
Electrodeposition of hydrated vanadium pentoxide on nanoporous carbon cloth for hybrid energy storage†
Juhan
Lee
 ab,
Sylvain
Badie
ab,
Pattarachai
Srimuk
ab,
Alexander
Ridder
ab,
Hwirim
Shim
ab,
Soumyadip
Choudhury
a,
Yoon-Chae
Nah
c and
Volker
Presser
*ab
ab,
Sylvain
Badie
ab,
Pattarachai
Srimuk
ab,
Alexander
Ridder
ab,
Hwirim
Shim
ab,
Soumyadip
Choudhury
a,
Yoon-Chae
Nah
c and
Volker
Presser
*ab
aINM – Leibniz Institute for New Materials, Campus D2 2, 66123 Saarbrücken, Germany. E-mail: volker.presser@leibniz-inm.de
bDepartment of Materials Science and Engineering, Saarland University, Campus D2 2, 66123 Saarbrücken, Germany
cInterdisciplinary Program in Creative Engineering, School of Energy, Materials and Chemical Engineering, Korea University of Technology and Education, 1600 Chungjeol-ro, Cheonan 31253, Republic of Korea
First published on 10th January 2018
Abstract
Electrodeposition is a simple and effective method for the synthesis of disordered hydrated vanadium pentoxide (V2O5·nH2O). For the synthesis of energy storage electrodes with high power performance, electrodeposition of hydrated V2O5 inside carbon micropores is particularly attractive to synergize electric-double layer formation and lithium ion intercalation. Here, we demonstrate that hydrated V2O5 can be effectively electrodeposited in carbon micropores of activated carbon cloth. Our study indicates that carbon pores larger than 1 nm are essential for the effective decoration with hydrated V2O5. A thermal treatment after the electrodeposition is often used to enhance the crystal structure of hydrated V2O5. However, thermal annealing of the hydrated vanadium pentoxide decorated activated carbon cloth under an oxygen-rich environment at high temperature (>330 °C) leads to a significant loss of pore volume, leading to a decreased electrochemical performance. At low annealing temperature (200 °C), the vanadium pentoxide electrodeposited activated carbon cloth electrode exhibits a maximum specific capacity of 137 mA h g−1 with stable cycle performance over 1600 cycles at a rate of 4C.
1. Introduction
Electrochemical energy storage systems such as secondary batteries and supercapacitors have been intensively and broadly investigated in the context of efficient energy management.1–3 Supercapacitors exhibit high power performance (>10 kW kg−1) with stable cycle performance (>10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles) but limited energy storage capacities (<30 W h kg−1); batteries provide significantly higher energy storage capacity (100–300 W h kg−1) but a much shorter cycling life compared to supercapacitors.3,4 The performance of supercapacitors and batteries has been improved by use of novel materials and system architectures.4 A particularly attractive approach to combine the benefits of both technologies is the use of hybrid materials and hybrid devices. This can be accomplished by applying different charge storage mechanisms of cathodes and anodes (hybrid devices) or introducing more than one charge storage mechanism into one electrode by use of hybrid materials.5,6
000 cycles) but limited energy storage capacities (<30 W h kg−1); batteries provide significantly higher energy storage capacity (100–300 W h kg−1) but a much shorter cycling life compared to supercapacitors.3,4 The performance of supercapacitors and batteries has been improved by use of novel materials and system architectures.4 A particularly attractive approach to combine the benefits of both technologies is the use of hybrid materials and hybrid devices. This can be accomplished by applying different charge storage mechanisms of cathodes and anodes (hybrid devices) or introducing more than one charge storage mechanism into one electrode by use of hybrid materials.5,6
Hybrid electrode materials capitalize on the combination of electric double-layer formation in nanoporous carbon and faradaic charge storage via charge transfer through reversible redox couples.1,7 Faradaic charge storage may utilize (i) redox-active electrolytes such as vanadium sulfate,8 potassium ferricyanide,5 and iodide systems,9,10 (ii) conducting polymers such as polyaniline,11 (iii) metal oxides such as MnO2, RuO2, and V2O5,12 or (iv) redox-active surface functionalities such as quinone-decorated carbonaceous materials.13 Faradaic redox reactions may exhibit clear redox peaks (i.e., battery-like behavior) or perform like a capacitor (also known as pseudocapacitance) which is characterized by the linear relationship between the accumulated charge and the electric potential of the electrode.14,15 Metal oxides like RuO2 and MnO2 are known as intrinsic pseudocapacitive materials since their capacitive features are independent of the particle size and morphology; in contrast, intercalation materials like V2O5 can be extrinsic pseudocapacitive by the design of their crystallite size and morphology.15–18
Due to the versatile design of the crystallite size, morphology, and phase as well as high theoretical specific charge storage capacity (up to 441 mA h g−1 when operating in the voltage window of 1.5–4 V, over 1500 F g−1), and the relatively high natural abundance of vanadium on Earth, vanadium pentoxide has become very attractive for electrochemical energy storage applications.12,15,19–21 In the case of lithium ion battery cathodes, bulk V2O5 shows several application limitations such as a poor cycle life time, low lithium diffusion kinetics, and high electric resistance.21,22 To overcome these limitations, various nano-engineered vanadium pentoxide materials have been developed such as nanotubes,23 nanowires,24 nanofibers,25 nanobelts,26 nanorods,27 microspheres,28 or hollow spheres.22 The most commonly used methods for vanadium pentoxide synthesis are the hydrothermal/solvothermal routes,22 electrospinning,29 sol–gel processes,30 polyol processes,31 electrodeposition,32,33 chemical vapor deposition,30,34 electrochemical activation,35 and atomic layer deposition.36 Among them, electrodeposition is particularly attractive due to its simplicity and flexibility, cost-effectiveness, and fast production rate.32,37–39 In most cases of electrodeposited V2O5 for lithium ion batteries, flat substrates have been applied such as platinum,40 indium tin oxide,36,38 polydopamine,41 and polyaniline;42 in addition, some studies have utilized structured substrates like carbon cloth39 and aluminum microrods.37
For higher power performance and stability, carbon materials such as carbon nanotubes,43 graphene,44 and onion-like carbons45 were used as conductive additives to nanostructured vanadium pentoxide electrodes. Recently, a new strategy has emerged by synthesizing a hybrid electrode for both double-layer formation and lithium ion intercalation.46 Yamada et al. reported lithium ion battery cathodes made from nanoporous V2O5/carbon composites by iterative V2O5 decoration via immersion of porous carbon in hydrosol with a maximum specific capacity of 107 mA h g−1 and a high rate performance up to 5 A g−1. Similar studies can also be found for supercapacitor applications: Ghosh et al. reported electrodeposited V2O5 on porous carbon nanofiber paper in 1 M KCl obtaining a maximum capacitance of 214 F g−1 (59 mA h g−1),47 and Wang et al. achieved 358 F g−1 (80 mA h g−1) in 2 M NaNO3 with a ternary composite of V2O5/carbon nanotube/activated carbon obtained from hydrothermal synthesis.48 A key challenge of such porous carbon/V2O5 hybrid electrodes is to overcome a low specific capacity; therefore, consistent and systematic studies are necessary to investigate the influence of the carbon pore structure and further optimization possibilities, for instance, implementation of a thermal annealing process. Particularly, activated carbon cloth (ACC) is an attractive candidate for the electrodeposition of vanadium pentoxide due to its binder-free feature and the simplicity for the electrode fabrication.
In this work, we electrodeposited hydrated vanadium pentoxide on activated carbon cloth with different activation degrees. The pore structure of the ACC electrodes will be presented in terms of the degree of activation before and after the electrodeposition process. The as-electrodeposited vanadium pentoxide is thoroughly investigated via X-ray photoelectron spectroscopy (XPS), X-ray diffraction (XRD), and Raman spectroscopy. The influence of the pore structure of the ACC electrodes will also be discussed in terms of electrochemical performances in a lithium battery half-cell as a cathode with 1 M LiClO4 in ethylene carbonate/dimethyl carbonate (1:1). Finally, we present the effects of annealing temperature on the electrochemical performance along with the porosity, the chemical composition, and the phase structure.
2. Experimental
2.1. Materials
We used microporous carbon cloth electrodes from Kynol which are refer red to as ACC-15 and ACC-20 as received (corresponding to the trademark names Kynol-507-15 and Kynol-507-20, respectively). More information about these electrode materials can be found in our previous work in ref. 49. ACC-20 was activated in flowing CO2 at 950 °C for 1 h with a gas flow rate of 200 cm3 min−1 and labeled ACC-20+. The physical activation process was carried out in a quartz tube furnace, where the inlet CO2 gas was passed through a water-filled flask before entering the furnace. The heating/cooling rate was 10 °C min−1. For the free-standing carbon film electrodes, type YP-80F activated carbon (AC) powder (Kuraray) was used with the aid of polytetrafluoroethylene (PTFE, Sigma-Aldrich) as a binder by following the procedure described in ref. 50. The final electrode material to binder mass ratio of the free-standing carbon film electrode was 95:5. The thickness of the PTFE-bound activated carbon (AC) electrode was measured to be 600 ± 50 μm. For the electrolyte, LiClO4 (battery grade, Sigma Aldrich) was dissolved in a 50:50 mixture of ethylene carbonate and dimethyl carbonate (EC/DMC; battery grade, BASF) while VOSO4 (97% purity, Sigma Aldrich) was mixed with deionized water. The pH value of the VOSO4 solution was adjusted by adding 10 M KOH solution.
2.2. Electrodeposition of vanadium oxides
Prior to electrodeposition, discs of porous carbon cloth with a diameter of 22 mm were soaked in 3 M VOSO4 solution (pH value of 1.8) for two days for wetting. Afterwards, the ACC working electrode (for electrodeposition) was assembled in a cell (ESI, Fig. S2†) with a double-stack of oversized AC electrodes (600 μm each) while keeping the mass ratio of working and counter electrodes at 1:7. As a separator, a double-stack of GF/A (Whatman) serves as an electrolyte reservoir while an additional Omnipore (Millipore, dry thickness 25 μm) separator was placed next to the working electrode. An Ag/AgCl reference electrode was mounted close to the counter electrode. The cell design used for the electrodeposition can be found in ref. 6 and 9. The electrodeposition was carried out via a VMP300 potentiostat/galvanostat (Bio-Logic) by applying ten galvanostatic cycles at a constant charging current (20 mA cm−2) at a cell voltage of 1.4 V with a subsequent resting time of 10 min while no discharging current was applied. After the tenth cycle, the cell was disassembled as the cell was charged at 1.4 V. Afterwards, the working electrode was collected and rinsed for three days to remove residual solute. Finally, the electrode was dried in a vacuum oven at 60 °C for three days. To understand the electrodeposition process, we used cyclic voltammetry with the same cell configuration as mentioned before. The potential of the working electrode (ACC) is scanned from 0 to 1 V vs. Ag/AgCl at a sweeping rate of 10 mV s−1.
2.3. Material characterization
For surface analysis of all carbon electrodes, nitrogen gas sorption analysis (GSA) with a Quantachrome Autosorb iQ system was carried out. Before measurement, the electrodes were degassed at 150 °C for ca. 20 h at a relative pressure of 0.1 Pa. Liquid nitrogen at −196 °C was employed as the experimental gas with relative pressure from 1 × 10−6 to 1.0 in 56 steps. The specific surface area (SSA) of the activated carbon (AC) and activated carbon cloth (ACC) electrodes was calculated both (i) by applying quenched-solid density functional theory (QSDFT) with a slit pore model51 and (ii) by use of the Brunauer–Emmett–Teller (BET) equation in the linear regime of the measured isotherms via ASiQwin-software 3.0.52,53The morphology of the carbon electrodes was examined by using a JEOL JSM 7500F field emission scanning microscope (FE-SEM) at an acceleration voltage of 3 kV. The chemical composition of the carbon electrodes was determined by energy dispersive X-ray spectroscopy (EDX) with an X-Max-150 detector (Oxford Instruments) attached to the SEM chamber. The spectra of ten spots were measured using an accelerating voltage range of 12 kV and averaged. High resolution electron transmission microscopy (TEM) and selected area electron diffraction (SAED) were performed using a JEOL JEM-2100F system, operating at 200 kV in a vacuum. The ground samples were dispersed and sonicated in ethanol for 1 min and drop casted on a carbon coated copper grid. For EDX mapping, a Thermo Scientific MC100021 detector attached to the TEM chamber was used, at 200 kV and an acquisition time of 10 min.
Small pieces of specimens were mounted on carbon tape and were investigated using an FEI-Dualbeam Versa 3D (FEI), equipped with a secondary electron detector, Everhard–Thornley-detector, an energy dispersive X-ray detector, and a gallium ion source. Single carbon fiber was sectioned vertically by the gallium ion source by stepwise decreasing the milling current from 15 nA to 100 pA to obtain a polished surface of the cross section. Prior to ion milling, a 30 nm thick Pt layer was deposited on the activated carbon fiber to avoid damage under the Ga ion beam. After obtaining a polished section, the sample stage was rotated to 45° to expose the sectioned area to the EDX detector. In the next step, energy dispersive X-ray (EDX) mapping of the sectioned surface of vanadium oxide coated activated carbon fibers was carried out with an Apollo XF detector and a DPP 4 analyzer at an accelerating voltage of 10 keV using EDAX Genesis software.
Raman spectroscopy was carried out with a Renishaw inVia Raman Microscope applying an Nd:YAG laser (532 nm). A grating of 2400 lines per mm, yielding a spectral resolution of approximately 1.2 cm−1, was used. The spot size on the sample was 1.2 μm with a numerical aperture of 0.9. Spectra were recorded for various carbon electrodes with an acquisition time of 30 s and ten accumulations.
X-ray diffraction (XRD) experiments were conducted using a D8 Advance (Bruker AXS) diffractometer with a copper X-ray source (Cu-Kα, 40 kV, 40 mA) and a nickel filter. All measurements were measured between 10 and 80° 2θ in a step size of 0.02° at a measurement time of 1 s per step. The samples were placed on a single sapphire crystal for the measurement. The diffractograms are corrected by the background removal function from DIFFRAC.EVA software (V4.2); the curvature and threshold parameters are set to 0.010 and 0.718, respectively.
X-ray photoelectron spectroscopy (XPS, K-ALPHA; Thermo Instruments Inc.) was used to determine the composition and chemical state of the samples. The XPS data were analyzed by XPS-Peak 4.1 program54 for the background subtraction (Shirley function) and the peak fitting with mixed Lorentzian–Gaussian curves (detailed peak fitting parameters in Table S1, ESI†). The atomic V5+/V4+ ratio is estimated based on the ratio of the fitted peak areas.
Thermogravimetric analysis (TGA) was carried out with a TG 209 F1 Libra system (Netzsch) in oxygen containing synthetic air (80% N2, 20% O2). The temperature was increased from 30 °C to 650 °C at a rate of 5 °C min−1.
For the electrochemical characterization, a VMP300 potentiostat/galvanostat (Bio-Logic) was used while the cells were stored at 25 ± 1 °C in a climate-controlled chamber. For the half-cell experiments, an oversized AC electrode (600 μm thickness, 12 mm diameter) was used as a counter electrode while the reference electrode was mounted on the side of the cell having close contact to both working and counter electrodes; the scheme of the cell design can be found elsewhere.55,56 In the case of aqueous solution, an Ag/AgCl reference (Bioanalytical Systems), GF/A glass fiber filter (Whatman), and platinum current collector were used. In the case of organic solution (1 M LiClO4 in EC/DMC with a 5:5 ratio), a piece of lithium metal serves as a reference electrode while carbon-coated aluminum foils (Zflo 2653, Exopack Technologies) and a GF/D glass fiber filter (Whatman) were applied as the current collector and separator, respectively. The aqueous electrolyte was injected by syringe vacuum backfilling after the cell assembly while the organic electrolyte was injected in an argon filled glovebox (MBraun Labmaster 130, O2 and H2O < 1 ppm) after drying the cell in a vacuum oven at 120 °C overnight. For the normalization of specific capacity, the total mass of the working electrode was considered. The C-rate was calculated by dividing the applied current by the measured maximum capacity.
3. Results and discussion
3.1. Electrodeposition of vanadium oxide on a porous carbon electrode
The binder-free continuous fiber network of the activated carbon cloth (as exemplified for ACC-15, Fig. 1a, inset) is an advantage for the electrodeposition process due to its low electric resistance compared to the PTFE-bound AC electrode (Table S1 in the ESI†).49 The activated carbon cloth (ACC) serves as a highly conductive substrate for the electrodeposition of vanadium oxides. Furthermore, the large total pore volume (>0.5 cm3 g−1, Table 1) of the ACC electrodes can provide a high surface area for the deposition of vanadium oxides while the narrow pore width can confine the size of vanadium oxides to the nanoscale which shortens the diffusion length for lithium ion intercalation.57 According to the nitrogen sorption analysis (Fig. 1a), ACC electrodes show clear plateaus over knee points in the pore size distribution (Fig. 1b). As the CO2 activation degree increases from ACC-15 to ACC-20+, the knee point shifts toward larger pore widths and the total pore volume increases. The volume-weighted average pore sizes (d50) of ACC-15, ACC-20, and ACC-20+ are 0.67 nm, 0.96 nm, and 1.16 nm, respectively.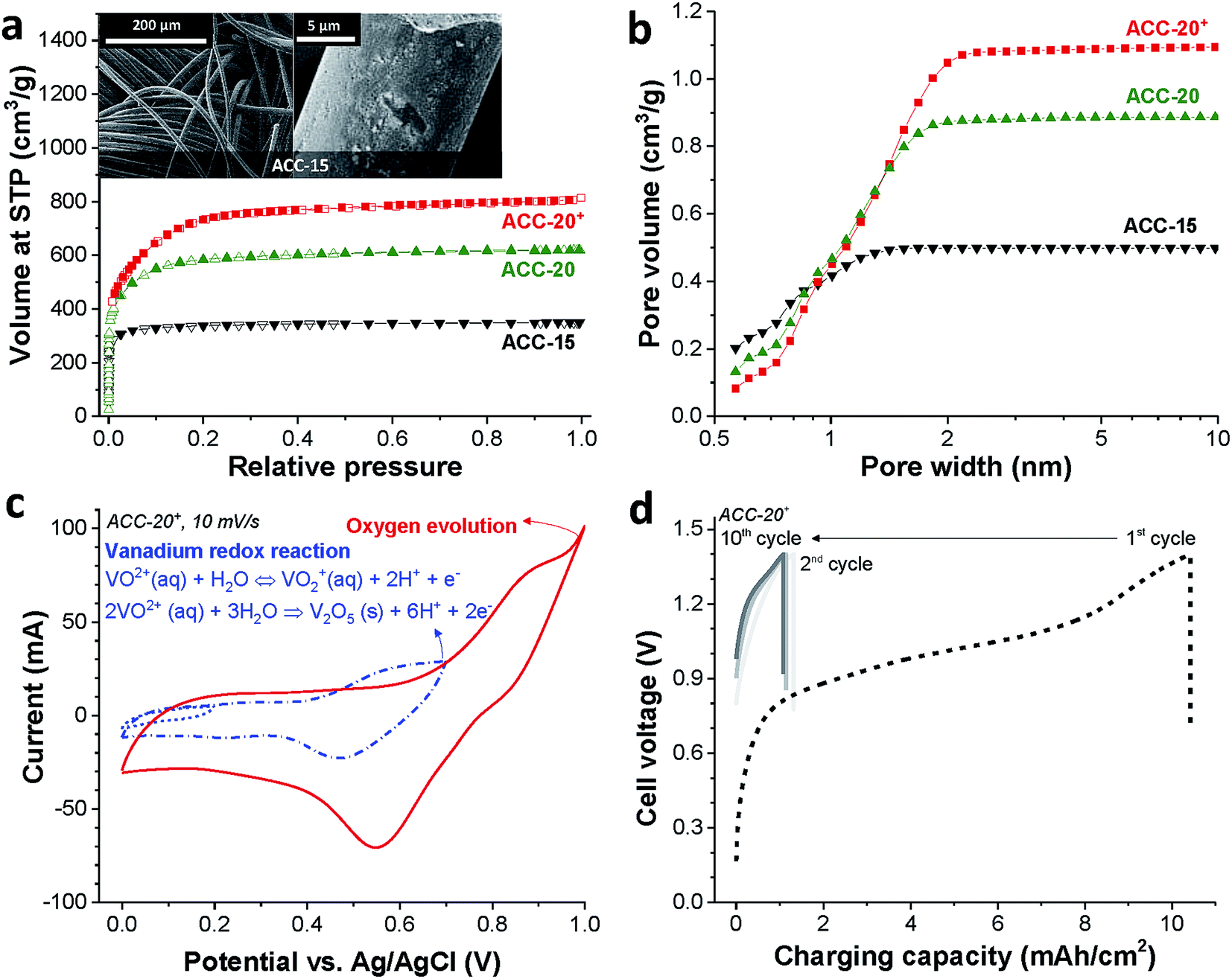 | ||
| Fig. 1 (a) Isotherms of the ACC electrodes obtained from nitrogen gas sorption analysis. The inset shows scanning electron micrographs of the ACC-15 electrode. (b) Cumulative pore size distribution of the ACC electrodes. (c) Cyclic voltammogram of the ACC-20+ electrode in 3 M VOSO4 aqueous solution (pH = 1.8). (d) Galvanostatic charge cycles at 20 mA cm−2 with a resting time of 10 min between cycles for electrodeposition of vanadium oxide. | ||
| Sample | SSA DFT (m2 g−1) | SSA BET (m2 g−1) | Average pore size (nm) | Total pore volume (cm3 g−1) |
|---|---|---|---|---|
| ACC-15 | 1450 | 1338 | 0.67 | 0.50 |
| ACC-20 | 1940 | 2209 | 0.96 | 0.89 |
| ACC-20+ | 2067 | 2571 | 1.16 | 1.10 |
The electrodeposition of vanadium oxide can be summarized in the following two equations:47
| 2VO2+(aq) + 3H2O → V2O5(s) + 6H+ + 2e− | (1) |
| VO2+(aq) + H2O ↔ VO2+(aq) + 2H+ + e− | (2) |
Eqn (2) is reversible, while the irreversible oxidation viaeqn (1) occurs with a narrow pH window around 1.8 at around +0.6 V vs. Ag/AgCl.39,40 The cyclic voltammogram of ACC-20+ (Fig. 1c) shows the reduction peak at +0.55 V vs. Ag/AgCl which can be identified by the redox activity viaeqn (2). The sharp current increase at the potential around +1.0 V vs. Ag/AgCl correlates with oxygen evolution, which can be observed from other ACC electrodes (ESI, Fig. S1a†).
Since the vanadium precipitation employs VO2+ following the redox reaction given in eqn (1),37,40 we applied galvanostatic charging without introducing a separate discharging period; instead, only a resting time of 10 min was applied between the charging cycles (Fig. 1d). As the cell potential was increased to 1.4 V, the potential of the working electrode increased to +0.85 V vs. Ag/AgCl while the working electrode potential decreases to 0.74 V vs. Ag/AgCl during the resting period (ESI, Fig. S1b†). The reproducible potential fluctuation of the working electrode in the range from +0.74 V to +0.85 V vs. Ag/AgCl assures that the working electrode potential was controlled even in a full cell configuration. This cell configuration allows the electrodeposition without the reference electrode, which can be easily degraded via cross contamination between the testing electrolyte and the electrolyte in the reference electrode.50
The strong decrease of the charging capacity between the first and the second cycle indicates the irreversible vanadium oxide formation (Fig. 1d) indicating the transition to vanadium pentoxide. With an increased cycle number, the irreversibility decreased continuously while the stagnation of the irreversibility can be seen in the tenth cycle as indicated by stabilized charging capacity over the ten cycles. The potential drop during the resting period (as seen from the vertical line in the graph) is most probably due to the diffusion of VO2+ ions.6,8 Similar electrochemical behavior was observed for the other porous carbon electrodes as can be seen in ESI, Fig. S1c and d.† For further material characterization after the electrodeposition, the ACC electrodes were labeled ACC-15-VOx, ACC-20-VOx, and ACC-20+-VOx to indicate that the vanadium oxides are introduced into the pristine ACC-15, ACC-20, and ACC-20+ electrodes, respectively.
3.2. Morphology and chemical composition
After electrodeposition, a partly coated surface on ACC-15-VOx can be seen in the scanning electron micrographs provided in Fig. 2a. In contrast to pristine ACC-15, a rough surface of vanadium pentoxide on ACC-15-VOx is observed which can be confirmed by the high vanadium and oxygen contents measured by EDX (Table 2). The surface morphology of ACC-15-VOx shows wrinkled features and thin layers of small crystalline vanadium oxides on the carbon fibers (ESI, Fig. S3a†). The crystalline domains cover the carbon phase as observed by TEM analysis (Fig. 3a). The SAED pattern (Fig. 3a, inset) of ACC-15-VOx exhibits diffuse rings and bright spots which are indicative of amorphous and crystalline features of the electrodeposited vanadium oxide, respectively. As the statistical approach for the SAED patterns indicates (Fig. S3d†), ACC-15-VOx exhibits both crystalline and amorphous vanadium oxide domains. The crystalline feature observed from ACC-15-VOx originates from the vanadium oxides deposited on the surface of the carbon fiber (Fig. S3a,† inset) while amorphous vanadium oxide was deposited in the carbon micropores inside the carbon fiber. In contrast, TEM images of ACC-20+-VOx show fewer crystalline domains and mainly amorphous carbon on the fiber surface as evidenced by the SAED pattern (Fig. 3b, inset) and XRD results (ESI, Fig. S4a and b†). After electrodeposition, ACC-20-VOx and ACC-20+-VOx exhibit mostly a smooth morphology on the fiber with the visible size of the pore entrance (Fig. 2, 3b and c, ESI†). Yet, the EDX results from ACC-20-VOx and ACC-20+-VOx indicate that the samples still contain a high amount of vanadium oxide (Table 2). Based on the TEM analysis, the lower presence of crystalline domains in ACC-20+-VOx than that in ACC-15-VOx suggests that vanadium oxide is mainly located in nano-domains inside the micro- and small mesopores, possibly with an amorphous structure. All samples show a comparable amount of oxygen of 1.6–2.5 mass%, as was shown in our previous work (ref. 49). Therefore, the domain size growth seems to be limited mostly by the carbon pore size.58 | ||
| Fig. 2 (a–c) Scanning electron micrographs of the ACC electrodes after the electrodeposition process. | ||
| Sample | C (mass%) | O (mass%) | V (mass%) | F (mass%) |
|---|---|---|---|---|
| ACC-15-VOx | 62.6 ± 6.5 | 23.0 ± 1.8 | 14.4 ± 4.9 | — |
| ACC-20-VOx | 78.8 ± 3.3 | 15.0 ± 2.8 | 5.9 ± 1.2 | — |
| ACC-20+-VOx | 74.3 ± 6.2 | 14.2 ± 2.8 | 11.5 ± 4.3 | — |
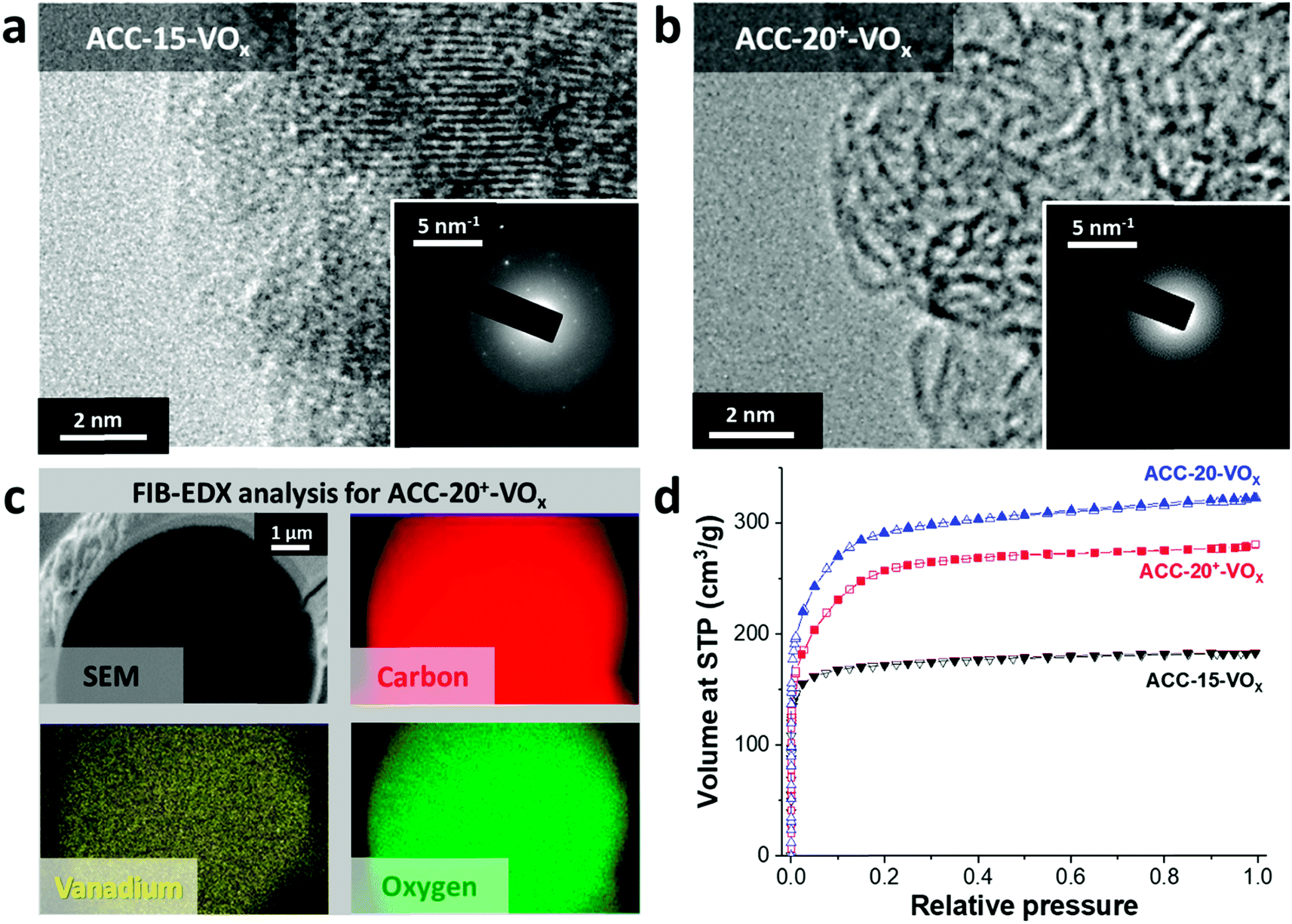 | ||
| Fig. 3 Transmission electron micrographs of (a) ACC-15-VOx and (b) ACC-20-VOx electrodes. (c) Energy dispersive X-ray spectrographs of a single fiber of the ACC-20+-VOx electrode. A single fiber was sectioned by a focused ion beam, and elemental mappings were recorded via EDX of the cross section. (d) Isotherms of the electrodeposited ACC electrodes obtained from nitrogen gas sorption analysis. | ||
The FIB-EDX results (Fig. 3c) show that the vanadium oxide is successfully deposited within the carbon fiber. In Fig. 3, only the results from ACC-20+-VOx are presented while the other carbon fiber samples showed similar results. To the best of our knowledge, no studies have reported the decoration of vanadium oxide inside carbon nanopores so far. Furthermore, as the GSA results reveal (Fig. 3d), the reduced pore volume compared to the isotherms before the electrodeposition process (Fig. 1a) indicates that vanadium oxide is successfully deposited on the samples. Yet, the obtained isotherms exhibit the characteristics of a large portion of microporosity at a small relative pressure. Therefore, a large fraction of micropores and mesopores is preserved while vanadium oxides mostly present inside the carbon fibers instead of being on the outer surface.
The Raman spectra (Fig. 4a) show that the ACC electrodes after the electrodeposition exhibit characteristic Raman shifts for vanadium oxides in the range from 100 to 1100 cm−1. The characteristic carbon D-mode (around 1342 cm−1) and G-mode (around 1605 cm−1) are also observed in the initial (uncoated) electrode. All ACC electrodes after electrodeposition show characteristic peaks for disordered vanadium pentoxides.59,60 The Raman peak at a high frequency around 1017 cm−1 (Fig. 4b) is representative of the terminal oxygen stretching mode (V![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) as also supported by the observation of bending vibrations at 412 cm−1 and 264 cm−1.60–62 The peak at 706 cm−1 corresponds to the double coordinated oxygen stretching mode (V2–O).36,60,62 The peak at 680 cm−1 indicates a V2–O stretching mode in a disordered V–O–V framework which further supports the disordered feature of the electrodeposited vanadium oxides. The peak at 512 cm−1 indicates the stretching mode of the triply coordinated oxygen (V3–O) which only appears in the chemical structure of vanadium pentoxide.60–62
O) as also supported by the observation of bending vibrations at 412 cm−1 and 264 cm−1.60–62 The peak at 706 cm−1 corresponds to the double coordinated oxygen stretching mode (V2–O).36,60,62 The peak at 680 cm−1 indicates a V2–O stretching mode in a disordered V–O–V framework which further supports the disordered feature of the electrodeposited vanadium oxides. The peak at 512 cm−1 indicates the stretching mode of the triply coordinated oxygen (V3–O) which only appears in the chemical structure of vanadium pentoxide.60–62
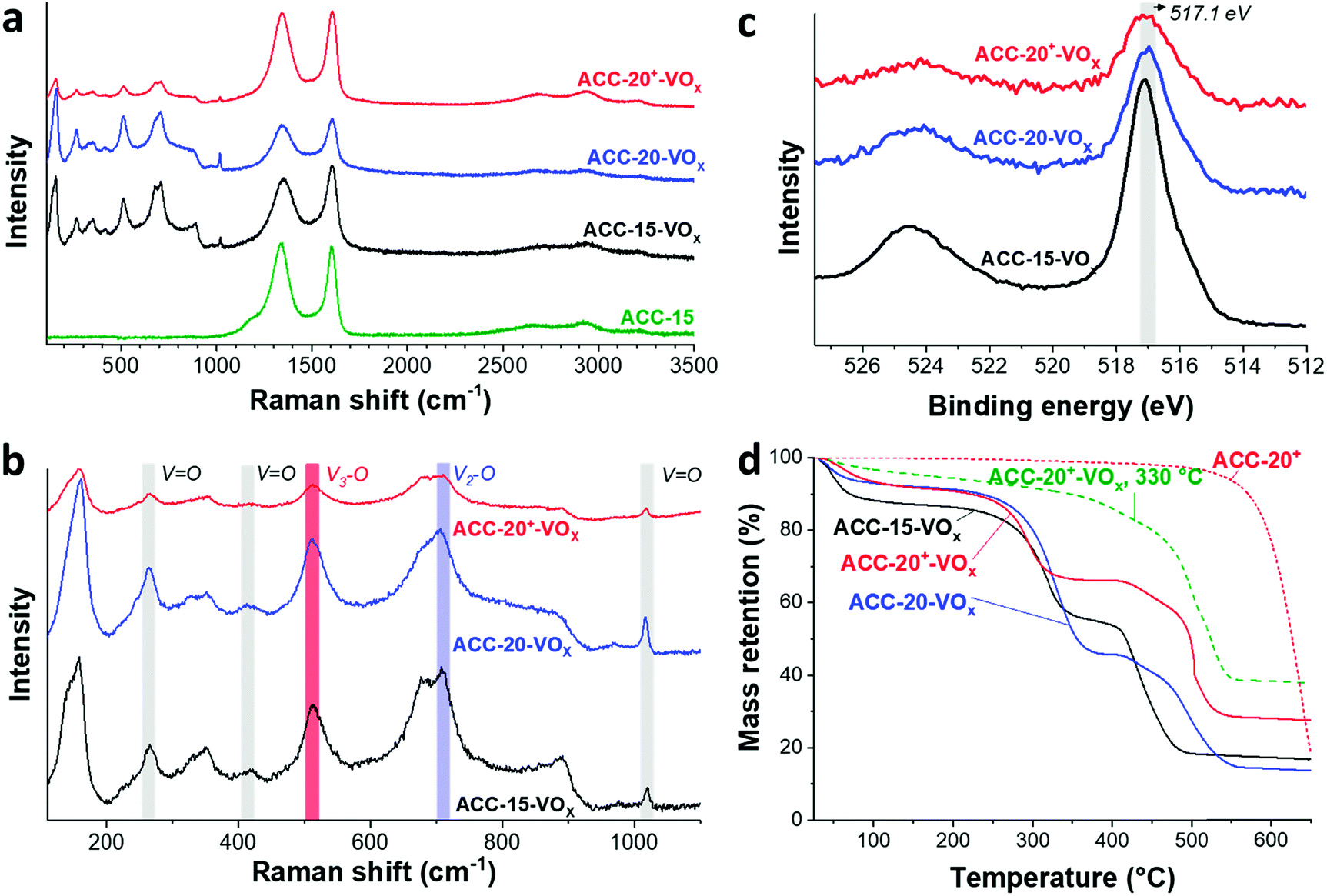 | ||
| Fig. 4 Raman spectra of the ACC-15 electrode material and the electrodeposited ACC electrodes in a full spectral range (a) and in the range from 100–1100 cm−1 (b). (c) XPS data of the electrodeposited ACC electrodes. (d) Thermal gravimetric analysis of the ACC electrodes before and after the electrodeposition of vanadium pentoxide as well as after annealing at 330 °C for 1 h. | ||
The presence of vanadium pentoxide is further confirmed by X-ray photoelectron spectra (Fig. 4c) exhibiting sharp peaks at 517.1 eV which can be assigned to the V 2p3/2 binding energy of the V5+ oxidation state. The spectrum shows the presence of V4+ (V5+/V4+ ratio of ca. 1.8), which possibly originates from the hydrated form of vanadium pentoxide; see also ESI, Fig. S4c.†63 Further evidence of the hydrated vanadium pentoxide was found by TGA (Fig. 4d). The rapid mass loss up to 100 °C corresponds to the removal of weakly bound (surface) water while the moderate mass loss from 100 °C to 200 °C corresponds to the removal of remaining crystal water.64,65
3.3. Electrochemical performance
Redox peaks of vanadium pentoxide are observed at +3.4 V, +3.2 V, and +2.4 V vs. Li/Li+ for the phase transition of α–ε, ε–δ, and δ–γ, respectively, via the following equations:28,66–68| ε-Li0.5V2O5 ↔ α-V2O5 + 0.5Li + 0.5e− | (3) |
| δ-LiV2O5 ↔ ε-Li0.5V2O5 + 0.5Li + 0.5e− | (4) |
| γ-Li2V2O5 ↔ δ-LiV2O5 + Li + e− | (5) |
Redox peaks at +2.95 V and +2.55 V vs. Li/Li+ have been reported for hydrated vanadium pentoxide aerogels, which also show a pronounced pseudocapacitive behavior (i.e., capacitor-like current response) in the potential range of 2.9–3.5 V.40,64,65,69,70 As shown in 1 M LiClO4 EC/DMC organic solution in the cyclic voltammograms for vanadium pentoxide deposited ACC electrodes (Fig. 5a), the specific current response of the ACC-15-VOx electrode shows two pairs of redox peaks at +2.6 V and +2.9 V vs. Li/Li+ as well as a large anodic and cathodic current distribution in the potential range from 3 V to 4 V without showing distinct peaks. The cyclic voltammogram of ACC-15-VOx is consistent with previous studies40,64,65,69,70 and aligns with an amorphous character of vanadium pentoxide or the presence of the hydrated structure V2O5·nH2O.
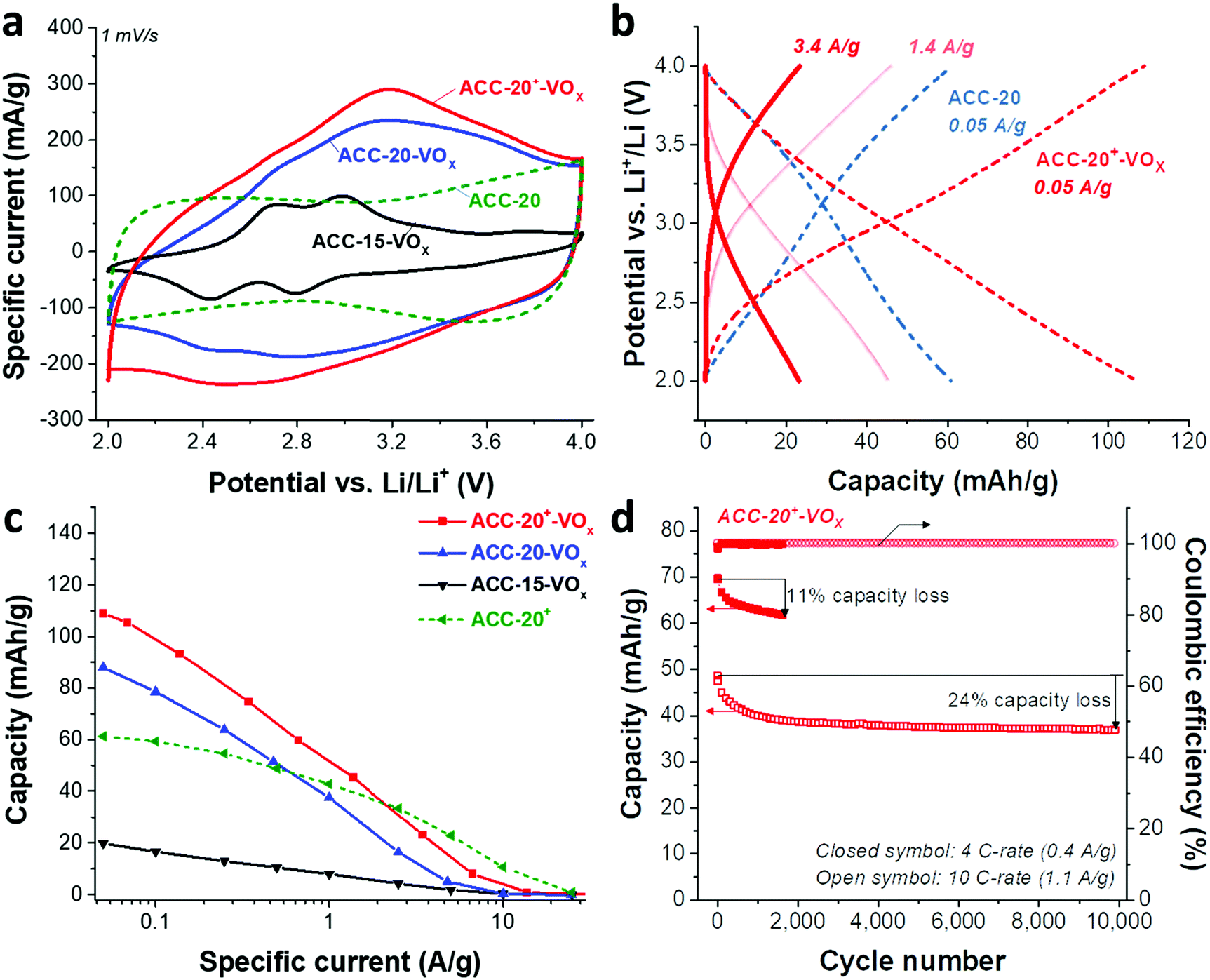 | ||
| Fig. 5 (a) Cyclic voltammograms from the 1st cycle recorded at 1 mV s−1, (b) galvanostatic cycling with potential limitation, and (c) specific capacities of the ACC-20+ electrode and the electrodeposited ACC electrodes. (d) Cycling stability of the ACC-20+-VOx electrode at 4C and 10 C-rate for 1600 and 10000 cycles, respectively. | ||
When the specific current of the ACC-15-VOx electrode is compared to the reference sample (ACC-20 electrode), we see that the contribution of electric double-layer capacitance (EDLC; ca. 100 mA g−1 throughout the potential range) is greatly reduced after electrodeposition while the clear redox peaks arise with a low current response (<100 mA g−1). Considering the narrow average pore size of 0.67 nm of the ACC-15 material (Fig. 1b), it seems that the ion electrosorption is hindered after the electrodeposition process. In contrast to the electrodeposited ACC-15 electrode, the electrodeposited ACC-20 (ACC-20-VOx) and ACC-20+ (ACC-20+-VOx) exhibit a higher current response (>100 mA g−1) while the broad peaks possibly originate from the redox reactions of hydrated vanadium pentoxide at +2.6 V and +2.9 V vs. Li/Li+. The contribution of electric double-layer formation in the micropores of ACC-20-VOx and ACC-20+-VOx possibly causes diffuse redox peaks in the cyclic voltammograms.
The specific capacities (Fig. 5c) of the different electrodes were quantified by galvanostatic charge/discharge cycling with potential limitation (Fig. 5b) and normalized to the total mass of the electrode. The highest capacity of 109 mA h g−1 (at 50 mA g−1, Table 3) was obtained from ACC-20+-VOx which also corresponds to the highest current response observed from the cyclic voltammetry (Fig. 5a). The maximum specific capacity of ACC-15-VOx and ACC-20-VOx was 20 mA h g−1 and 88 mA h g−1, respectively. The galvanostatic charge/discharge profile (Fig. 4b) of ACC-20+-VOx shows that the system is operating in a capacitor-like (pseudocapacitive) manner. The overall energy storage mechanism is most probably the combination of electric double-layer formation in the carbon nanopores and lithium intercalation to amorphous vanadium pentoxide. There remains a high charge storage contribution of electric double-layer formation when considering a large carbon content (72 mass% determined from TGA analysis, Table 3).
Given that the amount of vanadium pentoxide in ACC-15-VOx, ACC-20-VOx, and ACC-20+-VOx is 17 mass%, 13 mass%, and 28 mass%, respectively, the capacity of each electrode can be estimated by assuming the theoretical capacity of V2O5 of 294 mA h g−1 for the potential window from 2 V to 4 V vs. Li/Li+. For instance, the capacity of ACC-15-VOx can be estimated by considering the contribution of the electric double-layer capacity of 57 mA g−1 (measured value from ACC-15, Table 3) for the carbon content of 83 mass% and the contribution of lithium intercalation (theoretical capacity of 294 mA h g−1) for the vanadium pentoxide content of 17 mass%. The estimated value is about 97 ± 12 mA g−1 with a measurement error of ca. 5%. As seen in Table 3, the estimated value for ACC-15-VOx is much higher than the measured value of 20 mA h g−1. This further supports that the deposition of V2O5 in the carbon micropores hinders the effective electrodeposition process. Furthermore, the difference between the estimated and the measured value also indicates that the deposited vanadium oxides in the carbon fiber could not be effectively used for lithium intercalation since the measured value of 20 mA h g−1 is significantly below the specific capacity of 17 mass% of vanadium oxide (ca. 50 mA g−1). In the case of ACC-20-VOx and ACC-20+-VOx, the estimated and the measured specific capacitance are almost identical. These results indicate that the electrodeposited ACC-20-VOx and ACC-20+-VOx avoid detrimental pore blocking and allow a hybrid charge storage mechanism via electric double-layer formation in the carbon nanopores and concurrent lithium intercalation to vanadium pentoxide.
The best power handling performance among the electrodeposited ACC electrodes was observed for ACC-20+-VOx. At a high specific current of 1 A g−1, ACC-20+-VOx exhibits about 52 mA h g−1 which is even higher than that of the pure porous carbon electrode (41 mA h g−1, the ACC-20 electrode without electrodeposition). The high power handling performance of the ACC-20+-VOx electrode reflects fast ion adsorption/desorption (electric double-layer formation) in the carbon micropores,71 and a short diffusion length for the lithium ion intercalation. According to the FIB-EDX and GSA analysis of ACC-20+-VOx (Fig. 3C), hydrated vanadium pentoxide is believed to be deposited inside the carbon micropores while still preserving a large microporosity. Therefore, the size of the hydrated vanadium pentoxide domains will be below 2 nm. As the diffusion length for the lithium intercalation is in the range of a few nanometers, the power handling performance can be greatly enhanced.21,22,57
The cycling stability of ACC-20+-VOx (Fig. 5d) was tested at a rate of 4C (equal to 0.4 A g−1) and 10C (equal to 1.1 A g−1) via galvanostatic cycling in the potential range from 2 V to 4 V vs. Li. At a rate of 4C, the capacity retention of the ACC-20+-VOx electrode was about 89% after 1600 cycles while a capacity retention of about 76% was observed even at 10 C-rate after 10000 cycles. Compared to the reported capacity fading rate of vanadium pentoxide with carbon additives (0.2–0.4% after 100 cycles),19 the capacity fading rate for ACC-20+-VOx is very low (<0.1%, ESI, Fig. S6b†). This performance stability is possibly enabled by the high carbon content in the electrode and the confined vanadium pentoxide structure in the carbon nanopores.
3.4. Influence of annealing temperature
According to previous studies,44,64,65,72 vanadium pentoxide with a hydrated disordered structure can provide a higher lithium intercalation capacity than that of orthorhombic vanadium pentoxide because the water molecules increase the distance between the adjacent V2O5 layers. For hydrated disordered vanadium pentoxides, thermal annealing is often applied to reduce the level of crystal water while preserving the interlayer spacing.44,65 For further optimization, therefore, the as-electrodeposited ACC-20+-VOx electrode was annealed at 200 °C and 330 °C for 1 h in synthetic air (100 mL min−1 20% oxygen and 80% nitrogen) while the temperature was increased at 10 °C per min. The characteristic peaks from Raman spectra of the annealed and non-annealed electrodes (Fig. 6a) are attributed to vanadium pentoxide according to the characteristic peaks at 264 cm−1, 512 cm−1, 706 cm−1, and 1017 cm−1. The disappearance of the peak at 680 cm−1 after annealing at 330 °C indicates that the phase of hydrated disordered vanadium pentoxide is changed to the ordered state. The emergence of broad peaks at 7.8, 40.6, and 50.7° 2θ in the X-ray diffractogram for the electrode after annealing at 200 °C (Fig. 6b) indicates that the phase of the vanadium pentoxides starts to convert to a more ordered phase from its initial disordered phase. Particularly, the peak at 7.8° 2θ can be assigned to the (001) peak of hydrated vanadium pentoxide (Fig. S4d†).73 As the annealing temperature increases to 330 °C, distinct peaks can be identified as orthorhombic vanadium pentoxide (PDF #41-1426).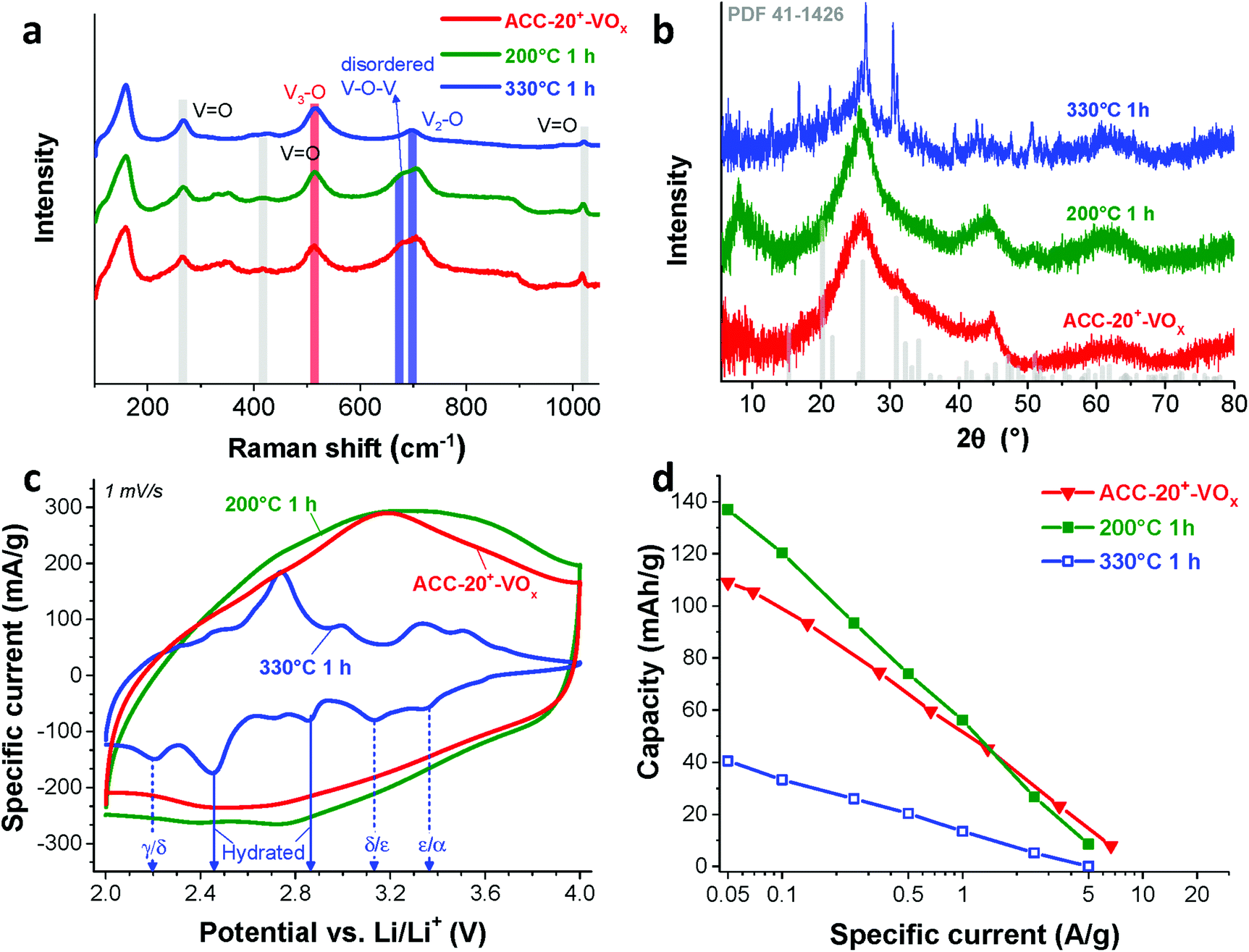 | ||
| Fig. 6 (a) Raman spectra, (b) X-ray diffractograms, and (c) cyclic voltammograms from the 1st cycle at 1 mV s−1, and (d) specific capacities of the non-annealed ACC-20+-VOx electrode and the annealed electrodes at 200 °C and 330 °C for 1 h. | ||
The phase transition from the hydrated disordered state to ordered orthorhombic can be further seen from the cyclic voltammograms (Fig. 6c, 1 mV s−1). In addition to the redox peaks observed for the hydrated disordered vanadium pentoxide (Fig. 5a), the electrode annealed at 330 °C exhibits the emergence of redox peaks which can be attributed to the ordered orthorhombic vanadium pentoxide viaeqn (3)–(5). According to the reduction peaks, the electrode after annealing at 330 °C shows hydrated disordered and ordered orthorhombic features. After annealing at 200 °C, no noticeable peaks were observed; however, the current response was higher than that of the non-annealed electrode which leads to a higher maximum capacity of 137 mA h g−1. The decreased capacity after annealing at 330 °C can originate from the collapse of the interlayer space via the removal of water crystals.44,65 The removal of water crystals is further supported by the low level of mass loss in the temperature regime from 30 °C to 200 °C for the electrode annealed at 330 °C compared to the non-annealed electrodes (Fig. 4d).
For hybrid electrodes where the charge storage combines electric-double layer formation on microporous carbon and the lithium intercalation into vanadium pentoxide, the integrity of the carbon pore structure must be critically assessed. After annealing at 200 °C for 1 h, no structural change can be seen in the scanning electron micrographs (Fig. 7a) as compared to that of the non-annealed electrode (Fig. 2c). In contrast, a huge change in the morphology is observed after annealing at 330 °C for 1 h (Fig. 7b and c). The thickness of the fiber became smaller, and the surface became significantly rough. Since carbon readily oxidizes above 250 °C in synthetic air (Fig. 4d), the structural change originates most probably from carbon oxidation during annealing at 330 °C. The loss of carbon mass further supports the carbon oxidation as evidenced by the increased mass of the vanadium oxide after annealing at 330 °C (Fig. 4d). Hence, the outer part of the carbon fiber seems to be oxidized resulting in a rough surface while the inner part maintained its fiber form (Fig. 7c). However, the oxidation of carbon led to a significant reduction of pore volume (Fig. 7d).
 | ||
| Fig. 7 Scanning electron micrographs of the ACC-20+-VOx electrodes after annealing at different temperatures for 1 h (a–c). (d) Nitrogen sorption isotherms of ACC-20+-VOx recorded at −196 °C and the annealed electrode at 330 °C for 1 h. | ||
As Wang et al. demonstrated,65 the reduced amount of water in the hydrated vanadium pentoxide plays a crucial role in cycle life time. An annealing temperature of 250 °C leads to the highest cycling stability by reducing the amount of crystal water while keeping the disordered crystal structure of vanadium pentoxide. During galvanostatic charge–discharge cycling (GCPL mode) at 4C from 4 V to 2 V vs. Li/Li+, the electrode annealed at 200 °C exhibits a very high stability (Fig. 8a) with a negligible capacity fading rate over 1600 cycles (<0.01% per cycle, Fig. 8b). Most probably, the stable performance originates from the reduced amount of water in the electrode while preserving the disordered structure (Fig. 6a–c) after annealing at 200 °C.
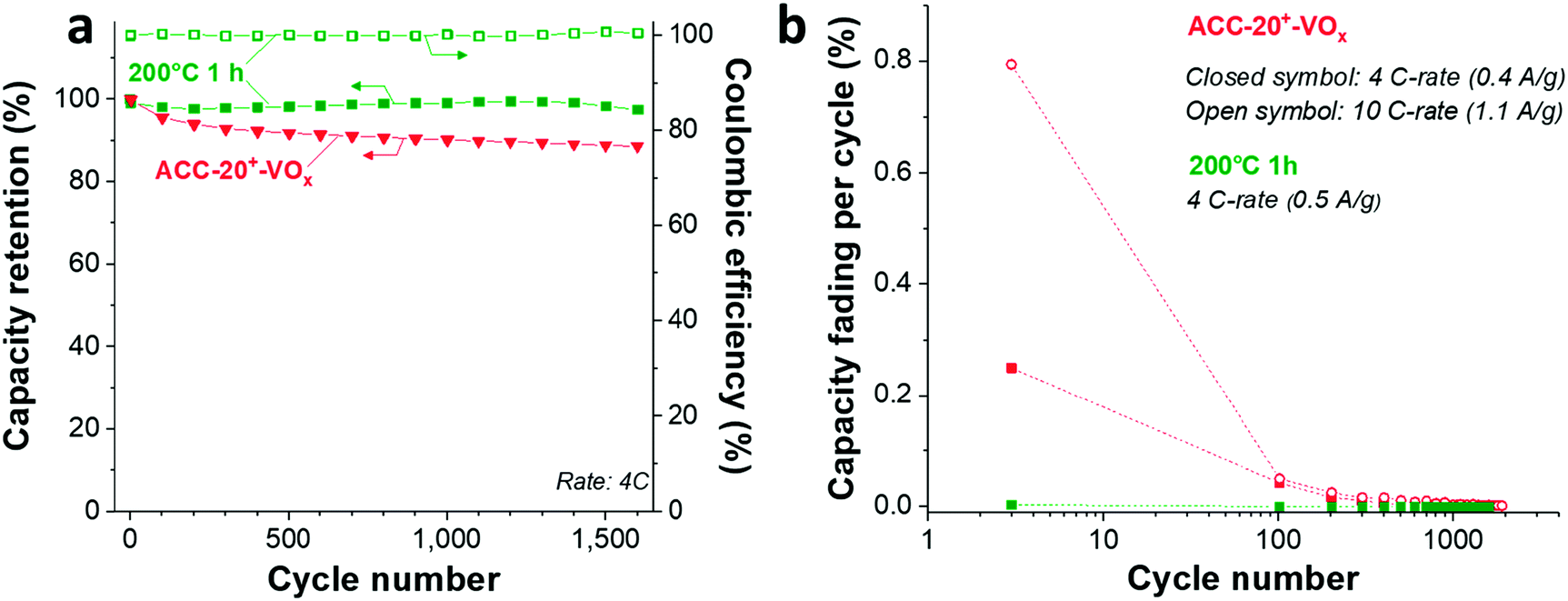 | ||
| Fig. 8 (a) Cycling stability of ACC-20+-VOx before and after annealing at 200 °C for 1 h. (b) Capacity fading per cycle as a rate (y-axis) is plotted as a function of cycle number (x-axis). The high fading rate at a low cycle number indicates that the performance degrades faster in the earlier cycles than in the later cycles. Dashed lines were added to guide the eye. | ||
4. Conclusions
In summary, hydrated disordered vanadium pentoxide was successfully electrodeposited in micropores of activated carbon cloth electrodes with various pore structures. We found that larger carbon pores (>1 nm) are essential for the effective deposition of vanadium pentoxide in the micropores. Activated carbon cloth only with pores <1 nm is not suitable since the deposition of vanadium pentoxide in the narrow nanopores significantly reduces the active surface area for the electric double-layer formation and hinders the effective lithium diffusion into the fiber core. Through annealing the electrodeposited activated carbon cloth with broad micropores (ACC-20+-VOx) at 200 °C for 1 h, a highest energy storage capacity of 137 mA h g−1 was achieved. Our study demonstrates that hydrated disordered vanadium pentoxide can be effectively electrodeposited in carbon micropores with sizes >1 nm for hybrid energy storage via electric double-layer formation and lithium intercalation. Further performance improvement can be obtained via thermal annealing, but the latter must be limited to around 200 °C to prevent carbon oxidization and loss of pore volume.Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. P. acknowledges financial support by the German Academic Exchange Service (DAAD award number 91579066). The authors thank Prof. Eduard Arzt (INM) and Prof. Haiwoong Park (Korea University of Technology and Education) for their kind and continuing support. The authors thank Öznil Budak, Benjamin Krüner, Aura Tolosa, Simon Fleischmann, Eunho Lim, Ingrid Grobelsek and Mesut Aslan (all at INM) for technical support and helpful discussions.References
- M. R. Lukatskaya, B. Dunn and Y. Gogotsi, Nat. Commun., 2016, 7, 12647 CrossRef PubMed.
- F. Béguin, V. Presser, A. Balducci and E. Frackowiak, Adv. Mater., 2014, 26, 2219–2251 CrossRef PubMed.
- L. Xia, L. Yu, D. Hu and G. Z. Chen, Mater. Chem. Front., 2017, 1, 584–618 RSC.
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef CAS PubMed.
- J. Lee, S. Choudhury, D. Weingarth, D. Kim and V. Presser, ACS Appl. Mater. Interfaces, 2016, 8, 23676–23687 CAS.
- J. Lee, B. Krüner, A. Tolosa, S. Sathyamoorthi, D. Kim, S. Choudhury, K.-H. Seo and V. Presser, Energy Environ. Sci., 2016, 9, 3392–3398 CAS.
- M. Salanne, B. Rotenberg, K. Naoi, K. Kaneko, P.-L. Taberna, C. Grey, B. Dunn and P. Simon, Nat. Energy, 2016, 1, 16070 CrossRef CAS.
- J. Lee, A. Tolosa, B. Krüner, N. Jäckel, S. Fleischmann, M. Zeiger, D. Kim and V. Presser, Sustainable Energy Fuels, 2017, 1, 299–307 CAS.
- B. Krüner, J. Lee, N. Jäckel, A. Tolosa and V. Presser, ACS Appl. Mater. Interfaces, 2016, 8, 9104–9115 Search PubMed.
- J. Lee, P. Srimuk, S. Fleischmann, A. Ridder, M. Zeiger and V. Presser, J. Mater. Chem. A, 2017, 5, 12520–12527 CAS.
- I. Shown, A. Ganguly, L.-C. Chen and K.-H. Chen, Energy Sci. Eng., 2015, 3, 2–26 CrossRef CAS.
- M. Zhi, C. Xiang, J. Li, M. Li and N. Wu, Nanoscale, 2013, 5, 72–88 RSC.
- M. Zeiger, D. Weingarth and V. Presser, ChemElectroChem, 2015, 2, 1117–1127 CrossRef CAS.
- T. Brousse, D. Bélanger and J. W. Long, J. Electrochem. Soc., 2015, 162, A5185–A5189 CrossRef CAS.
- V. Augustyn, P. Simon and B. Dunn, Energy Environ. Sci., 2014, 7, 1597–1614 CAS.
- V. Augustyn, J. Come, M. A. Lowe, J. W. Kim, P.-L. Taberna, S. H. Tolbert, H. D. Abruña, P. Simon and B. Dunn, Nat. Mater., 2013, 12, 518–522 CrossRef CAS PubMed.
- S. Fleischmann, M. Zeiger, N. Jäckel, B. Krüner, V. Lemkova, M. Widmaier and V. Presser, J. Mater. Chem. A, 2017, 5, 13039–13051 CAS.
- J. Lee, P. Srimuk, K. Aristizabal, C. Kim, S. Choudhury, Y. C. Nah, F. Mücklich and V. Presser, ChemSusChem, 2017, 10, 3611–3623 CrossRef CAS PubMed.
- Y. Yue and H. Liang, Adv. Energy Mater., 2017, 1602545, DOI:10.1002/aenm.201602545.
- S.-H. Ng, T. J. Patey, R. Buchel, F. Krumeich, J.-Z. Wang, H.-K. Liu, S. E. Pratsinis and P. Novak, Phys. Chem. Chem. Phys., 2009, 11, 3748–3755 RSC.
- X.-F. Zhang, K.-X. Wang, X. Wei and J.-S. Chen, Chem. Mater., 2011, 23, 5290–5292 CrossRef CAS.
- G. Li, Y. Qiu, Y. Hou, H. Li, L. Zhou, H. Deng and Y. Zhang, J. Mater. Chem. A, 2015, 3, 1103–1109 CAS.
- H. g. Wang, D. l. Ma, Y. Huang and X. b. Zhang, Chem.–Eur. J., 2012, 18, 8987–8993 CrossRef CAS PubMed.
- S.-L. Chou, J.-Z. Wang, J.-Z. Sun, D. Wexler, M. Forsyth, H.-K. Liu, D. R. MacFarlane and S.-X. Dou, Chem. Mater., 2008, 20, 7044–7051 CrossRef CAS.
- C. Ban, N. A. Chernova and M. S. Whittingham, Electrochem. Commun., 2009, 11, 522–525 CrossRef CAS.
- B. X. Li, Y. Xu, G. X. Rong, M. Jing and Y. Xie, Nanotechnology, 2006, 17, 2560–2566 CrossRef CAS PubMed.
- A. M. Glushenkov, V. I. Stukachev, M. F. Hassan, G. G. Kuvshinov, H. K. Liu and Y. Chen, Cryst. Growth Des., 2008, 8, 3661–3665 CAS.
- A. M. Cao, J. S. Hu, H. P. Liang and L. J. Wan, Angew. Chem., Int. Ed., 2005, 44, 4391–4395 CrossRef CAS PubMed.
- L. Mai, L. Xu, C. Han, X. Xu, Y. Luo, S. Zhao and Y. Zhao, Nano Lett., 2010, 10, 4750–4755 CrossRef CAS PubMed.
- S. Zhou, X. Yang, Y. Lin, J. Xie and D. Wang, ACS Nano, 2011, 6, 919–924 CrossRef PubMed.
- Y.-Z. Zheng, H. Ding, E. Uchaker, X. Tao, J.-F. Chen, Q. Zhang and G. Cao, J. Mater. Chem. A, 2015, 3, 1979–1985 CAS.
- K.-H. Kim, D.-K. Roh, I. K. Song, B.-C. Lee and S.-H. Baeck, J. Solid State Electrochem., 2010, 14, 1801–1805 CrossRef CAS.
- A. S. Etman, A. K. Inge, X. Jiaru, R. Younesi, K. Edström and J. Sun, Electrochim. Acta, 2017, 252, 254–260 CrossRef CAS.
- D. Kong, X. Li, Y. Zhang, X. Hai, B. Wang, X. Qiu, Q. Song, Q.-H. Yang and L. Zhi, Energy Environ. Sci., 2016, 9, 906–911 CAS.
- A. Moretti and S. Passerini, Adv. Energy Mater., 2016, 6, 1600868 CrossRef.
- D. Vernardou, A. Sapountzis, E. Spanakis, G. Kenanakis, E. Koudoumas and N. Katsarakis, J. Electrochem. Soc., 2013, 160, D6–D9 CrossRef CAS.
- D. Rehnlund, M. Valvo, K. Edström and L. Nyholm, J. Electrochem. Soc., 2014, 161, D515–D521 CrossRef CAS.
- J.-K. Lee, G.-P. Kim, I. K. Song and S.-H. Baeck, Electrochem. Commun., 2009, 11, 1571–1574 CrossRef CAS.
- M.-H. Bai, T.-Y. Liu, F. Luan, Y. Li and X.-X. Liu, J. Mater. Chem. A, 2014, 2, 10882–10888 CAS.
- E. Potiron, A. Le Gal La Salle, A. Verbaere, Y. Piffard and D. Guyomard, Electrochim. Acta, 1999, 45, 197–214 CrossRef CAS.
- K. Ma, X. Liu, Q. Cheng, P. Saha, H. Jiang and C. Li, J. Power Sources, 2017, 357, 71–76 CrossRef CAS.
- K.-I. Park, H.-M. Song, Y. Kim, S.-i. Mho, W. I. Cho and I.-H. Yeo, Electrochim. Acta, 2010, 55, 8023–8029 CrossRef CAS.
- X. Jia, L. Zhang, R. Zhang, Y. Lu and F. Wei, RSC Adv., 2014, 4, 21018–21022 RSC.
- Q. Liu, Z.-F. Li, Y. Liu, H. Zhang, Y. Ren, C.-J. Sun, W. Lu, Y. Zhou, L. Stanciu, E. A. Stach and J. Xie, Nat. Commun., 2015, 6, 6127 CrossRef PubMed.
- S. Fleischmann, N. Jäckel, M. Zeiger, B. Krüner, I. Grobelsek, P. Formanek, S. Choudhury, D. Weingarth and V. Presser, Chem. Mater., 2016, 28, 2802–2813 CrossRef CAS.
- H. Yamada, K. Tagawa, M. Komatsu, I. Moriguchi and T. Kudo, J. Phys. Chem. C, 2007, 111, 8397–8402 CAS.
- A. Ghosh, E. J. Ra, M. Jin, H.-K. Jeong, T. H. Kim, C. Biswas and Y. H. Lee, Adv. Funct. Mater., 2011, 21, 2541–2547 CrossRef CAS.
- Q. Wang, Y. Zou, C. Xiang, H. Chu, H. Zhang, F. Xu, L. Sun and C. Tang, Ceram. Int., 2016, 42, 12129–12135 CrossRef CAS.
- C. Kim, P. Srimuk, J. Lee, S. Fleischmann, M. Aslan and V. Presser, Carbon, 2017, 122, 329–335 CrossRef CAS.
- J. Lee, N. Jackel, D. Kim, M. Widmaier, S. Sathyamoorthi, P. Srimuk, C. Kim, S. Fleischmann, M. Zeiger and V. Presser, Electrochim. Acta, 2016, 222, 1800–1805 CrossRef CAS.
- G. Y. Gor, M. Thommes, K. A. Cychosz and A. V. Neimark, Carbon, 2012, 50, 1583–1590 CrossRef CAS.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CrossRef CAS.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
- G. Silversmit, D. Depla, H. Poelman, G. B. Marin and R. De Gryse, J. Electron Spectrosc. Relat. Phenom., 2004, 135, 167–175 CrossRef CAS.
- D. Weingarth, M. Zeiger, N. Jäckel, M. Aslan, G. Feng and V. Presser, Adv. Energy Mater., 2014, 4, 1400316 CrossRef.
- J. Lee, D. Weingarth, I. Grobelsek and V. Presser, Energy Technol., 2016, 4, 75–84 CrossRef CAS.
- Q. An, Q. Wei, L. Mai, J. Fei, X. Xu, Y. Zhao, M. Yan, P. Zhang and S. Huang, Phys. Chem. Chem. Phys., 2013, 15, 16828–16833 RSC.
- J. J. Niu, V. Presser, C. J. Karwacki and Y. Gogotsi, Mater. Express, 2011, 1, 259–266 CAS.
- C. Piccirillo, R. Binions and I. P. Parkin, Chem. Vap. Deposition, 2007, 13, 145–151 CrossRef CAS.
- S.-H. Lee, H. M. Cheong, M. J. Seong, P. Liu, C. E. Tracy, A. Mascarenhas, J. R. Pitts and S. K. Deb, Solid State Ionics, 2003, 165, 111–116 CrossRef CAS.
- S. Boukhalfa, K. Evanoff and G. Yushin, Energy Environ. Sci., 2012, 5, 6872–6879 CAS.
- C. Julien, G. Nazri and O. Bergström, Phys. Status Solidi B, 1997, 201, 319–326 CrossRef CAS.
- G. Teran-Escobar, J. Pampel, J. M. Caicedo and M. Lira-Cantu, Energy Environ. Sci., 2013, 6, 3088–3098 CAS.
- A. S. Etman, H. D. Asfaw, N. Yuan, J. Li, Z. Zhou, F. Peng, I. Persson, X. Zou, T. Gustafsson, K. Edstrom and J. Sun, J. Mater. Chem. A, 2016, 4, 17988–18001 CAS.
- Y. Wang, H. Shang, T. Chou and G. Cao, J. Phys. Chem. B, 2005, 109, 11361–11366 CrossRef CAS PubMed.
- Y. L. Cheah, N. Gupta, S. S. Pramana, V. Aravindan, G. Wee and M. Srinivasan, J. Power Sources, 2011, 196, 6465–6472 CrossRef CAS.
- A. Tranchant, R. Messina and J. Perichon, J. Electroanal. Chem. Interfacial Electrochem., 1980, 113, 225–232 CrossRef CAS.
- J. M. Cocciantelli, M. Ménétrier, C. Delmas, J. P. Doumerc, M. Pouchard, M. Broussely and J. Labat, Solid State Ionics, 1995, 78, 143–150 CrossRef CAS.
- K. West, B. Zachau-Christiansen, T. Jacobsen and S. Skaarup, Electrochim. Acta, 1993, 38, 1215–1220 CrossRef CAS.
- H. K. Park, W. Smyrl and M. Ward, J. Electrochem. Soc., 1995, 142, 1068–1073 CrossRef CAS.
- F. Beguin and E. Frackowiak, Supercapacitors: Materials, Systems and Applications, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2013 Search PubMed.
- X. Rui, J. Zhu, W. Liu, H. Tan, D. Sim, C. Xu, H. Zhang, J. Ma, H. H. Hng, T. M. Lim and Q. Yan, RSC Adv., 2011, 1, 117–122 RSC.
- Q. Wei, J. Liu, W. Feng, J. Sheng, X. Tian, L. He, Q. An and L. Mai, J. Mater. Chem. A, 2015, 3, 8070–8075 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7se00559h |
| This journal is © The Royal Society of Chemistry 2018 |