
Bio-inspired model of photosystem II: supramolecular assembly of an electron mediator into an SnO2 photoanode co-sensitized by a porphyrin photosensitizer and ruthenium molecular catalyst†
Yong
Na
 *,
Siwen
Miao
,
Li
Zhou
,
Peicheng
Wei
and
Yang
Cao
*,
Siwen
Miao
,
Li
Zhou
,
Peicheng
Wei
and
Yang
Cao
Department of Applied Chemistry, School of Chemistry and Chemical Engineering, Harbin Institute of Technology, Harbin 150001, P. R. China. E-mail: yongna@hit.edu.cn
First published on 2nd January 2018
Abstract
A bio-inspired electron transfer mediator was incorporated via a Zr4+ ion linkage into a photoanode assembled from a porphyrin photosensitizer displaying broad spectral coverage in the visible region and a ruthenium water oxidation catalyst. Incorporation of the electron mediator suppressed unwanted charge recombination and facilitated the transfer of charge from aqueous electrolyte to the photoanode, resulting in an increase in the photocurrent. A remarkable enhancement of faradaic efficiency for oxygen evolution upon introduction of the mediator was observed in the photoelectrochemical cell.
Visible light-driven splitting of water into H2 and O2 is of interest for solar energy conversion, for producing fuels, and for being utilized in a cyclic manner.1 Dye-sensitized photoelectrochemical cells (DS-PECs) for water splitting have attracted worldwide attention.2,3 The photoanode in a DS-PEC device is assembled from molecular dyes and water oxidation catalysts (WOCs) co-sensitized onto a mesoporous metal oxide electrode, where the dye sensitizer absorbs visible light, injects an electron from its excited state into the conduction band of the metal oxide, and subsequently transfers an oxidative equivalent to the catalyst to initiate water oxidation.4 So far, ruthenium tris-bipyridine complexes have been predominantly employed as light-harvesting units in DS-PEC applications, and the photoanodes based on these complexes have shown excellent performances in combination with 2,2′-bipyridine-6,6′-dicarboxylate (bda2−)-chelating ruthenium-based WOCs.3,5–9 However, the energy collection for the above photoanodes in the λ > 520 nm visible light region is poor, which may result in limits in solar-to-fuel conversion efficiency.9 Free base porphyrins and metalloporphyrins usually show a Soret band at a wavelength of 400–450 nm and Q bands at 500–650 nm, and hence display a broader spectral coverage than do ruthenium tris-bipyridine sensitizers.10 In this regard, high-potential electron-deficient porphyrin- and subporphyrin-sensitized photoanodes have very recently been developed for water oxidation, but the performances of the PEC devices based on these photoanodes have not been very promising.11–15 The poor performances may have been due to rapid charge recombination between the photo-injected electrons and the porphyrin radical cation likely caused by the relatively slow kinetics of the 4e−, 4H+ water oxidation reaction.12,16
Recently, a biomimetic model of a tyrosine–histidine pair that mediates electron transfer between the oxygen-evolving complex (OEC) and P680 in natural photosynthesis, has been developed and incorporated into a dye-sensitized photoanode by being covalently attached to a nanoparticle sized IrO2 catalyst for water oxidation, resulting in an acceleration of dye re-generation and a dramatic increase in photocurrent.16 In an attempt to further develop a porphyrin-sensitized photoanode in a DS-PEC application and design a new scheme to suppress the charge recombination, we herein assembled a photoanode by carrying out a stepwise co-sensitization of an SnO2 electrode with tetra(4-carboxyphenyl)-porphyrinato-tin(IV) (SnTPPC, 1), and ruthenium-based WOC with a silane-containing ligand, Ru(II)(bda)(4-picoline)L (L = 3-N-(pyridin-4-ylmethylene)aminopropyltriethoxysilane) (2). The carboxyl groups in SnTPPC provided binding sites for Zr4+ ion as a linkage to incorporate 2-(3,5-di-tert-butyl-2-hydroxyphenyl)-1H-benzimidazol-5(6)-carboxylic acid (3) as an electron mediator that suppressed the charge recombination during the photoreaction (Scheme 1).
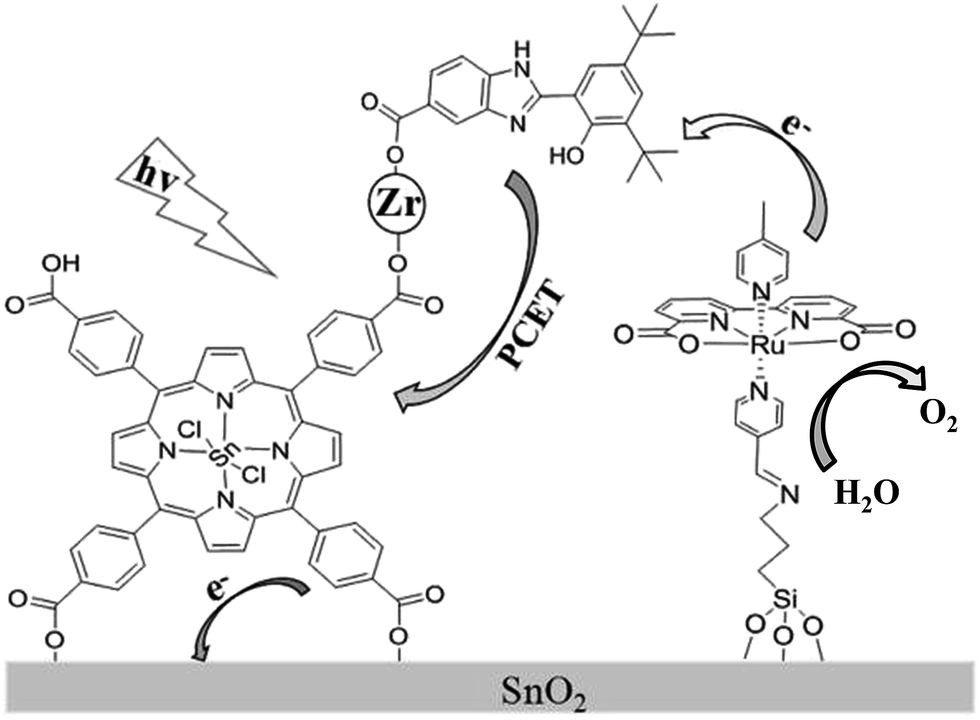 | ||
| Scheme 1 Schematic illustration of the photoanode assembled from porphyrin 1, RuWOC 2 and bio-inspired electron mediator 3. | ||
Published comparisons of the redox potentials of SnTPPC and Ru(bpy)32+ showed the oxidation potential of SnTPPC to be 0.13 V more positive than that of the commonly used Ru(bpy)32+, supporting a thermodynamically allowed transfer of an oxidative equivalent from the radical cation of photosensitizer 1 to catalyst 2 for water oxidation.5,17 But due to the photo-reducing capability of SnTPPC being 0.16 eV weaker than that of Ru(bpy)32+, and due to the conduction band of SnO2 being 0.3–0.5 eV more positive than that of TiO2, the mesoporous SnO2 electrode was chosen as the platform for molecular assembly.17,18 The rutile-structured SnO2 was synthesized using the hydrothermal method with tin(II) chloride as a starting material (see the ESI† for details). The broadness of the peaks in the X-ray diffraction spectrum of the synthesized sample indicated its nano-sized nature (20 nm), and the peaks were indexed to a pure cassiterite tetragonal phase (Fig. S1, ESI†). A mesoporous SnO2 film with a thickness of 10 μm was deposited onto an FTO glass substrate and then sensitized by 1, which was characterized by using UV-vis spectroscopy. The photosensitizer 1 in a methanol solution showed an intense absorption peak for the Soret band at a wavelength of 423 nm and three absorption peaks for the Q-bands at 517, 558 and 597 nm (Fig. S2, ESI†). Binding of 1 to SnO2 led to a slight red shift of the absorption peaks of 1 to 428, 521, 561 and 601 nm, respectively (Fig. S3, ESI†). In addition, all of the absorption peaks for 1 bound to SnO2 were much broader than those for 1 in the methanol solution, indicating the good solar spectral coverage displayed by the FTO/SnO2/1 electrode. This electrode was then immersed in a methanolic solution of catalyst 2 for co-sensitization. The amount of photosensitizer 1 on the FTO/SnO2/1 + 2 electrode was determined to be 5.6 × 10−8 mol cm−2 according to the absorbance of desorbed 1 from the FTO/SnO2/1 + 2 electrode in an aqueous solution of NaOH (0.05 M) at a wavelength of 421 nm (ε = 224![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1). Analysis of X-ray photoelectron spectroscopy (XPS) data demonstrated a 7:1 ratio of Cl to Ru, suggesting a 7:2 ratio of photosensitizer 1 to catalyst 2 on the film. The incorporation of 2 into the FTO/SnO2/1 electrode was further confirmed by carrying out cyclic voltammetry (CV) in the dark. As shown in Fig. 1, the FTO/SnO2/1 electrode was found to display no catalytic activity toward water oxidation even though its oxidation potential at 1.37 V (vs. Ag|AgCl) for 1 in a potassium phosphate-buffered aqueous solution (KPi buffer, pH = 6.86) was shown to be higher than the equilibrium potential of water oxidation. By contrast, an obvious increase in current intensity due to the catalytic water oxidation with an onset potential at 1.11 V (vs. Ag|AgCl) was observed after catalyst 2 was incorporated. A control experiment with the FTO/SnO2/2 electrode in a KPi buffer solution showed the first oxidation wave for 2 with peak potentials at Ep,ox = 0.60 V and Ep,red = 0.39 V (Fig. S4, ESI†), which might be attributed to the oxidation of the RuII catalyst to the RuIV species, but no catalytic wave was found in its cyclic voltammogram, probably due to the kinetically inhibited electron transfer at the electrode.19 Therefore, the catalysis of 2 toward water oxidation can be mediated by photosensitizer 1 when they are co-assembled on the surface of the FTO/SnO2 electrode. Based on a 1.99 eV value for the zero–zero energy (E00) of photosensitizer 1 in combination with the above CV results, we calculated the energy levels and proposed an electron-transfer process for the FTO/SnO2/1 + 2 electrode under light illumination as shown in Fig. 2a.
000 M−1 cm−1). Analysis of X-ray photoelectron spectroscopy (XPS) data demonstrated a 7:1 ratio of Cl to Ru, suggesting a 7:2 ratio of photosensitizer 1 to catalyst 2 on the film. The incorporation of 2 into the FTO/SnO2/1 electrode was further confirmed by carrying out cyclic voltammetry (CV) in the dark. As shown in Fig. 1, the FTO/SnO2/1 electrode was found to display no catalytic activity toward water oxidation even though its oxidation potential at 1.37 V (vs. Ag|AgCl) for 1 in a potassium phosphate-buffered aqueous solution (KPi buffer, pH = 6.86) was shown to be higher than the equilibrium potential of water oxidation. By contrast, an obvious increase in current intensity due to the catalytic water oxidation with an onset potential at 1.11 V (vs. Ag|AgCl) was observed after catalyst 2 was incorporated. A control experiment with the FTO/SnO2/2 electrode in a KPi buffer solution showed the first oxidation wave for 2 with peak potentials at Ep,ox = 0.60 V and Ep,red = 0.39 V (Fig. S4, ESI†), which might be attributed to the oxidation of the RuII catalyst to the RuIV species, but no catalytic wave was found in its cyclic voltammogram, probably due to the kinetically inhibited electron transfer at the electrode.19 Therefore, the catalysis of 2 toward water oxidation can be mediated by photosensitizer 1 when they are co-assembled on the surface of the FTO/SnO2 electrode. Based on a 1.99 eV value for the zero–zero energy (E00) of photosensitizer 1 in combination with the above CV results, we calculated the energy levels and proposed an electron-transfer process for the FTO/SnO2/1 + 2 electrode under light illumination as shown in Fig. 2a.
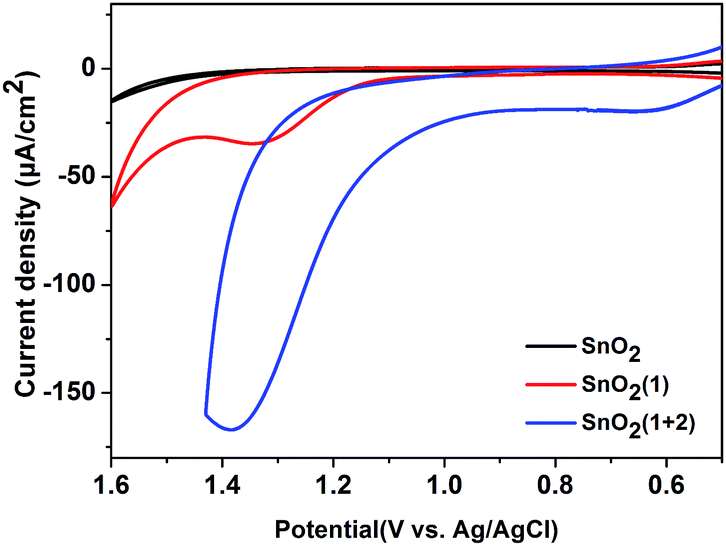 | ||
| Fig. 1 CVs of the FTO/SnO2 electrode (black), FTO/SnO2/1 electrode (red) and FTO/SnO2/1 + 2 electrode (blue) each in a KPi buffer solution (pH = 6.86) using a three-electrode system in the dark. | ||
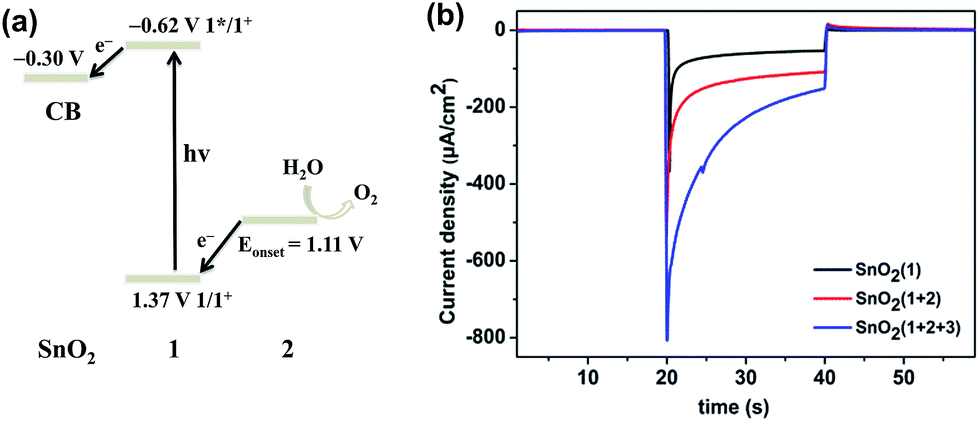 | ||
| Fig. 2 (a) Energy levels and proposed electron transfer processes in the present system. The potentials are referenced to the Ag|AgCl electrode. (b) I–T curves of the FTO/SnO2/1 photoanode (black), FTO/SnO2/1 + 2 photoanode (red) and FTO/SnO2/1 + 2 + 3 photoanode (blue) each in a KPi buffer solution (pH = 6.86) under illumination with 0.4 V (vs. Ag|AgCl) bias using a three-electrode system. | ||
The FTO/SnO2/1 + 2 electrode was employed as a photoanode that was fabricated into a DS-PEC device with Ag|AgCl (KClsat.) as the reference electrode, Pt foil as the counter electrode, and KPi buffer solution (pH = 6.86) as the electrolyte. Measurements of photocurrent at various applied potentials were first taken in order to determine an appropriate external bias for the present PEC device. Linear sweep voltammetry (LSV) with a light-illuminated FTO/SnO2/1 + 2 working electrode demonstrated that the current rapidly increased and then reached a maximum in the range 0.21 V to 0.46 V (vs. Ag|AgCl), and no further increase of the photocurrent was found at higher potentials (Fig. S5, ESI†). In contrast, there was no catalytic activity in the dark until the applied potential reached 0.8 V. The onset potential for water oxidation with FTO/SnO2/1 + 2 photoanode under light illumination negatively shifted to be 0.21 V (vs. Ag|AgCl) in comparison to that at 1.11 V in the dark, indicating that photosensitizer 1 can indeed inject an electron to the SnO2 film in action of light and subsequently transfer oxidative equivalent to catalyst 2 to initiate water oxidation. In view of the above LSV results, a bias potential of 0.4 V (vs. Ag|AgCl) for the present PEC device was adopted to promote electron transfer from the photoanode to cathode. As an experimental control, the FTO/SnO2/1 photoanode exhibited an initial photocurrent of 125 μA cm−2 under simulated sunlight illumination (AM 1.5G, 100 mW cm−2). Co-adsorption of catalyst 2 made significant contribution to the photocurrent transient of FTO/SnO2/1 + 2 photoanode, but the initial photocurrent of 400 μA cm−2 decreased rapidly to 110 μA cm−2 in less than 15 s (Fig. 2b red line), and the generation of oxygen after photo-assisted electrolysis for 100 s was confirmed by using a Clark-type oxygen electrode with the electrolyte near the photoanode as a sample (Fig. S6, ESI†). The steep decay of the photocurrent was probably caused by the charge recombination between the photo-injected electrons in SnO2 and the radical cation of 1 and/or the decomposition of 1 due to the slow reaction kinetics of water oxidation. As a result, the faradaic efficiency for oxygen evolution was only 28% as determined by quantification of the oxygen dissolved in the electrolyte using the Clark-type oxygen electrode.
Inspired by the work of the Mallouk group,16 we speculated that an electron mediator positioned between the photosensitizer 1 and catalyst 2 would promote the regeneration of the photosensitizer 1 and suppress the unwanted charge recombination. Accordingly, a bio-inspired electron mediator 3 containing the COOH group was prepared in order to combine it with the photosensitizer 1via a Zr4+ ion linkage, which had been proved to be an effective way to assemble components on an electrode.6,12 CV measurements of 1 and the esterified product of 3 (denoted as 3-COOCH3) in CH3CN were first taken to determine the thermodynamic feasibility of electron transfer from 3 to the radical cation of 1, in which a free energy ΔG of −0.33 eV was calculated according to the difference between the oxidation potential (Epc,ox) of 1 and that of 3-COOCH3 (Fig. S7, ESI†). The regeneration of 1 promoted by the electron mediator 3 during the photoreaction was then confirmed by performing surface photovoltage (SPV) spectroscopy, which has been established to provide the state of charge carrier on the surface of a material under light illumination.20 As shown in Fig. 3, the SnO2/1 film gave positive surface photovoltage signals, indicating that electrons were injected into the SnO2 film, leaving the positive charges (holes) accumulated at the surface of the film. The photovoltage intensity spectrum for the SnO2/1 film in the visible region was found to be in agreement with the absorption spectrum of photosensitizer 1 (Fig. S3, ESI†). This agreement suggested the photo-induced charge separation indeed resulted from photo-excitation of 1.20 When 3 was combined with 1via a Zr4+ ion linkage on the film, the intensity of the surface photovoltage for the SnO2/1 + 3 film increased, indicating more positive charges (holes) accumulated at the surface in comparison to that without 3.21 This result was mainly due to regeneration of 1, promoted by the electron mediator 3, enhancing the photoinduced charge separation between the electrons in the film and the holes on the surface of the film.
 | ||
| Fig. 3 SPV spectra of FTO/SnO2/1 (black) and FTO/SnO2/1 + 3 (red) photoanodes. | ||
Since a promotion of charge separation would be expected to benefit the performance of the photoanode, the electron mediator 3 was incorporated into the FTO/SnO2/1 + 2 electrode by making a Zr4+ ion connection with photosensitizer 1 (the ratio of 1 to 3 was ca. 5:4 based on X-ray photoelectron spectroscopy results). The possibility that water oxidation catalyzed by 2 could be mediated by incorporation of 3 was first investigated by taking CV measurements of the FTO/SnO2/1 + 2 + 3 electrode in a KPi buffer solution in the dark (Fig. S8, ESI†). Even though the redox event for 3 was not observed, which was consistent with the result from the Mallouk group, the FTO/SnO2/1 + 2 + 3 electrode displayed catalytic activity toward water oxidation with an onset potential at 1.01 V (vs. Ag|AgCl), which negatively shifted by nearly 100 mV in comparison to that of the FTO/SnO2/1 + 2 electrode without 3, suggesting that incorporation of 3 into the FTO/SnO2/1 + 2 photoanode could also facilitate the oxidation of catalyst 2 to a high-valence Ru-intermediate for water oxidation. As expected, photo-irradiation of the FTO/SnO2/1 + 2 + 3 photoanode indeed slowed down the decay of the photocurrent in comparison to that of FTO/SnO2/1 + 2 photoanode, and the initial photocurrent of 800 μA cm−2 decreased to 156 μA cm−2 in 20 s (Fig. 2b blue line), which was one and half times that without 3. After photo-assisted electrolysis for 100 s, small oxygen bubbles on the surface of the photoanode were clearly observed, but the amount of the produced oxygen was difficult to determine accurately (Fig. S9, ESI†).22 Hence, the faradaic efficiency for oxygen evolution was at least 61% (Fig. S10, ESI†), which was much higher than that without 3.
In order to get more insight into the function of the electron mediator 3, photoelectrochemical impedance spectroscopy (PEIS) was also used to investigate the charge transfer process at the photoanode-electrolyte interface.23 Nyquist plots were collected at a bias potential of 0.4 V under simulated sunlight illumination. As shown in Fig. 4, incorporation of the electron mediator 3 resulted in a smaller capacitive arc radius. Fitting the impedance spectrum to an appropriate equivalent circuit clearly showed that incorporation of 3 led to a decrease in Rct, meaning that the interface charge transfer rate was enhanced. As a result, the value of the photocurrent dramatically increased and the decay of the photocurrent was slowed down.
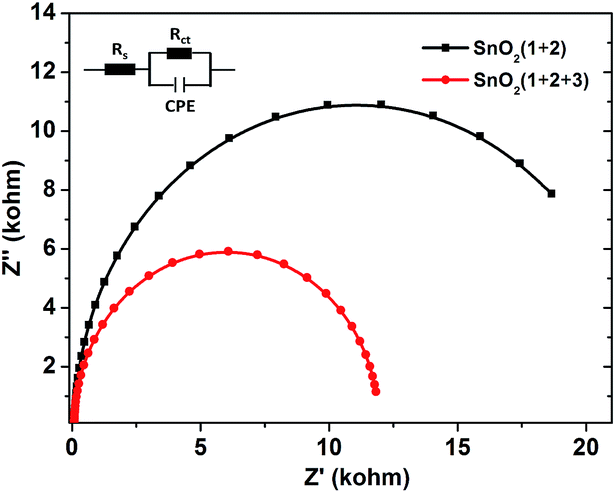 | ||
| Fig. 4 Nyquist plots of PEIS measurements with FTO/SnO2/1 + 2 (black) and FTO/SnO2/1 + 2 + 3 (red) photoanodes each in a KPi buffer solution (pH = 6.86) at a bias potential of 0.4 V under simulated sunlight illumination. | ||
In conclusion, a porphyrin 1-sensitized nano-structured SnO2 substrate with broad spectral response in the visible region has been successfully used in DS-PEC for water splitting by performing a co-sensitization of the photoanode with molecular catalyst 2. In addition, a new strategy for incorporating the bio-inspired electron mediator 3 was developed based on forming a Zr4+ ion connection. The electron mediator was shown to promote the regeneration of the photosensitizer and to facilitate water oxidation at the surface of the photoanode. Although the performance of the present assembly is not optimal at this stage, the strategy may be of value in solar fuel device applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21603046) and the Heilongjiang Postdoctoral Fund, China (LBH-Z14106).Notes and references
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729 CrossRef CAS PubMed.
- J. R. Swierk and T. E. Mallouk, Chem. Soc. Rev., 2013, 42, 2357 RSC.
- Z. Yu, F. Li and L. Sun, Energy Environ. Sci., 2015, 8, 760 CAS.
- M. K. Brennaman, R. J. Dillon, L. Alibabaei, M. K. Gish, C. J. Dares, D. L. Ashford, R. L. House, G. J. Meyer, J. M. Papanikolas and T. J. Meyer, J. Am. Chem. Soc., 2016, 138, 13085 CrossRef CAS PubMed.
- Y. Gao, X. Ding, J. Liu, L. Wang, Z. Lu, L. Li and L. Sun, J. Am. Chem. Soc., 2013, 135, 4219 CrossRef CAS PubMed.
- X. Ding, Y. Gao, L. Zhang, Z. Yu, J. Liu and L. Sun, ACS Catal., 2014, 4, 2347 CrossRef CAS.
- X. Ding, Y. Gao, L. Ye, L. Zhang and L. Sun, ChemSusChem, 2015, 8, 3992 CrossRef CAS PubMed.
- F. Li, K. Fan, L. Wang, Q. Daniel, L. Duan and L. Sun, ACS Catal., 2015, 5, 3786 CrossRef CAS.
- H. Li, F. Li, Y. Wang, L. Bai, F. Yu and L. Sun, ChemPlusChem, 2016, 81, 1056 CrossRef CAS.
- L. L. Li and E. W. Diau, Chem. Soc. Rev., 2013, 42, 291 RSC.
- G. F. Moore, J. D. Blakemore, R. L. Milot, J. F. Hull, H. Song, L. Cai, C. A. Schmuttenmaer, R. H. Crabtree and G. W. Brudvig, Energy Environ. Sci., 2011, 4, 2389 CAS.
- A. Nayak, R. R. Knauf, K. Hanson, L. Alibabaei, J. J. Concepcion, D. L. Ashford, J. L. Dempsey and T. J. Meyer, Chem. Sci., 2014, 5, 3115 RSC.
- J. R. Swierk, D. D. Méndez-Hernández, N. S. McCool, P. Liddell, Y. Terazono, I. Pahk, J. J. Tomlin, N. V. Oster, T. A. Moore, A. L. Moore, D. Gust and T. E. Mallouk, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 1681 CrossRef CAS PubMed.
- M. Yamamoto, L. Wang, F. Li, T. Fukushima, K. Tanaka, L. Sun and H. Imahori, Chem. Sci., 2016, 7, 1430 RSC.
- M. Yamamoto, Y. Nishizawa, P. Chabera, F. Li, T. Pascher, V. Sundström, L. Sun and H. Imahori, Chem. Commun., 2016, 52, 13702 RSC.
- Y. Zhao, J. R. Swierk, J. D. Megiatto Jr, B. Sherman, W. J. Youngblood, D. Qin, D. M. Lentz, A. L. Moore, T. A. Moore, D. Gust and T. E. Mallouk, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15612 CrossRef CAS PubMed.
- A. M. Manke, K. Geisel, A. Fetzer and P. Kurz, Phys. Chem. Chem. Phys., 2014, 16, 12029 RSC.
- M. K. Gish, A. M. Lapides, M. K. Brennaman, J. L. Templeton, T. J. Meyer and J. M. Papanikolas, J. Phys. Chem. Lett., 2016, 7, 5297 CrossRef CAS PubMed.
- M. V. Sheridan, B. D. Sherman, Z. Fang, K.-R. Wee, M. K. Coggins and T. J. Meyer, ACS Catal., 2015, 5, 4404 CrossRef CAS.
- Z. Liu, K. Pan, Q. Zhang, M. Liu, R. Jia, Q. Lü, D. Wang, Y. Bai and T. Li, Thin Solid Films, 2004, 468, 291 CrossRef CAS.
- D. Xu, Z. Fu, D. Wang, Y. Lin, Y. Sun, D. Meng and T. Xie, Phys. Chem. Chem. Phys., 2015, 17, 23924 RSC.
- Only trace amount of oxygen was observed during analysis of the sample in the headspace by gas chromatography.
- B. Klahr, S. Gimenez, F. Fabregat-Santiago, J. Bisquert and T. W. Hamann, Energy Environ. Sci., 2012, 5, 7626 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7se00515f |
| This journal is © The Royal Society of Chemistry 2018 |