
A noble metal-free photocatalytic system based on a novel cobalt tetrapyridyl catalyst for hydrogen production in fully aqueous medium†
N.
Queyriaux‡
a,
E.
Giannoudis‡
 b,
C. D.
Windle
b,
S.
Roy
a,
J.
Pécaut
c,
A. G.
Coutsolelos
*b,
V.
Artero
a and
M.
Chavarot-Kerlidou
*a
b,
C. D.
Windle
b,
S.
Roy
a,
J.
Pécaut
c,
A. G.
Coutsolelos
*b,
V.
Artero
a and
M.
Chavarot-Kerlidou
*a
aLaboratoire de Chimie et Biologie des Métaux, Univ. Grenoble Alpes, CNRS UMR 5249, CEA, 17 rue des martyrs, F-38054, Grenoble Cedex 9, France. E-mail: murielle.chavarot-kerlidou@cea.fr
bLaboratory of Bioinorganic Chemistry, Department of Chemistry, University of Crete, Voute Campus, 70013 Heraklion, Crete, Greece. E-mail: acoutsol@uoc.gr
cUniv. Grenoble Alpes, CEA, CNRS, INAC, SYMMES, UMR 5819 Equipe Chimie Interface Biologie pour l’Environnement la Santé et la Toxicologie, F-38054, Grenoble Cedex 9, France
First published on 7th December 2017
Abstract
The new cobalt tetrapyridyl complex 1(BF4)2 was characterized and assessed for hydrogen production in fully aqueous solution. Mechanistic information was gained thanks to a fast screening method, using chemical reductants and a Clark microelectrode. Optimal hydrogen production (443 TONs – 3 mL of H2) was achieved under visible light-driven conditions, in the presence of a noble metal-free photosensitizer, the water soluble porphyrin 2Cl4, and the ascorbate/tris-(2-carboxyethyl)phosphine (TCEP) sacrificial electron donor system.
In the context of artificial photosynthesis, the development of molecular photocatalytic systems for hydrogen production from sunlight and water is a highly active research area.1–4 Hydrogen is recognized as a promising alternative to fossil fuels in the context of the continually growing global energy demand. However, this carbon-free energy carrier needs to be produced via low-cost processes in order to be economically viable.5 This has prompted the development of noble metal-free molecular H2-evolving catalysts to provide an alternative to the use of rare and expensive noble metals such as platinum.6–8 Of particular interest are the recently reported polypyridyl and polyaminopyridyl cobalt complexes.9,10 These coordination spheres are known to efficiently accommodate the different redox states involved in the cobalt-based catalytic cycle. As a consequence, these catalysts display increased robustness and high efficiencies for electrocatalytic and photocatalytic H2 evolution under both aqueous and non-aqueous conditions, the former being a strategic choice for the targeted application. When switching to fully aqueous conditions for light-driven proton reduction, the choice of the light-harvesting unit becomes restricted compared to organic or mixed organic/aqueous conditions. While Ru(bpy)3Cl2 remains most widely employed,11–14 some rhenium complexes have also been shown to be effective.15 Both of them nevertheless rely on noble metals. Examples of noble metal-free molecular systems for photocatalytic H2 production active in pure water are even scarcer. Erythrosin dyes were successfully combined with nickel–thiolate catalysts, self-assembled in solution;16,17 however, this led to a complex mixture of structures, preventing any identification of the real catalytically active species, and limiting further developments and implementation of these catalysts. Two other studies reported very low activities for cobaloximes combined with either rose bengal or a tin porphyrin, in agreement with the known catalyst instability in aqueous solutions.18,19 The single reported example relying on the combination of a polypyridyl catalyst with the same tin porphyrin displayed no activity at all.19 The development of efficient molecular photocatalytic systems based on earth abundant elements and active in fully aqueous medium is therefore a key challenge in this field of solar fuel production.
Porphyrin photosensitizers have been extensively employed to study light-driven electron transfer processes, but surprisingly they found little use in photocatalytic water splitting.20 We recently reported original H2-evolving photocatalytic systems based on the water soluble porphyrin photosensitizer 2Cl4 (Fig. 1A) and cobaloxime catalysts, active under mixed organic/aqueous conditions.21,22 We describe here visible light-driven hydrogen production under fully aqueous conditions using a novel polypyridyl cobalt catalyst, 1(BF4)2, in combination with 2Cl4. Optimized performances are reached in water at pH 4.5 when TCEP is employed as the ultimate electron donor23 in combination with ascorbate as an electron relay.
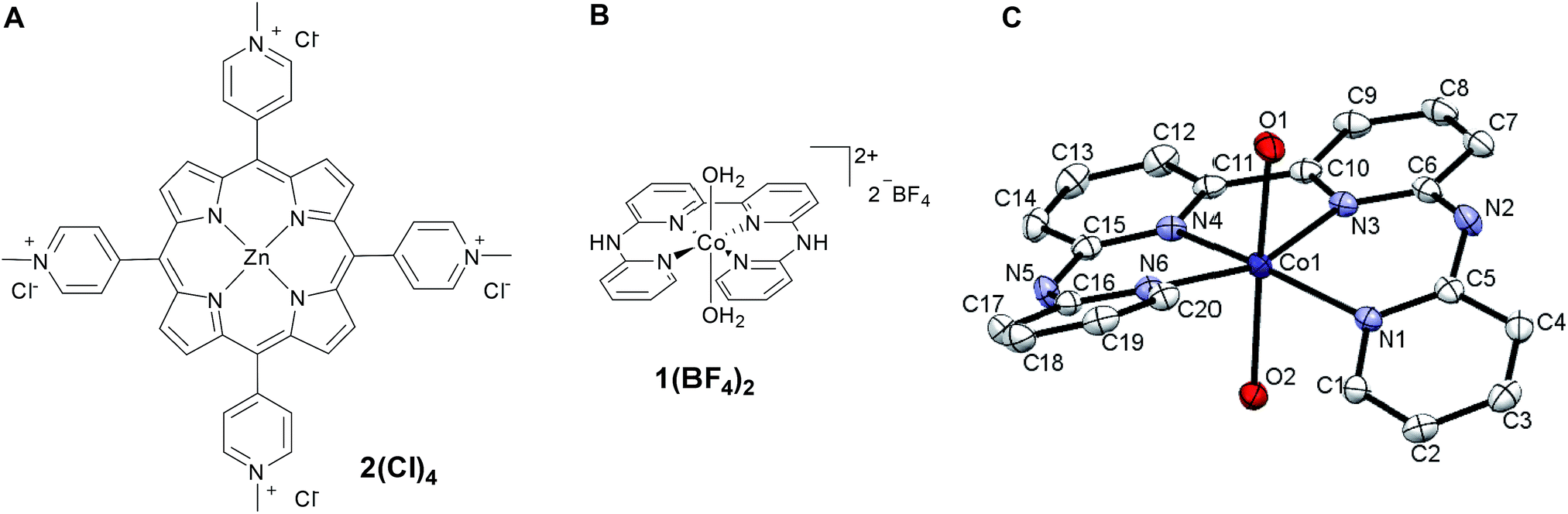 | ||
| Fig. 1 Schematic representations of water-soluble photosensitizer 2Cl4 (A) and catalyst 1(BF4)2 (B); X-ray structure of 1(BF4)2 (C), counter-ions and hydrogen atoms have been removed for clarity. | ||
Metalation of the 6,6′-bis-(2-aminopyridyl)-2,2′-bipyridine24,25 (bapbpy) tetrapyridyl ligand was achieved with Co(BF4)2·6H2O to yield the water-soluble complex 1(BF4)2 (Fig. 1B). Single crystals suitable for X-ray diffraction analysis were grown by slow evaporation of a H2O/THF solution of 1(BF4)2 (Fig. 1C). Complex 1(BF4)2 displays a distorted octahedral geometry, where the bapbpy ligand occupies the basal plane and two water molecules occupy the apical positions. Due to steric hindrance between hydrogen atoms in the ortho positions of the terminal pyridine rings, the ligand adopts a saddle-shaped conformation with a dihedral angle of 41.9774(12)°. Other relevant geometric features are summarized in Table S1.†
The cyclic voltammogram (CV) of a DMF solution of 1(BF4)2 (Fig. S1†) displays a relatively poorly defined wave at +0.12 V vs. Fc+/Fc, which is assigned to the CoIII/CoII oxidation process. Sweeping down towards cathodic potentials, a reversible process at −1.37 V vs. Fc+/Fc and an irreversible process at −1.99 V vs. Fc+/Fc are observed. These processes have been assigned to the CoII/CoI and bapbpy ligand-centred (CoI/CoI–L˙−) reductions, respectively, by comparison with the CV of the complex of the redox-inactive Zn2+ cation (Fig. S1†). In order to assess the hydrogen evolution activity of 1(BF4)2 in aqueous medium, the fast screening method we recently reported was first employed.26 It is based on the use of chemical reducing agents and a Clark microelectrode for H2 detection. Hydrogen production was observed in Tris–HCl buffer (pH 7), in the presence of a 20-fold excess of the europium salt EuII–DTPA (−1.14 V vs. NHE)26 (Fig. 2). 1(BF4)2 produced hydrogen with an overall conversion of 45(±5)%, corresponding to 4.5(±0.5) turnovers. It is worth noting that 1(BF4)2 did not generate hydrogen in the presence of the weaker reductant EuII–EGTA (−0.81 V vs. NHE),26 in agreement with the near-zero driving force for the reduction of 1(BF4)2 by EuII–EGTA (Table 1). The potential required for 1(BF4)2 to catalyse H2 evolution in aqueous medium at pH 7 is therefore located between −0.81 and −1.14 V vs. NHE, in agreement with the electrochemical process observed at −1.07 V vs. NHE in the linear sweep voltammogram of 1(BF4)2 recorded in 0.1 M Tris–HCl buffer pH 7 (Fig. S2†). This corresponds to an overpotential requirement of 650 mV (E°(H+/H2) = −0.42 V vs. NHE at pH 7) in line with related cobalt–polypyridyl catalysts.27,28
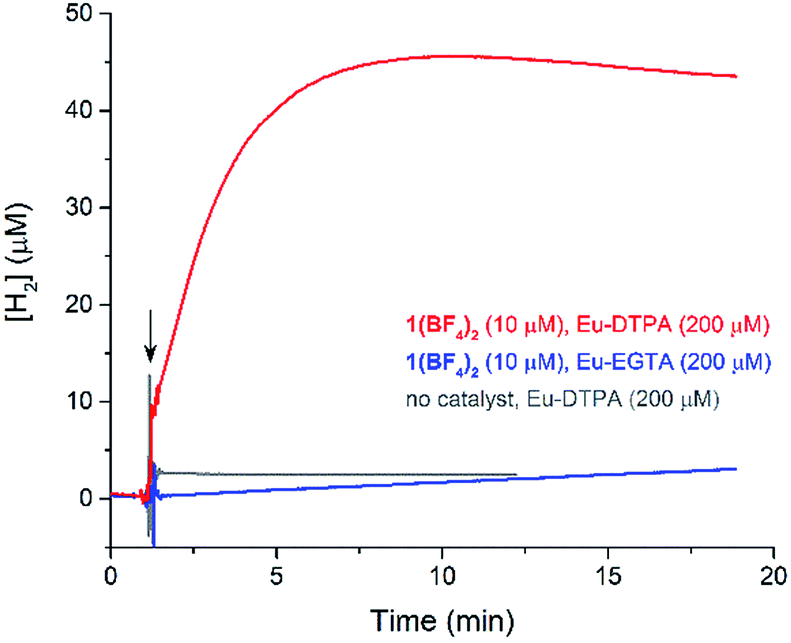 | ||
| Fig. 2 Traces of hydrogen production recorded by a Clark microelectrode upon the addition of an excess of europium reductants (200 μM, 20 equiv. with respect to 1(BF4)2) to an aqueous buffered solution of 1(BF4)2 (10 μM, pH 7). The arrow indicates the injection of the reductant. | ||
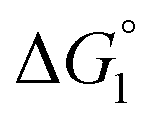 = driving force for the reduction of CoII to CoI;
= driving force for the reduction of CoII to CoI; 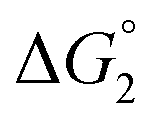 = driving force for the reduction of CoI to CoI–L˙−) for the different electron transfer processes. Potentials measured versus Fc+/Fc were converted to NHE using E(Fc+/Fc) = +0.57 V vs. NHE in DMF33
= driving force for the reduction of CoI to CoI–L˙−) for the different electron transfer processes. Potentials measured versus Fc+/Fc were converted to NHE using E(Fc+/Fc) = +0.57 V vs. NHE in DMF33
| CoII/CoI | CoI/L˙− | |
|---|---|---|
| 1(BF4)2 | −0.80 | −1.42 |
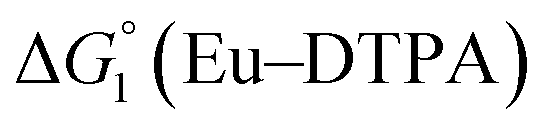
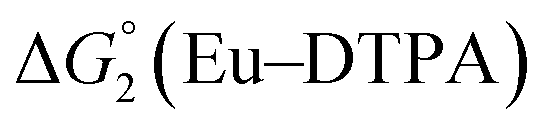
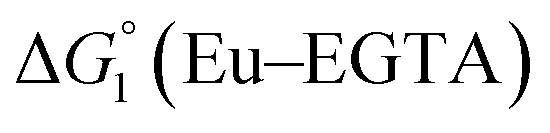
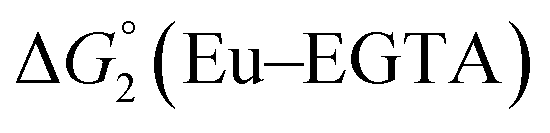
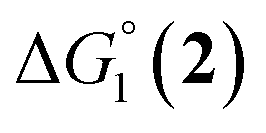
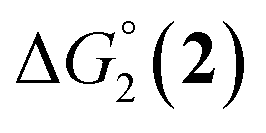
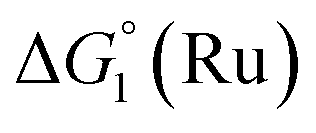
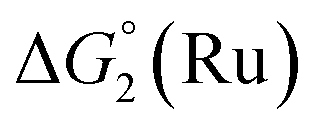
Different mechanisms can account for H2 evolution with cobalt polypyridyl catalysts, some involving participation of redox-active ligands such as the bipyridine moiety in 1(BF4)2.9 On the basis of the thermodynamic driving force ΔG° (Table 1), EuII–DTPA is not able to perform the second reduction on the catalyst; the formation of a CoI–L˙− catalytic intermediate can therefore be excluded. As a consequence, a first protonation step (chemical reaction, C step) should occur immediately after the CoII/CoI reduction (electron transfer, E step), yielding a cobalt–hydride CoIII–H intermediate. At this stage, two pathways have to be considered: (i) a second protonation step (C) leading to H2 production; (ii) a second reduction (E) yielding to a CoII–H hydride, from which hydrogen evolution occurs after protonation (C). When considering the first ECCE sequence,9 strong acidic conditions are usually required to protonate CoIII–H29 which is a weaker nucleophile compared to CoII–H. Such a pathway is therefore unlikely to be operative under these neutral conditions. An ECEC sequence9 thus appears most likely, as previously reported for related cobalt-based catalytic systems.30–32 Studies are currently in progress to provide a more detailed understanding of the catalytic mechanism at work with 1(BF4)2.
Our screening method was also employed under photochemical conditions, using Ru(bpy)3Cl2 as a photosensitizer (PS) in ascorbate buffer (pH 4).26 Hydrogen production was detected in solution, albeit at an extremely slow rate (Fig. S3†), with only 0.5 turnover numbers (calculated with regard to the amount of photosensitizer – TONPS) measured after one hour of irradiation. This lack of activity with Ru(bpy)3Cl2 was further confirmed by photocatalytic tests with gas chromatography quantification of the amount of hydrogen produced. Fig. 3 shows the amount of hydrogen produced over time upon irradiation with white LEDs of pH 4.5 aqueous solutions containing 1(BF4)2, ascorbic acid (AA) as a sacrificial electron donor and either Ru(bpy)3Cl2 or 2Cl4 (Fig. 1) as a photosensitizer. The initial H2 production rate is maintained for approximately 5 hours with 2Cl4 before starting to decline and finally ceasing after 24 hours of irradiation. The system containing 2Cl4/1(BF4)2 reached 18.5 TONPS compared to the <0.1 TONs of the Ru(bpy)3Cl2-based one. This observation is particularly interesting as most of the cobalt-based photocatalytic systems reported so far to be active in fully aqueous media rely on Ru(bpy)3Cl2 as a photosensitizer.9,11–14 Control experiments established that all three components together with light are required for hydrogen evolution (Table S2†). Moreover, mercury poisoning experiments were performed under vigorous stirring and showed no noticeable change in the H2 production rate (Table S2†), ruling out the formation of metallic nanoparticles as the active species during photocatalytic H2 evolution.
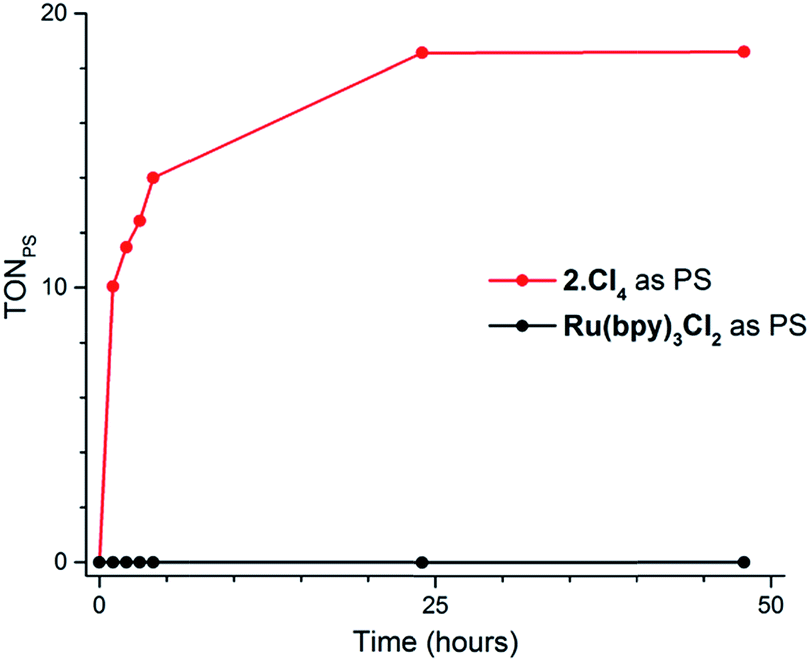 | ||
| Fig. 3 Plots of hydrogen production (TONPS) upon irradiation of pH 4.5 aqueous solutions containing 1(BF4)2 (4.9 × 10−4 M), AA (0.2 M) and Ru(bpy)3Cl2 (4.0 × 10−5 M) or 2Cl4 (4.0 × 10−5 M). | ||
The efficiency of the water-soluble zinc porphyrin photosensitizer compared to Ru(bpy)3Cl2 cannot be simply explained by thermodynamic considerations. Indeed, we previously established that the photocatalytic activity originates from the triplet excited state of 2Cl4.21 On the basis of the associated thermodynamic driving force (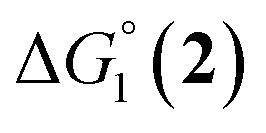 in Table 1), an oxidative quenching process of 2* by 1(BF4)2 can be discarded. Thus, hydrogen production with 2Cl4 is initiated via reductive quenching of PS* by AA, followed by thermal reduction of 1(BF4)2 by PS−. For the Ru-based photocatalytic system, reductive quenching by AA was previously established for a large number of related systems,11,12,36 although the oxidative quenching process by 1(BF4)2 is also slightly exergonic (
in Table 1), an oxidative quenching process of 2* by 1(BF4)2 can be discarded. Thus, hydrogen production with 2Cl4 is initiated via reductive quenching of PS* by AA, followed by thermal reduction of 1(BF4)2 by PS−. For the Ru-based photocatalytic system, reductive quenching by AA was previously established for a large number of related systems,11,12,36 although the oxidative quenching process by 1(BF4)2 is also slightly exergonic (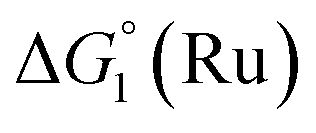 in Table 1). In both cases, a second reduction of the catalyst to form the CoI–L˙− species is thermodynamically disfavoured (
in Table 1). In both cases, a second reduction of the catalyst to form the CoI–L˙− species is thermodynamically disfavoured (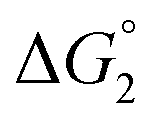 in Table 1). Electron delivery to the cobalt catalyst should therefore parallel chemical reduction by Eu–DTPA, as discussed above.
in Table 1). Electron delivery to the cobalt catalyst should therefore parallel chemical reduction by Eu–DTPA, as discussed above.
Hydrogen evolution previously reported under aqueous conditions with Ru(bpy)3Cl2 and AA relies on strongly different experimental conditions, with an excess of photosensitizer compared to the catalyst (up to 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1).10–14 By contrast, we employ here an excess of the catalyst, according to our previous studies.21,22 Under these conditions, the thermodynamically allowed oxidative quenching of the Ru excited state by 1(BF4)2 could occur and compete with the expected reductive quenching pathway. This could account for the striking difference of activity observed in favour of 2Cl4.
:1).10–14 By contrast, we employ here an excess of the catalyst, according to our previous studies.21,22 Under these conditions, the thermodynamically allowed oxidative quenching of the Ru excited state by 1(BF4)2 could occur and compete with the expected reductive quenching pathway. This could account for the striking difference of activity observed in favour of 2Cl4.
The 2Cl4/1(BF4)2 photocatalytic system was also assessed at pH 7 and pH 8, using triethanolamine (TEOA) as a sacrificial electron donor, all other parameters being unchanged. In all cases, no hydrogen could be detected after 24 hours of irradiation. This behaviour suggests that protonations at the cobalt centre are rate-determining steps for the catalytic process, in full agreement with our previous study employing 2Cl4 with a cobaloxime catalyst.21 When AA was employed under mixed organic/aqueous conditions, the activity was approximately four times lower.
Cessation of hydrogen production coincides with bleaching of the solution (Fig S4†). Photocatalytic activity could not be restored by addition of either the catalyst or photosensitizer, suggesting that both components undergo concomitant decomposition after some hours of irradiation. This was previously observed for other reductive quenching-based photocatalytic systems, where PS− degradation became competitive over time with electron transfer to the catalyst.37–39
Another parameter is known to hamper the hydrogen production activity. Accumulation of dehydroascorbic acid (DHA), the oxidized form of ascorbate produced during catalysis, was shown to self-inhibit the catalytic system developed by Alberto and co-workers;40 electron transfer from PS− to DHA indeed competes with reduction of the catalyst. To prevent this deleterious effect, regeneration of AA from DHA by tris-(2-carboxyethyl)phosphine (TCEP) as the ultimate electron donor has been reported to successfully increase the activity of ruthenium, rhenium and carbon nanodot-based photocatalytic systems.23,41,42
The same effect is observed for the 2Cl4/1(BF4)2 photocatalytic system. In the presence of TCEP (0.1 M) and AA (0.1 M), it reaches 443 TONs, compared to 19 in the absence of TCEP (Fig. 4 and Table S3†). This represents a 23-fold increase of activity and 3 mL (133 μmol) of H2 was produced under our experimental conditions. These results appear within the range of the H2-evolving performances previously reported for related Co catalysts under fully aqueous conditions;10–14 a straightforward comparison of numerical values such as TONs or amount of H2 produced is nevertheless prevented due to important differences in the experimental conditions and set-up (PS/Cat ratios, light sources…) employed.
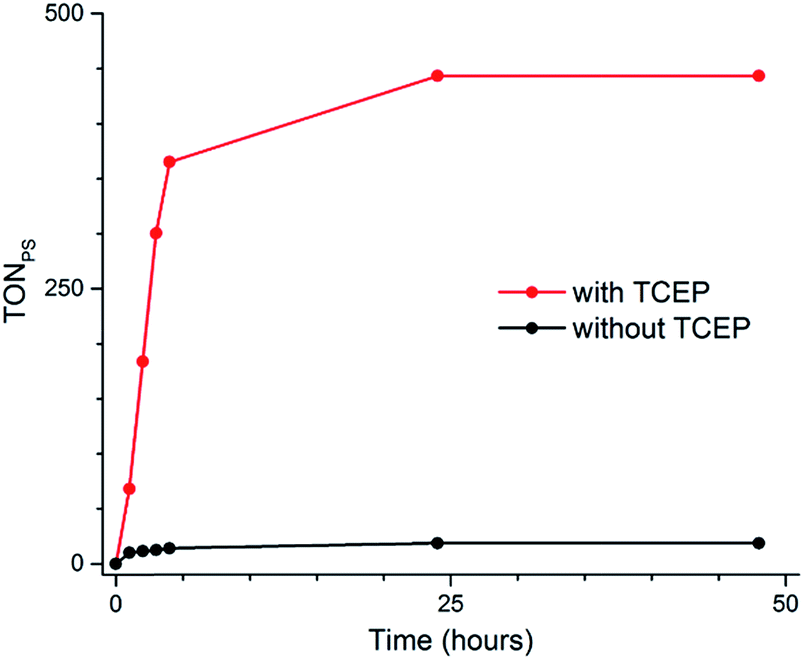 | ||
| Fig. 4 Plots of hydrogen production upon irradiation of pH 4.5 aqueous solutions containing 1(BF4)2 (4.9 × 10−4 M), 2Cl4 (4.0 × 10−5 M), and ascorbic acid (0.2 M) in the absence of TCEP (orange) or ascorbic acid (0.1 M) in the presence of TCEP (0.1 M) (blue). | ||
Conclusions
The new cobalt(II) complex based on the tetradentate 6,6′-bis-(2-aminopyridyl)-2,2′-bipyridine ligand, [Co(bapbpy)(H2O)2]2+ (12+), catalyzes H2 evolution in fully aqueous medium at pH 4.5. Hydrogen production was shown to occur within a potential range of −0.81 and −1.14 V vs. NHE, and a preliminary mechanistic analysis favours an ECEC mechanism. This novel catalyst could be combined with photosensitizers to design one of the rare photocatalytic systems for H2 evolution under visible light-driven conditions that is both efficient and based on earth-abundant elements. This system uses a water-soluble porphyrin as a dye, an approach that remains largely unexplored in that context.19 This work thus opens new perspectives regarding both novel catalytic platforms and water-soluble photosensitizers for the development of efficient and inexpensive photo-electrocatalytic devices based on earth-abundant elements for hydrogen production from sunlight and water.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Colette Lebrun (CEA/DRF/INAC-SyMMES) is acknowledged for the ESI-MS characterization of 1(BF4)2. This work was supported by the European Commission's Seventh Framework Program (FP7/2007–2013) under grant agreement no. 229927 (FP7-REGPOT-2008-1, Project BIOSOLENUTI) and no. 306398 (FP7-IDEAS-ERC, Project PhotocatH2ode), the COST Action CM1202 PERSPECT-H2O, the French National Research Agency (Labex Program, ArCANE, ANR-11-LABX-0003-01) and a Heraklitos grant from the Ministry of Education. Special research account of the University of Crete is also acknowledged.Notes and references
- E. S. Andreiadis, M. Chavarot-Kerlidou, M. Fontecave and V. Artero, Photochem. Photobiol., 2011, 87, 946 CrossRef CAS PubMed.
- T. S. Teets and D. G. Nocera, Chem. Commun., 2011, 47, 9268 RSC.
- W. T. Eckenhoff and R. Eisenberg, Dalton Trans., 2012, 41, 13004 RSC.
- S. Berardi, S. Drouet, L. Francas, C. Gimbert-Surinach, M. Guttentag, C. Richmond, T. Stoll and A. Llobet, Chem. Soc. Rev., 2014, 43, 7501 RSC.
- N. Armaroli and V. Balzani, ChemSusChem, 2011, 4, 21 CrossRef CAS PubMed.
- V. Artero, M. Chavarot-Kerlidou and M. Fontecave, Angew. Chem., Int. Ed., 2011, 50, 7238 CrossRef CAS PubMed.
- P. W. Du and R. Eisenberg, Energy Environ. Sci., 2012, 5, 6012 CAS.
- J. R. McKone, S. C. Marinescu, B. S. Brunschwig, J. R. Winkler and H. B. Gray, Chem. Sci., 2014, 5, 865 RSC.
- N. Queyriaux, R. T. Jane, J. Massin, V. Artero and M. Chavarot-Kerlidou, Coord. Chem. Rev., 2015, 304–305, 3 CrossRef CAS PubMed.
- D. Z. Zee, T. Chantarojsiri, J. R. Long and C. J. Chang, Acc. Chem. Res., 2015, 48, 2027 CrossRef CAS PubMed.
- W. K. C. Lo, C. E. Castillo, R. Gueret, J. Fortage, M. Rebarz, M. Sliwa, F. Thomas, C. J. McAdam, G. B. Jameson, D. A. McMorran, J. D. Crowley, M.-N. Collomb and A. G. Blackman, Inorg. Chem., 2016, 55, 4564 CrossRef CAS PubMed.
- R. S. Khnayzer, V. S. Thoi, M. Nippe, A. E. King, J. W. Jurss, K. A. El Roz, J. R. Long, C. J. Chang and F. N. Castellano, Energy Environ. Sci., 2014, 7, 1477 CAS.
- W. M. Singh, T. Baine, S. Kudo, S. Tian, X. A. N. Ma, H. Zhou, N. J. DeYonker, T. C. Pham, J. C. Bollinger, D. L. Baker, B. Yan, C. E. Webster and X. Zhao, Angew. Chem., Int. Ed., 2012, 51, 5941 CrossRef CAS PubMed.
- S. Varma, C. E. Castillo, T. Stoll, J. Fortage, A. G. Blackman, F. Molton, A. Deronzier and M.-N. Collomb, Phys. Chem. Chem. Phys., 2013, 15, 17544 RSC.
- A. Rodenberg, M. Orazietti, B. Probst, C. Bachmann, R. Alberto, K. K. Baldridge and P. Hamm, Inorg. Chem., 2015, 54, 646 CrossRef CAS PubMed.
- W. Zhang, J. Hong, J. Zheng, Z. Huang, J. Zhou and R. Xu, J. Am. Chem. Soc., 2011, 133, 20680 CrossRef CAS PubMed.
- J. Han, W. Zhang, T. Zhou, X. Wang and R. Xu, RSC Adv., 2012, 2, 8293 RSC.
- P. Zhang, M. Wang, J. Dong, X. Li, F. Wang, L. Wu and L. Sun, J. Phys. Chem. C, 2010, 114, 15868 CAS.
- L. Mintrop, J. Windisch, C. Gotzmann, R. Alberto, B. Probst and P. Kurz, J. Phys. Chem. B, 2015, 119, 13698 CrossRef CAS PubMed.
- K. Ladomenou, M. Natali, E. Iengo, G. Charalampidis, F. Scandola and A. G. Coutsolelos, Coord. Chem. Rev., 2015, 304–305, 38 CrossRef CAS.
- T. Lazarides, M. Delor, I. V. Sazanovich, T. M. McCormick, I. Georgakaki, G. Charalambidis, J. A. Weinstein and A. G. Coutsolelos, Chem. Commun., 2014, 50, 521 RSC.
- A. Panagiotopoulos, K. Ladomenou, D. Sun, V. Artero and A. G. Coutsolelos, Dalton Trans., 2016, 45, 6732 RSC.
- C. Bachmann, B. Probst, M. Guttentag and R. Alberto, Chem. Commun., 2014, 50, 6737 RSC.
- S. Bonnet, M. A. Siegler, J. S. Costa, G. Molnar, A. Bousseksou, A. L. Spek, P. Gamez and J. Reedijk, Chem. Commun., 2008, 5619 RSC.
- E. Molenbroek, N. Straathof, S. Duck, Z. Rashid, J. H. van Lenthe, M. Lutz, A. Gandubert, R. J. M. Klein Gebbink, L. De Cola and S. Bonnet, Dalton Trans., 2013, 42, 2973 RSC.
- S. Roy, M. Bacchi, G. Berggren and V. Artero, ChemSusChem, 2015, 8, 3632 CrossRef CAS PubMed.
- Y. Sun, J. P. Bigi, N. A. Piro, M. L. Tang, J. R. Long and C. J. Chang, J. Am. Chem. Soc., 2011, 133, 9212 CrossRef CAS PubMed.
- L. Tong, R. Zong and R. P. Thummel, J. Am. Chem. Soc., 2014, 136, 4881 CrossRef CAS PubMed.
- J. L. Dempsey, B. S. Brunschwig, J. R. Winkler and H. B. Gray, Acc. Chem. Res., 2009, 42, 1995 CrossRef CAS PubMed.
- A. Bhattacharjee, E. S. Andreiadis, M. Chavarot-Kerlidou, M. Fontecave, M. J. Field and V. Artero, Chem.–Eur. J., 2013, 19, 15166 CrossRef CAS PubMed.
- B. H. Solis and S. Hammes-Schiffer, Inorg. Chem., 2011, 50, 11252 CrossRef CAS PubMed.
- J. T. Muckerman and E. Fujita, Chem. Commun., 2011, 47, 12456 RSC.
- T. N. Huan, E. S. Andreiadis, J. Heidkamp, P. Simon, E. Derat, S. Cobo, G. Royal, A. Bergmann, P. Strasser, H. Dau, V. Artero and M. Fontecave, J. Mater. Chem. A, 2015, 3, 3901 CAS.
- K. Kalyanasundaram and M. Neumann-Spallart, J. Phys. Chem., 1982, 86, 5163 CrossRef CAS.
- A. Juris, V. Balzani, F. Barigelletti, S. Campagna, P. Belser and A. von Zelewsky, Coord. Chem. Rev., 1988, 84, 85 CrossRef CAS.
- C. V. Krishnan and N. Sutin, J. Am. Chem. Soc., 1981, 103, 2141 CrossRef CAS.
- T. Lazarides, T. McCormick, P. W. Du, G. G. Luo, B. Lindley and R. Eisenberg, J. Am. Chem. Soc., 2009, 131, 9192 CrossRef CAS PubMed.
- T. M. McCormick, B. D. Calitree, A. Orchard, N. D. Kraut, F. V. Bright, M. R. Detty and R. Eisenberg, J. Am. Chem. Soc., 2010, 132, 15480 CrossRef CAS PubMed.
- X. Wang, S. b. Goeb, Z. Ji, N. A. Pogulaichenko and F. N. Castellano, Inorg. Chem., 2011, 50, 705 CrossRef CAS PubMed.
- M. Guttentag, A. Rodenberg, R. Kopelent, B. Probst, C. Buchwalder, M. Brandstätter, P. Hamm and R. Alberto, Eur. J. Inorg. Chem., 2012, 2012, 59 CrossRef CAS.
- B. C. M. Martindale, E. Joliat, C. Bachmann, R. Alberto and E. Reisner, Angew. Chem., Int. Ed., 2016, 55, 9402 CrossRef CAS PubMed.
- S. Schnidrig, C. Bachmann, P. Müller, N. Weder, B. Spingler, E. Joliat-Wick, M. Mosberger, J. Windisch, R. Alberto and B. Probst, ChemSusChem, 2017, 10, 4570 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic details of 1(BF4)2; X-ray data; cyclic voltammograms; traces of hydrogen production by 1(BF4)2/Ru(bpy)3Cl2 recorded by a Clark microelectrode; tables of photocatalytic hydrogen production. CCDC 1564999. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7se00428a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |