
Mechanistic understanding of salt-assisted autocatalytic hydrolysis of cellulose†
Zhicheng
Jiang
ab,
Jiajun
Fan
 b,
Vitaliy L.
Budarin
b,
Duncan J.
Macquarrie
b,
Yang
Gao
b,
Tianzong
Li
b,
Changwei
Hu
*a and
James H.
Clark
*b
b,
Vitaliy L.
Budarin
b,
Duncan J.
Macquarrie
b,
Yang
Gao
b,
Tianzong
Li
b,
Changwei
Hu
*a and
James H.
Clark
*b
aKey Laboratory of Green Chemistry and Technology, Ministry of Education, College of Chemistry, Sichuan University, Chengdu, Sichuan 610064, China. E-mail: changweihu@scu.edu.cn
bGreen Chemistry Centre of Excellence, Department of Chemistry, University of York, York, YO10 5DD, UK. E-mail: james.clark@york.ac.uk
First published on 6th March 2018
Abstract
Depolymerisation of cellulose is a critical step for biomass-based bio-refining processes to produce valuable chemicals. Herein, we propose the mechanism of the promoting effect of NaCl on the cellulose hydrolysis process based on a systematic kinetic study involving variable temperature studies and the use of deuterated agents. It has been found that the presence of NaCl simultaneously enhances the generation of acidic products from cellulose decomposition and pushes the generated protons to the surface of cellulose, dramatically increasing surface acidity and facilitating the autocatalytic hydrolysis of cellulose. Cl− disrupted the intermolecular hydrogen bonding of cellulose, especially in the first surface layer. Thus, the solid cellulose chains were peeled off layer-by-layer, leading to an accelerated hydrolysis of cellulose by the adsorbed protons. Without the need for traditional acidic catalysts, this autocatalytic depolymerisation of cellulose in water, assisted by salt provides a practically viable route to the enhanced conversion of biomass to chemicals.
With the growth of economic and industrial development needed to supply an increasing population, alternative resources are urgently needed to supplement and eventually replace fossil resources.1,2 As a renewable and widespread resource, lignocellulosic biomass is a promising candidate and avoids competition with food supply.3–5 Cellulose is the largest component in lignocellulosic biomass and the most abundant polymer in nature, making it a great prospect for the production of biofuels and bio-based chemicals.6,7 The high degree of cellulose crystallinity is unfavourable for use with solvents and catalysts, and the poor heat transfer character of the solid cellulose also limits its application. Thus, overcoming the inherent recalcitrance of cellulose can make a more sustainable operation of the further biorefining process.8 Since the early 1920s, research has been focused on non-enzymatic cellulose hydrolysis,9 where acids are needed for the catalytic cleavage of the β-(1–4)glycosidic bonds.10 Conventional acid, heteropolyacids, and solid acid catalysts have been widely used for the hydrolysis of cellulose.11–13 The combination of strongly acidic catalysts and inorganic salts (especially chloride salts) has also been used to assist the degradation of cellulose.14,15 For example, NaCl–H3PO4 was used for cellulose conversion, where high ionic strength increased the proton concentration in the reaction system and, as a result, facilitated depolymerisation of cellulose improving the yield of levulinic acid.16 However, the use of strong acids has technical drawbacks such as corrosion of equipment and environmental problems from subsequent waste streams. Besides acids, metal chlorides and ionic liquids can also be solely used as the catalyst for cellulose conversion and to yield value-added products (e.g. HMF) sometimes with high selectivity.17–19 Recently, it has been found that NaCl can be used as an efficient promoter of cellulose depolymerisation in the absence of any acid, and the associated NaCl effect on the cleavage of hydrogen bonding of cellulose was discussed.20 Here, we report the effect of NaCl on pure microcrystalline cellulose conversion in a hydrothermal processing using microwave heating, a faster, more efficient and selective heating method. The mechanism of NaCl-promoted cellulose hydrolysis was investigated in detail via complete analysis of the liquid fractions and the post-treated solid samples, and reveals for the first time an autocatalytic process and the salt-assisted adsorption of H+ on the surface of cellulose. These results provide valuable information for the further enhancement and control of the selectivity of cellulose conversion to chemicals.
Initially, the effect of NaCl on the kinetics of hydrothermal conversion of cellulose was systematically investigated in the temperature range of 170 to 220 °C (Fig. 1) using an Anton Paar microwave reactor. Remarkably, there was no salt effect until over 180 °C which is in good agreement with a previous observation about cellulose alone.21 At 190 °C, the addition of NaCl showed only a very small effect on cellulose conversion. Increased reaction temperature led to an accelerated dissolution rate of cellulose and enlarged the difference between H2O and NaCl–H2O systems, reaching a 5-fold increase in the maximum reaction rate of cellulose hydrolysis after NaCl addition at 220 °C (Fig. 1B). The maximum reaction rates in both systems were observed at conversions of nearly 50%, and the corresponding activation energy of cellulose conversion was calculated accordingly (Fig. 1C). The activation energy we obtained using pure water was 240 kJ mol−1, very close to the literature value.22,23 Interestingly, the value increased to 310 kJ mol−1 using NaCl–H2O. The possible way that we could have increased reaction rates and achieved higher activation energy is through a substantial increase in the number of active centres. The two trends meet at a reaction temperature of 175 °C, consistent with the fact that we observed no salt effect with NaCl below 180 °C (Fig. 1C). It can be seen from Fig. 1 that the maximum conversion of cellulose is around 80% both in NaCl–H2O and H2O systems at higher reaction temperature; most of the remainder is solid residue biochar according to TGA analysis (Fig. S1, ESI†). Thus, the actual conversion of cellulose is even higher than that in Fig. 1A. However, to achieve the maximum conversion in the absence of salt, much longer reaction time was required.
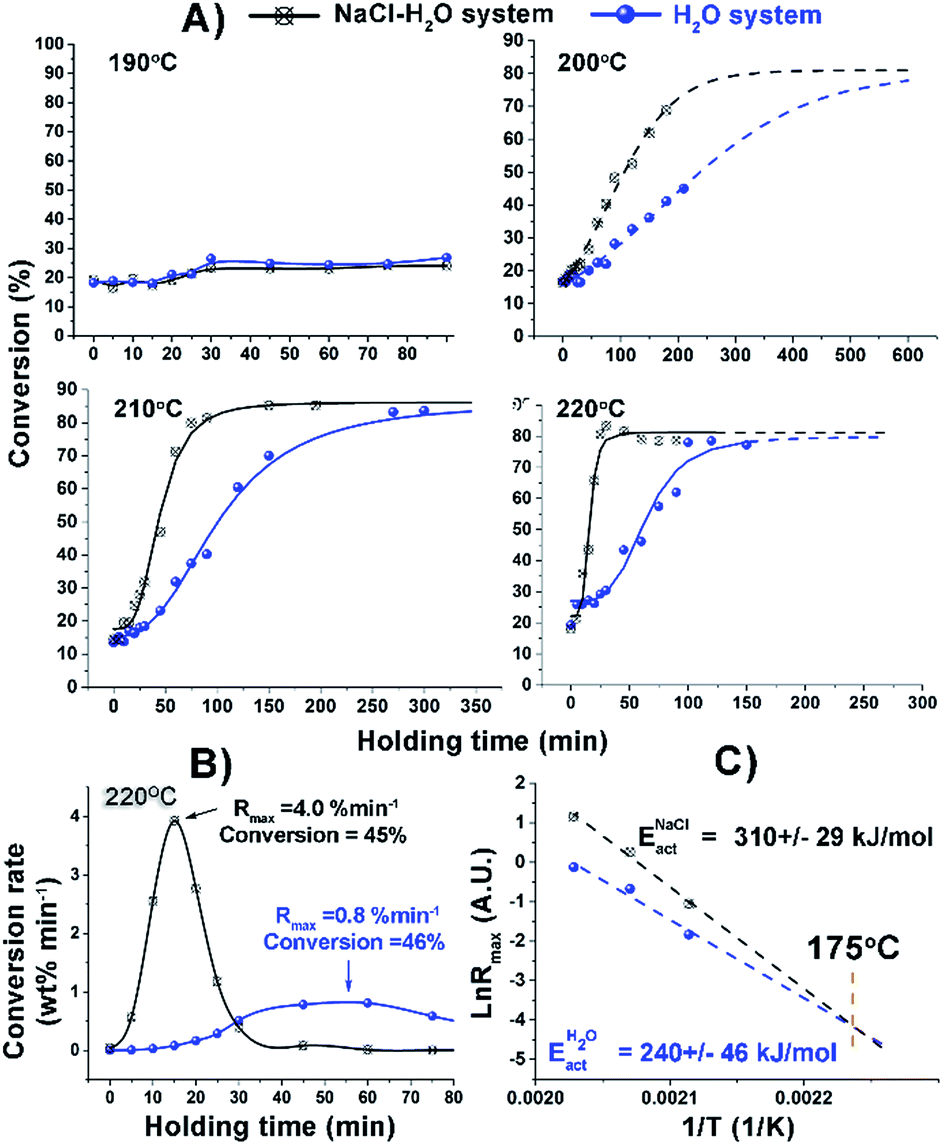 | ||
| Fig. 1 (A) Cellulose conversion in H2O and NaCl–H2O systems at different reaction temperatures; (B) conversion rate of cellulose in H2O and NaCl–H2O systems at 220 °C; (C) total activation energy. | ||
All kinetic traces obtained at temperatures above 190 °C both in the presence and absence of NaCl demonstrate clear S-shaped forms (Fig. 1A), which could be attributed to an autocatalytic process of cellulose depolymerisation, implying the formation of a catalytically active species during the depolymerisation process. The increased conversion rate of cellulose with the reaction proceeding (Fig. 1B) also supported the in situ generation of catalytic species. To help prove this, a control experiment was carried out where the hydrolysate formed after 30 minutes of a microwave depolymerisation was replaced by fresh NaCl–H2O. This resulted in a much reduced cellulose conversion (Fig. S2, ESI†). To try to identify new active species, HPLC analysis of the hydrolysates has been carried out. We found the major small molecular products of cellulose depolymerisation to be monosaccharides, HMF and carboxylic acids in both reaction systems in the temperature range of 190 to 220 °C (Fig. S3, ESI†). The analysis clearly showed that the addition of NaCl significantly increased the yield of glucose, formic and levulinic acids, while cellobiose and fructose yields were reduced (Fig. 2). Thus, NaCl enhances the cellobiose-to-glucose depolymerisation, fructose-to-HMF dehydration, HMF-to-levulinic acid and the formation of formic acid – all processes known to be catalysed by Brønsted acids.24–26 The much lower pH value of the liquid reaction fraction in the NaCl–H2O system (Table S1, ESI†) confirmed an increase in the concentration of acidic products. A control experiment showed that different inorganic salts could also promote the conversion of glucose to acids with NaCl being especially effective (Fig. S4, ESI†). This result is consistent with literature reports on the effects of salts on cellulose depolymerisation,27 and that acids can be obtained directly from sugars.26 NaCl has also been used to separate GVL from water, and the effects of added acid to the NaCl–H2O–GVL–cellulose biphasic system were studied but only at less than 180 °C, so any promoting effect of NaCl on autocatalysis of the depolymerisation of cellulose would not have been observed.28 We have studied this system at 220 °C (Fig. S5, ESI†). Lower rates of conversion of cellulose were achieved in the biphasic system compared to NaCl–H2O. This could be a result of the extraction of the acidic products into the GVL phase thus losing their catalytic effect on the cellulose depolymerisation. Our reaction times were kept short to minimize loss of cellulose through conversion to humin species. This biphasic system significantly increased the carbon content in the final liquid solution; less than 70% of the carbon could be detected based on the converted cellulose in the NaCl–H2O system. By reusing the solid residue, after only three cycles we achieved a total yield of chemical products in the GVL phase of 82% (mostly as 38.84 wt% of HMF and 29.91 wt% of levulinic acid).
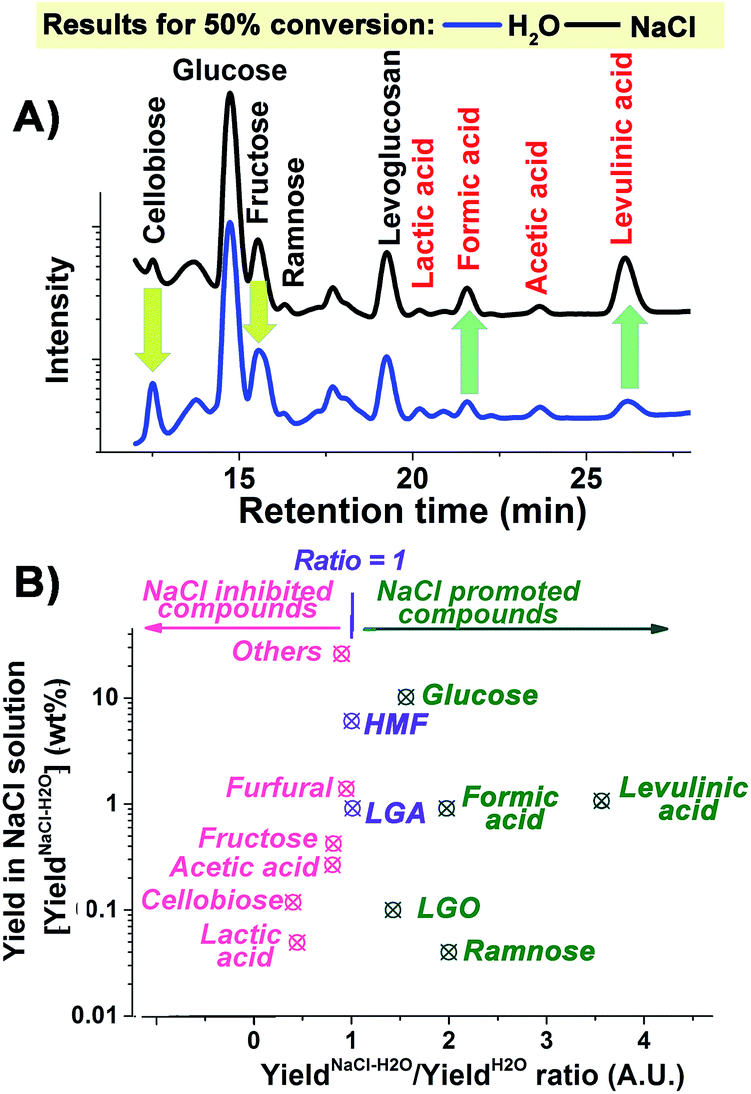 | ||
| Fig. 2 (A) HPLC signal traces and (B) NaCl effect of the products formed at 50% conversion of cellulose in H2O and NaCl–H2O systems at 210 °C. | ||
In the light of the above results, we focused our attention on formic acid as the most important active compound formed in the cellulose depolymerisation and on pH as a measure of the probable catalytic species (protons). The addition of NaCl reduced the pH of the reaction system even at room temperature (Fig. 3A); this can be attributed to a shift of the ionisation equilibrium of HCOOH by the salt effect.29–31 As we added cellulose into NaCl–HCOOH–H2O solution at room temperature, the pH value of the solution increased. A much smaller increase was observed in the absence of NaCl (Fig. 3A). Based on the observed changes in pH, we can conclude that NaCl causes greater than a fifteen fold increase in the amount of protons adsorbed on the surface of cellulose (Fig. 3B). Similar results were observed when using sulfuric acid instead of formic acid (Fig. S6, ESI†). These results strongly indicate that NaCl (and other salts) substantially enhances the adsorption of protons on the surface of cellulose.
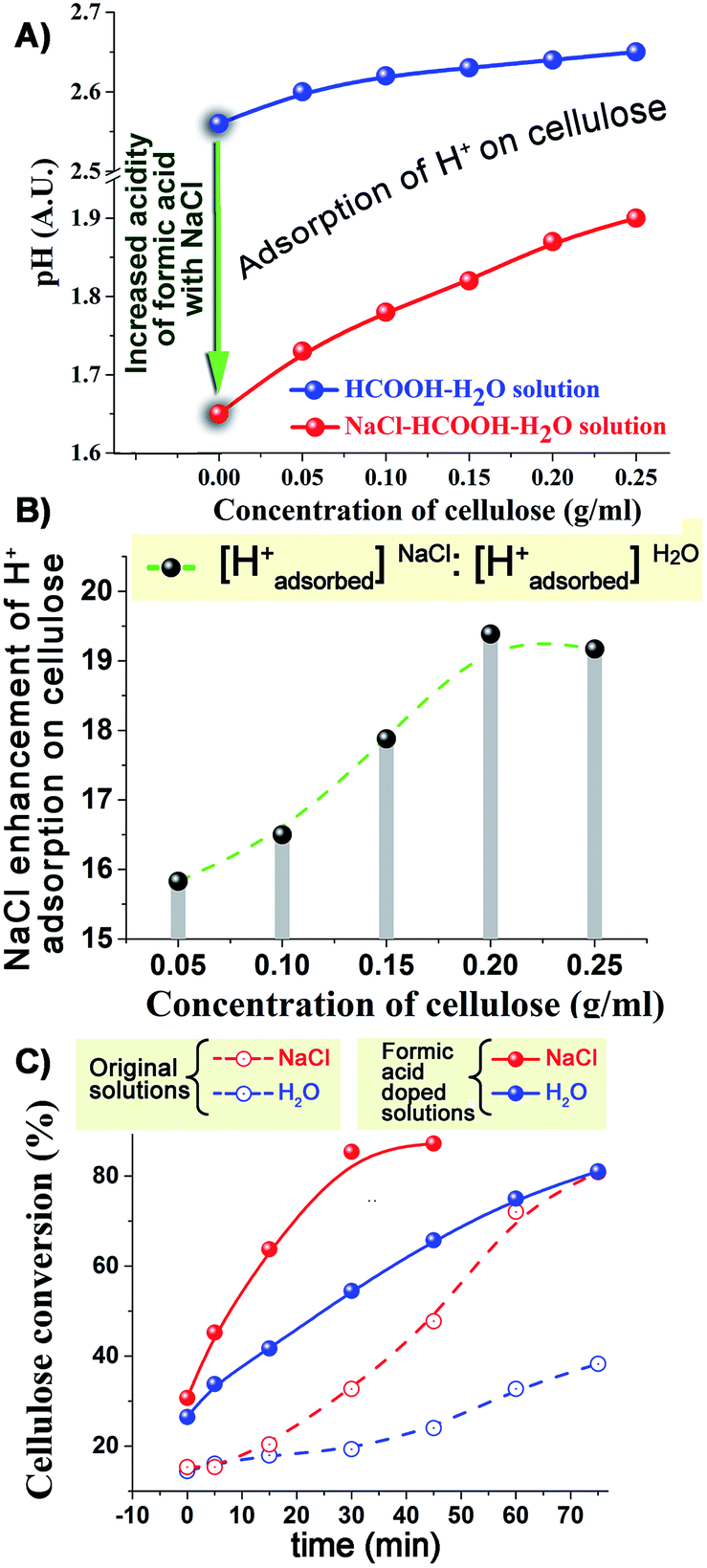 | ||
| Fig. 3 NaCl effect of (A) pH value and (B) H+ adsorption on cellulose surface with the different concentrations of cellulose in HCOOH–H2O solution at room temperature; (C) effect of the addition of formic acid on the conversion of cellulose in H2O and NaCl–H2O systems at 210 °C. | ||
HCOOH-doped solutions were also used to study the effects on the cellulose depolymerisation reaction at 210 °C (Fig. 3C). Because little acid was formed early in the reactions both using H2O and NaCl–H2O, the direct addition of HCOOH was observed to immediately accelerate the conversion of cellulose (in both systems). It is noteworthy that the conversion plots in the HCOOH-doped solutions were no longer S-shaped, further confirming that it is indeed acid that drives the autocatalytic hydrolysis of cellulose. The presence of NaCl in the HCOOH–H2O system further enhanced the rate of conversion of cellulose and led to less solid residues and more acidic products (Fig. S7 and S8, ESI†). Comparing with the pure H2O system, the increased cellulose conversion rate in the NaCl–HCOOH–H2O system was much higher than the sum of HCOOH-promoted plus NaCl-promoted rates. For example, after 15 min treatment, the increased cellulose conversion in NaCl–HCOOH–H2O, HCOOH–H2O and NaCl–H2O systems (compared to pure water) was 45.82%, 23.74% and 2.45%, respectively. This seems to confirm the suggestion that NaCl probably helps the adsorption of H+ on the surface of cellulose, accelerating the hydrolysis process.
Since chloride ions have been reported to break the hydrogen bonding of cellulose,32,33 a comparison of the results in HCl–H2O to those in HCl–NaCl–H2O was necessary. HCl–NaCl–H2O system was more favorable than the HCl–H2O system in the conversion of cellulose (Fig. S9 and S10, ESI†), showing that the effect of NaCl on cellulose conversion is not simply due to an addition of Cl− and H+ ion effects. NaCl-assisted adsorption of H+ ion on the surface of cellulose also contributed to the higher rate of conversion of cellulose in the HCl–NaCl–H2O system.
In order to gain more mechanistic understanding, we then studied the effect of deuterated formic acid (DCOOD). There was a clear H–D effect on the rate of cellulose depolymerisation in the absence of NaCl (Fig. 4A). In the acid electrolyte solution, DCOO− (or HCOO−) ions would disrupt the hydrogen bonding in cellulose and generate new hydrogen bonds with the cellulosic hydroxyl groups.34,35 A double layer would then be formed where the D+ (or H+) ions are accumulated on the negatively charged cellulose surface. D+-catalysed hydrolysis and decomposition of cellulose would happen in DCOOD–D2O solution, but the slowly generated H+ ions from cellulose would be heavily diluted in the deuterated reaction medium (Fig. 4D). Hence the hydrolysis of cellulose in DCOOD–D2O solution would be predominately D+-catalysed and hence slower than the completely H+-catalysed process. In the presence of NaCl, the rate of conversion of cellulose using HCOOH was higher than using DCOOD in the first 15 min of reaction, but unlike in the absence of NaCl, the rates then converged (Fig. 4B). In the first phase of the hydrolysis process, D+ also acted as the catalyst in DCOOD–NaCl–D2O solution, leading to lower rates of conversion. As the reaction proceeds, more and more H+ is generated from cellulose, which is then preferentially adsorbed and accumulated on the surface of cellulose (Fig. 4E). The high concentrations of NaCl would increase the charge density of the double layer and inhibit proton migration into the bulk solution. We then looked at the effects of pre-treatment of cellulose with H2O and D2O (210 °C for 30 min, leading to a strong OD band in the FTIR, Fig. S11, ESI†). The rates of conversion of these differently pretreated celluloses in the DCOOD–NaCl–D2O reaction system were significantly different (Fig. 4C). While the H2O-pretreated cellulose behaved essentially the same as the original cellulose (Fig. 4B, blue bar), the deuterated cellulose reacted more slowly, consistent with our proposed mechanism (Fig. 4F).
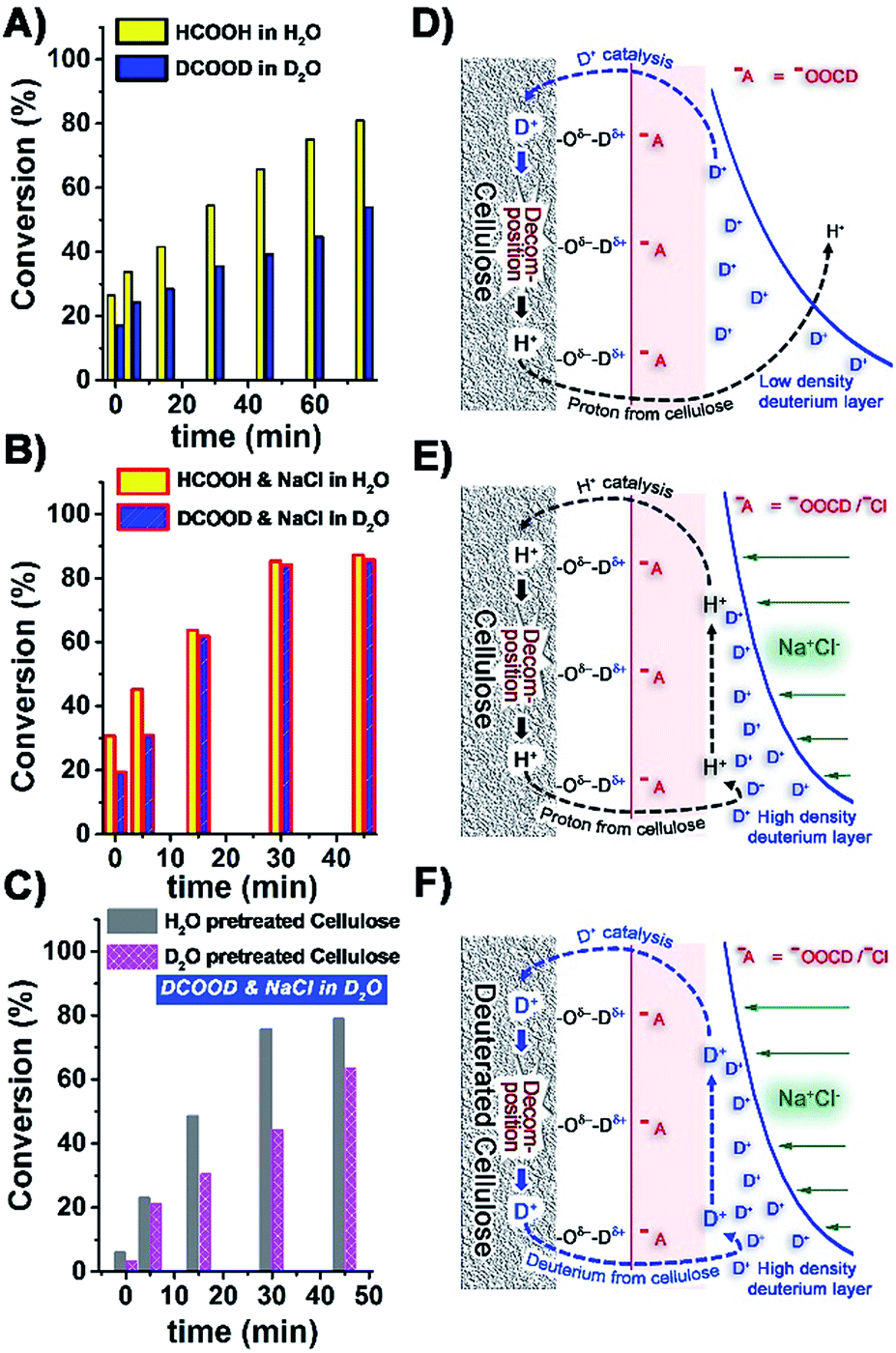 | ||
| Fig. 4 (A) Comparison of HCOOH and DCOOD catalysed conversion of cellulose at 210 °C; (B) comparison of HCOOH and DCOOD catalysed conversion of cellulose in the presence of NaCl at 210 °C; (C) comparison of the conversion of H2O-pretreated cellulose and D2O-pretreated cellulose in DCOOD–NaCl–D2O solution; (D) scheme for cellulose conversion in DCOOD–D2O solution; (E) scheme for cellulose conversion in DCOOD–NaCl–D2O solution; (F) scheme for deuterated cellulose conversion in DCOOD–NaCl–D2O solution. | ||
In order to investigate the conversion pathway of cellulose from the solid state into the liquid phase, cellulose and the solid residues after reaction were analysed by XRD, FT-IR and 13C solid-state NMR (Fig. 5, S12 to S14, ESI†). The crystallinity index (CI) values of the reaction residues are almost the same as that of the original cellulose even after half the cellulose has been converted both in the H2O system (60 min holding time) and NaCl–H2O system (15 min holding time). Solid state NMR and IR spectra also show no obvious change, especially in spectral regions characteristic of the ordered and disordered regions of cellulose.36 The small but potentially important changes in the IR bands characteristic of OHO intermolecular hydrogen bonds (ca. 1020 cm−1) of the cellulosic residues from the reaction with NaCl–H2O might indicate a partially disrupted hydrogen bonded structure (Fig. 5B). This could indicate that the cellulose structure is peeled off layer-by-layer rather than destruction of the bulk crystalline cellulose.20 At temperatures above 180 °C, the cellulose structure is softened, enabling Cl− ions to penetrate the first layer of cellulose and break the O(6)H⋯O(3) intermolecular hydrogen bonding (Fig. 5C).20,21 As more and more cellulose was converted, the reaction residues gradually became darker presumably due to the formation of biochar (including humins) from the carbonisation of the reaction intermediates,25 and in consequence, the CI decreased. The result of TGA analysis and the stronger IR bands at 1706 and 1604 cm−1 (associated with the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond stretching and furan ring vibrations) also suggest that hydrochars exist in the residues after prolonged reaction times (Fig. S1 and S13, ESI†).37,38
O bond stretching and furan ring vibrations) also suggest that hydrochars exist in the residues after prolonged reaction times (Fig. S1 and S13, ESI†).37,38
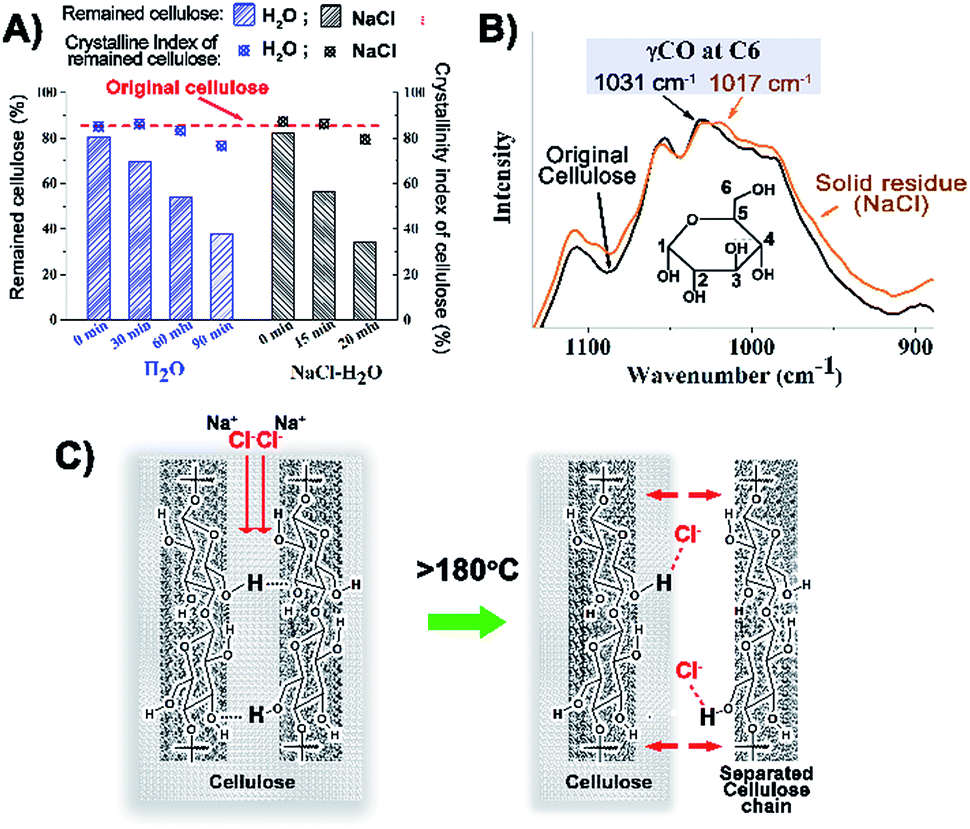 | ||
| Fig. 5 (A) Crystallinity index of cellulose before and after treatment in H2O and NaCl–H2O systems; (B) IR spectra of cellulose before and after treatment in the NaCl–H2O system; (C) scheme of the NaCl-assisted conversion of cellulose. | ||
Conclusion
Biomass naturally generates acidic species on depolymerisation and this can be enhanced by the presence of NaCl and other salts which are inevitably present in all forms of biomass. Our results throw considerable light on the effects of salts on the thermochemical depolymerisation of cellulose as well as show how a cheap and readily available mineral (e.g. in the form of sea water) can be used to help improve the resource efficiencies of lignocellulosic bio-refineries. The activity of the resulting protons towards cellulose is also enhanced by NaCl keeping the protons on the surface of the polysaccharide. Cl− erodes cellulose material layer-by-layer by breaking the intermolecular hydrogen bonding of cellulose. This is the first time this “NaCl-assisted adsorption of H+ on the surface of cellulose” effect has been proposed. Chloride ions can have the additional beneficial effect of disrupting the hydrogen bonding in the cellulose making it more amenable to depolymerisation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is financially supported by EPSRC for research grant no. EP/K014773/1, the Industrial Biotechnology Catalyst (Innovate UK, BBSRC, EPSRC) to support the translation, development and commercialisation of innovative industrial biotechnology processes (EP/N013522/1), National Natural Science Foundation of China (No. 21536007) and the 111 project (B17030). The authors would like to thank members of the Green Chemistry Centre of Excellence for their input and useful discussions. Zhicheng Jiang acknowledges support from China Scholarship Council (CSC No. 201501310005).Notes and references
- D. M. Alonso, S. G. Wettstein and J. A. Dumesic, Chem. Soc. Rev., 2012, 41, 8075–8098 RSC.
- Z. Jiang, H. Zhang, T. He, X. Lv, J. Yi, J. Li and C. Hu, Green Chem., 2016, 18, 4109–4115 RSC.
- P. Gallezot, ChemSusChem, 2008, 1, 734–737 CrossRef CAS PubMed.
- Y. Zhang, J. Pan, Y. Shen, W. Shi, C. Liu and L. Yu, ACS Sustainable Chem. Eng., 2015, 3, 871–879 CrossRef CAS.
- Z. Jiang and C. Hu, J. Energy Chem., 2016, 25, 947–956 CrossRef.
- E. M. Rubin, Nature, 2008, 454, 841–845 CrossRef CAS PubMed.
- A. Pichon, Nat. Chem., 2012, 4, 68–69 CrossRef CAS.
- A. M. Lopes and R. Bogel-Lukasik, ChemSusChem, 2015, 8, 947–965 CrossRef PubMed.
- W. Deng, J. R. Kennedy, G. Tsilomelekis, W. Zheng and V. Nikolakis, Ind. Eng. Chem. Res., 2015, 54, 5226–5236 CrossRef CAS.
- R. A. Antonopolis, H. W. Blanch, R. P. Freitas, A. F. Sciamanna and C. R. Wilke, Biotechnol. Bioeng., 1983, 25, 2757–2773 CrossRef PubMed.
- Y. B. Huang and Y. Fu, Green Chem., 2013, 15, 1095–1111 RSC.
- R. Rinaldi and F. Schuth, ChemSusChem, 2009, 2, 1096–1107 CrossRef CAS PubMed.
- J. Zhang, X. Liu, M. Sun, X. Ma and Y. Han, ACS Catal., 2012, 2, 1698–1702 CrossRef CAS.
- X. Li, Z. Fang, J. Luo and T. Su, ACS Sustainable Chem. Eng., 2016, 4, 5804–5813 CrossRef CAS.
- J. Potvin, E. Sorlien, J. Hegner, B. DeBoef and B. L. Lucht, Tetrahedron Lett., 2011, 52, 5891–5893 CrossRef CAS.
- K. Qin, Y. Yan, Y. Zhang and Y. Tang, RSC Adv., 2016, 6, 39131–39136 RSC.
- S. Dutta, S. De, M. I. Alam, M. M. Abu-Omar and B. Saha, J. Catal., 2012, 288, 8–15 CrossRef CAS.
- Y. Yang, C. Hu and M. M. Abu-Omar, Green Chem., 2012, 14, 509–513 RSC.
- Z. Zhang, J. Song and B. Han, Chem. Rev., 2017, 117, 6834–6880 CrossRef CAS PubMed.
- Z. Jiang, J. Yi, J. Li, T. He and C. Hu, ChemSusChem, 2015, 8, 1901–1907 CrossRef CAS PubMed.
- J. Fan, M. De bruyn, V. L. Budarin, M. J. Gronnow, P. S. Shuttleworth, S. Breeden, D. J. Macquarrie and J. H. Clark, J. Am. Chem. Soc., 2013, 135, 11728–11731 CrossRef CAS PubMed.
- M. J. Antal and G. Varhegyi, Ind. Eng. Chem. Res., 1995, 34, 703–717 CrossRef CAS.
- J. A. Conesa, J. A. Caballero, A. Marcilla and R. Font, Thermochim. Acta, 1995, 254, 175–192 CrossRef CAS.
- X. Zhang, B. B. Hewetson and N. S. Mosier, Energy Fuels, 2015, 29, 2387–2393 CrossRef CAS.
- R. Weingarten, W. C. Conner and G. W. Huber, Energy Environ. Sci., 2012, 5, 7559–7574 CAS.
- J. Tang, L. Zhu, X. Fu, J. Dai, X. Guo and C. Hu, ACS Catal., 2017, 7, 256–266 CrossRef CAS.
- J. G. Lynam, C. J. Coronella, W. Yan, M. T. Reza and V. R. Vasquez, Bioresour. Technol., 2011, 102, 6192–6199 CrossRef CAS PubMed.
- S. G. Wettstein, D. M. Alonso, Y. Chong and J. A. Dumesic, Energy Environ. Sci., 2012, 5, 8199–8203 CAS.
- M. R. Wright, An Introduction to Aqueous Electrolyte Solutions, Wiley, 2007 Search PubMed.
- Y. Yu, Z. Mohd Shafie and H. Wu, Ind. Eng. Chem. Res., 2015, 54, 5450–5459 CrossRef CAS.
- N. Akiya and P. E. Savage, Chem. Rev., 2002, 102, 2725–2750 CrossRef CAS PubMed.
- Y. Li, X. Liu, S. Zhang, Y. Yao, X. Yao, J. Xu and X. Lu, Phys. Chem. Chem. Phys., 2015, 17, 17894–17905 RSC.
- B. D. Rabideau and A. E. Ismail, Phys. Chem. Chem. Phys., 2015, 17, 5767–5775 RSC.
- Y. Liu, K. Thomsen, Y. Nie, S. Zhang and A. S. Meyer, Green Chem., 2016, 18, 6246–6254 RSC.
- A. Pinkert, K. N. Marsh, S. Pang and M. P. Staiger, Chem. Rev., 2009, 109, 6712–6728 CrossRef CAS PubMed.
- J. Fan, M. De bruyn, Z. Zhu, V. L. Budarin, M. J. Gronnow, L. D. Gomez, D. J. Macquarrie and J. H. Clark, Chem. Eng. Process., 2013, 71, 37–42 CrossRef CAS.
- M. Zhang, H. Yang, Y. Liu, X. Sun, D. Zhang and D. Xue, Nanoscale Res. Lett., 2012, 3, 38–42 CrossRef PubMed.
- S. Kang, X. Li, J. Fan and J. Chang, Ind. Eng. Chem. Res., 2012, 51, 9023–9031 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S13 and Tables S1 and S2. See DOI: 10.1039/c8se00045j |
| This journal is © The Royal Society of Chemistry 2018 |