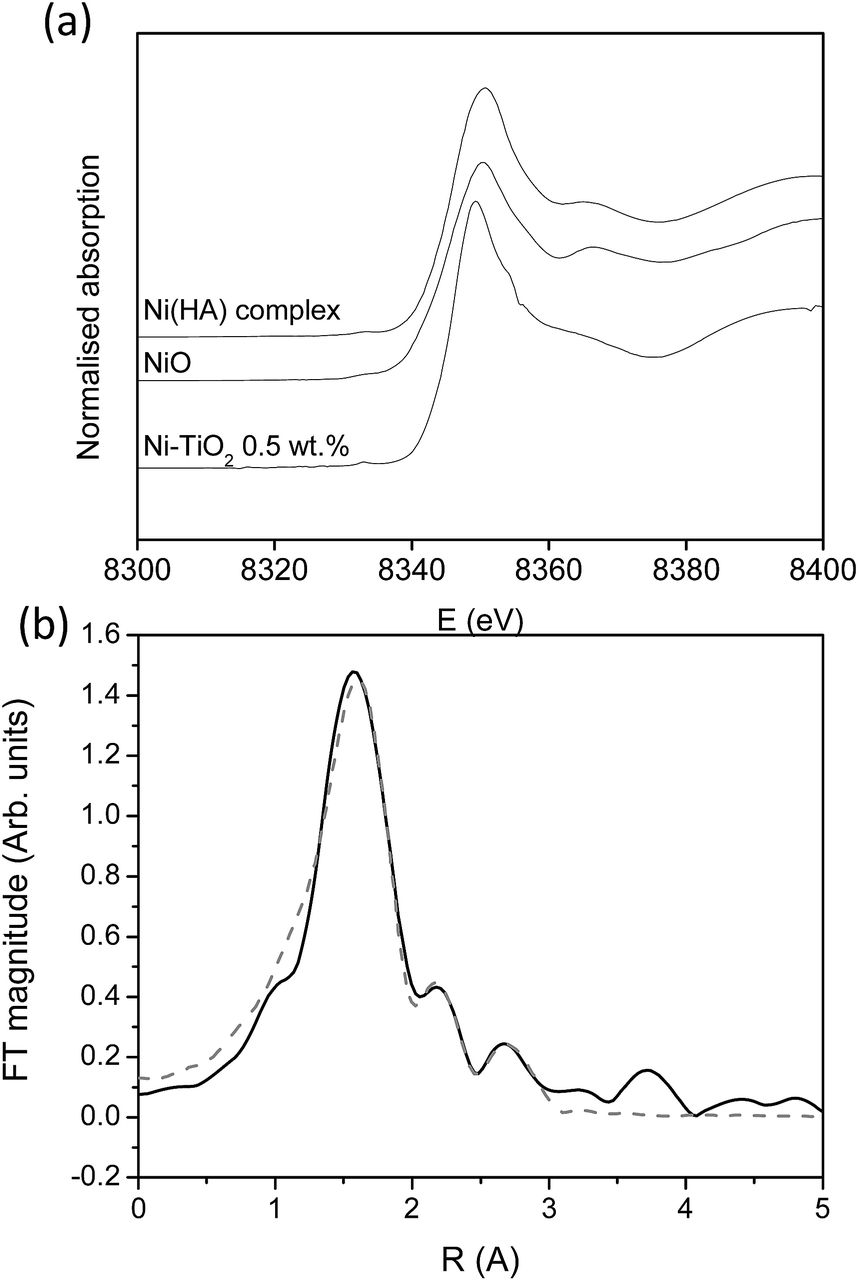 | ||
| Fig. 7 (a) Ni K-edge XANES spectra of the Ni-modified TiO2 sample and reference Ni2+ compounds with octahedral Ni coordination (NiO, Ni hyaluronate complex). (b) Fourier transform magnitude of the k2-weighted Ni EXAFS spectrum of the Ni modified TiO2 sample, calculated in the k range of 3.5–12 Å−1 and R range of 1.0–3.0 Å. Experiment – (solid line); best fit EXAFS model – (dashed line). | ||
In the Fourier transform Ni K-edge EXAFS spectrum of the Ni-modified TiO2 sample, contributions of Ni neighbour shells up to about 3 Å are resolved (Fig. 7b). The quantitative analysis of the EXAFS spectrum is performed with the IFEFFIT program package.31 Structural parameters of the local Ni neighbourhood (type and average number of neighbours, and the radii and Debye-Waller factor of neighbour shells) were quantitatively resolved from the EXAFS spectrum by comparing the measured EXAFS signal with a model signal, constructed ab initio with the FEFF6 program code32 from the set of scattering paths of the photoelectron in a tentative spatial distribution of neighbour atoms. The model comprised six oxygen atoms at the same distance in the first coordination shell and Ni and Ti atoms at two different distances in the second. The atomic species of neighbours are identified in the fit by their specific scattering factor and phase shift.
Three variable parameters for each shell of neighbours are introduced in the model: the shell coordination number (N), the distance (R) and the Debye–Waller factor (σ2). In addition, a common shift of energy origin ΔEo is also allowed to vary. The amplitude-reduction factor S20 is kept fixed at the value of 0.90. A very good agreement between the model and the experimental spectrum is found in the k range of 3.5–12.0 Å−1, and the R range from 1.0 Å to 3.5 Å (Fig. 7b). The list of best-fit parameters is given in Table 5.
| Ni neigh. | N | R [Å] | σ 2 [Å2] | R-factor |
|---|---|---|---|---|
| O | 5.9(8) | 2.04(1) | 0.008(2) | 0.0093 |
| Ti | 1.3(6) | 2.79(4) | 0.009(8) | |
| Ni | 1.2(7) | 2.97(4) | 0.009(8) |
Ni cations are found to be coordinated to six oxygen atoms in the first coordination shell at 2.04 Å, and, on average, to one Ti and one Ni neighbour at larger distances. The Ni–O–Ti and Ni–O–Ni bridges detected in EXAFS analysis suggest that Ni2+ cations are partially attached to the surface of the photocatalytically active TiO2 nanoparticles, and partially agglomerated with NiO nanoparticles.
To further elucidate the nature of such species, EPR spectra were recorded at 20 K. It showed the formation of oxygen radicals (O2−˙) and Ni2+ centres upon visible light irradiation, depicted as the difference in Fig. S9.† A broad signal at the low magnetic field was ascribed to very small Ni nanoparticles, with a geff of 2.28.33 These can be observed since Ni-particles with a size significantly less than 50 nm (indicated also with TEM) may have non-stoichiometric nature resulting in a significant portion of Ni3+ and thus exhibit paramagnetism.34 The presence of disorder in such clusters causes the reduction in size-dependent exchange interactions between two neighbouring Ni2+ ions mediated through oxygen ions and induces anisotropy of Ni3+. Upon irradiation, the presence of e− from TiO2 increased the interactions in the nickel surrounding thus increasing the Ni2+ signal, together with O2˙− (g = 2.002). Ti3+ centres weren't seen since these are already present in some proportion in the parent (non-irradiated) materials, as indicated by the EELS from TEM. From the observation of these separate techniques we named the particles on the surface metal-oxo-nanoclusters.
3.4 Mechanisms of photocatalytic activity
Due to their electronic nature transition metals are capable of changing their oxidation states.35 This is also the main benefit with regard to their contribution to solar light photoactivity, and thus they serve as co-catalysts on the surface of titania. First, if a part of UV is present in irradiation (which in solar irradiation accounts for ca. 4% (ref. 36)) TiO2 forms photogenerated charge carriers: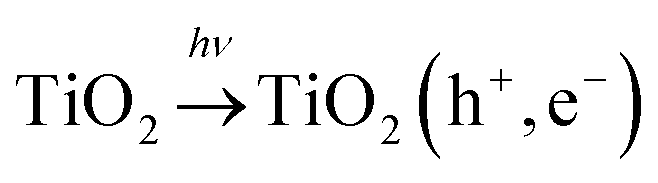 | (4) |
Later the fate of charge carriers depends on their successful migration to the surface and reactions there. Nanoclusters are reduced by the photogenerated e− which leaves more h+, which do not recombine, for oxidation reactions:
| Mn+ + e− → M(n − 1)+ | (5) |
However, the same reduced metal sites on the surface can act as h+ traps:
| M(n − 1)+ + h+ → Mn+ | (6) |
In order to avoid such cyclic phenomena, the nanoclusters have to be well separated spatially, which is achieved by their low loading (see above). An additional reason for such low loadings to be optimal is the avoidance of shading effect in which species on the surface of TiO2 would limit the penetration of light to titania nanoparticles.
With regard to the mechanism of clusters, iron, for example, gets reduced to Fe2+ and then serves as the oxygen reduction catalyst:2
| 4Fe2+ + O2 + 4H+ → 4Fe3+ + 2H2O | (7) |
| 4Fe2+ + O2 + H2O → 4Fe3+ + 4OH− | (8) |
Copper which is reduced on the other hand has similar co-catalyst capabilities:
| 2Cu+ + O2 + 2H+ → 2Cu2+ + H2O2 | (9) |
| 3Cu+ + O2 + 4H+ → 2Cu2+ + Cu3+ + 2H2O | (10) |
| Cu+ + O2(ads) → Cu2+ + O2˙− | (11) |
Zinc can promote photoactivity by creating additional ˙OH and O2˙−.37 For instance:
| Zn+ + O2(ads) → Zn2+ + O2˙− | (12) |
| Zn2+ + OH− → Zn+ + ˙OH | (13) |
In the presence of Mn-clusters on the surface of TiO2, the following reactions are possible:38
| Mn2+ + ecb− → Mn + | (14) |
| Mn+ + O2(ads) → Mn2+ + O2˙− | (15) |
| Mn3+ + OH− → Mn2+ + ˙OH | (16) |
However, it should be noted that Mn+ and Mn3+ are relatively unstable, compared to Mn2+, which can have an effect on trapping the photogenerated e− (Mn3+) and h+ (Mn+). This is probably the reason why manganese showed only minor results in the improvement of photoactivity.
We hypothesise that surface nickel species can react and help the photocatalytic process as follows:
| Ni2+ + 2e− → Ni (−0.26 V) | (17) |
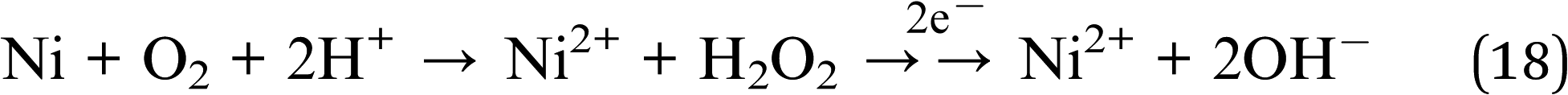
| Ni(OH)2 + 2e− → Ni + 2OH− (−0.72 V) | (19) |
Nussbaum et al. doped BiOCl with Fe and Nb and argued that the reduction potential of the doped metals should be relatively low (Nb5+/Nb4+ is 0.35 V) and the location of the 4+ level is close to the O2/˙O2− level.39 This facilitates the reduction of oxygen to superoxide, thus back-flipping the 4+ state into the 5+ (for Nb at least). The mechanism is arguably particularly important under solar light, where the main active species are superoxides. Thus, in Fig. 8, 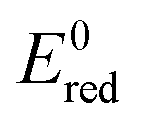 for single e− (or 2 in the case of Ni) transfer are plotted on the right side. It can be seen that Zn, Co and Ni potentials are close to the O2/O2˙− level. This could lead to the above described back-flipping mechanism. However, it must be noted that those Ni cations that are bound to the surface of TiO2 could change the valence band of TiO2 and hence improve the absorption of visible light.
for single e− (or 2 in the case of Ni) transfer are plotted on the right side. It can be seen that Zn, Co and Ni potentials are close to the O2/O2˙− level. This could lead to the above described back-flipping mechanism. However, it must be noted that those Ni cations that are bound to the surface of TiO2 could change the valence band of TiO2 and hence improve the absorption of visible light.
 | ||
Fig. 8 Schematic diagram showing the band positions of different bulk metal oxides. The values of band positions were obtained from ref. 21. Arrows indicate the thermodynamically feasible transfer of e− from the CB and h+ from the VB of TiO2 and thus paths to the recombination of charge carriers. On the right, 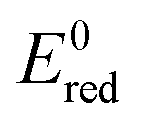 (in V vs. NHE) for the single-electron transfer of the selected transition metal ions in water are plotted. Values for (in V vs. NHE) for the single-electron transfer of the selected transition metal ions in water are plotted. Values for 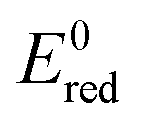 are from ref. 40. are from ref. 40. | ||
On the other hand, Li et al.5 argued that metal oxides whose band edges are bracketed by TiO2 edges lead to the transfer of photogenerated charge carriers from TiO2 which then recombine thus decreasing the overall activity of the materials. Hence, for Co VB and CB positions are bracketed by the EVB and ECB of TiO2, which favours higher recombination of photogenerated charge carriers, thus reducing the activity under solar light. Interestingly, although the XRD, UV-VIS and XANES data clearly show that, in our case, the structures do not resemble the oxide structure of nanoclusters; it seems that the bracketing of VB and CB increases the recombination in metal-oxo-nanoclusters apart from Ni and Zn. In this study, nanoclusters are present as highly dispersed species on the surface of TiO2, which are partially connected to Ti via oxygen bridges and partially forms M–O–M dimers or an amorphous MxOx structure. The very small size of clusters facilitates electron transfer from the anatase surface, which in turn decreases the recombination if the above-mentioned constraints are fulfilled.
The second factor that limits the activity improvement is the potential of one electron reduction of ˙OH to H2O set at 2.31 V (vs. NHE). Holes created at levels lower than that do not have redox potential to contribute to activity significantly. Hence, if a hole is transferred to a metal oxide which has a low E0red, the latter probably serves to increase the e−–h+ separation at best or to increase the recombination of the charge carriers, which is more probable at higher loadings.
The valence band of ZnO (−7.39 eV) is just at the level of TiO2 (−7.46 eV) which favours the transfer of h+ to Zn clusters. Hence, zinc is mostly active at oxidation reactions, which was clearly shown in the TPA test that follows the oxidation product of the model compound. Interestingly, nickel also showed improvement in oxidation photoactivity, although its EVB (7.5 eV) is slightly lower than EVB (TiO2). On the other hand in the reduction reaction Zn acted as a charge trap site for photogenerated e− from TiO2, which resulted in slower reaction kinetics. The same phenomena can explain lower solar activity toward reduction concerning Fe-, Cu- and Co-modified TiO2 films, again, due to the bracketing of the CB and VB of the transition metal oxides by the bands of TiO2, which results in metal oxides that act as recombination centres.
The experiments conducted by using a UV-cutoff filter showed that UV-excitation is still the dominant force driving the chosen reactions; this makes sense since the TPA oxidation proceeds via direct oxidation with holes.19 However, without the suitable band positions of the metal-oxo-nanoclusters, such charge carriers would only result in recombination at clusters. The latter was evident in the case of Mn-modification; its suitable CB-band position allowed for improved reduction of Rz ink, while the VB of MnO (−7.09 eV) is significantly lower than that of TiO2, which makes such cluster traps for h+, hence limiting the oxidation of TPA to HTPA via holes. On the other hand, the EPR experiment showed that Ni-oxo-nanoclusters are centers for generating O2˙− radicals, while h+ from TiO2 are free to oxidize the reactants.
Hence, if such materials are to be used in aqueous media, the degradation of organic molecules would then proceed via direct attack by h+ and via ˙OH; the former was experimentally proven by the TPA test. The superoxide radicals generated at the metal-oxo-nanoclusters would also add to the final degradation activity. The limiting factors are the aforementioned VB positions of the respective metal oxides and the spatial distribution of metal-oxo-nanoclusters on the TiO2 surface.
4. Conclusions
We demonstrated that the enhancement of the solar-light activity of TiO2 by the deposition of metal-oxo-nanoclusters on the surface depends strongly on the reduction potential of such species. The small size of metal-oxo-nanoclusters, synthesised in this study, allows dynamic acceptance/release of e− from TiO2VB; this is governed mainly via the position of the reduction potential of such species as close as possible to the redox potential of O2/O2˙− (−0.33 V vs. NHE). If this condition is fulfilled, the contribution of such species is still limited by the position of the valence band of their corresponding oxides, since these are also present on the surface of TiO2. We proved this by monitoring the formation of ˙OH, where the oxides of deposited metals whose bands were bracketed by TiO2 bands resulted in a decreased solar-light activity, probably due to the higher recombination of e−–h+, for which such clusters offered additional trapping sites. Additionally, the metal grafting shifted the absorption region in films toward lower energies and increased scattering in the films in the 400–450 nm region. This resulted in higher efficiencies to harvest solar light with a consequence of even higher photoactivity.Ni and Zn showed a winning combination of redox potential and band positions of TiO2 supported metal-oxo-nanoclusters for enhanced solar-light activity. However, Ni was the only metal which had a positive effect on solar photoactivity in both oxidation and reduction reactions. The results presented here open a new thinking paradigm for designing surface modification of semiconductors to enhance their solar-light photoactivity.
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The authors gratefully acknowledge the financial support of the Slovenian Research Agency (ARRS) through the Programmes P1-0021, P1-0134 and the Project N1-0028. Nives Vodišek from Laboratory for Environmental and Life Sciences, University of Nova Gorica is acknowledged for conducting TPA tests. Access to synchrotron radiation facilities of ELETTRA (beamline XAFS, project 20160239) is acknowledged. Giuliana Aquilanti and Luca Olivi of ELETTRA are acknowledged for expert advice on beamline operation. Pegie Cool acknowledges support from the Fund for Scientific Research-Flanders, Belgium (FWO-Flanders; project nr. G038215N).References
- H. Irie, K. Kamiya, T. Shibanuma, S. Miura, D. a. Tryk, T. Yokoyama and K. Hashimoto, J. Phys. Chem. C, 2009, 113, 10761–10766 CAS.
- H. Yu, H. Irie, Y. Shimodaira, Y. Hosogi, Y. Kuroda, M. Miyauchi and K. Hashimoto, J. Phys. Chem. C, 2010, 114, 16481–16487 CAS.
- D. M. Tobaldi, C. Piccirillo, N. Rozman, R. C. Pullar, M. P. Seabra, A. S. Škapin, P. M. L. Castro and J. A. Labrincha, J. Photochem. Photobiol., A, 2016, 330, 44–54 CrossRef CAS.
- M. Miyauchi, H. Irie, M. Liu, X. Qiu, H. Yu, K. Sunada and K. Hashimoto, J. Phys. Chem. Lett., 2015, 7, 75–84 CrossRef PubMed.
- G. Li, N. M. Dimitrijevic, L. Chen, T. Rajh and K. a. Gray, J. Phys. Chem. C, 2008, 112, 19040–19044 CAS.
- G. S. Pozan, M. Isleyen and S. Gokcen, Appl. Catal., B, 2013, 140–141, 537–545 CrossRef CAS.
- D. T. Sawyer and J. S. Valentine, Acc. Chem. Res., 1981, 14, 393–400 CrossRef CAS.
- B. Srikanth, R. Goutham, R. Badri Narayan, A. Ramprasath, K. P. Gopinath and A. R. Sankaranarayanan, J. Environ. Manage., 2017, 200, 60–78 CrossRef CAS PubMed.
- A. Šuligoj, U. L. Štangar, A. Ristić, M. Mazaj, D. Verhovšek and N. N. Tušar, Appl. Catal., B, 2016, 184, 119–131 CrossRef.
- C. M. Malengreaux, S. Douven, D. Poelman, B. Heinrichs and J. R. Bartlett, J. Sol-Gel Sci. Technol., 2014, 71, 557–570 CrossRef CAS.
- D. Riemer, IEEE Trans. Compon., Hybrids, Manuf. Technol., 1985, 8, 474–480 CrossRef.
- Urad Republike Slovenije za intelektualno lastnino, SI 23585 A, 2012, 14.
- M. Kete, E. Pavlica, F. Fresno, G. Bratina and U. Lavrenčič Štangar, Environ. Sci. Pollut. Res., 2014, 21, 11238–11249 CrossRef CAS PubMed.
- U. Černigoj, U. L. Štangar, P. Trebše, U. O. Krašovec and S. Gross, Thin Solid Films, 2006, 495, 327–332 CrossRef.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquerol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603–619 CrossRef CAS.
- E. P. Barrett, L. G. Joyner and P. P. Halenda, J. Am. Chem. Soc., 1951, 73, 373–380 CrossRef CAS.
- A. B. Murphy, Sol. Energy Mater. Sol. Cells, 2007, 91, 1326–1337 CrossRef CAS.
- U. Černigoj, M. Kete and U. Lavrenčič Štangar, Catal. Today, 2010, 151, 46–52 CrossRef.
- A. Mills, J. Hepburn, D. Hazafy, C. O'Rourke, J. Krýsa, M. Baudys, M. Zlamal, H. Bartkova, C. E. Hill, K. R. Winn, M. E. Simonsen, E. G. Søgaard, S. C. Pillai, N. S. Leyland, R. Fagan, F. Neumann, C. Lampe and T. Graumann, J. Photochem. Photobiol., A, 2013, 272, 18–20 CrossRef CAS.
- A. Mills, J. Wang and D. F. Ollis, J. Catal., 2006, 243, 1–6 CrossRef CAS.
- O. Pliekhova, I. Arčon, O. Pliekhov, N. N. Tušar and U. L. Štangar, Environ. Sci. Pollut. Res., 2017, 24, 12571–12581 CrossRef CAS PubMed.
- A. Mills, C. O'Rourke, K. Lawrie and S. Elouali, ACS Appl. Mater. Interfaces, 2014, 6, 545–552 CAS.
- A. Mills, N. Wells and C. O'Rourke, J. Photochem. Photobiol., A, 2016, 330, 86–89 CrossRef CAS.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CrossRef CAS.
- Y. Xu and M. A. A. Schoonen, Am. Mineral., 2000, 85, 543–556 CrossRef CAS.
- H. Kisch and D. Bahnemann, J. Phys. Chem. Lett., 2015, 6, 1907–1910 CrossRef CAS PubMed.
- C. F. Bohren and D. R. Huffman, Absorption and Scattering of Light by Small Particles, Wiley, 2008 Search PubMed.
- E. Tratar Pirc, I. Arčon, A. Kodre and P. Bukovec, Carbohydr. Res., 2004, 339, 2549–2554 CrossRef CAS PubMed.
- M. Marinšek, J. P. Gomilšek, I. Arčon, M. Čeh, A. Kodre and J. Maček, J. Am. Ceram. Soc., 2007, 90, 3274–3281 CrossRef.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- J. J. Rehr, R. C. Albers and S. I. Zabinsky, Phys. Rev. Lett., 1992, 69, 3397–3400 CrossRef CAS PubMed.
- F. Alonso, P. Riente, J. A. Sirvent and M. Yus, Appl. Catal., A, 2010, 378, 42–51 CrossRef CAS.
- J. B. Yi, J. Ding, Y. P. Feng, G. W. Peng, G. M. Chow, Y. Kawazoe, B. H. Liu, J. H. Yin and S. Thongmee, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 2–6 Search PubMed.
- T. L. Brown, H. E. LeMay and B. E. Bursten, Chemistry: the Central Science, Pearson Education, Inc., Boston, 12th edn, 2012 Search PubMed.
- Z. Zou, J. Ye, K. Sayama and H. Arakawa, Nature, 2001, 414, 625–627 CrossRef CAS PubMed.
- M. A. Rauf, M. A. Meetani and S. Hisaindee, Desalination, 2011, 276, 13–27 CrossRef CAS.
- S. D. Sharma, K. K. Saini, C. Kant, C. P. Sharma and S. C. Jain, Appl. Catal., B, 2008, 84, 233–240 CrossRef CAS.
- M. Nussbaum, N. Shaham-Waldmann and Y. Paz, J. Photochem. Photobiol., A, 2014, 290, 11–21 CrossRef CAS.
- D. R. Lide, CRC Handbook of Chemistry and Physics, CRC Press, New York, 85th edn, 2004 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ta07176k |
| This journal is © The Royal Society of Chemistry 2018 |