
Bifunctional electrocatalysts for Zn–air batteries
E.
Davari
 and
D. G.
Ivey
*
and
D. G.
Ivey
*
Department of Chemical and Materials Engineering, University of Alberta, Edmonton, Alberta, Canada T6G 1H9. E-mail: divey@ualberta.ca
First published on 7th November 2017
Abstract
Rechargeable metal–air batteries have recently attracted significant attention because of their high theoretical energy output and low cost. Among metal–air batteries, Zn–air batteries (ZABs) have garnered renewed interest as one of the most viable future options to power energy grids and electric vehicles. However, the slow oxygen reduction reaction (ORR) and oxygen evolution reaction (OER) kinetics represent a limiting factor for the energy conversion efficiency of state-of-the-art ZABs. The focus of this review is the latest advances related to the development of non-precious metal catalysts. Four groups of bifunctional catalysts used in primary/secondary ZABs are reviewed. These include transition metal oxides (single/mixed-metal oxides, spinels and perovskites), transition metals, carbon-based materials and precious metals/alloys. ZAB electrochemistry and challenges originating from individual components of the system, such as the Zn electrodes, electrolytes and separators, are also outlined. In addition, the design and architecture of the air-electrode are discussed for future development of ZABs.
1. Introduction
The demand for energy is increasing in modern society and finding alternatives to fossil fuels is a necessary step towards a low-carbon economy and reducing environmental pollution. According to the World Resources Institute, 61.4% of total greenhouse gas emissions come from the energy sector.1 In recent years, renewable energy sources such as wind, hydroelectric, solar, biomass and geothermal have been increasingly utilized. However, wind and solar power are constrained by climate conditions. Also, pumped hydroelectricity, which is one of the preferred ways of producing electrical energy, is limited by its site-specific nature and the high cost of large scale operation. Therefore, finding a safe, reliable and efficient way to store energy from these renewable and sustainable sources is an urgent need. Electrochemical conversion technologies, such as batteries with high power density, are among the key energy storage devices for the next generation of electronics and green vehicles.2–4 Batteries are closed systems that contain all the necessary components for their operation. The oxidation and reduction reactions (redox) occur due to the potential difference between two opposite electrodes (anode and cathode) in a conductive medium (electrolyte). Therefore, batteries have always been of great interest in the electronics market, and recently the transportation sector, to reduce emissions from fossil fuels.During the last few decades rechargeable batteries such as Pb-acid, Li-ion, redox flow, Na–S and metal–air batteries have been widely investigated.5 Since 1990, Li-ion batteries have been implemented in electronic appliances due to their high energy efficiency, high energy density and long cycle life. However, Li-ion batteries face many challenges, such as a volume change during the charge–discharge process and safety problems.6–8
Among various battery types, metal–air batteries hold great promise for future energy applications. Metal–air batteries offer the highest theoretical energy density (Fig. 1) among all rechargeable battery types.9 This is due to the use of O2 in air at the cathode enabling a significant reduction in cathode size and the overall weight of the battery. During operation of a metal–air battery, the cathode consumes O2 continuously from the air as an active material. The anode material is a metal such as Zn, Al, Mg, Li, Fe or Ca. Among all metal–air batteries, Li–air batteries have the highest theoretical specific energy of 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 140 W h kg−1, which is very close to that of gasoline (12200 W h kg−1). The first successful attempt to design a rechargeable metal–air battery was a study by Abraham et al.10 They constructed a Li–air battery composed of a Li+ conductive organic polymer electrolyte membrane sandwiched between a thin Li metal foil anode and a thin carbon composite as the cathode (air electrode). In 2006, Li–air batteries were revised and modified by Bruce et al.11 and their study initiated more extensive research in this field.
140 W h kg−1, which is very close to that of gasoline (12200 W h kg−1). The first successful attempt to design a rechargeable metal–air battery was a study by Abraham et al.10 They constructed a Li–air battery composed of a Li+ conductive organic polymer electrolyte membrane sandwiched between a thin Li metal foil anode and a thin carbon composite as the cathode (air electrode). In 2006, Li–air batteries were revised and modified by Bruce et al.11 and their study initiated more extensive research in this field.
 | ||
| Fig. 1 Theoretical and practical specific/volumetric energy density of various types of rechargeable batteries.3,9 | ||
The properties of electrochemical metal–air cells are listed in Table 1.12 Aluminum, Mg, Li and Ca anodes have the highest specific energy. Lithium–air batteries have major limitations such as higher cost, an unstable anode which reacts with the electrolyte and safety issues.13 For Al and Mg, rapid corrosion occurs in aqueous electrolytes due to more negative electrode potentials.2,3 Therefore, they only can be used in mechanically rechargeable metal–air batteries by refueling the system with fresh metal at the anode and electrolyte.14
| Anode | Electrochemical equivalent of metal (A h g−1) | Theoretical cell voltage (V) | Practical cell voltage (V) | Theoretical specific energy of the metal (kW h kg−1) | Valence change |
|---|---|---|---|---|---|
| Li | 3.8 | 3.4 | 2.4 | 13.3 | 1 |
| Al | 2.9 | 2.7 | 1.6 | 8.1 | 3 |
| Mg | 2.2 | 3.0 | 1.4 | 6.8 | 2 |
| Ca | 1.3 | 3.4 | 2.0 | 4.6 | 2 |
| Fe | 0.9 | 1.2 | 1.0 | 1.2 | 2 |
| Zn | 0.8 | 1.6 | 1.2 | 1.3 | 2 |
| Cd | 0.4 | 1.2 | 0.8 | 0.6 | 2 |
Of the common types of batteries, zinc–air batteries (ZABs) have the highest energy density and lowest cost. In addition, compared with fuel cells, ZAB anode and cathode designs are less stringent and there is no need for hydrogen production and storage.15,16
The performance of ZAB technologies greatly depends on a pair of sluggish electrochemical reactions at the cathode, i.e., the oxygen reduction reaction (ORR) and the oxygen evolution reaction (OER). Improving the activity and stability of ORR and OER can be achieved by using precious metals as electrocatalysts. Platinum-based electrocatalysts are ORR active, but OER activity is limited due to the formation of an oxide layer at high potentials. Conversely, Ir or Ru-based electrocatalysts are effective OER catalysts, but not as active as Pt for ORR.17 This issue, along with the prohibitive cost and scarcity of precious metals, represents a grand challenge for ZABs. Therefore, the development of a single high-performance, non-precious metal electrocatalyst with simultaneous ORR and OER activity (bifunctional) is critically needed as an alternative to precious metals. In this review, catalysts are categorized into four different groups: noble or precious metals, non-precious transition metals/transition metal oxides, transition metals and carbon-based materials. The review bridges the gap between previous reviews dedicated to bifunctional (ORR/OER) electrocatalyst development and ZAB performance. The review also provides a recent snapshot of the fast-developing field of bifunctional catalysts for rechargeable ZABs over the last few years.
2. Zn–air batteries (ZABs)
A comparison between different properties of metal–air batteries shows that ZABs are one of the most promising metal–air systems due to two main reasons. First, Zn is an abundant metal that has been used by human culture throughout history. World Zn resources are estimated to be 1.8 gigatons and 200 megatons were economically available in 2008.13,18 Second, Zn is a low cost and abundant metal (Zn – US $2 per kg vs. Li – US $8 per kg) which can be fully recycled.18Fig. 2 shows the relationship between metal price and relative abundance.19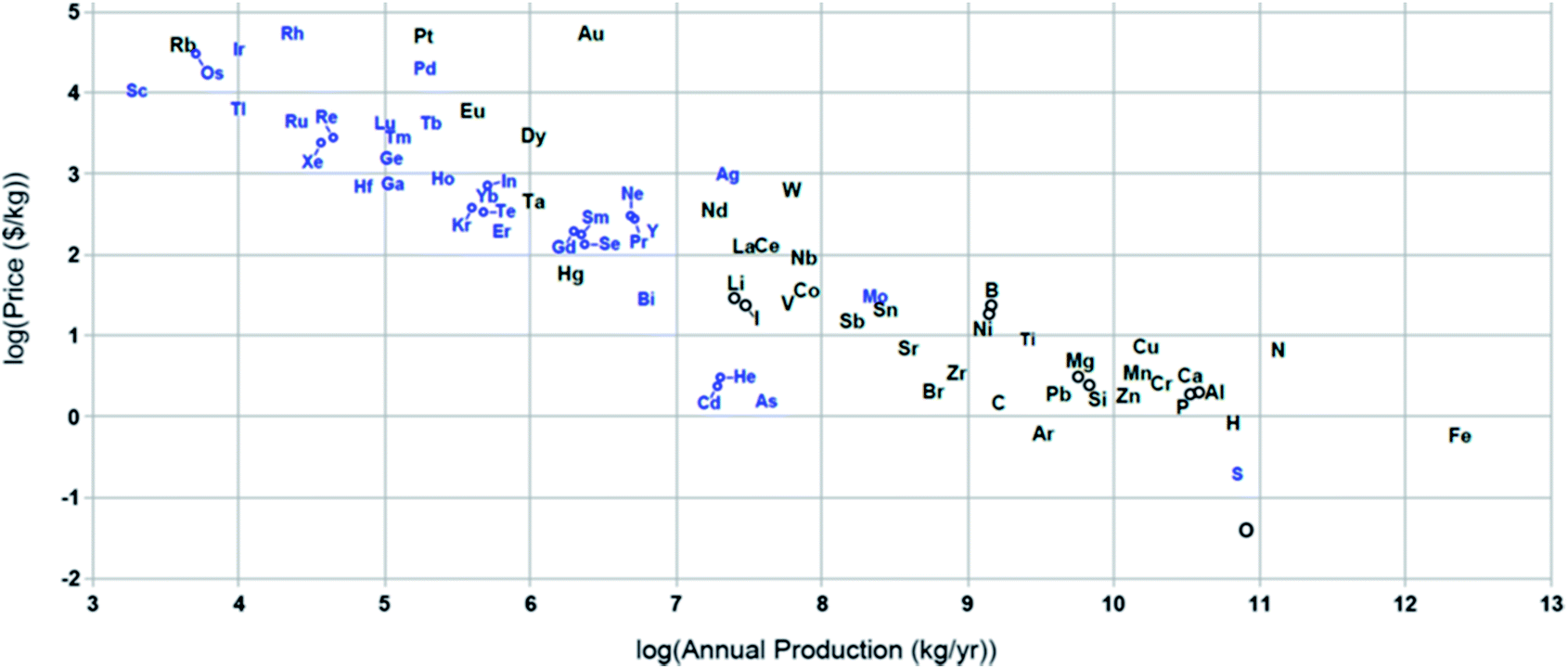 | ||
| Fig. 2 Price of elements (in $ per kg) vs. their annual production (in kg per year).19 | ||
ZABs are composed of three main parts: a Zn anode, an alkaline electrolyte (usually concentrated KOH) and an air cathode (a carbon substrate with a catalytic layer). All the components will be further discussed in detail in the following paragraphs. There are two types of ZABs, classified into primary and secondary or rechargeable.2,14 Primary ZABs have a long and stable storage life. A sealed battery will only experience 2% capacity decay after one year of storage. ZABs are available in many sizes and voltages (1.14, 1.4 and 3.0 V cells). Similar to other primary batteries, they may be combined in series to produce a battery with a higher voltage. Primary ZABs contain high power and energy in a small package, so they are very practical for applications where small size and high performance is critical. They are typically used in hearing aids, buoys and railroad applications.20
ZABs can be recharged mechanically. In mechanically rechargeable Zn–air cells, the consumed Zn is physically removed from the battery and replaced with a fresh electrode. Spent Zn is processed separately to metallic Zn. Such ZABs can be used for grid storage applications. These batteries were developed for the first time by Electric Fuel Limited and used in power military devices in the late 1960s. However, their short active lifetime, intermittent operation and the emergence of Li batteries with simpler operating systems reduced the scientific interest in developing mechanically rechargeable ZABs.2,21
Advances in materials science and engineering offer new opportunities for the design of electrically rechargeable ZABs. Most of the materials employed in primary ZABs are also used in electrically rechargeable ZABs. Although some companies (as listed in Table 2) have commercialized rechargeable ZABs, there is still much room for improvement. They suffer from serious basic technological problems.16 The main issue in the anode is low utilization efficiency due to passivation, corrosion and dendritic growth of Zn during recharge. For the cathode, the main problem is the sluggish and high overpotential oxygen reactions. Extensive studies have been done on ZABs, covering the challenges either of the individual components or the overall system.13–16,22,29–33 As an example, Evans et al.34,35 studied the irreversible agglomeration and shape change of the Zn-electrode in a rechargeable ZAB.
| Company | Battery type | Features and performance | Ref. |
|---|---|---|---|
| Fluidic energy | Secondary | Ionic salts plus additives as the electrolyte, coupled with nanostructured Zn as the anode | 23 and 24 |
| EOS | Secondary | Neutral chloride electrolyte (Zn/ZnCl2, NH4Cl, H2O/O2); charge and discharge potentials at 2.1 V and 0.9 V; 2700 cycles with no degradation | 21 |
| Revolt | Mechanically rechargeable | Liquid Zn slurry which flows through tubes that function as air cathode; 200 cycles at 100 mA cm−2 | 25–27 |
| EDF France | Secondary | Faradaic efficiency is 96% at 30 mA cm−2; energy efficiency is 70%; energy density is 100 W h kg−1; cycle life is 90 cycles 15 min per cycle | 28 |
Numerical modelling is a very useful tool for defining the critical design parameters for battery applications. Various numerical studies have been performed on ZABs, such as Zn electrode thickness/morphology and dissolution,36–38 gas–liquid interface movement39,40 and the growth of O2 bubbles during recharging.41 Numerical methods provide valuable insights into the challenges in a ZAB system before performing time-consuming experiments. For example, a model used in a study by Deiss et al.42 investigated the charge–discharge behavior of a ZAB. It predicted that OH− depletion is the major limiting factor for OER overpotential at high current charging and GDL pore plugging is a major limiting factor for ORR during long-term cycling. Schröder et al.39 developed a technique to monitor the movement of the three phase boundary using a finite volume method.
Air electrode design and the reaction mechanisms are the most complex part of a rechargeable ZAB and by far the dominating factors affecting the performance of this system. Reactions at the cathode during discharging and charging (ORR/OER) involve complex steps and the exact mechanisms remain elusive in most cases. Normally, the integration of numerical and experimental studies is the most precise way to explain the ORR and OER mechanisms. In this regard, many studies have been dedicated to investigating the reaction mechanisms and response of a diverse group of materials as electrocatalysts for ORR/OER.12,31
2.1. ZAB architecture and components
The structure of a rechargeable ZAB is schematically shown in Fig. 3 and is composed of a Zn electrode, an air electrode and an alkaline electrolyte.43 The air electrode is divided into two layers: a gas diffusion layer (GDL) and a catalytic layer. Upon discharge, Zn oxidizes to Zn(OH)42− at the negative electrode (Reaction (1)). Supersaturation of Zn(OH)42− results in the formation of ZnO which is in the form of a white insulating powder (Reaction (2)). At the positive electrode, O2 diffuses through the GDL due to pressure differences between the outside and inside of the cell. Oxygen is reduced at the surface of the electrocatalyst (Reaction (3)). | ||
| Fig. 3 Schematic of a rechargeable ZAB and reactions.43 | ||
The key process of the ZAB cathode is a three-phase reaction (gas–liquid–solid). During discharge, the catalyst (solid) accelerates the reduction of O2 (gas) to hydroxyl ions in the electrolyte (liquid). During charge, oxygen evolves at the positive electrode and Zn is electroplated at the negative electrode to complete the cell.
Dendritic growth of Zn during recharging can occur due to the inability of zincate ions to fully return to the same locations during recharge; this is one of the important challenges in the operation of a rechargeable ZAB. Another issue that arises during cycling is the sensitivity of the highly alkaline electrolyte to CO2 concentration from the feed air. As presented in Reaction (5), CO2 from the feed air reacts with the KOH electrolyte to form carbonates. Carbonates can precipitate and clog the pores of the air electrode and limit air access for ORR. The current challenges in rechargeable ZABs and the fast-growth of Li-ion batteries in the market have limited attention to developing rechargeable ZABs. However, a few groups, listed in Table 2, have successfully developed long cycle life secondary ZABs for commercial application.
The discharge processes in the anode and cathode of a ZAB are described by the following chemical reactions ((1)–(5)):
Anode:
| Zn(s) + 4OH(aq)− → Zn(OH)4(aq)2− + 2e− (E° = −1.25 V vs. SHE) | (1) |
| Zn(OH)4(aq)2− → ZnO(s) + 2OH(aq)− + H2O(aq) | (2) |
Cathode:
| O2(g) + 2H2O(aq) + 4e− → 4OH(aq)− (E° = +0.401 V vs. SHE) | (3) |
Overall reaction:
| 2Zn(s) + O2(g) → 2ZnO(s) (cell potential: 1.65 V) | (4) |
| 2KOH(aq) + CO2(g) → K2CO3(s) + H2O(aq) | (5) |
The reversible potential for ZABs is 1.65 V (Reaction (4)). However, depending on the load (applied current density), the practical voltage can drop to <1.4 V. The origin of this drop is largely due to the high overpotential at the air electrode during ORR and OER. Thus, rechargeable ZABs usually have a low round-trip energy efficiency of <55–65% (battery load: 1–50 mA cm−2), which depends on the applied current density.
2.2. Zn anode
Zinc is employed in various forms including Zn-foil, Zn-fibers or Zn-powder. Zinc is an abundant metal, relatively inexpensive and non-toxic (Fig. 2). The specific energy density of Zn is 1084 W h kg−1 and the power density demonstrated by primary ZABs is 300 W h kg−1.44 Zinc has a low electrical resistivity of 5.95 μΩ cm−1. Although a high Zn surface area is preferred due to better inter-particle contact, self-corrosion of the Zn electrode becomes more significant with smaller particle size.45–48 Zinc oxidation accompanied by the hydrogen evolution reaction (HER) on the electrode surface will have a negative effect on Zn, the electrolyte and the sealing structure.22 Many strategies have been developed to retard the HER and increase the corrosion resistance of the Zn anode. Table 3 summarizes recent studies on the modification of the Zn electrode during the past few years.| Modification | Materials | Effects | Ref. |
|---|---|---|---|
| Alloying | Hg | Improved electrical conductivity, adverse environmental problems, inhibits corrosion by 40% | 2 and 16 |
| Pb, Cd, Ni, Sn, Bi | High HER overvoltage, improved discharge due to addition of an electronically conductive material, increases capacity of Zn | 2, 16, 55 and 56 | |
| 7.5 wt% Ni and 2.5 wt% In | Reduced dendrite formation, increased HER overpotential | 2, 16, 55 and 56 | |
| Coating | 25 wt% Al2O3 | Suppressed HER by preventing direct exposure of Zn to the electrolyte, 50% longer discharge time in 9 M KOH | 57 and 58 |
| 0.1 wt% Li2O–2B2O3 | Increased discharge capacity (1.7 A h), prevents Zn surface from facing KOH | 59 | |
| Neodymium conversion film | Stabilizes cycle life of Zn | 15 | |
| ZnO with polypyrene (PPy) | Restricts dendrite growth, superior cycling stability, stable charge voltage plateau | 60 | |
| Ni and Bi | Decreased corrosion current, decreased HER rate, shifts HER overpotential to more negative value | 61 | |
| Addition of corrosion inhibitors to the electrolyte | Anions of organic, phosphoric, tartaric, succinic and citric acids | Absorbs at sites of rapid dendrite growth, increased HER overpotential | 2, 15 and 16 |
| Phosphoric acid esters (GARAC RA 600) | |||
| Modified polyethylene glycol (PEG) | |||
| Increasing surface area | Fibrous | Provides 20% more capacity at high discharge currents (70 A h), 90% material utilization at 1/7C discharge rate | 48 |
| 3D wired sponge | Uniform deposition of discharged products, low internal resistance, dendrite-free discharge cycling at depth of discharge of 20% | 62 | |
| Addition of binders | Na2SiO4 | Higher conversion efficiency due to porous nature of silica binder, fixes metal particles, provides easy access to electrolyte | 44 |
| Cross-linked polyacrylic acid polymers | Even distribution of Zn deposit, dendrite growth decreased | 2, 15, 16 and 62 | |
| 2 wt% Super P carbon black | High specific discharge capacity (776 mA h g−1), improved power density (20 mW cm−2) | 47 |
Another factor that limits the overall performance of ZABs is Zn dendrite growth during charge–discharge. Several reports have been published regarding the electrodeposition mechanism for Zn; these have investigated the effects of electrolyte concentration and the amount of available H2O molecules.15 For example, the addition of metal oxides, such as Bi2O3, Ti2O3 and In2O3 with higher HER overpotentials, to the electrolyte enhances the conductivity, improves current distribution and promotes the formation of a thin, compact Zn deposit during recharge.22
It is important to note that Zn corrosion can be avoided using hydrophilic or hydrophobic room temperature ionic liquids (RTILs) (non-aqueous electrolytes). As discussed further in Section 2.4, the absence of protons in the electrolyte is an advantage for ZABs using RTILs. The type of RTIL determines the reaction mechanism, kinetics and Zn redox cyclability. Also, smooth Zn deposition and less dendrite formation have been observed in several reports.49,50 More information can be found in the review by Xu et al.51
2.3. Air electrode: gas diffusion layer (GDL)
Since 1932, after the invention of wax-treated carbon electrodes by Heise and Schumacher,52 a GDL has been used as the cathode material for ZABs. Fig. 4 shows a typical structure of the air electrode used in a ZAB. The GDL acts as an interface between the gas flow and the electrocatalyst, directing the electrolyte to active sites. The side of the GDL facing the electrolyte is normally covered with a catalytic layer to provide a reaction zone at the three-phase boundary.53 Typical GDLs consist of a bilayer structure including a macro-porous layer and a micro-porous layer (MPL). The mechanical properties (response to compression, bending and shear), as well as the thermal and electrical properties, are governed by the fibrous backing material of the GDL. The MPL facilitates catalyst deposition and improves the pore size distribution. Polytetrafluoroethylene (PTFE) loaded on both sides adjusts the degree of hydrophobicity depending on the type of application (e.g., polymer electrolyte membrane fuel cells and metal–air batteries).54 GDLs are commonly fabricated by wet-laying of chopped polyacrylonitrile (PAN)-based carbon fibers; this is a highly scalable process.63 The degree of hydrophobicity is different on either side of the GDL. The side in contact with the electrolyte contains a thin catalytic layer, which is usually spray-coated or directly deposited. This side is mildly hydrophobic with a smaller pore structure. The side in contact with the gas (e.g., pure O2 or air) is highly hydrophobic with a larger pore size to allow gas to diffuse into the cell. Generally, carbon blacks suffer from catastrophic corrosion at very high potential during OER (>0.7 V vs. SHE). Corrosion of the GDL can reduce the reaction site density, leading to battery performance degradation. Therefore, increasing the corrosion resistance and porosity level and decreasing the thickness while maintaining the mechanical properties are common challenges in developing advanced GDLs.64 | ||
| Fig. 4 General structure of air electrode in a ZAB. | ||
Another challenge is related to the formation and maintenance of a stable three-phase boundary at the air electrode. The electrolyte may slowly flood the air electrode, leading to an increased diffusion path for O2 into the structure and possibly the subsequent failure of the air electrode.
In general, the hydrophobicity of the PTFE layer is progressively reduced in a highly alkaline solution, which results in movement of three phase reaction sites towards the back side of the GDL away from the electrocatalysts (air side). Flooding of the system results in increased ohmic resistance and a loss in the power density and efficiency. Flooding of the air electrode is also caused by carbonate by-products (Reaction (5)) which clog the pores of the GDL, hindering electrolyte access to the triple-phase reaction zone.22 The maximum working temperature of a ZAB is 80 °C; at this temperature the viscosity and the surface tension of the electrolyte is reduced which results in easier liquid infiltration in the GDL structure. Ventilation and cooling of the battery reduce the risk of the liquid infiltration.65,66
2.4. Electrolyte
Secondary ZABs utilize a highly alkaline electrolyte, typically KOH, NaOH or LiOH. KOH has a higher conductivity than NaOH (30 wt% KOH: ∼0.6 S cm−1 (ref. 67) vs. NaOH: ∼0.2 S cm−1 (ref. 68) at room temperature).69 Moreover, K2CO3, the reaction product of KOH with air (Reaction (5)) has better solubility than Na2CO3. The concentration of KOH is optimized at 26–30 wt% (6–7 M), i.e., the highest electrical conductivity. Higher concentrations increase the viscosity and the possibility of ZnO formation (Reaction (1)). Based on the Nernst equation:| E = E° − (RT/nF)(lnQc) |
484.56 C mol−1) and Qc is the concentration of the products divided by the concentration of the reactants in a reaction.4 The electrode potential shifts about −0.83 V when the pH value of the electrolyte is changed from 0 to 14, which significantly affects the electric field at the electrode–electrolyte interface.2,15
Despite the desirable properties of KOH, its usage in ZAB raises several technical issues. Carbon corrosion is one of the major problems in concentrated alkaline electrolytes, which may proceed via the following reaction:
| C(s) + 6OH(aq)− → CO3(aq)2− + 3H2O(l) + 4e− | (6) |
Continuous oxidation of carbon results in a brown colored electrolyte, due to the formation of carboxylic acids, such as mellitic acid and humic acid. As a result, reducing the pH of the electrolyte to a more neutral value can remediate the corrosion problem associated with highly alkaline electrolytes. Chloride-based electrolytes are preferred due to their good conductivity and the absence of electrolyte carbonation. Sumboja et al.70 used a chloride-based electrolyte in a ZAB consisting of a cathode with a directly grown MnOx catalyst. The rechargeable ZAB operated with no charge–discharge potential loss for 90 days at 1 mA cm−2. An acidic medium is an inappropriate electrolyte for ZABs due to the severe reaction of hydrogen ions with Zn metal. Also, some electrocatalytic materials, transition metal oxides in particular, are not stable in strongly acidic conditions.
Two major disadvantages of alkaline electrolytes are: (1) carbonate precipitation (Reaction (5)) which clogs the GDL pores and (2) the high sensitivity to temperature and humidity especially during long-term charge–discharge cycling.71 To circumvent the carbonate issue, supplying purified air, employing O2 selective permeable membranes, using CO2 absorbents (e.g., soda lime, LiOH and LiOH–Ca(OH)2) and circulating the electrolyte are effective countermeasures that have been studied. The addition of K2CO3 is another possibility to mitigate the negative impact of CO2. Increasing the amount of CO32− ions in the electrolyte decreases the carbonation reaction rate (Reaction (5)).72 The issue of temperature and humidity sensitivity can be resolved by strategies such as using a solid state electrolyte with an ionic conductivity of 10−2–10−3 S cm−1.73–75 Solid-state electrolytes can be used as both an ion conducting media and a separator, simplifying the design of ZABs and increasing cycle life.76
Gel polymers have attracted much attention as electrolytes for rechargeable ZABs. They are formed by incorporating a liquid electrolyte into a polymeric matrix. The conduction mechanism in polymer gels is similar to the one observed in liquid electrolytes, with the advantage of a solid structure. The most common gel-type electrolytes are poly-methylmethacrylate (PMMA), poly-vinylidene fluoride (PVdF), poly-ethylene oxide (PEO), polyacrylonitrile (PAN), polyvinyl alcohol (PVA) and polyvinyl acetate.22,62,77–79 Efficient, flexible ZABs have been developed by utilizing a PVA gel, which functions as both electrolyte and separator between the air electrode and Zn film. The porous gelled PVA membrane enhances electrolyte accessibility to the surface of the electrode under bending conditions as well as accommodates the electrolyte through capillary forces. A ZAB has been cycled 120 times at a charge–discharge rate of 150 A L−1 with a high volumetric energy density of 250 W h L−1 and gravimetric energy density of 581 W h kg−1.80 Another study developed lightweight functionalized cellulose nanofibers applied as a conductive hydroxide solid state electrolyte for a highly flexible rechargeable ZAB. Fig. 5 shows a schematic diagram of the ZAB device integrated with a bandage. The device, wrapped around an index finger, powered a light emitting diode (LED) under bending conditions. High water retention and stability of natural cellulose nanofibers were observed with no power density fading under bending conditions.81
 | ||
| Fig. 5 Schematic diagram of a flexible solid state ZAB device integrated with a bandage (left). The flexible device wrapped around an index finger powers a red LED under bending conditions (right).81 | ||
Recently, a new rechargeable ZAB has been developed using a molten Li0.87Na0.63K0.50CO3 eutectic electrolyte with added NaOH. The battery operates at 550 °C with reversible deposition/dissolution of Zn. There is no need for catalysts in the air electrode (Ni-foam). The performance is stable for 110 cycles with average charge–discharge potentials of 1.43 V and 1.04 V, respectively.82
Hydrophilic or hydrophobic room temperature ionic liquids (RTILs) (non-aqueous electrolytes) have been evaluated as alternatives to aqueous electrolytes.83–88 The benefits and drawbacks of RTILs are summarized in a recent comprehensive review by Xu et al.51 Improved cyclability, slower corrosion rates, suppressed Zn dendritic growth and fewer carbonate by-products during charge–discharge cycling have been demonstrated.15 However, the performance of ZABs using RTILs is inferior to batteries using KOH. There are still several challenges facing the use of RTILs and considerable work needs to be done before they are commercially viable. The challenges include the formation of insoluble metal peroxide/superoxides when using smaller cation salts (irreversible ORR), high viscosity of RTILs (i.e., limited GDL wettability), rapid voltage drop and low maximum power density.16,22
2.5. Separator
Separators are fine porous structures between the Zn electrode and the air electrode. They are made of non-woven polymers, such as polyethylene (PE), PVA, polyolefin (PO) and polypropylene (PP). Separator materials serve to retard Zn dendrite growth and play an important role in determining the transport of species between negative and positive electrodes. The basic requirement for a suitable separator for ZABs includes an appropriate pore size, high ionic conductivity, high stability, high adsorption capacity and high electrical resistivity in alkaline solutions. Celgard® 5550, which is a tri-layered structure (PP/PE/PP) coated with surfactants for rapid electrolyte wetting, has been commonly used as the separator in recent ZAB studies.16,22,81One challenge in using separators is that the porous structure of separators allows Zn2+ ions to migrate towards the air electrode, which will decrease the capacity of the battery. The use of anion exchange membranes, which are selective to the passage of ions, has been proposed to overcome this problem.10,21 For example, Celgard® 5550 was coated by an ionic liquid induced copolymer in a study by Hwang et al.89 The coating reduced the pore size, thereby limiting the migration of Zn2+ ions toward the cathode. The ZAB battery exhibited a 281% increase in lifetime and 1.4% higher initial energy efficiency.
In a recent study by Zarrin et al.,90 freestanding graphene oxide (GO) was functionalized with 1-hexyl-3-methylimidazolium chloride (HMIM) molecules and used as a flexible electrolyte membrane in a solid-state thin ZAB. Compared with the commercial polymeric membrane (Celgard® 5550), HMIM/GO had 3.8 times higher OH− conductivity (0.064 ± 0.0021 S cm−1 at 30% RH and room temperature), which minimized the sensitivity of the ZAB to operating conditions.
3. Air electrode oxygen reaction and mechanisms
The main purpose of the air electrode is to facilitate OER and ORR during charge and discharge, respectively. These reactions include a series of complex electron transfer reactions. In general, there are three adsorption modes for an O2 molecule on the surface of a metal or metal oxide catalyst: the Griffiths model, side-on mode (Yeager model) and the end-on mode (Pauling model) (Fig. 6).12 In the Griffiths model, the π orbitals of the O2 molecules interact laterally with empty d-orbitals of the metal ions. The O–O bond is subsequently weakened, with an incremental increase in its length. If the interaction is sufficiently strong, it will lead to the dissociative adsorption of the O–O bond. The reactions may be followed by the reduction of the metal atom to regenerate the catalyst site. In the Pauling mode, the O2 molecules interact with an end-on position on the electrode surface. Like the Griffiths mode, the π orbitals of O2 molecules interact with the empty d-orbitals of the catalyst. However, only partial charge transfer occurs, which results in the formation of peroxides and superoxides. The Yeager mode requires two adsorption sites with partially filled d-orbitals for bonding with π-orbitals of O2 molecules. The adsorption mode depends on the metal/metal oxide surface and its electronic structure, which affect the nature of corresponding active sites. The original concept of active sites can be traced to the studies of Taylor and Boudart.91 In a recent study, it was shown that preferred oxygen adsorption occurs with the side-on mode on the exposed edges of graphene nanoparticles (NPs) as opposed to adsorption on the basal plane (end-on mode).92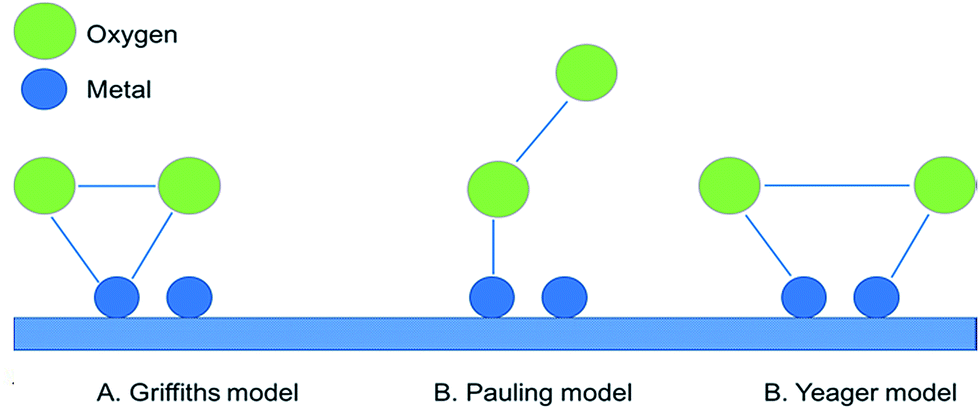 | ||
| Fig. 6 Possible configurations of O2 molecule interaction with a metal or metal oxide (M). | ||
The kinetics and mechanisms of ORR/OER are a function of many experimental factors including selection of the cathode material (e.g., electrocatalysts), the electrolyte, the concentration of active species (e.g., peroxides and hydroxides) and electrode design. The mechanisms of ORR/OER in precious metals and metal oxides have been extensively studied in the last few years.93–95 Advanced computational methods have enabled researchers to predict which catalysts will have more activity towards ORR/OER. These predictions help save time and costs in developing proper catalysts. The d-band center theory, suggested by Nørskov,96 has played an important role in the studies of catalysts, and the famous volcano plot (catalyst activity vs. oxygen binding energy) has been used to predict activity and selectivity of ORR/OER catalysts.
ORR multistep reactions proceed by the generation of *–OH, *![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and *–OOH as intermediates, which are briefly given in the following reactions:94
O and *–OOH as intermediates, which are briefly given in the following reactions:94
| O2(g) + 2H2O(aq) + 4e− → 4OH(aq)− (E° = 0.40 V vs. SHE) four-electron pathway | (7) |
| O2(g) + 2H2O(aq) + 2e− → HO2(aq)− + OH(aq)− (E° = −0.07 V vs. SHE) two-electron pathway | (8) |
| HO2(aq)− + H2O(aq) + 2e− → 3OH(aq)− (E° = 0.87 V vs. SHE) | (9) |
ORR may proceed via a four-electron pathway (Reaction (7)) or a two-electron pathway on the surface of an electrocatalyst (Reaction (8)). The number of electrons transferred can be obtained from rotating disk electrode (RDE) and rotating ring disk electrode (RRDE) tests, which reveal the ORR mechanism for selected materials.
Recent experimental results and mathematical simulation using density functional theory (DFT) on different materials have shown that the ORR pathway is a structure sensitive reaction depending on the surface geometry, electronic structure and O2 adsorption sites. For example, a recent DFT study by Busch et al.94 showed that the reversibility of ORR/OER is determined by the sequence of four coupled proton/electron transfer mechanisms (* denotes active sites):
* + H2O(aq) ![[left over right harpoons]](https://www.rsc.org/images/entities/char_21cb.gif) *–OH + H(aq)+ + e− *–OH + H(aq)+ + e− | (10) |
| *–OH(aq) *O + H(aq)+ + e− | (11) |
| *O + H2O(aq) *–OOH(aq) + H(aq)+ + e− | (12) |
| *–OOH(aq) * + H(aq)+ + e− + O2(g) | (13) |
The potential for each individual step results from the difference in binding energy of the different intermediates (i.e., *–OH, *O and *–OOH) before and after a charge transfer. The major obstacle in finding good bifunctional catalysts is the interdependence of the binding energy of the intermediate in the first and the third electron transfer step, i.e., *–OH and *–OOH (Reactions (10) and (13)). Ideally, the energy difference between each of the four intermediates should be 1.23 eV. However, the minimum average energy for Reactions (10) and (12) is at least 1.6 eV, which results in ∼0.4 V overpotential for both OER and ORR. The scaling relations between the *–OH and *–OOH binding energies prevent any compound with a single site to be both ORR and OER active. As Fig. 7 illustrates, active ORR catalysts are at the top of the ORR pyramid (blue) whereas active OER catalysts are at the tip of the inverted OER pyramid (green). Therefore, a bifunctional catalyst which is active towards both would fall in the forbidden region (the gap between the two cross section red triangles in Fig. 7).
 | ||
| Fig. 7 Two pyramids which depict the best achievable potential for ORR (blue) and the potential determining step for OER (green) as a function of the free energies for Reactions (10)–(13). The constraint set by the constant offset of 3.2 eV between *–OH and *–EOOH is represented by the red plane. The red plane cuts the two pyramids creating two separate volcanoes for OER and ORR which are darkened.94 | ||
It is now generally accepted that precious metals facilitate four electron pathways, whereas the two-electron pathway is primarily observed for carbonaceous materials.14 For transition metals and oxides, the surface area, specific crystal structure and composition determine the reversibility and the oxygen reduction pathways.14,94
A brief summary of the ORR/OER mechanisms for non-aqueous solutions is provided here. The first step for ORR in both protic (with dissociable H+ and able to donate hydrogen) and aprotic (no accessible H+) RTILs is the one electron reduction of oxygen to superoxide (O2˙−) (Reaction (14)). In aprotic RTILs superoxide formation terminates further electron transfer. In protic RTILs, the superoxide reacts with the proton and generates a radical which is further reduced to peroxide and hydrogen peroxide (Reactions (15)–(18))97
| O2(g) + e− → O2˙− | (14) |
| O2˙− + H(aq)+ → HO2˙− | (15) |
| HO2˙− + e− → HO2(aq)− | (16) |
| HO2˙− + O2˙− → HO2(aq)− + O2(g) | (17) |
| HO2(aq)− + H(aq)+ → H2O2(aq) | (18) |
| H2O2(aq) + 2H(aq)+ → 2H2O(aq) | (19) |
| 2H2O(aq) → O2(g) + 4H(aq)+ + 4e− | (20) |
Since the process involves only one electron transfer, the completion of four electron transfer depends on (1) the air electrode used (e.g., Pt or glassy carbon) and (2) overpotential. For example, on a clean Pt surface, hydrogen peroxide is readily reduced to water (Reaction (19)). During OER (reverse process) the oxidation of water regenerates the oxygen as shown in Reaction (20).51,97
4. Bifunctional catalysts for ZAB air electrodes
As mentioned previously, a major challenge in developing a highly efficient and stable rechargeable ZAB is overcoming the slow kinetics of ORR and OER. The main function of an efficient bifunctional electrode is to promote ORR/OER in a single layered structure. Reviews on cathode electrocatalysts can be found in previous studies.14,18,29,31–33,95,98–101 Reviews by Cheng et al.14 Jörissen,31 Lee et al.32 and Wang et al.102 mainly focused on the bifunctional activity of different groups of materials for the air electrode of different types of metal–air batteries. The review by Feng et al.18 covers the electrocatalytic activity and synthesis methods of non-precious metal catalysts for fuel cell systems. The review by Hamdani et al.99 provides a good summary of Co3O4 and Co-based spinel oxide bifunctional electrocatalysts. A recent review by Fu et al.17 has covered various ZAB cell configurations, as well as the various components of a ZAB including some discussion on air electrode catalysts. The current study covers the electrocatalytic activity, stability and synthesis techniques of bifunctional catalysts used in ZABs. In this section, bifunctional catalyst materials are divided into several groups: noble metals and alloys, transition metals, transition metal oxides (spinels and perovskites) and carbon-based materials. Some examples of high performance catalysts and ZABs are presented for each category.4.1. Noble metals and alloys
Noble metals, such as Pt and Ir, are well-known benchmark catalysts for ORR and OER, respectively. While they offer the advantage of high catalytic activity and electrical conductivity, their high cost and scarcity are serious concerns. Noble metal-based catalysts are normally well dispersed on a high surface area carbon support (e.g., carbon black, carbon nanotubes (CNTs) and graphene). As shown in Fig. 2, Pt is one of the most expensive and least abundant metals. Three main strategies have been taken to reduce the cost of precious metal electrocatalysts, including using less expensive alternatives such as Ag, alloying with highly ORR/OER active low cost metals/metal oxides or nanostructuring noble metals to minimize the mass required. Table 4 presents a summary of recently developed precious metal-based bifunctional electrocatalysts used in ZABs. Several examples of recent studies are provided in the next paragraphs.| Catalyst material | Secondary (S)/primary (P) ZAB battery characteristics | ORR and OER catalyst characteristics | Ref. |
|---|---|---|---|
| a ND: not determined; Op: onset potential; Lcd: limiting current density; E1/2: half-wave potential; PD: power density; Sc: specific capacity; Sed: specific energy density; n: number of electrons transferred. SWCNT: single wall carbon nanotubes; MCNA: mesoporous carbon nanofiber array; NR: nanorods; rGO: reduced graphene oxide; NP: nanoparticles; SC: single crystal. | |||
| 10 wt% Ag/C | (P) PD: 34 mW cm−2 at 35 °C | n = 4 for larger particles, n = 2 for smaller particles | 106 |
| Ag–Cu | (S) Echarge: 2.04 V; Edischarge: 1.1 V; efficiency: 53.09% | ORROp: −0.19 V, OEROp: 0.55 V vs. SCE; n = 3.9 | 115 |
| (P) PD: 85.8 mW cm−2 at 100 mA cm−2; Sc: 72 mA h g−1 | |||
| NPAg–SWCNT | (P) Sc: 515 mA h g−1; Sed: 300 W h kg−1 | ND | 107 |
| Ordered Pd3Pb/C–NiCo2O4 | (S) Echarge − Edischarge: 0.72 V (first cycle); 0.86 V (135th cycle); 4 h per cycle at 10 mA cm−2 | ORRop: 1.03 V vs. RHE ORRE1/2: 0.92 V vs. RHE ORRLcd: −6.5 mA cm−2; n = 4 | 111 |
| (P) Sc: 574 mA h g−1; Sed: ∼710 W h kg−1 at 10 mA cm−2 | |||
| CuPt–nanocages | (P) 253.8 mA cm−2 at 1 V; Sc: 560 mA h g−1; Sed: 728 W h kg−1 at 20 mA cm−2 | ORROp: 0.95 V vs. RHE ORRLcd: −6.25 mA cm−2 | 110 |
| RuO2–MCNA | (S) initial Echarge: <1.9 V and Edischarge: ∼1.25 V at 4 mA cm−2; 100 cycles at 1200 s per cycle | ORRE1/2: 0.8 V vs. RHE; n = 4 | 116 |
| AgCu–nanoalloys | (S) Echarge: 2.2 V Edischarge: 1.08 at 10 mA cm−2; +0.01 V increase after 200 cycles | ORRLcd: 5.9 mA cm−2; n = 3.9 | 112 |
| (P) PD: 86.3 mW cm−2; discharge current: 60 mA cm−2 at 1 V | |||
| AgCu | (S) efficiency: 56.4% at 20 mA cm−2; 4 h per cycle | ORROp: −0.15 V vs. SCE ORRLcd: −25.6 mA cm−2 at 0.8 V | 115 |
| (P) PD: 86.5 mW cm−2 at 100 mA cm−2 | |||
| C–N/Ag | (P) Edischarge: 1.2 V at current density of 10 mA cm−2 (stable for 25 h) | ORRop: 0.93 V vs. RHE; ORRLcd: −6.26 mA cm−2; n = 4 | 105 |
| LaMnO3–NR/rGO | (S) Echarge − Edischarge: 0.86 V (first cycle); 0.95 V (80th cycle); 1 h per cycle at 25 mA cm−2 | ORRop: 0.9186 V vs. RHE; ΔE = EOER (at 10 mA cm−2) − EORR (at 3 mA cm−2) = 1.06 V | 109 |
| SC–CoO/NRs | (S) Echarge − Edischarge: 1.4 V (first cycle) at 5 mA cm−2 | ORRop: 0.98 V vs. RHE, ORRLcd: −12 mA cm−2 in 1 M KOH | 114 |
Silver at only 1% of the cost of Pt is one of most cost effective precious metals. Silver-based catalysts are usually coupled with MnO2 and carbon supports (e.g., single walled carbon nanotubes (SWCNTs) and carbon blacks) as ORR/OER active composites.103,104
A noble metal, N-doped biomass C–Ag composite has been synthesized using soymilk as a precursor material for nitrogen and carbon. The sample was heat treated at 900 °C and showed an ORR onset potential of 0.93 V vs. RHE, which is identical to 20 wt% Pt/C.105 Rechargeable ZABs with only 10 wt% Ag/C (25.9 nm Ag particle size) as the electrocatalyst demonstrated a power density of 34 mW cm−2 at 35 °C.106 Although the electrocatalytic results were not compared with Pt/C, the power density was shown to increase to 72 mW cm−2 at 80 °C, which is close to the ZAB operating temperature. In a different study, a novel GDL was designed using Ag–NPs supported on SWCNTs as a cathode of a ZAB. The weight and thickness of the GDL were reduced to 0.005 mg cm−3 and 0.05 mm, respectively, for AgNP–SWCNTs vs. 0.79 mg cm−3 and 0.49 mm for conventional carbon-based electrodes with comparable performance.107 It was also shown that Ag-based catalysts had better OER (i.e., more negative OER onset potential) performance compared with Pt/C in highly alkaline electrolytes. The low OER performance of Pt/C was attributed to the accumulation of oxygenated species, which cover the surface and decrease the active surface area.108 As an alternative to carbon, transition metal oxides integrated with Ag nanoparticles have been investigated in recent studies. For example, LaMnO3 perovskite nanorods/reduced graphene oxide (LMO-NR/rGO) decorated with Ag nanoparticles were studied as a bifunctional catalyst. The catalyst was synthesized via a simple hydrothermal method. Silver nanoparticles were homogenously overlain between the sheets of rGO, providing high conductivity and rapid charge transfer.109 Alloying precious metals with non-precious metals such as Cu and Pb is also one of the strategies to reduce the cost and increase the long-term stability of ZABs.110–114 For example, structurally ordered Pd3Pb/C catalysts exhibit ∼2–4 times higher mass activity relative to the traditional baseline Pt/C catalyst. The activity enhancement of Pd3Pb/C originates from the higher number of active sites created by varying the Pd–Pd bond distance and modification of the electron configuration. The corresponding ZAB exhibited excellent long-term stability with only a 0.14 V overpotential increase after 560 h of cycling.111
In another study, CuPt-nanocages (NCs), an intermetallic structure, with particle sizes of ∼20 nm were prepared by a solvothermal method. Open faceted hollow geometric CuPt–NCs with an ordered atomic arrangement provided facile O2 molecular accessibility and high numbers of active sites within the structure. Applying CuPt–NCs in a ZAB led to superior specific capacity and energy density (560 mA h gZn−1 and 728 W h kgZn−1, at a discharge rate of 20 mA cm−2) in comparison with the performance of a system with Pt/C as the cathode (480 mA h gZn−1 and 624 W h kgZn−1, at a discharge rate of 20 mA cm−2).110 Recently, dendrite shaped Ag–Cu developed by a galvanic displacement reaction was suggested as an active bifunctional catalyst. A facile preparation technique, carbon-free structure and high stability were touted as advantages of this catalyst.115 Also, DFT analysis of a similar catalyst (Ag–Cu nanoalloy), fabricated using pulsed layer deposition (PLD), showed an increase in oxygen adsorption energy from −0.86 eV for pure Ag clusters to −1.36 eV for AgCu (Cu-shell) clusters. This facilitated the thermodynamics of ORR by lowering the adsorption energy.112,113
Coating the carbon support with precious metals/metal oxides has been reported in a recent report. Using a crab shell template, RuO2-coated ordered mesoporous carbon nanofiber arrays (MCNA) were developed. The uniform RuO2 coating provides efficient ORR/OER active sites and long cycle life (160 h at 4 mA cm−2) in a rechargeable ZAB (Fig. 8).116 However, controllable fabrication of precious metals on metal oxides or carbon materials with atomically matched interfaces is a significant challenge. In addition, recently developed hybrid precious/non-precious metal catalysts were in powder form and suffered from detachment from the supports. To meet these challenges, Pt nanoparticles have been deposited epitaxially onto single crystal CoO nanorods by a magnetron sputtering technique. Significant ORR performance and durability enhancement were observed in a ZAB due to the strong synergistic interaction between the Pt nanoparticles and single crystal CoO nanorods.114
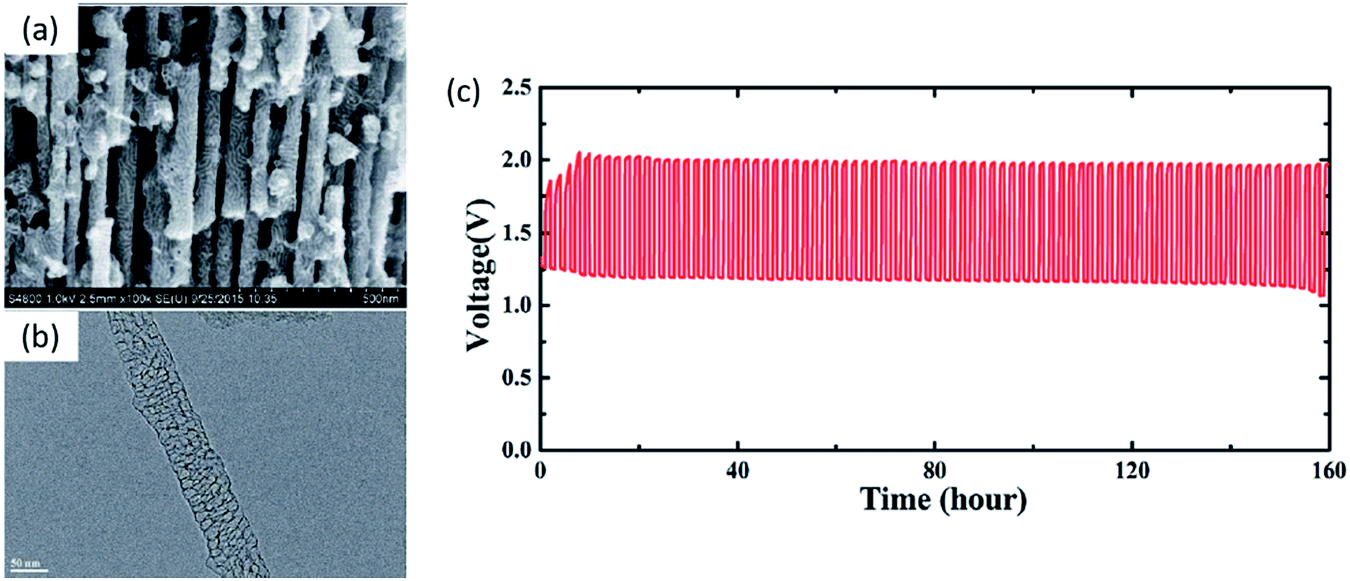 | ||
| Fig. 8 (a) SEM and (b) TEM images of RuO2-coated MCNAs. (c) Charge–discharge cycling curves for a ZAB with RuO2-coated MCNA catalyst (current density of 4 mA cm−2 and 1 h per cycle).116 | ||
4.2. Transition metal oxides
Compared with precious metals, non-precious metal catalysts are more desirable for applications such as fuel cells and batteries due to their abundance and low cost.117 Among them, transition metal oxides have been extensively investigated in the form of single, binary or ternary oxides. Spinels and perovskites are two specific oxide structures with transition metal cations present in different oxidation states. Because of their unique electronic structure, spinels and perovskites are promising electrocatalysts and have been widely investigated in recent years. Reactions (21)–(25) (“M” denotes a transition metal) summarize the ORR catalytic process for transition metal-oxides. Based on this theory, the competition between O2−/OH− displacement (Reaction (21)) and OH− regeneration (Reaction (23)) on the surface of transition-metal ions determines the rate-limiting steps for ORR in alkaline solutions.118| Mm+–O2− + H2O + e− → M(m−1)+–OH− + OH− | (21) |
| O2 + e− → O2− (ads) | (22) |
| M(m−1)+–OH− + O2− (ads) → Mm+–O–O2− + OH− | (23) |
| Mm+–O–O2− + H2O + e− → M(m−1)+–O–OH− + OH− | (24) |
| M(m−1)+–O–OH− + e− → Mm+–O2− + OH− | (25) |
| Catalyst material | Secondary (S)/primary (P) battery characteristics | ORR and OER catalyst characteristics | Ref. |
|---|---|---|---|
| a Op: onset potential; Lcd: limiting current density; Cd: current density; PD: power density; Sc: specific capacity; Sed: specific energy density; N/A: not applicable; n: number of electrons transferred. NP: nanoparticles; LDH: layered double hydroxide; CNT: carbon nanotubes; GO: graphene oxide; rGO: reduced graphene oxide; IL: ionic liquid. | |||
| MnOx nanowire-Ketjenblack carbon | (P) PD: ∼190 mW cm−2 | ORRop: −0.05 V vs. Hg/HgO ORRLcd: −7.5 mA cm−2 at 3200 rpm | 130 |
| MnO2–Ketjenblack carbon | (P) PD: 133.17 mA cm−2 at 188.51 mA cm−2 | N/A | 152 |
| MnO2–Ag NP | (S) Echarge: 2.6 V and Edischarge: 1.1 V at 5 mA for 90 h | ORRop: −0.06 V vs. Ag/AgCl; ORRLcd: −1.9 mA cm−2; OERcd: 11.61 mA cm−2; average n = 3.7 | 135 |
| NiFe LDH/CNT–CoO/N–CNT | (S) Echarge: 1.95 V; Edischarge: 1.25 V at 20 mAcm−2 for 200 h; efficiency: ∼65% | N/A | 147 |
| (P) PD: 265 mW cm−2; current density at 1 V: 200 mA cm−2; Sc: ∼570 mA h g−1; Sed: 700 W h kg−1 | |||
| NCNT/CoO–NiO–NiCo | (S) Echarge − Edischarge = 0.75 V at 20 mA cm−2; 600 s per cycle for 100 cycles | ORRop: 0.97 V; EOER: 0.27 V vs. RHE at 10 mA cm−2; n = 3.85 | 153 |
| (P) Sc: 545 mA h g−1; Sed: 615 W h kg−1 at 35 mA cm−2 | |||
| rGO–IL/Mn3O4 | (P) PD: 120 mW cm−2 at 200 mA cm−2 | ORRop: −0.1 V vs. Hg/HgO, ORRLcd: −0.22 mA at −0.45 V; average n = 3.5 | 134 |
| Co(OH)2–N–rGO | (S) Echarge − Edischarge = 1.2–1.3 V at 15 mA cm−2 for 50 h; efficiency: 46% | ΔEOER–ORR = 0.87 V; n: ∼3.6; EOER: 1.68 V at 10 mA cm−2; EORR: 0.66 vs. RHE at −3 mA cm−2 | 139 |
| CoOx/XC–N | (P) PD: 110 mW cm−2 at 160 mA cm−2 | ORRop: 0.86 V vs. RHE n ∼ 3.7 | 138 |
| NCNT/CoxMn1−xO | (S) Echarge − Edischarge = 1.18 V at 50 mA cm−2; Echarge − Edischarge = 0.57 V at 7 mA cm−2 | ORRop: 0.086 V vs. RHE; ORRLcd: −5.5 vs. RHE; OERcd at 0.3 V vs. RHE: 4.6 mA cm−2 | 144 |
| (P) Sc: 581 mA h g−1; Sed: 695 W h kg−1 at 7 mA cm−2 | |||
| MnO2–NCNT | (S) Echarge − Edischarge = 1.6 V at 10 mA cm−2 300 s per cycle | ORRcd = −4 mA cm−2 at −0.8 V vs. Ag/AgCl; OERcd: ∼39 mA cm−2 | 127 |
| Ni–MnOx/C | (S) Edischarge: 0.96 V at 100 mA cm−2 | ORRop: −0.063 V vs. Hg/HgO ORRcd: 0.94 mA; n = 3.82 | 122 |
| (P) PD: 122 mW cm−2 | |||
| MnO2/C–NiO/NiOOH | (S) faradiac efficiency: 96% at 30 mA cm−2; energy efficiency: 70% Sed: 100 W h kg−1 cycle life: 90 cycles at 15 s per cycle | N/A | 28 |
| (P) Sc: 819.8 mA h g−1 | |||
| N–NiFe–LDH | (P) PD: 0.55 mW cm−2 at 0.65 V | E OER: 0.23 V vs. Ag/AgCl at 10 mA cm−2 | 149 |
| CuFe | (P) PD: 212 mW cm−2 | n = 3.7–3.9 | 154 |
| MnO2 | (S) Echarge: 2.04 V; Edischarge: 1.27 V; at chargeCd: 7.5 mA cm−2 at 4 h per cycle; dischargeCd: 15 mA cm−2 at 2 h per cycle | ND | 133 |
| Co3O4/MnO2–CNTs | (S) Echarge − Edischarge = 1.0 V at 10 mA cm−2 after 504 cycles (84 h) 10 min per cycle | ORRop: 0.87 V vs. RHE, ORRLcd: −5.6; OERop: 1.59 V vs. RHE; OERcd: 30 mA cm−2; n = 3.83 | 155 |
| (P) PD: 340 mA cm−2 at 0.75 V; Sc: 775 mA h g−1; Sed: 992 W h kg−1 | |||
| NiFeO@MnOx | (S) Echarge − Edischarge = 0.74 V at 10 mA cm−2 | ΔEOER (5 mA cm−2)–ORR (3 mA cm−2) = 0.792 V; n = 3.9 | 156 |
| (P) PD: 81 mW cm−2 at 100 mA cm−2 | |||
The low conductivity of MnOx catalysts is one of the factors limiting their utilization as an air electrode catalyst for ZABs. To overcome this problem, conductive carbon materials such as carbon black (Vulcan XC-72), CNTs and graphene are commonly added to MnOx as the catalyst support.134 Another approach is to increase the conductivity of MnOx by incorporating conductive species such as Ag–NPs135,136 or other ORR/OER active transition metal oxides catalysts such as Co3O4.137
Other bifunctional catalysts that have been extensively studied are transition metal oxides such as Co-oxide/hydroxide NPs supported on graphene, graphene oxide and carbon blacks.138–143 It has been proposed, by a DFT study, that the origin of CoOx/C ORR reactivity relies on the gap between the highest occupied molecular orbitals (HOMO) and the lowest unoccupied molecular orbitals (LUMO) of the crystalline surface of Co(II). A small gap implies low kinetic stability but high electrochemical activity to extract the electrons from the HOMO to the LUMO.138 Using the same numerical technique, it was shown that hexagonal Co(OH)2/N-doped reduced graphene oxide (rGO) nanoplates outperformed CoO and Co3O4 with a ΔE (energy gap between the HOMO and LUMO) of 0.87 V. This means that the Co–OH bond is neither too strong nor too weak, which results in enhanced bifunctional activity of Co(OH)2.139
Recently, Liu et al.144 developed highly active bifunctional catalysts, fabricated by a non-surfactant assisted hypothermal method, consisting of non-spinel Co–Mn oxide supported by N-doped CNTs (NCNT/CoxMn1−xO).144 A rechargeable ZAB with a low charge–discharge gap (0.57 V) and high stability was reported. The authors suggested that graphitized nanocarbon, such as CNTs, can mitigate carbon oxidation and increase the corrosion resistance resulting in long-term stability.
One of the most interesting discoveries in this group of electrocatalysts is the development of layered double hydroxide (LDH) materials. LDHs are conducting hydroxide clay materials with the formula [M1−xIIMxIII(OH)2][(An−)x·mH2O], where MII is a divalent cation such as Ni2+, MIII is a trivalent cation such as Fe3+ and An− is an anion such as CO32−. Ni–Fe–CO32− LDH materials have electronic and ionic conductivities of 5 × 10−3 and 6 × 10−6 S cm−1, respectively.145 Also, the presence of CO32− will inhibit carbonate precipitation at the triple phase boundary in a rechargeable ZAB. Various Ni–Fe mixed compounds have been synthesized and reported in the last few years.146–148 For example, a rechargeable ZAB in a tri-electrode configuration was designed using a CoO/N–CNT hybrid as the ORR catalyst and Ni–Fe–LDH as the OER catalyst for the cathode. High ORR and OER activities were attributed to the strong coupling effect between nanoscale inorganic particles and the conducting CNT substrate. A peak power density of 265 mW cm−2 and a current density of ∼200 mA cm−2 at 1 V were reported, which is a significant improvement in performance for a primary ZAB in comparison with previous reports. It is worth noting that the air electrode electrocatalysts were loaded on two separate electrodes for charge (CoO/N–CNT) and discharge (Ni–Fe–LDH), respectively. Improved activity and efficiency were also confirmed for a rechargeable ZAB (∼65% at 20 mA cm−2) using a tri-electrode configuration. No significant voltage change and no obvious degradation were discerned during both the charge and discharge segments after 200 h cycling at 20 mA cm−2 in this configuration.147 Ni–Fe–LDH was also used as an anion exchange membrane (AEM)-type electrode to prevent the formation of K2CO3 (Reaction (5)).148 The AEM was placed at the interface between the alkaline electrolyte and catalyst layer of the air electrode. Applying an AEM suppressed the permeation of K+ cations from the electrolyte into the air electrode and inhibited the formation of carbonate. The other advantage of Ni–Fe–LDH is its ability to sense the external electric field through a color change. In a recent study by Chen et al.,149 a N-doped Ni–Fe–LDH film was prepared by a facile one-step chemical bath deposition method. Nickel and Fe salts were used as precursors and hexamethylenetetramine (HMT, C6H12N4) was used as the structural template as well as the nitrogen source. Nickel foam was used as the substrate. A well-crystallized 3-dimensional microporous material composed of numerous thin nanolayers (∼0.8 nm) was vertically grown on a framework. As shown in Fig. 9, the pristine color of Ni–Fe–LDH is gray-silver, which changed to dark black during the OER process for a potential scan from 0 to 1 V vs. RHE (reversible hydrogen electrode). Interestingly, the color can recover to the original state during the reverse scan from 1 to 0 V. The origin of the color change corresponds to the oxidation of Ni species (Reaction (19)) at ∼0.45 V during charge and the reverse reaction at ∼0.25 V during discharge.149
| Ni(OH)2 + OH− → NiOOH + H2O+ e− | (26) |
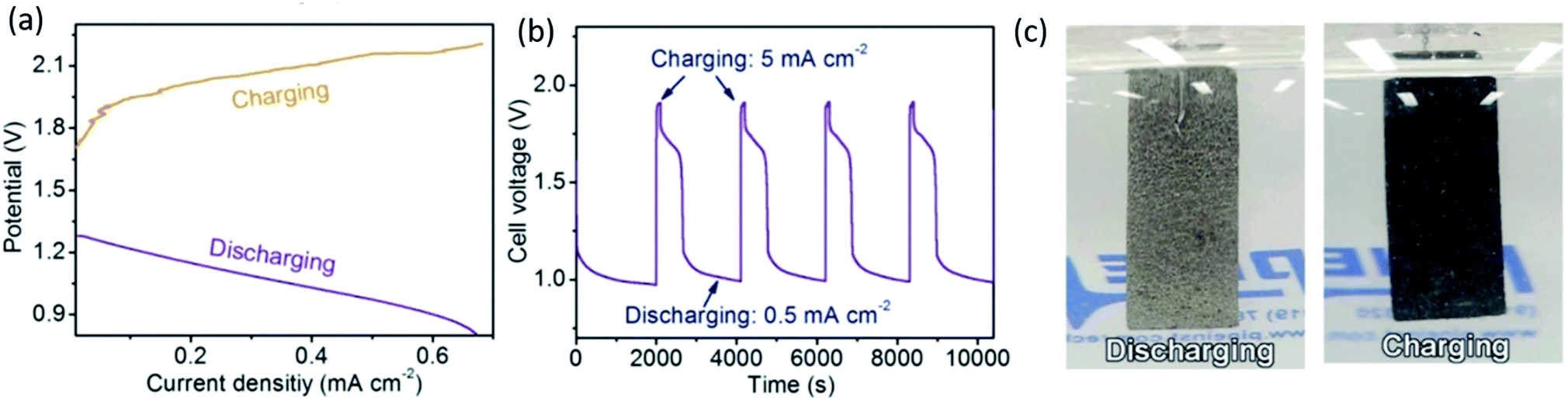 | ||
| Fig. 9 (a) Charge and discharge polarization (V–i) curves for Ni–Fe–LDH at 5 mV s−1. (b) Cycling performance of the battery at a charging current density of 5 mA cm−2 for 200 s and discharging current density of 0.5 mA cm−2 for 2000 s. (c) Optical images during charging and discharging processes.149 | ||
Titanium oxide materials have also been studied recently. Magneli phase materials such as TixO2x−1 (4 < x < 10) have been explored as electrode materials due to their high electrical conductivity (1000 S cm−1) and corrosion resistance in aggressively alkaline/acidic solutions.150 TiO2 (anatase) is formed by one-step vapor hydrolysis of TiCl4. Anatase showed a small polarization of 8 mV at 150 mA cm−2 and the catalyst showed steady performance during discharge (1.05 V).151
The above examples, as well as the summary provided in Table 5, indicate that transition metal-based electrocatalysts have potential to achieve widespread practical application in ZABs.
Both DFT and experimental analysis demonstrated the phase dependence of ORR and OER activities for the corresponding spinel structures. The effect was attributed to the different oxygen binding ability on the surface of each phase. CoMnO–B outperformed CoMnO–P in ORR and, since OER is the reverse process, CoMnO–P demonstrated higher OER activity.167 The hypothermal microwave technique is also a rapid and scalable method for the synthesis of highly homogenous transition metal oxides nanoparticles, especially spinels.168,169
Due to the low electrical conductivity of most spinel structures, integration of spinels with nanocarbon supports provides a more conductive network and facilitates charge transfer during ORR and OER. Moreover, nanocarbon materials enhance dispersion uniformity for the metal oxide, prevent agglomeration of the metal oxide particles and provide additional accessible surface area.161,170 Gelatin-coated Ketjenblack carbon can be transformed into N-doped carbon that can function as both bifunctional catalysts and conducting agents to improve the applicability of porous MnxCo3−xO4.165
Hybrid spinel-based bifunctional catalysts and carbon-based materials such as N-doped rGO, CNTs, carbon nanofibers and carbon black have been recently studied and the activity was attributed to the synergistic coupling of nanocarbons and oxide materials.171 The following are a few examples showing the effects of the addition of conductive nanocarbon supports to spinel structures.
Prabu et al.157,172 investigated several groups of spinel-based materials as bifunctional air electrodes for ZABs, such as 1-dimensional NiCo2O4,173 CoMn2O4/rGO and CoMn2O4/N–rGO. Strong coupling between Co2MnO4 and rGO provided a notable improvement in electrochemical activity with high stability. The details of secondary and primary ZABs for this group are summarized in Table 6.
| Catalyst material | Secondary (S)/primary (P) battery characteristics | ORR and OER catalyst characteristics | Ref. |
|---|---|---|---|
| a Op: onset potential; Lcd: limiting current density; PD: power density; Sc: specific capacity; Sed: specific energy density; N/A: not applicable; n: number of electrons transferred; E1/2: half-wave potential; E1/2, Ej10: OER current density at 10 mA cm−2. N–rGO: N-doped reduced graphene oxide; NP: nanoparticles; CNF: carbon nanofibers; NVC: N-doped Vulcan carbon; CNT: carbon nanotubes, 3DRGO: 3 dimensional reduced graphene oxide. | |||
| Co2MnO4–N–rGO | (S) Echarge − Edischarge = 1.25 V at 75 mA cm−2; round-trip efficiency = 58% (1st cycle) | N/A | 157 |
| Co3O4–NVC | (S) Echarge: 2.1 V and Edischarge:: 1.1 V at 20 mA | ORRop: −0.08 V vs. Ag/AgCl; ORRLcd: 4.6 mA cm−2 at −0.8 V, OEROp: 0.55 V vs. Ag/AgCl and OERCd: 30 mA cm−2 at 0.9 V; n = 4 | 182 |
| (P) PD = 33 mW cm−2 at 53 mA cm−2 | |||
| Dual phase-MnCo2O4/N–rGO | (S) Echarge: 2 V and Edischarge: 1.25 V at 5 mA cm−2 | ORRop: −0.09 V vs. Ag/AgCl; ORRLcd 5.53 mA cm−2 at −0.8 V; OERop: 0.59 V vs. Ag/AgCl, OERCd: 10.85 mA cm−2 at 0.8 V; n = 4 | 161 |
| NiCo2O4–CNT | (S) Echarge − Edischarge = 0.75 V at 510 mA cm−2 | ORRop: 0.934 V vs. RHE; ORRLcd: 7.2 mA cm−2 at 0.2; OERCd: 16 mA cm−2 at 1.7 V vs. RHE | 170 |
| (P) PD: 320 mW cm−2 at 210 mA cm−2 (1 V) | |||
| Co3O4 NC/N–CNT | (S) Echarge: 2.16 V and Edischarge: 1.14 V at 20 mA cm−2 | ORRop: −0.044 V vs. SCE; ORRLcd: 3.55 mA cm−2; OERCd: 20 mA cm−2 at 0.802 V vs. SCE | 181 |
| Co3O4 nanodisks | (S) Echarge: 2.2 V and Edischarge: 1 V at 50 mA | N/A | 180 |
| Co3O4 nanowires | (S) Echarge: 2 V and Edischarge: 0.98 V at 50 mA | N/A | 179 |
| Spherical Co3O4/NGr-24 h | (P) PD: 190 mA cm−2; Sc: 590 mA h gZn−1 | ORRop: 0.03 V vs. Hg/HgO; n = 3.7 | 178 |
| MnCoFeO4–NrGO | (S) Echarge − Edischarge = ∼1.2 V | ORRop: 0.91 V vs. RHE; n = 3.8; EOER − EORR = 0.93 V | 175 |
| MnO2/Co3O4 | (S) Echarge − Edischarge = ∼1 V at 15 mA cm−2; +5% after 60 cycles | ORRop: 1.05 V vs. RHE; ORRLcd: 3.4 mA cm−2; OERCd: 2.2 mA cm−2 at 1.8 V vs. RHE | 159 |
| (P) PD: 33 mA cm−2 at 50 mA cm−2 | |||
| CoMnO–P | (P) Sed: 255 W h kg−1 at 20 mA | ORRop: −0.08 V vs. Hg/HgO; ORRLcd: −43.2 mA mg−1; n = 3.7 | 167 |
| MnOx–Co3O4/C | (P) PD: 97 mW cm−2 at 0.97 V | ORRop: 0.73 V vs. RHE; ORRLcd: 21.8 mA mg−1 | 183 |
| Co NP–CNF | (S) Echarge − Edischarge = 0.85 V at 20 mA cm−2; +0.1 V change after 55 cycles 1 h per cycle | E OER: 0.64 V at 2 mA cm−2vs. Ag/AgCl; E1/2 ORR: −0.188 V vs. Ag/AgCl; n = 4 | 171 |
| (P): PD: 125 mW cm−2 at 81 mA cm−2 | |||
| Cubic-CoMn2O4/C | (S) discharge potential change −8.5% after 155 cycles at 400 s per cycle | ORRop: 0.95 V vs. RHE; ORRLcd: −5.7 mA cm−2; n = 3.91 | 184 |
| (P) Sc: ∼500 mA h g−1; Sed: ∼650 W h kg−1 at 10 mA cm−2 | |||
| NiO–Co3O4 Co/Ni: 9/1 | (P) PD: 100.1 mW cm−2 at 148.3 mA cm−2; discharge capacity: 579.5 mA h g−1 at 50 mA cm−2 | N/A | 185 |
| Co3O4@3DRGO | (S) initial Echarge − Edischarge = 1.3 V at 2 mA cm−2 | ΔE = Ej10 − E1/2 = 0.95 V | 186 |
| CuCo2O4/N–CNT | (P) Sc: 817.4 mA h g−1, Sed: 653.9 W h kg−1 | ORRop: −0.05 V vs. SCE; ORRLcd: −4.9 mA cm−2 | 187 |
| (S) initial Echarge − Edischarge = 0.37 V; +0.2 V after 20 cycles | |||
A highly stable bifunctional catalyst was developed using NH3-treated, N-doped macro/meso-porous carbon as the ORR active component. Co3O4, directly grown on Ni foam, was active as the OER component.174 The ORR and OER catalysts were decoupled in a ZAB set up similar to the tri-electrode design studied by Li et al.174 The ZAB was cycled for 800 h at a current density of 10 mA cm−2 with only a 4.5% decrease in voltaic efficiency (60% efficiency after 800 h of cycling) (Fig. 10). The battery performance was unprecedented in terms of the cycle life for all spinel-based bifunctional catalysts reported during the last few years.
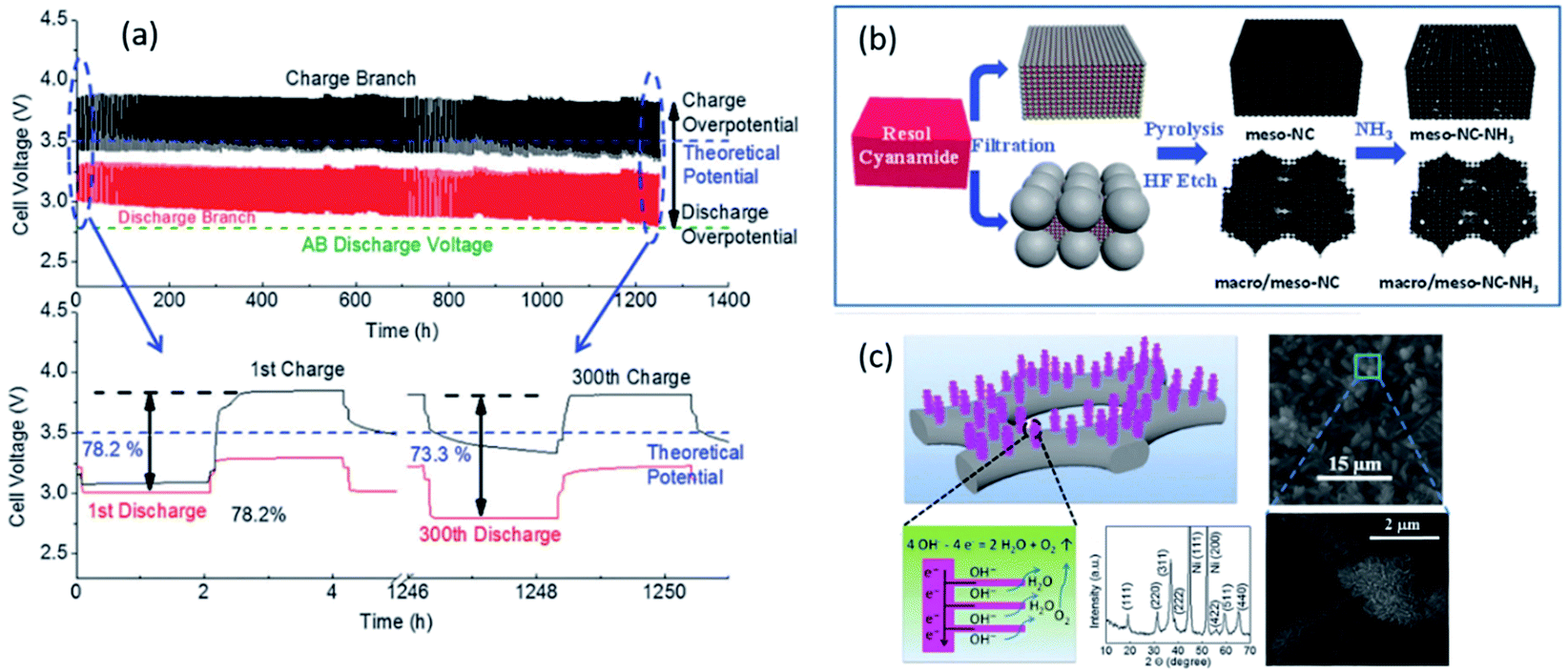 | ||
| Fig. 10 (a) Discharge and charge voltage profiles of ZAB with developed hierarchical structure. Co3O4 was directly grown on Ni foam at a current density of 10 mA cm−2. (b) Schematic illustration of the synthesis route for macro/meso–NC–NH3. (c) Schematic representation of Co3O4 microtrepangs grown on Ni-foam and OH− splitting on the branches of Co3O4 microtrepangs.171 XRD pattern shows pure spinel Co3O4.174 | ||
The bifunctional activity of Fe3O4-based spinels has also been well studied.169,175,176 Manganese and Co were co-substituted in a spinel structure of Fe3O4 NPs and integrated with N-doped-rGO (MnCoFeO4–NrGO). The catalyst was prepared using a hypothermal technique and showed high stability under ZAB conditions for 16.7 hours. Enhanced electronic conductivity of the cubic spinel Fe3O4 by doping with Mn and Co, as well as the coupling of MnCoFeO4 with N–rGO nanosheets, improved the cyclability of the MnCoFeO4–NPs.175 Another hypothermal technique was used in a different study to develop Co3O4 NPs with various morphologies such as cubic, blunt edge cubic and spherical particles. The catalyst was supported on N-doped graphene.177 Rotating ring-disk electrode (RRDE) results showed that spherical Co3O4 significantly outperformed the other tested morphologies, with performance close to that of Pt/C. Calculated specific capacity and power density were reported as ∼590 mA h gZn−1 and 190 mA cm−2, respectively.178
The recent work of Lee et al.179,180 was focused on Co3O4 spinel nanocrystals (nanodisks and nanowires). The current density decay rate at 1 V vs. the saturated calomel electrode (SCE) was lower for Co3O4 nanocapsules/N-doped-CNTs in comparison with Co3O4 nanocapsule/Vulcan carbon after 1000 cycles. This was attributed to the higher stability of N-doped-CNTs than Vulcan carbon and strong coupling between N-doped-CNTs and Co3O4 nanocapsules.181 In a different study, a power density of 33 mW cm−2 was achieved using N-doped Vulcan carbon/Co3O4 at a current density of 53 mA cm−2. However, the ZAB malfunctioned after 15 days of cycling (discharged at a constant current of 20 mA over 4 h and then charged at a constant current of 10 mA over 8 h). The malfunction was attributed to the partial loss of electrolyte, catalyst detachment from the air electrode and corrosion of carbon black.182
Yuan et al.188 prepared a non-carbon Ni/Co-based spinel (NixCo2−xO4) using a sol–gel technique. Low ORR/OER overpotentials and an electron transfer number of four were reported. The non-carbon based catalyst reached a maximum current density of 56 mA cm−2 during discharge and 62 mA cm−2 during charge. More complex spinel-type electrocatalysts, such as LaMn2−xCoxO4, LaMn2−xFexO4 and LiMn2−xCox−2Fex−2O4, were also investigated. They were synthesized by an improved citric precursor method. LaMn1.6Co0.4O4 was found to be the most active compound towards OER and ORR with an energy efficiency of 60–70% at 100 mA cm−2.189
A summary of ZAB performance with the spinel based catalysts mentioned above, as well as other recent studies on this group of materials, can be found in Table 6.
Perovskites are more stable and corrosion resistant compared with spinels (AB2O4). Partial substitution of other cations in A and B sites enhances the electrical conductivity, chemical stability, oxygen adsorption properties and catalytic activities. Mixing perovskites with high surface area carbon black, graphene and/or CNTs helps to overcome the relatively low electrical conductivity of these structures. It has been noted in several studies that the ORR activity of perovskites is mostly attributed to the B site cations compared with the A site cations.118,192
Simulation studies on this category of materials suggest that there is a correlation between doping and catalytic activity due to modification of the electronic structure, as opposed to morphological variations. In fact, the 3d-electron number of B site ions (anti-bonding electron occupation of B–O) can influence B–O2 interaction strength. Further studies on various La-based perovskites (Fig. 11) showed that the intrinsic ORR activity as a function of eg-orbital filling of B ions produces a volcano shape plot with a voltage span of 0.25 V. Transition metal oxide perovskites with an eg-filling of slightly below one showed the highest ORR activity, whereas perovskites with an eg value occupancy slightly above one were a better OER catalyst.118 The influence of the ratio between A and B site cations has been the subject of many studies. CNTs and α-MnO2 nanorods were integrated in a LaNiO3 perovskite. The study showed ratio ranges of x:y from 1:1 to 3:2 and w:z from 1:2 to 3:2 for [(a-MnO2)x (LaNiO3)y]w/(CNTs)z (where x and y represent molar ratios and w and z represent weight ratios) result in the highest conductivity and bifunctional activity for these catalysts.193 The bifunctional activity of La0.7Sr0.3Co1−xFexO3 (x = 0.1–0.4) has also been investigated, using cyclic voltammetry (CV) and linear sweep voltammetry (LSV). It was shown that a composition of La0.7Sr0.3Co0.7Fe0.3O3 is the best catalyst in this family. According to anodic and cathodic polarization curves, the smallest potential gap (0.82 V) at 100 mA cm−2 was achieved using La0.7Sr0.3Co1−xFexO3.194
 | ||
| Fig. 11 Role of eg electrons on ORR activity of La-based perovskite oxides.118 | ||
Perovskites can be doped with a wide range of aliovalent cations, which results in different catalytic properties. Cation doping increases the ability to catalyze ORR and the concentration of the dopant plays a very important role.195 For example, studies on Ca and Co doping of LaMnO3 perovskites suggest increased ORR activity when Mn3+ is replaced by Ca2+ or Co2+ ions. This effect is explained by the creation of a large number of oxygen vacancies which facilitate the diffusion of O2 within the crystal structure.196–198 Remarkable improvement was achieved by simply mixing an N-doped component polypyrrole (pPy) with a perovskite-based catalyst (NdBa0.25Sr0.75Co2O5.9).199 Oxygen is adsorbed on pPy (Py+·O2−). This was confirmed by the mass gain of pPy-deposited quartz crystal in an oxygen saturated media as well as through DFT analysis. In the next step, the partially charged oxygen (O2δ−) was transferred to the active site of the metal oxide to form O2−–M or O2δ−–M. This was followed by the same mechanisms for ORR on transition metal oxides (Reactions (14)–(18)).
Lanthanum-based perovskites have received significant attention as active bifunctional catalysts.200–205 Among all La-based perovskites, research has primarily focused on the composition La1−xAxMO3−δ (where A = Ca, Sr, Mn, Co or Ba and M = Co, Ni, Mn, Fe or Ir). Some examples of recent studies on this group of electrocatalysts are provided in the next paragraphs.
Table 7 summarizes in more detail the battery and catalytic characteristics of perovskite-based electrocatalysts. LaNiO3 is a highly OER active catalyst, while CNTs show excellent ORR activity. Nitrogen-doped CNT/LaNiO3 is referred to as a core-corona bifunctional catalyst (CCBC). Excellent charge–discharge stability and activity were achieved with a hybrid material of LaNiO3 (core) and N-doped CNTs (corona) in a rechargeable ZAB. After a full range of degradation tests (long-term charge–discharge cycling), CCBC-2 fabricated using 2 ml of precursor had 3 and 13 times greater ORR and OER current, respectively, compared with state-of-the-art Pt/C.206
| Catalyst material | Secondary (S)/primary (P) battery characteristics | ORR and OER catalyst characteristics | Ref. |
|---|---|---|---|
| a Op: onset potential; Lcd: limiting current density; PD: power density; Sc: specific capacity; Sed: specific energy density; N/A: not applicable; n: number of electrons transferred. CNT: carbon nanotubes; NP: nanoparticles; LDH: layered double hydroxide; LMCO: lanthanum manganese cobalt oxide; RGO: reduced graphene oxide. | |||
| LaNiO3–NCNT | (S) Edischarge: 0.94 V; Echarge: 2.33 V after 12 h at 24.5 A g−1 | ORRLcd: −3.00 mA cm−2 at −0.3 V; OERCd = 20.5 mA cm−2 at 1 V vs. Ag/AgCl | 206 |
| α-MnO2–LaNiO3/CNTs | (S) PD: 55.1 mA cm−2 at 81 mA cm−2; Edischarge: 1.191 V; Echarge: 2.048 V after 12 h C–D at 24.5 A g−1 | n ∼ 4 | 193 |
| La0.7Sr0.3Co0.7Fe0.3O3 | (S) Echarge − Edischarge = 0.82 V at 100 mA cm−2 | N/A | 194 |
| LaMnO3/LaNiO3 | (S) ORRop: 900–1000 mV more positive LaNiO3 electrode at 300 mA cm−2 | N/A | 192 |
| La0.9Ca0.1CoO3 | (S) Echarge − Edischarge = 0.89 V at 50 mA cm−2 | N/A | 207 |
| LMCO/NCNT | (S) Echarge = 2.2 V and Edischarge = 1 V at 18 mA cm−2 | ORROp: −0.11 V SCE and OERCd: 27 mA cm−2 | 208 |
| LaNiO3 NP–NCNT | (S) Echarge = 2.05 V and Edischarge = 0.94 V at 50 mA | N/A | 209 |
| La0.7Sr0.3MnO3 | (S) Echarge − Edischarge = 1.06 V at 100 mA cm−2 | N/A | 210 |
| La2NiO4 doped with Sr2+ and Ca2+ | (S) Echarge − Edischarge = 1.51 V at 75 mA cm−2 | ORROp: 0.91 V vs. RHE and OERCd: 27 mA cm−2 | 195 |
| LaMn0.9Co0.1O3–graphene | (P) initial Edischarge: 1.186 V at 20 mA cm−2 | ORRLcd: −4.5 mA cm−2; n = 3.872 | 196 |
| La0.7(Ba0.5Sr0.5)0.3Co0.8Fe0.2O3−δ | (S) initial Echarge − Edischarge = 0.75 V; final Echarge − Edischarge = 1 V at 10.5 mA cm−2; 5 min per cycle for 100 cycles | ORROp: 0.72 V vs. RHE; ORRLcd: −5.9 mA cm−2; OER overpotential at 10 mA cm−2: 370 mV | 211 |
| La(Co0.71Ni0.25)0.96O3−δ | (S) initial Echarge − Edischarge = 0.791 V at 5 A g−1; battery efficiency: 67.8% | OER overpotential at 10 mA cm−2: 324 mV | 212 |
| (P) discharge capacity: 705 mA h g−1; Sed: 710 mW h g−1 | |||
| LaMnO3 | (S) initial Echarge − Edischarge = 1.63 V at 25 mA cm−2; 600 s per cycle | N/A | 200 |
| (P) Sed: 885 W h kg−1 at 25 mA cm−2; discharge voltage of 1.18 V | |||
| La0.8Ca0.2MnO3 | (S) initial Echarge − Edischarge = 1.15 V; final Echarge − Edischarge = 1 V at 50 mA cm−2 | N/A | 198 |
| (P) PD: 98 mW cm−2 at 168.5 mA cm−2 | |||
| LaTi0.65Fe0.35O3−δ | (S) initial Echarge − Edischarge = 0.57 V; final Echarge − Edischarge = 0.83 V at 5 A g−1; 5 min per cycle for 60 cycles; round-trip efficiency before and after: 69% vs. 59% | ORROp: 0.92 V vs. RHE, n = 4 | 213 |
| (P) Sc: 440 mA h g−1; flat discharge: 1.16 V for 12 h | |||
| LaCoO3 and LaMnO3 | (S) Echarge: 2.1 V; Edischarge: 1.22 V after 150 cycles | N/A | 204 |
| (P) PD: 70–77 mW cm−2 at 140 mA cm−2 | |||
| LaNiO3–nanorods/RGO | (S) Echarge: 2.068 V; Edischarge: 1.096 V after 97 cycles (13 h) | ORROp: −0.185 V vs. Hg/HgO; n = 3.5; OEROp: 0.586 V vs. Hg/HgO; ΔE = Ej10 − E1/2 = 1.183 V | 205 |
| (P) PD: 2.85 W g−1 | |||
Perovskite based La1−xCaxCoO3 (0.2 < x < 0.4) (LCCO) catalysts were prepared by a citrate method. The partial substitution of La3+ by Ca2+ (B site cation) increased the BET (Brunauer–Emmett–Teller) surface area as well as the bifunctional activity. The optimum Ca-doping concentration for LaCoO3 was evaluated as x = 0.1 (La0.9Ca0.1CoO3) due to its homogeneity in the material and high current density.207
The carbon substrate was replaced by LaNiO3 in a recent study, to alleviate the carbon corrosion issue in ZABs. LaNiO3 is an active perovskite for OER. LaMnO3 was loaded on a LaNiO3 substrate as an active ORR catalyst using a reverse micelle method. The LaMnO3/LaNiO3 composite showed improved bifunctional activity and stability over that of a carbon-based LaMnO3 catalyst.192
As previously mentioned, making perovskite nanoparticles through traditional synthesis routes is a great challenge. Recently, 50 nm La0.7(Ba0.5Sr0.5)0.3Co0.8Fe0.2O3−δ particles were synthesized through a complex polymerization method. Rhombohedral LaCoO3 nanoparticles were incorporated on the surface of Ba0.5Sr0.50Co0.8Fe0.2O3−δ. Lanthanum concentration and calcination temperature were controlled to influence oxide defect chemistry and particle growth. A five-fold increase in OER current density was observed in comparison with the IrO2 baseline.211 Another alternative to traditional synthesis routes is electrospinning. Electrospinning is a simple technique, which has recently been used to develop nanosized perovskites. PAN acted as a source of porous carbon with high nitrogen content, apart from being an electrospinning medium.212,213 Also, the process is simple and low cost to prepare one dimensional interconnected structures such as nanotubes, nanofibers and nanobelts. Electrospinning was used in a recent study by Vignesh et al.212 for synthesis of La(Co0.71Ni0.25)0.96O3−δ NPs. The particles had a nanotubular structure based on SEM observations, with outer and inner diameters of 100–150 nm and 50–70 nm, respectively. The OER overpotential was 324 mV at 10 mA cm−2, which is 56 mV more negative than baseline IrO2. Electrochemical analysis indicated the presence of Ni in octahedral sites, which alter eg-filling (<1) and weaken the bonding strength of OH− species on the perovskite structure, leading to good OER activity.
The above examples of mixed transition metals oxides (spinels and perovskites) show that the rich defect properties of this group of electrocatalyst materials make their structures flexible and adaptable for developing advanced air electrodes for future rechargeable ZABs.
4.3. Transition metals
A combination of transition metals and carbon-based materials has been proposed to endow the hybrids with favorable electrocatalytic activity for both ORR and OER. However, the multistep synthesis procedure is one of the limitations for large scale commercialization of this group of materials.There is growing interest in FeCo, FeNi and NiCo alloy nanoparticle catalysts for ZAB applications.214,224,229,230 Studies show that the alloys have high stability and bifunctional activity. Table 8 summarizes the battery performance (both secondary and primary ZABs) of this group of alloys and others.
| Catalyst material | Secondary (S)/primary (P) battery characteristics | ORR and OER catalyst characteristics | Ref. |
|---|---|---|---|
| a Op: onset potential; Lcd: limiting current density; E1/2: half-wave potential; Ej10: OER current density at 10 mA cm−2; PD: power density; Sc: specific capacity; Ed: energy density; Sed: specific energy density; N/A: not applicable; n: number of electrons transferred. NC: N-doped carbon; PFC: porous fibrous carbon; SCCF: simple-cubic carbon framework; BMZIF: bimetallic zeolitic imidazolate frameworks; NSPC: nitrogen- and sulfur-doped porous carbon; UFR: urea-formaldehyde resin; Cu–N©C: Cu(I)–N sites embedded in a graphene matrix. | |||
| FeCo@NC | (S) Edischarge: 1.25 V; Echarge: 2 V after 20 h at 10 mA cm−2 | ORROp: 0.94 V vs. RHE ORRLcd: −4.82 mA cm−2; n = 4, OEROp = 1.45 V vs. RHE, OERCd: 10 mA cm−2 at 1.49 vs. RHE | 214 |
| (P) PD: 136 mW cm−2; Sc: 763 mA h g−1 at 1.275 V | |||
| PFSA–Fe3.5Ni | (P) PD: 55.1 mA cm−2 at 81 mA cm−2; Edischarge: 1.191 V; Echarge: 2.048 V after 12 h C–D at 24.5 A g−1 | n ∼ 4 | 193 |
| MoSe2/MoO3 | (S) Edischarge: 1.17 V; Echarge: 1.4 V for 130 cycles & ∼7 days (1 mA cm−2) | ORROp: 0.93 V vs. RHE; OEROp = 1.2 V vs. RHE | 215 |
| (P) PD: 250 mW cm−2 at 182 mA cm−2 (0.8 V) | |||
| NiCo/PFC aerogels | (S) battery efficiency of 56.7% after 600 h cycling at 10 mA cm−2 | ORROp: 0.92 V vs. RHE; n = 3.89, ΔE = Ej10 − E1/2 = 0.86 V vs. RHE | 216 |
| Fe–N–SCCF | (P) current density: 205 mA cm−2 at 1.0 V and PD: 300 mW cm−2 | ORROp: 1.03 V vs. RHE; n = 3.9–4.0 | 217 |
| Fe/C/N@BMZIF | (S) Edischarge: 1.11 V; Echarge: 1.96 V for 100 cycles & ∼17 days (10 mA cm−2) | ORROp: 0.96 V vs. RHE; n = 3.85–4.0, OERCd: 10 mA cm−2 at 1.64 vs. RHE; ΔE = Ej10 − E1/2 = 0.79 V | 218 |
| (P) PD: 235 mA cm−2 at 307 mA cm−2 | |||
| FeNC-850 | (P) circulated electrolyte; discharge time of 23 h at 50 mA cm−2; PD: 186 mW cm−2 | ORROp: 0.91 V vs. RHE; n = 4.0, ORRLcd: −5.97 mA cm−2 | 219 |
| Co9S8/NSPC | (S) initial Edischarge: 1.15 V; Echarge: 2.11 V at 50 mA cm−2 | ΔE = Ej10 − E1/2 = 0.92 V vs. RHE; n = 3.94 | 220 |
| C–Fe–UFR | (S) stable charge–discharge for 34 h at 10 mA cm−2 | ORROp: 0.86 V vs. RHE; Ej10: 1.68 vs. RHE | 221 |
| (P) PD: 142 mA cm−2 at 0.65 V; Sc: 438 mA h g−1 at 50 mA cm−2 | |||
| Poly-iron–phthalocyanine | (P) PD: 192 mA cm−2 at 1.6 V | ORROp: 0.94 V vs. RHE; ORRLcd: −5.58 mA cm−2; n = 3.47 | 79 |
| Cu–N©C | (P) PD: 210 mW cm−2, current density: 142 mA cm−2 at 1 V, Edischarge: 1.16 V after 100 h at 20 mA cm−2 | n = 3.37, ORRLcd: −6.7 mA cm−2 | 222 |
| CoFe@NCNTs | (P) PD: 150 mW cm−2 at 1.45 V; Sc: 808 mA h g−1 | ORROp: 0.84 V vs. RHE; OEROp: 1.45 V vs. RHE; OERCd: 10 mA cm−2 at 1.68 vs. RHE ORRLcd: −5.58 mA cm−2; n = 4 | 223 |
| (S) initial Edischarge: 1.25 V at 10 mA cm−2 | |||
| Ni–Co@ND–C | (S) Echarge − Edischarge = 1 V after 60 h cycling at 10 mA cm−2 | E OER − EORR = 0.71 V | 224 |
| Co@NCNT | (S) stable up to 16 h at 10 mA cm−2 | ORRE1/2: 0.828 V vs. RHE; ORRLcd: −6.3 mA cm−2; n = 3.95–3.99, 429 mV overpotential at OERCd: 10 mA cm−2 | 225 |
| (P) PD: 138.82 mW cm−2 at 59.02 mA cm−2 | |||
| Fe–histidine 700 | (S) Echarge − Edischarge = 0.95 V after 50 h cycling at 20 mA cm−2 | ORR ORR: 1.02 V vs. RHE; n = 3.97; ΔE = Ej10 − E1/2 = 0.81 V | 226 |
| (P) 212 mW cm−2; Sc: 813 mA h g−1 Ed: 960 W h kg−1 | |||
| Ni3Fe/N–C | (S) Echarge − Edischarge = 0.98 V after 105 cycle 420 h cycling at 10 mA cm−2 | ORROp: 0.90 V vs. RHE ΔEOER (10 mA cm−2)−ORR (3 mA cm−2) = 0.84 V, n = 3.75–3.87 | 156 |
| (P) Sc: 528 W h kg−1; Ed: 634 W h kg−1 | |||
| Co–N/CNT | (P) PD: 300 mW cm−2 at 1.2 V; stability of 8 h at 50 mA cm−2 | E 1/2: 0.91 V vs. RHE in 1 M KOH | 227 |
| Fe–N/C-700 | (P) Sc: 682 mA h g−1 at 20 mA cm−2 and Ed: 787 W h kg−1 | ORROp: 0.96 V vs. RHE, ORRLcd: −5.8 mA cm−2; n = 4.02 | 228 |
Nickel- and Co-based bifunctional electrocatalysts (standalone metallic or NiCo/oxide-hydroxides) have attracted considerable attention due to their high electrical conductivity.153,231
A NiCo alloy coupled with a small fraction of its oxides with N-doped CNTs as the catalyst support (NCNT/CoO–NiO–NiCo) exhibited excellent bifunctional activity and stability for ORR-OER in primary and rechargeable ZABs. Under ambient air conditions, the primary ZAB showed a high energy density of 615 W h kgzn−1 at a current density of 20 mA cm−2. Also, a smaller charge–discharge potential gap (0.86 V) was achieved compared with NCNT/CoO–Co and NCNT/CoO–NiO–NiCo catalysts in a rechargeable ZAB. This is due to the synergistic effect between the monometallic Co-based (ORR active) and Ni-based (OER active) electrocatalysts.232
Furthermore, Co and Fe and their oxides have been reported to have high bifunctional activity.233 Copper-Fe-NP alloys encapsulated within graphitic carbon layers are highly active and durable ORR catalysts. DFT calculations revealed a complex ORR mechanism and a possible synergistic effect due to alloying. The results showed that the CuFeinner surface facilitated ORR at a lower overpotential. High resolution transmission electron microscopy (HRTEM) after 1000 continuous cycles showed no change in the crystal structure of the pristine CuFe particles, whereas the graphitic carbon layers were completely transformed into amorphous carbon. The carbon layer was also reported to increase the corrosion resistance of the core catalyst and acted like a protective layer.154
In a recent study by Xiong and Ivey, CoFe alloys were directly applied on GDL using a simple cathodic electrodeposition technique. The catalyst exhibited high OER stability and activity with similar battery efficiency compared with Pt/Ru-CB.229,234
A hybrid material derived from the perfluorosulfonic acid (PFSA)/PTFE copolymer with an optimum ratio of Fe to Ni has been studied as a candidate material for ZABs. PFSA–Fe3.5Ni exhibits the highest ORR activity among PFSA-derived hybrid materials. High ORR activity is attributed to the carbon and the FeNi species are considered as the OER catalyzing sites. A power density of 262 mW cm−2 was obtained in a primary ZAB at 0.77 V, which is superior to commercial 40 wt% Pt/C.235
Transition metal chalcogenides (TMCs) with high electrocatalytic activities for OER/ORR have also been considered as bifunctional catalysts for rechargeable ZABs.220 Among various TMCs, Group VIB dichalcogenides including WS2/WSe2, MoS2 and MoSe2 have shown excellent performance due to their tunable band structures and good electronic conductivity. In a study by Karfa et al.,215 a combination of MSe2 and MO3 (M = Mo or W) was developed as a robust and highly efficient electrocatalyst. Four different MoSe2/MoO3 and WSe2/WO3 shapes, including marigold-like flowers, small rod-like agglomerated flowers, rhombic dodecahedra and nanorod nanohybrids were synthesized by varying the reaction temperatures. Of these, the flower-shaped MoSe2/MoO3–2 nanohybrids with thin and small rod-like petals showed the best bifunctional performance. No obvious change in the current was observed after charge–discharge cycling for ∼7 days, indicating the high stability of the as-prepared air electrode. Nickel and Co nanoparticles embedded in carbon based materials have been a focus of the many researchers.225,236 Ni–Co nanoparticles anchored on porous fibrous carbon aerogels (NiCo/PFC) demonstrated excellent performance in a rechargeable ZAB. As explained by authors, the advantage of using PFE aerogel as opposed to other carbon supports is the large open spaces between the neighboring carbon nanofibers and nanopores, which enables the active species to diffuse to/from the Ni/Co nanoparticles (Fig. 12a). Also, the uniform distribution of covalently anchored catalytic particles suppresses agglomeration/dissolution of the nanoparticles. The structure is highly durable in a rechargeable ZAB with an efficiency of 56.7% after 300 cycles (600 h) at 10 mA cm−2 (Fig. 12b).216
 | ||
| Fig. 12 (a) Schematic illustration of the advantages of NiCo/PFE aerogels as electrocatalysts. (b) Charge–discharge voltage profile of a rechargeable ZAB with NiCo/PFC aerogel catalysts at a current density of 10 mA cm−2.216 | ||
Recently, metal–organic frameworks (MOF) have been widely used as a composite precursor, enabling the fabrication of microporous metal–N–carbon catalysts by direct pyrolysis.217,225 The composite material is derived from pyrolyzing different precursor materials such as bimetallic zeolitic imidazolate frameworks (BMZIFs),218 Fe–histidine complex,225 2-methylimidazole/Co-nitrate,225 Zn nitrate hexahydrate (Zn(NO3)2·6H2O)/2-methylimidazole,237 Prussian blue (PB),219 iron-chelated urea-formaldehyde resin hydrogel,221 b-cyclodextrin/Co-nitrate,225p-phenylenediamine/o-phenylenediamine225 and Fe-1,4-bis(1H-1,3,7, 8-tetraazacyclopenta (1) phenanthren-2-yl) benzene (btcpb).228
A pyrolysis-based route was used to develop Ni3Fe nanoparticles embedded in porous N-doped carbon sheets.17 The SEM image in Fig. 13a shows good dispersion of Ni3Fe particles on carbon sheets. The thickness of the sheets is about 41 nm and the particle diameter is estimated at 39.7 nm. EDX mapping reveals a homogeneous distribution of both Ni and Fe (Fig. 13b). The overall oxygen electrode performance was evaluated by the difference in potential between the OER current density at 10 mA cm−2 and the ORR current density at −3 mA cm−2 (ΔE = Ej10 − Ej3). The lowest ΔE value was achieved for Ni3Fe/N–C (0.84 V) for the studied catalysts (Fig. 13c).156
 | ||
| Fig. 13 (a) SEM SE image of Ni3F/N–C (white and red arrows reveal the uniform distribution of Ni3Fe particles and nanopores, respectively). (b) STEM-image and corresponding EDX maps of Ni and Fe on the Ni3F/N–C sheets. (c) Overall polarization curves for Ni3Fe/N–C sheets, Ni3Fe/C sheets, IrO2 and Pt/C catalysts within the ORR and OER potential window (Inset: the value of ΔE for the four catalysts (ΔE = Ej10 − Ej3)).17 | ||
M–N4 macrocycles (porphyrin and phthalocyanine (Pc)) are emerging electrocatalysts due to their highly porous structure and low-cost synthesis techniques. Since M–N4 based electrocatalysts have poor stability and conductivity, many approaches including pyrolysis, covalent grafting with carbon-based materials and polymerization have been employed to resolve this issue. Porous two dimensional (2D) poly-iron–phthalocyanine (PFe–Pc) based oxygen reduction electrocatalysts were created with a simple solid-state chemical reaction without pyrolysis.79 SiO2 nanoparticles were used to preserve the Fe–N4 structure during the polymerization process and to assist in the formation of a porous structure. One of the obstacles of using Fe–N4 is that the mass loading is often less than 0.2 wt%. Increasing the mass loading leads to the formation of Fe nanoparticles. Cu–N4 has been proposed as an alternative. However, Cu(II) is inert towards ORR due to its saturated coordination. Therefore, reducing Cu(II) to Cu(I) is needed which was done using dicyandiamide in a recent report. Dicyandiamide is a widely used graphene precursor and its decomposition species has a strong reducing agent which is used to reduce Cu(II)–N.222
4.4. Carbon-based bifunctional catalysts/catalyst support
Bifunctional catalytic activity has been studied in various carbon-based materials, such as carbon black, graphene/GOs,238–240 reduced GOs,186 graphene quantum dots,241 CNTs76,242,243 and nanodiamond.244 The details of secondary and primary ZABs for this group are summarized in Table 9.| Catalyst materials | Secondary (S)/primary (P) battery characteristics | ORR and OER catalyst characteristics | Ref. |
|---|---|---|---|
| a Op: onset potential; Lcd: limiting current density; PD: power density; Sc: specific capacity; Sed: specific energy density; N/A: not applicable, n: number of electrons transferred; E1/2: half-wave potential. PoPD: pyrolyzed o-phenylenediamine; ND: N-doped; PMF: phenol–melamine–formaldehyde; SWCNT: single wall carbon nanotube; PDA: polydopamine; PCN: P-doped graphitic-C3N4; CFP: carbon-fiber paper; macro/meso–NC–NH3: NH3-activated N-doped macro/mesoporous carbon; NCNF: nanoporous carbon nanofiber film; NC: N-doped carbon; GC: graphitic carbon; CNS: carbon nanocrystal; HGF: holey graphene framework; VA: vertically aligned; GF: graphene foam. | |||
| Meso/micro-PoPD | (P) Sc: 630 mA h g−1 for 100 h | ORRop: 0.84 V vs. RHE ORRE1/2: 0.85; n = 3.97 | 238 |
| Eggplant derived 2D carbon sheets | (S) Edischarge: 1.23 V at 5 mA cm−2 | ORRop: 0.069 V vs. Ag/AgCl; ORRLcd: −6.09 mA cm−2 at 0.65 V vs. Ag/AgCl; n = 4 | 261 |
| (P) Sc: 669 mA h g−1 | |||
| N/P doped mesoporous carbon | (S) PD: 55 mW cm−2; stable operation for 240 h at 2 mA cm−2 | N/A | 250 |
| (P) Sc: 735 mA h g−1; Sed: 835 W h g−1 | |||
| ND-hollow mesoporous carbon | (S) Echarge: 2.13 V; Edischarge: 1.24 V at 2 mA cm−2 | ORRop: −0.05 V vs. Hg/HgO; ORRLcd: −4.95 at −0.7 V; n = 4 | 95 |
| Fe and N-doped graphene nanosheets | (P) PD: 61 mW cm−2 | ORRop: −0.023 V vs. SCE; n = 3.79 to 3.99 | 252 |
| N-Doped gelatin with Ketjenblack carbon | (P) PD: 193 mW cm−2 | n = 3.7–3.85 | 260 |
| N-Doped carbon fibers like | (P) PD: 194 mW cm−2 | Kinetic current: 6.85 mA cm−2 at −0.15 V; n = 3.7–3.8; peroxide yield: 13% at −0.4 V vs. Hg/HgO | 262 |
| Graphene oxide and PMF | (P) PD: 70 mW cm−2 at 100 mA cm−2; Sc: 400 mA g−1 | ORRop: −0.1 V vs. SCE; n = 3.4–3.8 | 239 |
| N-Doped CNT | (P) PD: 69.5 mWcm−2 at 78.6 mA cm−2 | N/A | 263 |
| SWCNT | (P) discharge capacity: 375 mA h g−1 at 0.25 mA | N/A | 242 |
| Co–PDA–C | (S) Echarge: 2.15 V Edischarge: 1.21 V at 2 mA cm−2; +0.23 V increase in gap after 500 h | ORRpp: 0.783 V vs. RHE; ORRE1/2: 0.767 V; EOER: 1.601 V at 2 mA cm−2vs. RHE; n ∼ 4 | 264 |
| N-D dope nondiamond | (S) Echarge:: 2.4 V Edischarge: 1 V at 16 mA cm−2; 300 s per cycle | ORRpp: −0.02 V vs. SCE; ORRE1/2: −0.18 V; n ∼ 3.96 | 244 |
| (P) PD: 24.8 mW cm−2 | |||
| PCN–CFP | (S) Echarge: 2.46 V; Edischarge: 1.05 V at 10 mA cm−2; 5 min per cycle | ORRop: 0.94 V vs. RHE; OERop: 1.53 V vs. RHE | 265 |
| Macro/meso–NC–NH3 | (S) Echarge − Edischarge = 0.7 V at 10 mA cm−2; 200 cycles at 4 h per cycle | ORRLcd: −6.6 mA cm−2 at −0.4 V SCE | 264 |
| NCNF | (S) initial Echarge: 1.93 V; Edischarge: 1.20 V at 10 mA cm−2; 5 min per cycle in air | ORRop: 0.97 V vs. RHE; ORRLcd: −4.7 mA cm−2 at 1600 rpm; n = 4; OERop: 1.84 V | 266 |
| (P) PD: 185 mW cm−2; Sed: 776 W h kg−1; Sc: 626 mA h g−1 | |||
| Graphene quantum dots | (P) PD: 70 mW cm−2 at 100 mA cm−2 | ORRop: −0.13 V vs. Ag/AgCl | 241 |
| S-Doped graphene fiber | (S) Echarge − Edischarge = 0.8 V at 1 mA cm−2; 148 cycles (20 h) | ORRop: −0.02 V vs. Ag/AgCl; ORRLcd: −125 mA cm−2; OERop: 0.29 V vs. Ag/AgCl; OERLcd: 497.8 mA cm−2 | 267 |
| (P) PD: 300 mW cm−2 at 0.6 V | |||
| NC@GC | (S) voltage gap increase ∼0.07 V at 20 mA cm−2 after 30 cycles (1 h per cycle) | ORRop: 0.98 V vs. RHE; n = 3.69–4; EOER: 1.57 V vs. RHE at 10 mA cm−2 | 237 |
| (P) constant discharge potential of 1.29 V for 140 h at 5 mA cm−2 | |||
| P,S doped-CNS | (S) initial Echarge: 2.02 V Edischarge: 1.22 V at 25 mA cm−2 | ORRop: 0.97 V vs. RHE, ORRLcd: −6.7 mA cm−2, OERop: 1.26 V vs. Ag/AgCl | 256 |
| (P) PD: 198 mA cm−2, Sc: 830 mA h g−2, Sed: 970 Wh kg−1 (stable for 210 h) | |||
| N,S-Doped hierarchically porous carbon | (S) Echarge − Edischarge = 0.77 V at 10 mA cm−2, 85 mV potential loss after 55 h (300 cycle 11 min per cycle) | ORRop: 0.99 V vs. RHE, ORRLcd: −5.8 mA cm−2, n = 3.86–3.96 | 257 |
| (P) PD: 151 mW cm−2 at 72 mA cm−2 | |||
| Graphene-like nanobubble/nanosheet hybrid | (P) PD: 201 mW cm−2 at 0.7 V; Sc: 767 mA h g−1 | ORRop: 0.971 V vs. RHE, n = 3.98 | 258 |
| N–CNTs–HGF | (S) initial Echarge − Edischarge = 1.5 V at 5 mA cm−2 | ORRop: 1.08 V vs. RHE | 76 |
| (P) PD: 8.5 mW cm−2 at 17.5 mA cm−2; Sc: 635 mA h gZn−1; Sed: 614 W h kgZn−1 | |||
| VA–CNTs/GF | (S) Echarge − Edischarge = 0.85 V at 2 mA cm−2 after 40 h | ORRop: −0.19 V vs. Ag/AgCl, ORRLcd: 48 mA g−1 at −1.0 vs. Ag/AgCl; n = 3.9 | 243 |
| B,N-Doped carbon | (S) initial: Echarge: 2.19 V Edischarge: 1.16 V at 2 mA cm−2 | ORRop: 0.894 V vs. RHE; ORRLcd: −4.73 mA cm−2, OERop: 1.38 V vs. RHE; OERLcd: 4.94 mA cm−2 at 1.6 V | 268 |
| (P) Edischarge: 1.14 V and 25% coulombic efficiency | |||
Two important factors determine the performance of carbon-based electrocatalysts, i.e., surface functionality and surface area. Carbon blacks (e.g., Vulcan XC-72) are the most common catalyst supports for ZAB applications.245 A study by Eom et al.246 investigated the effect of carbon black types with different surface areas. It was shown that the air cathodes consisting of mesoporous and macroporous carbon blacks, such as Darco G-60 N, have the highest power density (353 mW cm−2) at 0.71 V. However, the calculated BET surface area of Darco G-60 N was lower compared with other types of carbon blacks (e.g., Vulcan XC-72). This indicates that high surface area is not the only factor affecting catalytic performance. In general, mesoporous/microporous carbon is a desirable ORR active electrocatalyst because it provides numerous active sites for favorable and efficient mass transport during charge–discharge cycling.247
Another important factor is optimizing the degree of hydrophobicity of mesoporous/microporous carbon particles for catalyst supports. Thick hydrophobic carbon layers as a catalyst support may block the electrolyte, limit the mobility of Zn ions and increase the overall interfacial resistance.242
A study by Li et al.248 showed high ZAB performance using CNTs with a diameter of 10 nm as the catalyst support due to high durability (up to 100 cycles/300 s per step) and a discharge power density of 66.3 mW cm−2 at 105.75 mA cm−2. A study on the effect of the surface area of Ketjenblack vs. graphitized carbon and activated Vulcan XC-72 as the catalyst support for La0.6Ca0.4CoO3 bifunctional catalysts showed that Ketjenblack ED-600JD, with the highest surface area (1300 m2 g−1), has the best ORR activity with a potential of −0.2 V vs. Hg/HgO at 100 mA cm−2. It was suggested that homogenously dispersed, high surface area carbon supports provide shorter diffusion pathways for peroxide ions resulting in smaller total resistance.249
Recent reports have been highly focused on doping nitrogen into pristine graphene, graphene oxide and CNTs. N-doping of these carbon structures creates pyrrolic, pyridinic and graphitic C–N bonds which increase the number of active sites.250–252 Graphitic N can provide electrons to the π-conjugated system which leads to increased nucleophile strength for the neighboring carbon rings. This will enhance the probability of O2 adsorption. Pyridinic N can accept electrons from adjacent carbon atoms and facilitate the adsorption of water oxidation intermediates which results in higher OER activity.253 Doping of N into carbon networks is commonly performed by pyrolysis of various precursors such as vitamin B5,254 pyrrole/melamine,255 aminoguanidine,256 trithiocyanuric acid257 and keratin.258 It was found that charge delocalization in N-doped carbon facilitates the Yeager model for oxygen chemisorption over the Pauling model, which could effectively weaken the O–O bond (Fig. 6).259 Nitrogen is an n-type carbon dopant, meaning that N donates electrons to carbon and creates an uneven charge distribution. Also, N-doped carbon materials are more disordered than their undoped analogues.260
For example, novel, highly active mesoporous N-doped, carbon-based ORR catalysts were developed using a hard-templating synthesis technique (polymerization of o-phenylenediamine (oPD)) in the presence of a silica colloid. The process was followed by the pyrolysis of the oPD/silica composite at high temperature. The resulting high surface area (1280 m2 g−1) catalyst, denoted as microporous/mesoporous pyrolyzed-oPD, showed a remarkable ORR onset potential of 0.85 V vs. RHE and stability of up to 100 h in a ZAB. It was found that the catalytic activity of pyrolyzed-oPD derived catalysts was largely influenced by NH3 activation time (N-doping time). The ORR activity improved with increasing N-doping time, plateauing after 15 min. Based on quantum theory calculations, quaternary nitrogen in the graphene planes reduces the energy barrier for oxygen adsorption by inducing a non-uniform electron distribution in short C–N bonds. Therefore, the dissociation and adsorption of oxygen is facilitated.238 In a different study, eggplant-derived 2D mesoporous carbon microsheets with high surface area (1051 m2 g−1) were fabricated by simple carbonization and activation steps. The catalyst was applied in a rechargeable ZAB to evaluate its bifunctional activity. The results showed high stability for 62 h after charge–discharge cycling at 5 mA cm−2.261
In another study, N-doped hydrophobic Ketjenblack carbon was derived from gelatin and tested in a ZAB cell at 25 and 50 mA cm−2. The peak power density of the cell was 5.3% higher than the Pt/C baseline.
Doping with a second heteroatom such as B, S or P can also modulate the electronic properties and surface polarities for further improvement of carbon-based electrocatalytic activity.239,250,260,263,265,269,270 The reason for enhanced ORR activity after doping with heteroatoms is described as being due to partial positive charges remaining on the heteroatom centers, which act as active sites for O–O dissociation. Three-dimensional N–P-co-doped mesoporous nanocarbon foam was synthesized in a one-step process involving the pyrolysis of a polyaniline aerogel in the presence of phytic acid. The electron transfer number obtained after RRDE testing was 3.85 with a small ratio of peroxide species (<8%). The bifunctional activity of the catalysts was evaluated in primary and rechargeable ZABs. The primary ZAB was operated with no significant discharge potential drop after 30 h at 5 mA cm−2. Integrating two Zn–air button cell batteries in series generated an open circuit potential of ∼2.8 V to power LEDs. First-principle DFT calculations were performed to investigate the possible active sites on the doped structures, providing a minimum ORR overpotential. In most cases, OER and ORR occurred near the edges of the graphene basal plane. Based on volcano plots for ORR, N-doped sites showed lower overpotential (0.44 V) in comparison with P-doped sites (0.47 V) and N–P-co-doped sites (0.47 V). On the other hand, N–P-co-doped graphene was identified as the most OER active with the lowest overpotential of 0.39 V.95
Dopamine (DA) has also been used as a carbon and N-doping source in the development of active ORR/OER carbon-based electrocatalysts. DA can be readily self-polymerized at room temperature to form polydopamine (PDA) on a substrate. Also, DA possesses OH− chelating groups, which can contain metal ions during polymerization and form metal/PDA composites.227
Very recently, researchers designed a material with a 3-dimensional, carbon-based architecture as a flexible electrode for wearable electronics.242,265,266,271 High mechanical integrity and cycling stability were observed in P-doped graphitic-C3N4 directly grown on carbon-fiber paper (PCN-CFP) prepared through a hypothermal process. Different folded and rolled-up forms of the air-electrode were studied using ORR/OER polarization testing, as shown in Fig. 14. LSV curves of the folded PCN-CFP before and after 3000 cycles exhibited negligible current loss. The highly rolled up structure preserved 87.8% and 92.7% of the initial ORR and OER currents, respectively.265 Also, ZABs based on nonporous carbon nanofiber films (NCNFs) were integrated in series to power commercial light emitting diodes (LED, 3V) worn on a human hand, demonstrating the possibility of ZABs as a flexible power source in wearable optoelectronics.266
 | ||
| Fig. 14 (a) LSV current for PCN-CFP in different folded and rolled up forms. (b) Charge–discharge cycling curves using PCN-CFP and Pt-CFP directly as the air electrode (inset: ZAB configuration schematic).265 | ||
Sulfur-doped graphene-Idli (S-DGF) was prepared using a microwave technique and utilized as a metal-free bifunctional catalyst. With a high surface area of 400 m2 g−1, the S-DGF catalyst had an ORR onset potential of −0.02 V vs. Ag/AgCl and an OER onset potential of 0.29 V vs. Ag/AgCl. However, ZAB charge–discharge is only reported for 1 mA cm−2, which is a lower current density than that for previously reported carbon-based catalysts.267
Novel metal-free electrocatalysts have been developed using 2 different MOFs as a core–shell material. The resulting shell material is highly graphitic carbon (GC, carbonized from ZIF-67) and the core material is N-doped carbon (NC, carbonized from ZIF-8) (NC@GC). Fig. 15a shows a TEM image of NC@GC. The NC structure is amorphous with high N content, whereas GC is highly crystalline and contains Co-nanoparticles which function as a catalyst booster. The charge–discharge potential difference increased only 0.07 V after 40 h cycling in a rechargeable ZAB operated at 20 mA cm−2 (Fig. 15b).237
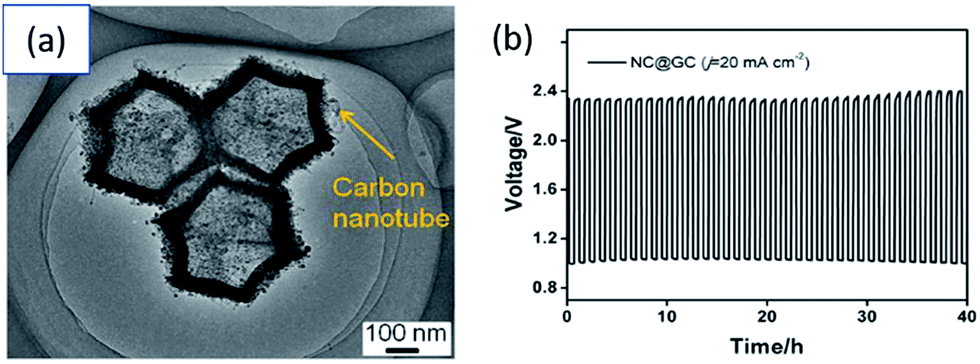 | ||
| Fig. 15 (a) TEM bright field image of NC@GC. (b) Cycling performance of ZAB using NC@GC at 20 mA cm−2.237 | ||
5. Progress and outlook
This review has highlighted recent progress in the design and electrochemistry of bifunctional electrocatalysts for primary/secondary ZABs. To sum up, there are still major challenges that exist in this field; however, promising progress has been made over the past few decades. The number of publications dedicated to secondary/primary ZABs derived from Scopus and Google Scholar databases is summarized in Fig. 16, which shows that the number of scientific reports, journal papers and patents has increased by ∼80% since 1996. | ||
| Fig. 16 The number of publications addressing the challenges in primary/secondary ZABs between 1996 and 2016 (the numbers are provided from the Scopus and Google Scholar databases). | ||
In general, it is much easier to handle secondary ZABs relative to other metal–air batteries such as Li–air batteries.272,273 All the components of ZABs are moderately stable towards moisture and all the reactions can be carried out under ambient air conditions. Therefore, the manufacturing process for ZABs is less stringent and cheaper than that for Li–air batteries. Although there has been progress, many aspects of ZABs are not fully understood and extensive investigation is still required in this field.
Rechargeable ZABs can be used in bi-electrode or tri-electrode configurations, which can dramatically affect the cycle life of the battery. Bi-electrodes are structurally compact with limited cycle life. The catalytic layer is normally a multilayered structure, where the hydrophilic side of the GDL (facing the electrolyte) is used for OER catalysts and the ORR catalysts are oriented towards the air side. Using N-doped graphene and CNTs can improve the electrical conductivity of the air electrode. Also, as discussed in Section 4.2.1, catalysts such as Ni–Fe–CO32−–LDH can prevent carbonate precipitation on the air electrode. However, there is also a high probability of catalyst detachment from GDL during recharge at high potentials due to the generation of O2 bubbles.
The tri-electrode mode, on the other hand, is structurally complex but can significantly improve the battery's cycle life.226,256 Some examples are provided in Sections 4.2.1 and 4.2.2. The charging process does not occur on the third electrode, so O2 bubbling will not damage the ORR catalysts.
Also, the third electrode is usually a Ni or Ni alloy foil, which is a suitable OER catalyst. Furthermore, carbon corrosion can be avoided because the air electrode operates at lower potential. As a result, the performance of the ZAB is mainly determined by the stability and activity of the applied ORR catalyst.22,147
Sectionalized deposition of OER and ORR catalysts is another strategy to minimize the physical interference of catalytic function. For example, selective electrodeposition of MnO2 and Co3O4 onto 3D porous nickel foam has been studied using an agarose gel-mediated technique. In this method, selective electrodeposition can take place on desired areas of the substrate, thus preventing crossover of metal ions through the porous nickel foam structure.274
Amendola et al.21 investigated a horizontal configuration for rechargeable ZABs. As shown in Fig. 17, the air electrode is at the top and Zn is at the bottom of the cell. The electrolyte is contained and sealed by the frame. An airflow tunnel is provided between the Zn electrode and the air electrode. In this specific configuration, gravity helps in settling the discharge product as an even layer on the Zn electrode so there may be no need for a separator. This configuration is suitable for stationary devices requiring a large amount of energy density. The battery cycle life is reported as 2700 cycles with no performance degradation.
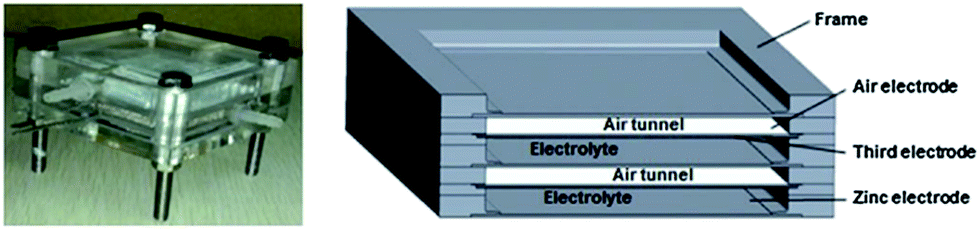 | ||
| Fig. 17 The structure of a rechargeable ZAB arranged in a horizontal configuration.22 | ||
The use of a flowing electrolyte as opposed to a static electrolyte is another design modification to prevent the growth of Zn dendrites and promote the formation of an even layer during charging. In addition, heat and generated gas bubbles are removed, which should increase the charge/discharge rate of the battery.22 A recent study by Yang et al.219 showed that discharge time (at 50 mA cm−2) (Fig. 18a) can be extended to about 23 h until Zn is fully consumed. Pumping the electrolyte, as shown in Fig. 18b, prevents passivation of the Zn electrode and a rapid drop in the discharge potential.
 | ||
| Fig. 18 (a) Discharge curve for ZAB cell with circulating electrolyte. (b) Schematic of a ZAB cell with pump-circulated electrolyte.219 | ||
Finally, although batteries have been utilized for different applications for quite some time, it is only in the last decades that a fundamental understanding of the structure–property relationships that govern performance has been achieved. This was instrumental in the development of Li-ion batteries based on intercalation reactions in 1991, which quickly led to the dominance of Li-ion batteries in the portable electronics market.13 However, further expansion into the field of stationary storage is a challenge for Li-ion technology due to concerns regarding Li availability and cost. Similar challenges exist for Li–air batteries as well. Battery technology based on Zn, which is abundant and economical, may be a solution for generalized applications in large-scale storage. ZAB batteries, once commercialized, are projected to cost ∼$160 kW h−1 or almost half the cost of Li-ion batteries (∼$300 kW h−1).32 However, commercialization of ZABs is heavily dependent on the performance-to-cost ratio of bifunctional catalysts. Recent studies have been mostly focused on the design, crystal structure and composition of catalysts and there is a lack of studies on electrochemical durability and long-term cyclability. Furthermore, the synthesis routes for high-performance hybrid bifunctional catalysts can be costly and time consuming. Reducing the number of processing steps and engineering one-pot synthesis strategies will be more desirable in the future.32,166 ZABs are the only type of metal–air batteries that are found in current applications. Traditional primary ZABs have only penetrated a niche market; e.g., hearing aids and buoys, as a result of their limited power density.20 With recent improvements in power capability, due to the development of novel catalysts and battery designs, rechargeable ZABs are viable for numerous applications, specifically for stationary devices. It is difficult to predict the rate of success, but it is clear that with continued dedicated research and breakthroughs, ZABs should be able to command a larger role in future battery technology.
6. Conclusions
There has been ongoing progress in the area of Zn–air batteries (ZABs) over the past few years. They offer benefits in terms of energy density, environmental considerations and safe operation to store and generate clean electricity for both stationary and mobile devices. Although there have been developments in this field, many aspects of ZAB are not fully understood. The most critical problem in ZABs originates from the slow electrochemistry of the oxygen reduction reaction (ORR) and oxygen evolution reaction (OER). In this review, four categories of ORR/OER catalyst materials, termed “bifunctional”, ranging from transition metal oxides (single/mixed-metal oxides, spinels and perovskites) to transition metals, carbon-based materials and precious metals/alloys, as well as the architecture of air electrode, have been surveyed for use in primary/secondary ZABs. Although the Zn anode, separator and electrolyte were not emphasized in this review, it is critical to develop a rational design considering the challenges for each component of a rechargeable ZAB structure. The performance of ZABs is highly affected by durability and activity of innovative bifunctional electrocatalysts, the degree of hydrophobicity/hydrophilicity of the catalyst substrate, the conductivity and composition of the electrolyte and the utilization efficiency of the Zn anode. Overcoming the remaining challenges will require ongoing research efforts from different disciplines.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge funding support from the Natural Sciences and Engineering Research Council (NSERC) of Canada.References
- Y. Keho, Energy Policy, 2016, 91, 233–246 CrossRef.
- V. Caramia and B. Bozzini, Mater. Renew. Sustain. Energy, 2014, 3, 28–40 CrossRef.
- D. Linden and T. B. Reddy, Linden's handbook of batteries, McGraw-Hill, New York, 4th edn, 2011 Search PubMed.
- A. J. Bard and L. R. Faulkner, Electrochemical Method, John Wiley & Sons, New York, NY, 2nd edn, 2001 Search PubMed.
- F. Beck and P. Rüetschi, Electrochim. Acta, 2000, 45, 2467–2482 CrossRef CAS.
- W. Tahil, The Zinc Air Battery and the Zinc Economy: A Virtuous Circle, 2007 Search PubMed.
- J. R. Owen, Chem. Soc. Rev., 1997, 26, 259–267 RSC.
- V. Etacheri, R. Marom, R. Elazari, G. Salitra and D. Aurbach, Energy Environ. Sci., 2011, 4, 3243–3262 CAS.
- H. D. Yoo, E. Markevich, G. Salitra, D. Sharon and D. Aurbach, Mater. Today, 2014, 17, 110–121 CrossRef CAS.
- K. M. Abraham and Z. Jiang, J. Electrochem. Soc., 1996, 143, 1–5 CrossRef CAS.
- T. Ogasawara, A. Débart, M. Holzapfel, P. Novák and P. G. Bruce, J. Am. Chem. Soc., 2006, 128, 1390–1393 CrossRef CAS PubMed.
- K. Kinoshita, Electrochemical Oxygen Technology, Wiley, New York, 2nd edn, 1992 Search PubMed.
- Q. Xu and T. Kobayashi, Advanced materials for clean energy, Taylor a Francis Group, LLC, Boca Raton, 2015 Search PubMed.
- F. Cheng and J. Chen, Chem. Soc. Rev., 2012, 41, 2172–2192 RSC.
- J.-S. Lee, S. T. Kim, R. Cao, N.-S. Choi, M. Liu, K. T. Lee and J. Cho, Adv. Energy Mater., 2011, 1, 34–50 CrossRef CAS.
- Y. Li and H. Dai, Chem. Soc. Rev., 2014, 43, 5257–5275 RSC.
- G. Fu, Z. Cui, Y. Chen, Y. Li, Y. Tang and J. B. Goodenough, Adv. Energy Mater., 2017, 7, 1–8 Search PubMed.
- Y. Feng and N. Alonso-Vante, Phys. Status Solidi, 2008, 245, 1792–1806 CrossRef CAS.
- M. Shao, Q. Chang, J.-P. Dodelet and R. Chenitz, Chem. Rev., 2016, 116, 3594–3657 CrossRef CAS PubMed.
- R. M. Dondelinger, Biomed. Instrum. Technol., 2004, 38, 100–110 Search PubMed.
- S. Amendola, US Pat., 0021303 A1, 2012.
- P. Pei, K. Wang and Z. Ma, Appl. Energy, 2014, 128, 315–324 CrossRef CAS.
- D. Wolfe, M. I. Us and P. B. Johnson, US Pat., 0321970 A1, 2012, pp. 1–20.
- Z. Yang, J. Zhang, M. C. W. Kintner-Meyer, X. Lu, D. Choi, J. P. Lemmon and J. Liu, Chem. Rev., 2011, 111, 3577–3613 CrossRef CAS PubMed.
- T. Burchardt and M. Lanfranconi, US Pat., 0027664 A1, 2011.
- T. Burchardt and M. Lanfranconi, US Pat., 0027666 A1, 2011.
- T. Burchardt and R. F. Ngamga, US Pat., 0199055 A1, 2011.
- G. Toussaint, P. Stevens, L. Akrour, R. Rouget and F. Fourgeot, ECS Trans., 2010, 28, 25–34 CAS.
- B. Wang, J. Power Sources, 2005, 152, 1–15 CrossRef CAS.
- V. Neburchilov, H. Wang, J. J. Martin and W. Qu, J. Power Sources, 2010, 195, 1271–1291 CrossRef CAS.
- L. Jörissen, J. Power Sources, 2006, 155, 23–32 CrossRef.
- D. U. Lee, P. Xu, Z. Cano, A. Ghorbani Kashkooli, M. G. Park and Z. Chen, J. Mater. Chem. A, 2016, 4, 7107–7134 CAS.
- J. Fu, Z. P. Cano, M. G. Park, A. Yu, M. Fowler and Z. Chen, Adv. Mater., 2017, 29, 1–34 Search PubMed.
- T. Huh, G. Savaskan and J. W. Evans, J. Appl. Electrochem., 1992, 22, 916–921 CrossRef CAS.
- J. C. Salas-Morales and J. W. Evans, J. Appl. Electrochem., 1992, 22, 909–915 CrossRef.
- K. Wang, P. Pei, Z. Ma, H. Xu, P. Li and X. Wang, J. Power Sources, 2014, 271, 65–75 CrossRef CAS.
- K. Wang, P. Pei, Z. Ma, H. Chen, H. Xu, D. Chen and X. Wang, J. Mater. Chem. A, 2015, 3, 22648–22655 CAS.
- S. Siahrostami, V. Tripković, K. T. Lundgaard, K. E. Jensen, H. A. Hansen, J. S. Hummelshøj, J. S. G. Mýrdal, T. Vegge, J. K. Nørskov and J. Rossmeisl, Phys. Chem. Chem. Phys., 2013, 15, 6416 RSC.
- D. Schröder, V. Laue and U. Krewer, Comput. Chem. Eng., 2016, 84, 217–225 CrossRef.
- D. Schröder, T. Arlt, U. Krewer and I. Manke, Electrochem. Commun., 2014, 40, 88–91 CrossRef.
- K. Wang, P. Pei, Z. Ma, H. Chen, H. Xu, D. Chen and H. Xing, J. Power Sources, 2015, 296, 40–45 CrossRef CAS.
- E. Deiss, F. Holzer and O. Haas, Electrochim. Acta, 2002, 47, 3995–4010 CrossRef CAS.
- J. Stamm, A. Varzi, A. Latz and B. Horstmann, J. Power Sources, 2017, 360, 136–149 CrossRef CAS.
- M. Hilder, B. Winther-Jensen and N. B. Clark, Electrochim. Acta, 2012, 69, 308–314 CrossRef CAS.
- J. F. Parker, E. S. Nelson, M. D. Wattendorf, C. N. Chervin, J. W. Long and D. R. Rolison, ACS Appl. Mater. Interfaces, 2014, 6, 19471–19476 CAS.
- J. F. Parker, C. N. Chervin, E. S. Nelson, D. R. Rolison and J. W. Long, Energy Environ. Sci., 2014, 7, 1117–1124 CAS.
- M. N. Masri and A. A. Mohamad, J. Electrochem. Soc., 2013, 160, A715–A721 CrossRef CAS.
- X. G. Zhang, J. Power Sources, 2006, 163, 591–597 CrossRef CAS.
- G. A. Giffin, J. Mater. Chem. A, 2016, 4, 13378–13389 CAS.
- Z. Liu, P. Bertram and F. Endres, J. Solid State Electrochem., 2017, 21, 2021–2027 CrossRef CAS.
- M. Xu, D. G. Ivey, Z. Xie and W. Qu, J. Power Sources, 2015, 283, 358–371 CrossRef CAS.
- G. W. Heise and A. Schumacher, US Pat., 1890178 A, 1922.
- Y. Takeshita, S. Fujimoto and M. Sudoh, ECS Trans., 2013, 50, 3–12 CrossRef.
- D. B. Zhou and H. Vander Poorten, Electrochim. Acta, 1995, 40, 1819–1826 CrossRef CAS.
- C. W. Lee, K. Sathiyanarayanan, S. W. Eom, H. S. Kim and M. S. Yun, J. Power Sources, 2006, 160, 161–164 CrossRef CAS.
- C. W. Lee, K. Sathiyanarayanan, S. W. Eom and M. S. Yun, J. Power Sources, 2006, 160, 1436–1441 CrossRef CAS.
- S. M. Lee, Y. J. Kim, S. W. Eom, N. S. Choi, K. W. Kim and S. B. Cho, J. Power Sources, 2013, 227, 177–184 CrossRef CAS.
- K. Wongrujipairoj, L. Poolnapol, A. Arpornwichanop, S. Suren and S. Kheawhom, Phys. Status Solidi, 2016, 2, 1–6 Search PubMed.
- Y. Da Cho and G. T. K. Fey, J. Power Sources, 2008, 184, 610–616 CrossRef.
- W. Gan, D. Zhou, J. Zhao and L. Zhou, J. Appl. Electrochem., 2015, 45, 913–919 CrossRef CAS.
- H. S. Kim, Y. N. Jo, W. J. Lee, K. J. Kim and C. W. Lee, Electroanalysis, 2015, 27, 517–523 CrossRef CAS.
- R. Othman, W. J. Basirun, A. H. Yahaya and A. K. Arof, J. Power Sources, 2001, 103, 34–41 CrossRef CAS.
- R. Schweiss, C. Meiser, T. Damjanovic, I. Galbiati and N. Haak, SGL Gr., 2016, 1–10 Search PubMed.
- W. H. Zhu, B. A. Poole, D. R. Cahela and B. J. Tatarchuk, J. Appl. Electrochem., 2003, 33, 29–36 CrossRef CAS.
- Y. Xu, X. Xu, G. Li and Z. Zhang, Int. J. Electrochem., 2013, 8, 11805–11813 CAS.
- M. Maja, C. Orecchia, M. Strano, P. Tosco and M. Vanni, Electrochim. Acta, 2000, 46, 423–432 CrossRef CAS.
- R. J. Gilliam, J. W. Graydon, D. W. Kirk and S. J. Thorpe, Int. J. Hydrogen Energy, 2007, 32, 359–364 CrossRef CAS.
- A. V. Wolf, Aqueous Solutions and Body Fluids, Harper and Row, New York, 1966 Search PubMed.
- A. A. Mainar, O. Leonet, M. Bengoechea, B. Iker, I. de Meatza, A. Kvasha, A. Guerfi and J. A. Blázquez, Int. J. Energy Res., 2016, 40, 1032–1049 CrossRef CAS.
- A. Sumboja, X. Ge, G. Zheng, F. W. T. Goh, T. S. A. Hor, Y. Zong and Z. Liu, J. Power Sources, 2016, 332, 330–336 CrossRef CAS.
- M. Sato, M. Ohta and M. Sakaguchi, Electrochim. Acta, 1990, 35, 945–950 CrossRef CAS.
- D. Schröder, N. N. Sinai Borker, M. König and U. Krewer, J. Appl. Electrochem., 2015, 45, 427–437 CrossRef.
- N. Vassal, E. Salmon and J. F. Fauvarque, Electrochim. Acta, 2000, 45, 1527–1532 CrossRef CAS.
- C. C. Yang and S. J. Lin, J. Power Sources, 2002, 112, 497–503 CrossRef CAS.
- C. C. Yang, S. J. Lin and S. T. Hsu, J. Power Sources, 2003, 122, 210–218 CrossRef CAS.
- Z. Cheng, Q. Fu, C. Li, X. Wang, J. Gao, M. Ye, Y. Zhao, L. Dong, H. Luo and L. Qu, J. Mater. Chem. A, 2016, 4, 18240–18247 CAS.
- A. A. Mohamad, J. Power Sources, 2006, 159, 752–757 CrossRef CAS.
- D. Gelman, B. Shvartsev and Y. Ein-Eli, Top. Curr. Chem., 2016, 374, 1–42 CrossRef CAS PubMed.
- A. Ganesan, S. C. Abbas, J. Lv, K. Ding, Q. Liu, D. D. Babu, Y. Huang, X. Jia-Fang, M. Wu and Y. Wang, Dalton Trans., 2016, 46, 1803–1810 Search PubMed.
- J. Fu, D. U. Lee, F. M. Hassan, Z. Bai, M. G. Park and Z. Chen, Adv. Mater., 2015, 27, 5617–5622 CrossRef CAS PubMed.
- J. Fu, J. Zhang, X. Song, H. Zarrin, X. Tian, J. Qiao, L. Rasen, K. Li and Z. Chen, Energy Environ. Sci., 2016, 9, 663–670 CAS.
- S. Liu, W. Han, B. Cui, X. Liu, F. Zhao, J. Stuart and S. Licht, J. Power Sources, 2017, 342, 435–441 CrossRef CAS.
- M. Kar, B. Winther-Jensen, M. Armand, T. J. Simons, O. Winther-Jensen, M. Forsyth and D. R. MacFarlane, Electrochim. Acta, 2016, 188, 461–471 CrossRef CAS.
- M. Kar, B. Winther-Jensen, M. Forsyth and D. R. MacFarlane, Phys. Chem. Chem. Phys., 2014, 16, 10816–10822 RSC.
- M. Kar, B. Winther-Jensen, M. Forsyth and D. R. MacFarlane, Phys. Chem. Chem. Phys., 2013, 15, 7191–7197 RSC.
- T. J. Simons, P. C. Howlett, A. A. J. Torriero, D. R. Macfarlane and M. Forsyth, J. Phys. Chem., 2013, 117, 2662–2669 CrossRef CAS PubMed.
- M. Xu, D. G. Ivey, W. Qu and Z. Xie, J. Power Sources, 2014, 252, 327–332 CrossRef CAS.
- M. Xu, D. G. Ivey, Z. Xie, W. Qu and E. Dy, Electrochim. Acta, 2013, 97, 289–295 CrossRef CAS.
- H. J. Hwang, W. S. Chi, O. Kwon, J. G. Lee, J. H. Kim and Y. G. Shul, ACS Appl. Mater. Interfaces, 2016, 8, 26298–26308 CAS.
- H. Zarrin, S. Sy, J. Fu, G. Jiang, K. Kang, Y. S. Jun, A. Yu, M. Fowler and Z. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 25428–25437 CAS.
- G. A. Somorjai, K. R. McCrea and J. Zhu, Top. Catal., 2002, 18, 157–166 CrossRef CAS.
- H. T. Chung, J. H. Won and P. Zelenay, Nat. Commun., 2013, 4, 1–5 Search PubMed.
- F. Calle-Vallejo, J. I. Martínez and J. Rossmeisl, Phys. Chem. Chem. Phys., 2011, 13, 15639–15643 RSC.
- M. Busch, N. B. Halck, U. Kramm, S. Siahrostami, P. Krtil and J. Rossmeisl, Nano Energy, 2016, 29, 126–135 CrossRef CAS.
- J. Zhang, Z. Zhao, Z. Xia and L. Dai, Nat. Nanotechnol., 2015, 10, 444–452 CrossRef CAS PubMed.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- M. Kar, T. J. Simons and D. R. Macfarlane, Phys. Chem. Chem. Phys., 2014, 16, 18658–18674 RSC.
- E. Fabbri, A. Habereder, K. Waltar, R. Kötz and T. J. Schmidt, Catal. Sci. Technol., 2014, 4, 3800–3821 CAS.
- M. Hamdani, R. N. Singh and P. Chartier, Int. J. Electrochem. Sci., 2010, 5, 556–577 CAS.
- G. Liu, X. Li, J.-W. Lee and B. N. Popov, Catal. Sci. Technol., 2011, 1, 207–217 CAS.
- A. Morozan, B. Jousselme and S. Palacin, Energy Environ. Sci., 2011, 4, 1238–1254 CAS.
- Z.-L. Wang, D. Xu, J.-J. Xu and X.-B. Zhang, Chem. Soc. Rev., 2014, 43, 7746–7786 RSC.
- G. Q. Zhang, X. G. Zhang and Y. G. Wang, Carbon, 2004, 42, 3097–3102 CrossRef CAS.
- F. P. Hu, X. G. Zhang, F. Xiao and J. L. Zhang, Carbon, 2005, 43, 2931–2936 CrossRef CAS.
- Z. Zhang, H. Li, J. Hu, B. Liu, Q. Zhang, C. Fernandez and Q. Peng, J. Alloys Compd., 2017, 694, 419–428 CrossRef CAS.
- J.-J. Han, N. Li and T.-Y. Zhang, J. Power Sources, 2009, 193, 885–889 CrossRef CAS.
- T. Wang, M. Kaempgen, P. Nopphawan, G. Wee, S. Mhaisalkar and M. Srinivasan, J. Power Sources, 2010, 195, 4350–4355 CrossRef CAS.
- M. Chatenet, L. Genies-Bultel, M. Aurousseau, R. Durand and F. Andolfatto, J. Appl. Electrochem., 2002, 32, 1131–1140 CrossRef CAS.
- J. Hu, Q. Liu, L. Shi, Z. Shi and H. Huang, Appl. Surf. Sci., 2017, 402, 61–69 CrossRef CAS.
- V. M. Dhavale and S. Kurungot, ACS Catal., 2015, 5, 1445–1452 CrossRef CAS.
- Z. Cui, H. Chen, M. Zhao and F. J. DiSalvo, Nano Lett., 2016, 16, 2560–2566 CrossRef CAS PubMed.
- X. Wu, F. Chen, Y. Jin, N. Zhang and R. L. Johnston, ACS Appl. Mater. Interfaces, 2015, 7, 17782–17791 CAS.
- Y. Jin, F. Chen, Y. Lei and X. Wu, ChemCatChem, 2015, 7, 2377–2383 CrossRef CAS.
- C. Meng, T. Ling, T. Ma, H. Wang, Z. Hu, Y. Zhou and J. Mao, Adv. Mater., 2017, 29, 1–7 Search PubMed.
- Y. Jin and F. Chen, Electrochim. Acta, 2015, 158, 437–445 CrossRef CAS.
- Z. Guo, C. Li, W. Li, H. Guo, X. Su, P. He, Y. Wang and Y. Xia, J. Mater. Chem. A, 2016, 4, 6282–6289 CAS.
- H. Osgood, S. V. Devaguptapu, H. Xu, J. Cho and G. Wu, Nano Today, 2016, 11, 601–625 CrossRef CAS.
- J. Suntivich, H. A. Gasteiger, N. Yabuuchi, H. Nakanishi, J. B. Goodenough and Y. Shao-Horn, Nat. Chem., 2011, 3, 546–550 CrossRef CAS PubMed.
- P. Li, C. Hu, T. Lee, W. Chang and T. Hai, J. Power Sources, 2014, 269, 88–97 CrossRef CAS.
- P. Li, C. Hu, H. Noda and H. Habazaki, J. Power Sources, 2015, 298, 102–113 CrossRef CAS.
- Z. Wei, W. Huang, S. Zhang and J. Tan, J. Power Sources, 2000, 91, 83–85 CrossRef CAS.
- Q. Wu, L. Jiang, L. Qi, E. Wang and G. Sun, Int. J. Hydrogen Energy, 2014, 39, 3423–3432 CrossRef CAS.
- T.-H. Yang, S. Venkatesan, C.-H. Lien, J.-L. Chang and J.-M. Zen, Electrochim. Acta, 2011, 56, 6205–6210 CrossRef CAS.
- G. Q. Zhang and X. G. Zhang, Electrochim. Acta, 2004, 49, 873–877 CrossRef CAS.
- Y.-J. Huang and W.-S. Li, J. Inorg. Mater., 2013, 28, 341–346 CrossRef CAS.
- Y. Huang, Y. Lin and W. Li, Electrochim. Acta, 2013, 99, 161–165 CrossRef CAS.
- Z. Chen, A. Yu, R. Ahmed, H. Wang and H. Li, Electrochim. Acta, 2012, 69, 295–300 CrossRef CAS.
- D. L. Liu, J. N. Cheng and J. J. Han, Adv. Mater. Res., 2013, 724–725, 808–812 CrossRef.
- Y. L. Cao, H. X. Yang, X. P. Ai and L. F. Xiao, J. Electroanal. Chem., 2003, 557, 127–134 CrossRef CAS.
- J. Lee, G. S. Park, S. T. Kim, R. Cao and M. Liu, Nano Lett., 2011, 11, 5362–5366 CrossRef CAS PubMed.
- E. Davari, A. D. A. D. Johnson, A. Mittal, M. Xiong and D. G. D. G. Ivey, Electrochim. Acta, 2016, 211, 735–743 CrossRef CAS.
- E. Davari and D. G. Ivey, MRS Online Proc. Libr., 2015, 1777, 1–6 CrossRef CAS.
- A. Sumboja, X. Ge, F. W. T. Goh, B. Li, D. Geng, T. S. A. Hor, Y. Zong and Z. Liu, Chempluschem, 2015, 80, 1341–1346 CrossRef CAS.
- J.-S. Lee, T. Lee, H.-K. Song, J. Cho and B.-S. Kim, Energy Environ. Sci., 2011, 4, 4148–4154 CAS.
- F. W. T. Goh, Z. Liu, X. Ge, Y. Zong, G. Du and T. S. A. Hor, Electrochim. Acta, 2013, 114, 598–604 CrossRef CAS.
- Q. Wu, L. Jiang, L. Qi, L. Yuan, E. Wang and G. Sun, Electrochim. Acta, 2014, 123, 167–175 CrossRef CAS.
- X. Xie, Z. Ma, X. Ma, Q. Ren and V. M. Schmidt, J. Electrochem. Soc., 2007, 154, 733–738 CrossRef.
- J. Liu, L. Jiang, Q. Tang, E. Wang, L. Qi, S. Wang and G. Sun, Appl. Catal., B, 2014, 148–149, 212–220 CrossRef CAS.
- Y. Zhan, G. Du, S. Yang, C. Xu, M. Lu, Z. Liu and J. Y. Lee, ACS Appl. Mater. Interfaces, 2015, 7, 12930–12936 CAS.
- Z. Jiang, Z.-J. Jiang, T. Maiyalagan and A. Manthiram, J. Mater. Chem. A, 2016, 4, 5877–5889 CAS.
- J. Song, C. Zhu, S. Fu, Y. Song, D. Du and Y. Lin, J. Mater. Chem. A, 2016, 4, 4864–4870 CAS.
- X. Zhang, R. Liu, Y. Zang, G. Liu and G. Wang, Chem. Commun., 2016, 52, 5946–5949 RSC.
- Z. Wang, B. Li, X. Ge, F. W. T. Goh, X. Zhang, G. Du, D. Wuu, Z. Liu, T. S. Andy Hor, H. Zhang and Y. Zong, Small, 2016, 12, 2580–2587 CrossRef CAS PubMed.
- X. Liu, M. Park, M. G. Kim, S. Gupta, X. Wang, G. Wu and J. Cho, Nano Energy, 2016, 20, 315–325 CrossRef CAS.
- G. B. B. Varadwaj and V. O. Nyamori, Nano Res., 2016, 9, 3598–3621 CrossRef CAS.
- M. Gong and H. Dai, Nano Res., 2015, 8, 23–39 CrossRef CAS.
- Y. Li, M. Gong, Y. Liang, J. Feng, J.-E. Kim, H. Wang, G. Hong, B. Zhang and H. Dai, Nat. Commun., 2013, 4, 1805–1822 CrossRef PubMed.
- D. Kubo, K. Tadanaga, A. Hayashi and M. Tatsumisago, J. Mater. Chem. A, 2013, 1, 6804–6809 CAS.
- S. Chen, J. Duan, P. Bian, Y. Tang and R. Zheng, Adv. Energy Mater., 2015, 5, 1500936–1500943 CrossRef.
- X. Li, A. L. Zhu, W. Qu, H. Wang, R. Hui, L. Zhang and J. Zhang, Electrochim. Acta, 2010, 55, 5891–5898 CrossRef CAS.
- J. Zhang and W. Chen, Adv. Mater. Res., 2012, 347–353, 3621–3625 CAS.
- M. C. Wu, T. S. Zhao, H. R. Jiang, L. Wei and Z. H. Zhang, Electrochim. Acta, 2016, 222, 1438–1444 CrossRef CAS.
- X. Liu, W. Liu, M. Ko, M. Park, M. G. Kim and P. Oh, Adv. Funct. Mater., 2015, 25, 5799–5808 CrossRef CAS.
- G. Nam, J. Park, C. Min, O. Pilgun, P. Suhyeon, K. Min Gyu, J. Cho, P. Neojung, J. Cho and J. Lee, ACS Nano, 2015, 9, 6493–6501 CrossRef CAS PubMed.
- X. Li, N. Xu, H. Li, M. Wang, L. Zhang and J. Qiao, Green Energy Environ., 2017, 2, 316–328 CrossRef.
- Y. Cheng, S. Dou, J.-P. Veder, S. Wang, M. Saunders and S. P. Jiang, ACS Appl. Mater. Interfaces, 2017, 9, 8121–8133 CAS.
- M. Prabu, P. Ramakrishnan, H. Nara, T. Momma, T. Osaka and S. Shanmugam, ACS Appl. Mater. Interfaces, 2014, 6, 16545–16555 CAS.
- D. Wang, X. Chen, D. G. Evans and W. Yang, Nanoscale, 2013, 5, 5312–5315 RSC.
- G. Du, X. Liu, Y. Zong, T. S. A. Hor, A. Yu and Z. Liu, Nanoscale, 2013, 5, 4657–4661 RSC.
- M. Prabu, P. Ramakrishnan and S. Shanmugam, Electrochem. Commun., 2014, 41, 59–63 CrossRef CAS.
- X. Ge, Z. Zhang, S. H. Lim, B. Li, X. Wang and Z. Liu, ACS Appl. Mater. Interfaces, 2014, 6, 12684–12691 CAS.
- T. Maiyalagan, K. A. Jarvis, S. Therese, P. J. Ferreira and A. Manthiram, Nat. Commun., 2014, 5, 3943–3950 Search PubMed.
- F. Cheng, J. Shen, B. Peng, Y. Pan, Z. Tao and J. Chen, Nat. Chem., 2011, 3, 79–84 CrossRef CAS PubMed.
- K.-N. Jung, S. M. Hwang, M.-S. Park, K. J. Kim, J.-G. Kim, S. X. Dou, J. H. Kim and J.-W. Lee, Sci. Rep., 2015, 5, 1–10 Search PubMed.
- J. S. Lee, G. Nam, J. Sun, S. Higashi, H. W. Lee, S. Lee, W. Chen, Y. Cui and J. Cho, Adv. Energy Mater., 2016, 6, 1–6 CAS.
- Z.-Q. Liu, H. Cheng, N. Li, T. Y. Ma and Y.-Z. Su, Adv. Mater., 2016, 28, 3777–3784 CrossRef CAS PubMed.
- F. Cheng, J. Shen, B. Peng, Y. Pan, Z. Tao and J. Chen, Nat. Chem., 2011, 3, 79–84 CrossRef CAS PubMed.
- D. Kundu, B. D. Adams, V. Duffort, S. H. Vajargah and L. F. Nazar, Nat. Energy, 2016, 1, 16119 CrossRef CAS.
- L. Hadidi, E. Davari, D. G. Ivey and J. G. C. Veinot, Nanotechnology, 2017, 28, 1–10 CrossRef PubMed.
- C. Ma, N. Xu, J. Qiao, S. Jian and J. Zhang, Int. J. Hydrogen Energy, 2016, 41, 9211–9218 CrossRef CAS.
- B. Li, X. Ge, F. W. T. Goh, T. S. A. Hor, D. Geng, G. Du, Z. Liu, J. Zhang, X. Liu and Y. Zong, Nanoscale, 2015, 7, 1830–1838 RSC.
- M. Prabu, P. Ramakrishnan and S. Shanmugam, Electrochem. Commun., 2014, 41, 59–63 CrossRef CAS.
- M. Prabu, K. Ketpang and S. Shanmugam, Nanoscale, 2014, 6, 3173 RSC.
- L. Li, C. Liu, G. He, D. E. Fan and A. Manthiram, Energy Environ. Sci., 2015, 8, 3274–3282 CAS.
- Y. Zhan, C. Xu, M. Lu, Z. Liu and J. Y. Lee, J. Mater. Chem. A, 2014, 2, 16217–16223 CAS.
- J. M. Ang, Y. Du, B. Y. Tay, C. Zhao, J. Kong, L. P. Stubbs and X. Lu, Langmuir, 2016, 32, 9265–9275 CrossRef CAS PubMed.
- S. K. Singh, V. M. Dhavale and S. Kurungot, ACS Appl. Mater. Interfaces, 2015, 7, 21138–21149 CAS.
- S. K. Singh, V. M. Dhavale and S. Kurungot, ACS Appl. Mater. Interfaces, 2015, 7, 21138–21149 CAS.
- D. U. Lee, J. Y. Choi, K. Feng, H. W. Park and Z. Chen, Adv. Energy Mater., 2014, 4, 1–5 Search PubMed.
- D. U. Lee, J. Scott, H. W. Park, S. Abureden, J. Y. Choi and Z. Chen, Electrochem. Commun., 2014, 43, 109–112 CrossRef CAS.
- D. U. Lee, M. G. Park, H. W. Park, M. H. Seo, X. Wang and Z. Chen, ChemSusChem, 2015, 8, 3129–3138 CrossRef CAS PubMed.
- T. An, X. Ge, T. S. A. Hor, F. W. T. Goh, D. Geng, G. Du, Y. Zhan, Z. Liu and Y. Zong, RSC Adv., 2015, 5, 75773–75780 RSC.
- Y. Wang, X. Ma, L. Lu, Y. He, X. Qi and Y. Deng, Int. J. Hydrogen Energy, 2013, 38, 13611–13616 CrossRef CAS.
- C. Li, X. Han, F. Cheng, Y. Hu, C. Chen and J. Chen, Nat. Commun., 2015, 6, 7345–7353 CrossRef CAS PubMed.
- G. Li, M. A. Mezaal, K. Zhang and L. Lei, Int. J. Electrochem. Sci., 2015, 10, 5395–5404 CAS.
- M. Zhao, X. Li, L. Song, D. He and Z. Zhang, ChemCatChem, 2016, 8, 2808–2816 CrossRef CAS.
- H. Cheng, M. Li, C. Su, N. Li and Z. Liu, Adv. Funct. Mater., 2017, 27, 1–10 Search PubMed.
- X.-Z. Yuan, W. Qu, X. Zhang, P. Yao and J. Fahlman, ECS Trans., 2013, 45, 105–112 CrossRef.
- N. Li, X. Yan, Y. Jin, S. Li and B. Lin, J. Appl. Electrochem., 1999, 29, 1351–1354 CrossRef CAS.
- R. E. Carbonio, C. Fierro, D. Tryk, D. Scherson and E. Yeager, J. Power Sources, 1988, 22, 387–398 CrossRef CAS.
- A. S. Bhalla, R. Guo and R. Roy, Mater. Res. Innovations, 2000, 4, 3–26 CrossRef CAS.
- M. Yuasa, M. Nishida, T. Kida, N. Yamazoe and K. Shimanoe, J. Electrochem. Soc., 2011, 158, A605–A610 CrossRef CAS.
- H. Ma and B. Wang, RSC Adv., 2014, 4, 46084–46092 RSC.
- S. Ahn, K. Kim, H. Kim, S. Nam and S. Eom, Phys. Scr., T, 2010, 139, 1–4 Search PubMed.
- K.-N. Jung, J.-H. Jung, W. B. Im, S. Yoon, K.-H. Shin and J.-W. Lee, ACS Appl. Mater. Interfaces, 2013, 5, 9902–9907 CAS.
- J. Hu, L. Wang, L. Shi and H. Huang, J. Power Sources, 2014, 269, 144–151 CrossRef CAS.
- J. Hu, L. Wang, L. Shi and H. Huang, Electrochim. Acta, 2015, 161, 115–123 CrossRef CAS.
- G. Li, M. A. Mezaal, R. Zhang, K. Zhang, W. Liu and L. Lei, Int. J. Electrochem. Sci., 2015, 10, 8412–8422 CAS.
- D.-G. Lee, S. H. Kim, S. H. Joo, H.-I. Ji, H. Tavassol, Y. Jeon, S. Choi, M.-H. Lee, C. Kim, S. K. Kwak, G. Kim and H.-K. Song, Energy Environ. Sci., 2016, 10, 523–527 Search PubMed.
- Y. Lee, P. Peng, W. Chang and C. Huang, J. Taiwan Inst. Chem. Eng., 2014, 45, 2334–2339 CrossRef CAS.
- X. Wang, P. J. Sebastian, M. A. Smit, H. Yang and S. A. Gamboa, J. Power Sources, 2003, 124, 278–284 CrossRef CAS.
- A. Weidenkaff, S. G. Ebbinghaus and T. Lippert, Chem. Mater., 2002, 14, 1797–1805 CrossRef CAS.
- N. Wu, W. Liu and S. Su, Electrochim. Acta, 2003, 48, 1567–1571 CrossRef CAS.
- C. Zhu, A. Nobuta, I. Nakatsugawa and T. Akiyama, Int. J. Hydrogen Energy, 2013, 8, 13238–13248 CrossRef.
- J. Hu, Q. Liu, Z. Shi, L. Zhang and H. Huang, RSC Adv., 2016, 6, 86386–86394 RSC.
- Z. Chen, A. Yu, D. Higgins, H. Li and H. Wang, Nano Lett., 2012, 12, 1946–1952 CrossRef CAS PubMed.
- S.-W. Eom, S.-Y. Ahn, I.-J. Kim, Y.-K. Sun and H.-S. Kim, J. Electroceram., 2009, 23, 382–386 CrossRef CAS.
- D. U. Lee, M. G. Park, H. W. Park, M. H. Seo, V. Ismayilov, R. Ahmed and Z. Chen, Electrochem. Commun., 2015, 60, 38–41 CrossRef CAS.
- D. U. Lee, H. W. Park, M. G. Park, V. Ismayilov and Z. Chen, ACS Appl. Mater. Interfaces, 2015, 7, 902–910 CAS.
- S. W. Eom, S. Y. Ahn, C. W. Lee, Y. K. Sun and M. S. Yun, Solid State Phenom., 2007, 124–126, 1055–1058 CrossRef CAS.
- J. Jung, M. Risch, S. Park, M. G. Kim, G. Nam, H. Jeong, Y. Shao-horn and J. Cho, Energy Environ. Sci., 2016, 9, 176–183 CAS.
- A. Vignesh, M. Prabu and S. Shanmugam, ACS Appl. Mater. Interfaces, 2016, 8, 6019–6031 CAS.
- M. Prabu, P. Ramakrishnan and P. Ganesan, Nano Energy, 2015, 15, 92–103 CrossRef CAS.
- P. Cai, S. Ci, E. Zhang, P. Shao, C. Cao and Z. Wen, Electrochim. Acta, 2016, 220, 354–362 CrossRef CAS.
- P. Karfa, R. Madhuri, P. K. Sharma and A. Tiwari, Nano Energy, 2017, 33, 98–109 CrossRef CAS.
- G. Fu, Y. Chen, Z. Cui, Y. Li, W. Zhou, S. Xin, Y. Tang and J. B. Goodenough, Nano Lett., 2016, 16, 6516–6522 CrossRef CAS PubMed.
- B. Wang, X. Wang, J. Zou, Y. Yan, S. Xie, G. Hu, Y. Li and A. Dong, Nano Lett., 2017, 17, 2003–2009 CrossRef CAS PubMed.
- M. Wang, T. Qian, J. Zhou and C. Yan, ACS Appl. Mater. Interfaces, 2017, 9, 5213–5221 CAS.
- J. Yang, J. Hu, M. Weng, R. Tan, L. Tian, J. Yang, J. Amine, J. Zheng, H. Chen and F. Pan, ACS Appl. Mater. Interfaces, 2017, 9, 4587–4596 CAS.
- H. Zhong, K. Li, Q. Zhang, J. Wang, F. Meng, Z. Wu, J. Yan and X. Zhang, NPG Asia Mater., 2016, 8, e308 CrossRef CAS.
- F. Meng, H. Zhong, J. Yan and X. Zhang, Nano Res., 2017, 1–12 CAS.
- H. Wu, H. Li, X. Zhao, Q. Liu, J. Wang, J. Xiao, S. Xie, R. Si, F. Yang, S. Miao, X. Guo, G. Wang and X. Bao, Energy Environ. Sci., 2016, 9, 3736–3745 CAS.
- P. Cai, Y. Hong, S. Ci and Z. Wen, Nanoscale, 2016, 8, 20048–20055 RSC.
- M. Wang, Y. Liu, K. Zhang, F. Yu, F. Qin, J. Fang, Y. Lai and J. Li, RSC Adv., 2016, 6, 83386–83392 RSC.
- K. Zhou, G. Chen, J. Liu, Z.-P. Zhang, P. Sun, W.-Z. Zhang, F. Niu, W. Zhang and J. Liang, RSC Adv., 2016, 6, 90069–90075 RSC.
- Y. Ding, Y. Niu, J. Yang, L. Ma, J. Liu, Y. Xiong and H. Xu, Small, 2016, 12, 5414–5421 CrossRef CAS PubMed.
- Y. Liu, F. Chen, W. Ye, M. Zeng, N. Han, F. Zhao, X. Wang and Y. Li, Adv. Funct. Mater., 2017, 27, 1606034 CrossRef.
- Z. K. Yang, L. Lin and A. W. Xu, Small, 2016, 12, 5710–5719 CrossRef CAS PubMed.
- M. Xiong and D. G. Ivey, Electrochem. Commun., 2017, 75, 73–77 CrossRef CAS.
- C. Su, H. Cheng, W. Li, Z. Liu, N. Li and Z. Hou, Adv. Energy Mater., 2017, 7, 1–12 Search PubMed.
- N. Ma, X. Yang, X. She, L. Zhang, Z. Peng, X. Yao and D. Yang, J. Mater. Chem. A, 2016, 4, 6376–6384 CAS.
- X. Liu, M. Park, M. G. Kim, S. Gupta, G. Wu and J. Cho, Angew. Chem., Int. Ed., 2015, 54, 9654–9658 CrossRef CAS PubMed.
- S. Luo and D. B. Zhou, J. Electrochem. Soc., 2014, 161, 23–27 CrossRef.
- M. Xiong and D. G. Ivey, J. Electrochem. Soc., 2017, 164, A1012–A1021 CrossRef CAS.
- H. Wu, J. Wang, G. Wang, F. Cai, Y. Ye, Q. Jiang, S. Sun, S. Miao and X. Bao, Nano Energy, 2016, 30, 801–809 CrossRef CAS.
- Y. Hao, Z. Lu, G. Zhang, Z. Chang, L. Luo and X. Sun, Energy Technol., 2017, 2, 1–8 Search PubMed.
- Z. Wang, Y. Lu, Y. Yan, T. Y. P. Larissa, X. Zhang, D. Wuu, H. Zhang, Y. Yang and X. Wang, Nano Energy, 2016, 30, 368–378 CrossRef CAS.
- H.-W. Liang, X. Zhuang, S. Brüller, X. Feng and K. Müllen, Nat. Commun., 2014, 5, 4973 CrossRef CAS PubMed.
- Y. Sun, C. Li and G. Shi, J. Mater. Chem., 2012, 22, 12810–12816 RSC.
- L.-L. Tian, J. Yang, M.-Y. Weng, R. Tan, J.-X. Zheng, H.-B. Chen, Q.-C. Zhuang, L.-M. Dai and F. Pan, ACS Appl. Mater. Interfaces, 2017, 9, 7125–7130 CAS.
- M. Wang, Z. Fang, K. Zhang, J. Fang, F. Qin, Z. Zhang, J. Li, Y. Liu and Y. Lai, Nanoscale, 2016, 8, 11398–11402 RSC.
- N. Gupta, T. Toh, M. W. Fatt, S. Mhaisalkar and M. Srinivasan, J. Solid State Electrochem., 2012, 16, 1585–1593 CrossRef CAS.
- X. Cai, B. Y. Xia, J. Franklin, B. Li, X. Wang, Z. Wang, L. Chen, J. Lin, L. Lai and Z. Shen, J. Mater. Chem. A, 2017, 5, 2488–2495 CAS.
- Y. Liu, S. Chen, X. Quan, H. Yu, H. Zhao, Y. Zhang and G. Chen, J. Phys. Chem. C, 2013, 117, 14992–14998 CAS.
- E. Davari and D. G. Ivey, J. Appl. Electrochem., 2017, 47, 815–827 CrossRef CAS.
- S.-W. Eom, C.-W. Lee, M.-S. Yun and Y.-K. Sun, Electrochim. Acta, 2006, 52, 1592–1595 CrossRef CAS.
- L. Hadidi, E. Davari, M. Iqbal, T. K. T. Purkait, D. G. Ivey and J. G. C. Veinot, Nanoscale, 2015, 7, 20547–20556 RSC.
- P. C. Li, C. C. Hu, T. H. You and P. Y. Chen, Carbon, 2017, 111, 813–821 CrossRef CAS.
- S. Müller, F. Holzer, H. Arai and O. Haas, J. New Mater. Electrochem. Syst., 1999, 2, 227–232 Search PubMed.
- D. Higgins, Z. Chen, D. U. Lee and Z. Chen, J. Mater. Chem. A, 2013, 1, 2639–2645 CAS.
- D. U. Lee, H. W. Park, D. Higgins, L. Nazar and Z. Chen, J. Electrochem. Soc., 2013, 160, F910–F915 CrossRef CAS.
- B. J. Kim, D. U. Lee, J. Wu, D. Higgins, A. Yu and Z. Chen, J. Phys. Chem. C, 2013, 117, 26501–26508 CAS.
- H. Cui, Z. Zhou and D. Jia, Mater. Horiz., 2017, 4, 7–19 RSC.
- L. Wang, C. Yang, S. Dou, S. Wang, J. Zhang, X. Gao, J. Ma and Y. Yu, Electrochim. Acta, 2016, 219, 592–603 CrossRef CAS.
- S. H. Wu, P. C. Li and C. C. Hu, Mater. Chem. Phys., 2016, 183, 551–560 CrossRef CAS.
- S. S. Shinde, C.-H. Lee, A. Sami, D.-H. Kim, S. U. Lee and J.-H. Lee, ACS Nano, 2016, 11, 347–357 CrossRef PubMed.
- Z. Pei, H. Li, Y. Huang, Q. Xue, Y. Huang, M. Zhu, Z. Wang and C. Zhi, Energy Environ. Sci., 2017, 10, 742–749 CAS.
- J. Zhang, H. Zhou, X. Liu, J. Zhang, T. Peng, J. Yang, Y. Huang and S. Mu, J. Mater. Chem. A, 2016, 4, 15870–15879 CAS.
- Q. Li, R. Cao, J. Cho and G. Wu, Adv. Energy Mater., 2014, 4, 1–19 Search PubMed.
- G. Nam, J. Park, S. T. Kim, D. Shin, N. Park, Y. Kim, J.-S. Lee and J. Cho, Nano Lett., 2014, 14, 1870–1876 CrossRef CAS PubMed.
- B. Li, D. Geng, X. S. Lee, X. Ge, J. Chai, Z. Wang, J. Zhang, Z. Liu, T. S. A. Hor and Y. Zong, Chem. Commun., 2015, 51, 8841–8844 RSC.
- G. S. Park, J. S. Lee, S. T. Kim, S. Park and J. Cho, J. Power Sources, 2013, 243, 267–273 CrossRef CAS.
- S. Zhu, Z. Chen, B. Li, D. Higgins, H. Wang and H. Li, Electrochim. Acta, 2011, 56, 5080–5084 CrossRef CAS.
- B. Li, Y. Chen, X. Ge, J. Chai, X. Zhang, T. S. A. Hor, G. Du, Z. Liu, H. Zhang and Y. Zong, Nanoscale, 2016, 8, 5067–5075 RSC.
- T. Y. Ma, J. Ran, S. Dai, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2015, 54, 4646–4650 CrossRef CAS PubMed.
- Q. Liu, Y. Wang, L. Dai and J. Yao, Adv. Mater., 2016, 28, 3000–3006 CrossRef CAS PubMed.
- S. Patra, R. Choudhary, E. Roy, R. Madhuri and P. K. Sharma, Nano Energy, 2016, 30, 118–129 CrossRef CAS.
- Y. Qian, Z. Hu, X. Ge, S. Yang, Y. Peng, Z. Kang, Z. Liu, J. Y. Lee and D. Zhao, Carbon, 2017, 111, 641–650 CrossRef CAS.
- M. Wu, J. Qiao, K. Li, X. Zhou, Y. Liu and J. Zhang, Green Chem., 2016, 18, 2699–2709 RSC.
- G. S. Park, J.-S. Lee, S. T. Kim, S. Park and J. Cho, J. Power Sources, 2013, 243, 267–273 CrossRef CAS.
- F. Meng, H. Zhong, D. Bao, J. Yan and X. Zhang, J. Am. Chem. Soc., 2016, 138, 10226–10231 CrossRef CAS PubMed.
- G. Girishkumar, B. McCloskey, A. C. Luntz, S. Swanson and W. Wilcke, J. Phys. Chem. Lett., 2010, 1, 2193–2203 CrossRef CAS.
- Y.-C. Lu, B. M. Gallant, D. G. Kwabi, J. R. Harding, R. R. Mitchell, M. S. Whittingham and Y. Shao-Horn, Energy Environ. Sci., 2013, 6, 750–768 CAS.
- G. P. Kim, H. H. Sun and A. Manthiram, Nano Energy, 2016, 30, 130–137 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2018 |