
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStabilising fleeting intermediates of stilbene photocyclization with amino-borane functionalisation: the rare isolation of persistent dihydrophenanthrenes and their [1,5] H-shift isomers†
Yong-gang
Shi‡
a,
Soren K.
Mellerup‡
b,
Kang
Yuan
b,
Guo-Fei
Hu
a,
Francoise
Sauriol
b,
Tai
Peng
a,
Nan
Wang
*a,
Pangkuan
Chen
*a and
Suning
Wang
 *ab
*ab
aBeijing Key Laboratory of Photoelectronic/Electrophotonic Conversion Materials, School of Chemistry, Beijing Institute of Technology, Beijing 100081, P. R. China
bDepartment of Chemistry, Queen's University, Kingston, Ontario K7L 3N6, Canada. E-mail: sw17@queensu.ca
First published on 27th March 2018
Abstract
The key intermediate, 4a,4b-dihydrophenanthrene (DPH), involved in the photocyclization of stilbene and derivatives is known to be unstable, and is therefore poorly characterized/understood. We have found that functionalising stilbenes with NMe2 and BMes2 groups can greatly enhance the stability of 4a,4b-DPHs, allowing quantitative isolation and full characterization of these rare species. Furthermore, we discovered that the new amino-borane decorated 4a,4b-DPHs can undergo thermal [1,5] H sigmatropic shift, forming isomers 4a,10a-DPHs. Both 4a,4b-DHPs and 4a,10a-DHPs are stable towards air and moisture, while only the former were found to undergo oxidative dehydrogenation upon irradiation at 365 nm under air, yielding brightly blue/green fluorescent NMe2 and BMes2 functionalised phenanthrene analogues. Control studies established that the trans-Mes2B–Ph–NMe2 unit is responsible for the stability of these isolated 4a,4b-DHPs and their [1,5]-H shift isomers.
Introduction
The field of organic photochemistry consists of many interesting and unique chemical transformations mediated by the absorption and dissipation of light. Of the many well-studied reactivities within this realm of chemistry,1 the photocyclization of stilbene and its analogues is a prominent one.2 These reactions proceed via electrocyclic ring-closure of the cis-isomer (Z) according to the Woodward–Hoffman rules3 to give trans-4a,4b-dihydropheanthrene derivatives (4a,4b-DHPs, Scheme 1), which are highly unstable and either revert back to their cis-stilbene form or readily oxidize to their respective phenanthrenes.4 Photochemical cyclization of stilbenes and derivatives is therefore a straightforward strategy for preparing highly diverse phenanthrenes. Despite the critical role of 4a,4b-DHPs in the formation of their phenanthrene products, little is known of the nature of these photochemically generated intermediates due to their short lifetime/poor stability. As a result, isolated and well-characterized DPHs are extremely rare and seldom observed.5,6 In fact, nearly all of the previously known DHPs could not be isolated or obtained as pure substances due to competing side reactions and/or their poor stability toward ambient conditions. | ||
| Scheme 1 Key species involved in stilbene photocyclization and donor–acceptor derivatives investigated in this work. | ||
We have discovered recently that a photoswitchable olefin system (e.g. E in Scheme 1) containing a donor–acceptor (D–A; Mes2B–p-Ph–NMe2) unit can produce the rare 4a,4b-DHP species via the Z isomer quantitatively upon irradiation, which can be isolated and are stable for days under air at ambient temperature. Furthermore, we observed that the amino-borane decorated 4a,4b-DHPs can undergo a thermal [1,5] sigmatropic H migration, forming the previously unknown 4a,10a-DHP isomers quantitatively, which are also air stable and do not dehydrogenate to the phenanthrene derivatives under any conditions. Under aerobic, photolytic conditions, the new 4a,4b-DHPs convert to phenanthrene derivatives in 40% yield, comparable to previously reported photochemical generation of phenanthrene and derivatives from stilbenes and analogues.7 The remarkably high and unprecedented stability of the new DHPs is very unusual. The results of our study show that the stability of DHPs bearing para-substituted D–A systems is a general phenomenon, with varying aryl substituents exhibiting similar photoreactivity. Alterations to the D–A system (i.e. removal of the donor/acceptor or differing connectivity patterns) resulted in molecules that only display trans–cis isomerisation with irradiation. This role of the amino-borane8 functionalities represents a new facet of such D–A systems, broadening their utility in chemical syntheses and materials applications. The details of our investigation and several mechanistic aspects of the new donor–acceptor appended photocyclization systems are presented herein.
Results and discussion
Syntheses and structures of 1a–9a
The BMes2 and/or NMe2 appended stilbene analogues 1a–9a were prepared in high yields according to the procedures shown in Scheme 2. The ylide reagents were synthesized according to the relevant literature.9–13BO-1 was prepared according to a procedure we developed recently.14BO1′ and BO1m were prepared according to procedures outlined in the ESI.† All compounds were fully characterized by 1H, 13C and 11B NMR, HRMS spectroscopic analysis. The 11B chemical shifts of these compounds are between ∼70–75 ppm, typical of triaryl-boranes.15 Additionally, with the exception of 2a and 8a, the structures of these compounds were all determined by single-crystal X-ray diffraction (Fig. 1 and ESI†). All nine compounds have a trans-structure with respect to the olefin bond that is persistent in solution unless irradiated by UV light. The two possible trans-rotamers, a and b are shown in Scheme 2. In the crystal lattice, these compounds adopt the trans-rotamer-a structure exclusively, attributable to the reduced steric interactions between the BMes2 group and the olefin unit in this conformation. The crystal structures of E-1a and E-9a are shown in Fig. 1 as representative examples (see ESI† for the others). The NMe2 group in these molecules is coplanar with the phenyl ring. One important feature of the D–A-functionalised stilbene E-1a is that the benzene ring containing the donor and acceptor groups has a quinoid-like structure caused by the push–pull substituents.21b,d Similar C–C bond length variations in the benzene ring of the D–A system were found for E-3a, E-4a, E-6a and E-7a, which agree well with those calculated by DFT for the parent molecule Mes2B–Ph–p-NMe2 (see ESI†). As a consequence of the quinoid-like structure, the Mes2B–Ph–p-NMe2 ring has a lower aromaticity compared to that of benzene as evidenced by the NICS values calculated by DFT: NICS(0) and NICS(1) for Mes2B–Ph–p-NMe2 are −7.58 and −9.36, respectively, while those for benzene are −9.86 and −11.20, respectively. These bonding features are in contrast to the structure of E-9a, where meta-substitution of the D–A system across the phenyl ring yields bond lengths more similar to those of benzene (Fig. 1). | ||
| Scheme 2 The synthetic routes of compounds E-1a–E-9a investigated in this work. | ||
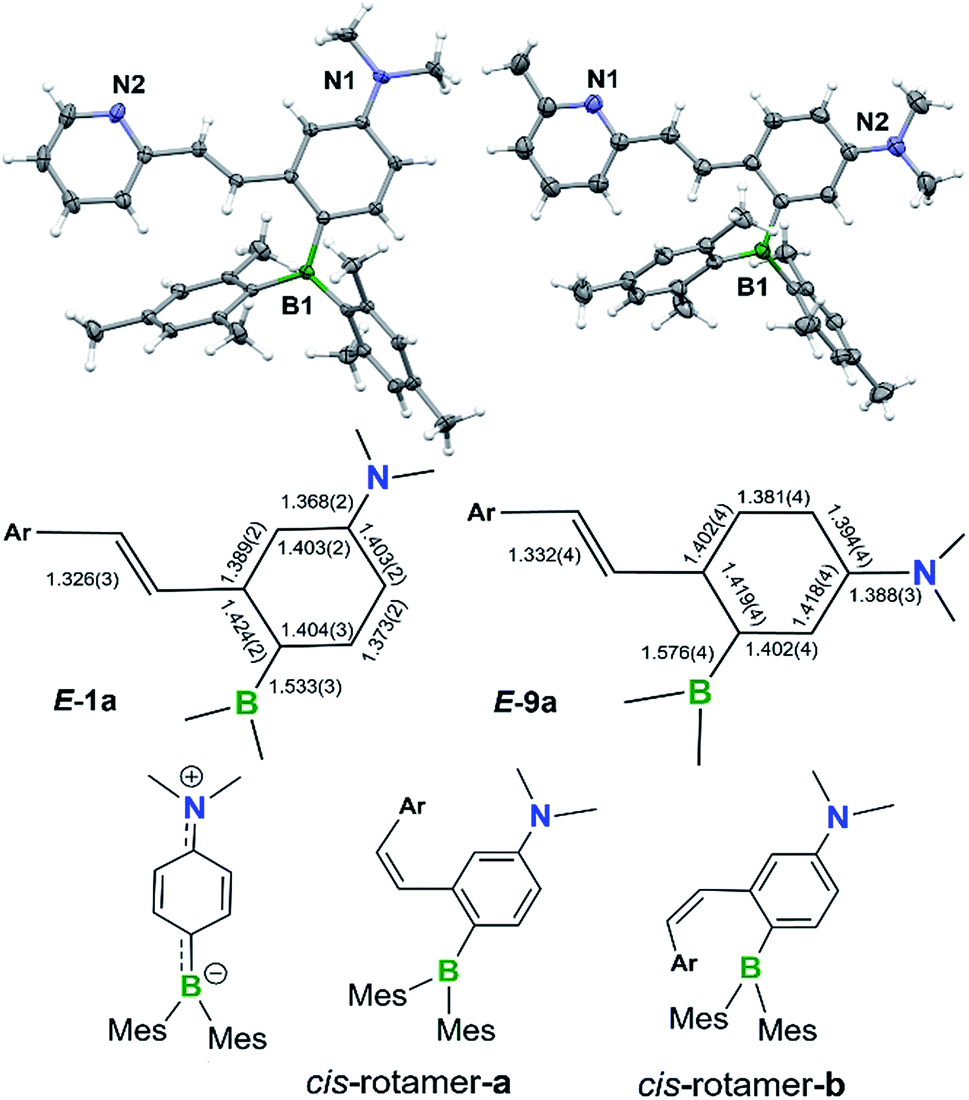 | ||
| Fig. 1 Top: crystal structures of E-1a (left) and E-9a (right) with 35% thermal ellipsoids with the key bond lengths (Å) shown in the schematic representation of their structures. Bottom: the structures of the cis-rotamers and the quinoid resonance form of the para-substituted D–A unit. | ||
The cis-isomers of compounds 1a–9a also have two possible rotamers a and b shown in Fig. 1 with the cis-rotamer-a being favoured due to its reduced steric congestion compared to cis-rotamer-b. DFT computational data indicate that cis-rotamer-a is ∼3–5 kcal mol−1 higher in energy than that of the respective trans-rotamer-a for 1a–9a.
Photoreactivity of E-1a–E-9a and the isolation of DHPs 1b–4b
Compounds E-1a–E-4a and E-6a–E-8a have a light-yellow colour with intense absorption bands at λmax = ∼360–380 nm (ε = ∼17000 to 28000 M−1 cm−1) in THF. They are all brightly fluorescent, emitting a sky-blue colour (λem = ∼460–480 nm) in THF under UV light, with ΦFL = 0.26 to 0.41 (see Table 1). TD-DFT computational data shows that the S0 → S1 transition for these molecules primarily involves HOMO and LUMO orbitals (∼90%) with a large oscillator strength. The HOMO is located mainly on the NMe2-phenyl portion while the LUMO spreads over the entire molecule with a large contribution from the boron atom. In contrast, the acceptor-only molecule E-5a is colourless with an absorption band at λmax = ∼300 nm, and is weakly blue emissive (λem = 450 nm, ΦFL = 0.06 in THF). The meta-D–A substituted molecule E-9a has a significantly red shifted absorption and emission band with a greater emission efficiency (λem = 536 nm, ΦFL = 0.65), compared to those of E-1a–E-8a (see Table 1 and ESI†).| Compound | Absorption in THF λabs (nm) (ε, M−1 cm−1) | Fluorescence in THF | |
|---|---|---|---|
| λ em (nm) | Φ FL | ||
| a Determined with an absolute QY spectrometer. | |||
| E-1a | 315 (35457), 384 (19155) | 483 | 0.39 |
| 1b | 414 (21282) | 464 | 0.03 |
| 1c | 422 (24494) | 435 | 0.06 |
| 1d | 305 (16521), 374 (16133) | 480 | 0.49 |
| E-2a | 318 (32368), 382 (17130) | 484 | 0.40 |
| 2b | 415 (22839) | 462 | 0.01 |
| 2c | 422 (28856) | 471 | 0.03 |
| 2d | 306 (17036), 375 (15802) | 480 | 0.54 |
| E-3a | 321 (27864), 379 (22173) | 476 | 0.41 |
| 3b | 418 (24133) | 469 | 0.01 |
| 3c | 424 (29914) | 468 | 0.03 |
| 3d | 314 (20179), 383 (15967) | 480 | 0.41 |
| E-4a | 315 (37129), 382 (22122) | 478 | 0.43 |
| 4b | 417 (22283) | 434 | 0.01 |
| 4c | 423 (27366) | 472 | 0.02 |
| 4d | 313 (17469), 378 (14129) | 480 | 0.49 |
| E-5a | 304(27602) | 450 | 0.06 |
| E-6a | 315 (39642), 380 (27831) | 473 | 0.37 |
| 6d | 272 (19697), 294 (15720), 374 (13113) | 480 | 0.45 |
| E-7a | 325 (34151), 380 (21960) | 485 | 0.26 |
| E-8a | 318 (42807), 357 (32632) | 463 | 0.38 |
| E-9a | 333 (18568), 362 (18454), 420 (6833) | 536 | 0.65 |
Despite their highly emissive nature (excluding E-5a) and congested structures, E-1a–E-9a all undergo photoisomerisation upon irradiation at 365 nm under nitrogen in benzene or THF, which were monitored by NMR, UV-vis and fluorescence spectroscopy. The photoreactions of E-1a–E-4a are similar while those of E-5a–E-9a are quite different. The discussion will focus on the phototransformation of compounds E-1a–E-4a first, using E-2a as a representative example. As shown by the time-lapsed 1H NMR spectra of E-2a (1 mg of E-1a in 0.6 mL of C6D6, Fig. 2), within minutes of irradiation at 365 nm, Z-2a (cis-rotamer-a) forms and becomes the dominating species. The 3J olefin proton coupling constant decreases from 16.0 Hz in E-2a to 12.4 Hz in Z-2a, which agrees with previous reports in the literature.16 In addition, a new set of peaks that were assigned to the DHP 2b also begin to appear in the spectrum. After about 2 hours of irradiation, E-2a and Z-2a are fully and cleanly converted to the bright yellow compound 2b. The characteristic NMR features of 2b are the two peaks at 3.87 and 3.68 ppm, respectively, which have a typical AB splitting pattern with a 3J coupling constant of 20.7 Hz. 2D-NOESY NMR experiments established that these two H atoms are arranged in a trans-configuration with respect to one another (Ha and Hb, Fig. 2; see also Fig. S6 in the ESI†). Ha is further coupled to the adjacent Hc (3J = 4.7 Hz) and Hd (5J = 2.5 Hz) atoms. The formation of a trans-4a,4b-DHP (2b) from the photocyclization of Z-2a is in agreement with a conrotatory electrocyclization, as predicted by the Woodward–Hoffman rules. Due to the different substituents on the alkenyl moiety, 2b represents the very first example of a 4a,4b-DHP where coupling of the inner H atoms is directly observed. Interestingly, NMR tracking indicated that compound 2b is air-stable for days in solution at ambient temperature and does not dehydrogenate to the expected phenanthrene product (see Fig. S29 in the ESI†). As such, 2b can be isolated and studied as a pure compound, which is exceedingly rare for 4a,4b-DHP systems. Nonetheless, efforts to obtain single-crystals of 2b for X-ray diffraction analysis have not been successful.
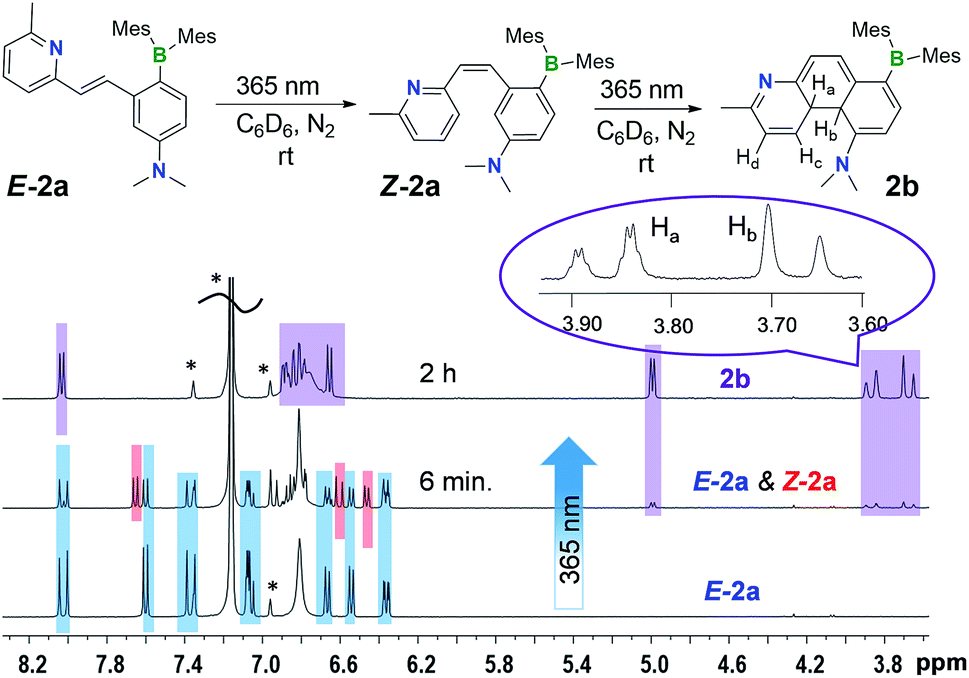 | ||
| Fig. 2 1H NMR spectral change (the aliphatic region is omitted for clarity) of E-2a (1 mg in 0.6 mL of C6D6) with 365 nm irradiation at ambient temperature under N2. The representative peaks for each species are colour-coded (blue: E-2a; red: Z-2a; purple: 2b). The solvent peak and the spin sidebands are indicated by *. | ||
The transformation of E-2a to 2b can also be tracked by UV-vis and fluorescence spectroscopy. In the UV-vis spectra, a clear sequential spectral change was observed (Fig. 3). Irradiation first causes the absorption band of E-2a at 318 nm to decrease and a hypsochromic shift of the band at 382 nm to 368 nm is observed with clear isosbestic points. This portion of spectral change is attributed to the formation of Z-2a, which is in agreement with previously reported trans to cis-isomerisation of stilbenes and derivatives.4 TD-DFT computational data also confirmed the hypsochromic shift (385 to 360 nm) of the first vertical excitation band from E-2a to Z-2a. The Z-2a absorption band decreases with irradiation time, accompanied by the rise of a low energy band at 415 nm, which is assigned to 2b. In the fluorescence spectra, the emission peak of E-2a decreases sharply in intensity as it is converted to Z-2a and 2b. Compound 2b is weakly blue fluorescent with λem = 462 nm and ΦFL = 0.03. TD-DFT data indicated that the S0 → S1 vertical excitation in 2b is a HOMO to LUMO transition (100%) localized on the DHP unit with a high oscillator strength and little contributions from the boron unit (see ESI†).
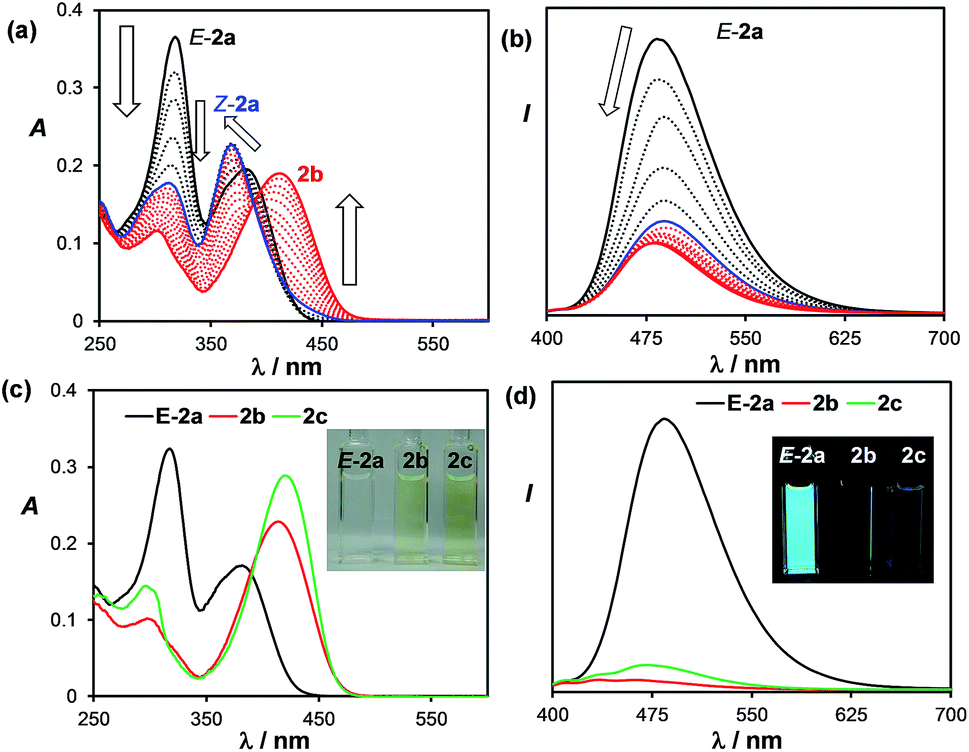 | ||
| Fig. 3 Top: UV-vis (a) and fluorescence (b) spectral change of E-2a in THF upon irradiation at 365 nm (the final spectrum corresponds to ∼80% formation of 2b. Bottom: the UV-vis (c) and fluorescence (d) spectra of isolated E-2a, 2b, and 2c in THF (1.0 × 10−5 M). Inset: photographs showing the absorption and emission colours and relative intensity of E-2a, 2b, and 2c in THF. | ||
NMR, UV-vis and fluorescence spectral tracking showed that with 365 nm irradiation, E-1a, E-3a and E-4a all undergo a similar transformation as that of E-2a, forming the corresponding DHPs 1b, 3b and 4b quantitatively via the cis-stilbene intermediate (Z). The 3JHa–Hb coupling constants (20.5–20.7 Hz) in 1b, 3b and 4b are similar to that of 2b (see ESI†). Like 2b, compound 1b is also stable for days in solution under ambient conditions. For 3b and 4b, after one day of standing under air at ambient temperature, a small amount of their [1,5] H-shifted products, 3c and 4c, were observed by NMR, which will be discussed in the following section. Again, 1b, 3b and 4b do not undergo dehydrogenation under ambient conditions. Similar to 2b, these three compounds are weakly fluorescent with λem = 460–470 nm and ΦFL = 0.01–0.03. The photophysical data of 1b–4b are summarized in Table 1.
The unusually high stability of 1b–4b is not fully understood, although we postulate that the kinetic barrier for their reversal back to Z-1a–Z-4a is likely very high. DFT optimized structures show H-bond interactions between the amino nitrogen atom and nearby H atoms such as the inner Hb in 1b–4b, which may contribute to the stability of these DHPs. The coexistence of the BMes2 and NMe2 group in 1b–4b appears to be essential for their formation and stability, as the previously reported17 analogue of 1a that lacks the BMes2 group does not produce a stable DHP, while that which lacks the NMe2 group (E-5a) is photoreactive but does not form a DHP at all (see Fig. S19 in the ESI†). The products of E-5a phototransformation have not been determined and are beyond the scope of this current investigation. The other key factor for the stability of 1b–4b is the presence of an alternative isomerisation pathway for these molecules, namely a thermal [1,5] H migration, leading to the generation of much more stable isomers 1c–4c which are described in the next section.
Compound E-6a behaves similarly to compounds E-1a–E-4a, but only partially converts to the cyclized compound 6b at 365 nm, which undergoes decomposition with further irradiation. Irradiation at 410 nm converts 6b back to Z-6a. Due to its poor stability, 6b was not isolated. In contrast to E-1a–E-4a, compounds E-7a–E-9a only undergo trans–cis isomerisation upon irradiation at 365 nm, reaching their photostationary states with approximately 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of the E vs. Z isomers in solution, and no observable cyclization (see ESI†). In E-8a, the positions of the NMe2 and BMes2 groups are switched compared to those in E-1a. The lack of photocyclization observed for E-8a may be attributed to the steric congestion imposed by the bulky BMes2 that prevents C–C bond formation at the adjacent carbon atom. In E-9a, the NMe2 and BMes2 groups are meta to each other, as opposed to para in the other derivatives. The lack of photocyclization for E-9a further confirms the necessity of the para-D–A system (and its resulting quinoidal structure) in the phototransformation.
:1 ratio of the E vs. Z isomers in solution, and no observable cyclization (see ESI†). In E-8a, the positions of the NMe2 and BMes2 groups are switched compared to those in E-1a. The lack of photocyclization observed for E-8a may be attributed to the steric congestion imposed by the bulky BMes2 that prevents C–C bond formation at the adjacent carbon atom. In E-9a, the NMe2 and BMes2 groups are meta to each other, as opposed to para in the other derivatives. The lack of photocyclization for E-9a further confirms the necessity of the para-D–A system (and its resulting quinoidal structure) in the phototransformation.
[1,5] H migration of 1b–4b and the isolation of 4a,10a-DHPs 1c–4c
An interesting observation we made is that compounds 1b–4b can undergo further structural change, producing isomers 1c–4c, respectively, either after an extended period of irradiation at 365 nm or heating. Careful control of the experimental conditions indicated that this new transformation is a thermal process. Heat generated from the photo reactor during irradiation is sufficient to drive this rearrangement reaction in some of the DHPs. The thermal isomerisation of 2b is described here as a representative example.The 1H NMR spectra in Fig. 4 show that 2b transforms to 2c quantitatively upon heating in a benzene solution at 120 °C for ∼24 hours (1 mg in 0.6 mL of C6D6). The structural assignment of 2c was accomplished first by 1D and 2D NMR spectroscopic analyses, which indicated that 2c is an analogue of cis-4a,10a-DHP. The chemical shifts of the two inner H atoms, Ha and Hb, appear at 3.51 and 3.57 ppm, respectively, with a 3J coupling constant of 7.6 Hz, much smaller than that observed in 2b. The Ha atom is further coupled to Hc and Hd with 3JHa–Hc ≈ 3JHa–Hb ≈ 7.6 Hz and 3JHa–Hd = 2.0 Hz. Compound 1b undergoes a similar transformation, forming 1c quantitatively at 120 °C, with a similar 1H NMR spectral change as observed for the 2b to 2c conversion (see ESI†). Surprisingly, compounds 1c and 2c are stable under air for days, and can even be purified by column chromatography under air. The crystal structures of 1c and 2c were determined by single-crystal X-ray diffraction analyses, which confirmed the cis-4a,10a-DHP structure for both. The structure of 2c is shown in Fig. 5 and that of 1c is provided in the ESI.† Although various H migrations of 4a,4b-DHPs and derivatives were described previously6,18 and implicated in some phototransformations of stilbenes, no H migrated species/intermediates have been isolated and fully characterized to date. Compounds 1c and 2c are therefore the first examples of structurally characterized 4a,10a-DHPs. Formally, the transformation of 1b–2b to 1c–2c can be described as a [1,5] H migration.
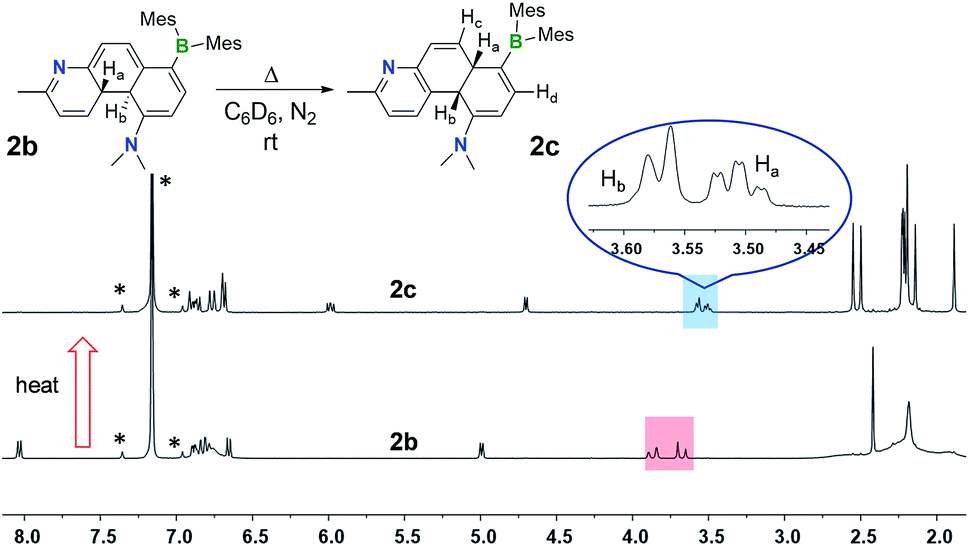 | ||
| Fig. 4 1H NMR spectra showing the clean conversion of 2b to 2c in C6D6 upon heating at 120 °C for 24 hours. The solvent peak and its spin satellite peaks are indicated by *. | ||
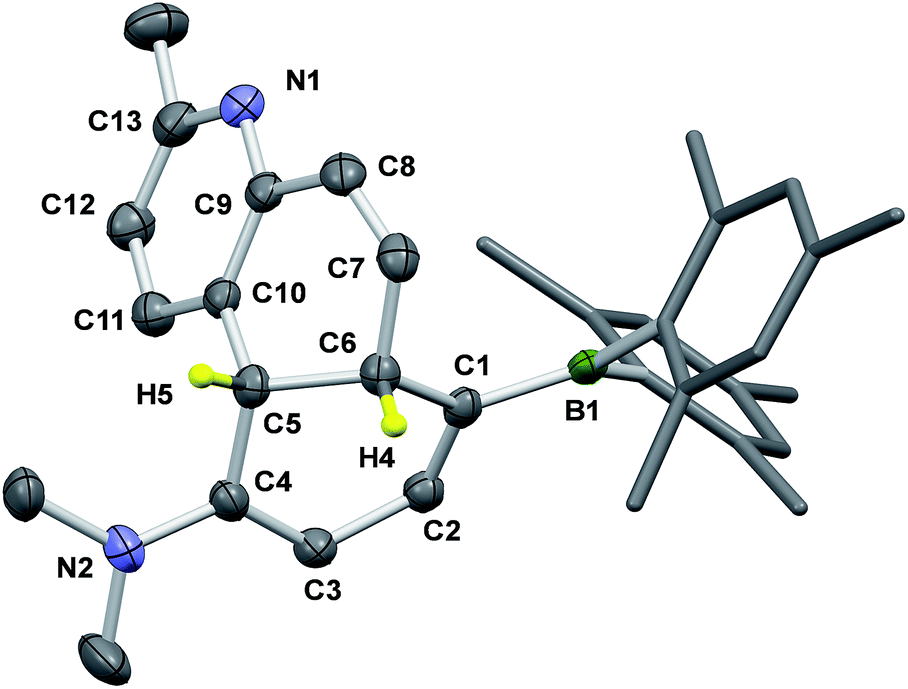 | ||
| Fig. 5 The crystal structure of 2c with the key atoms being labelled and 35% thermal ellipsoids. H atoms except the two inner ones are omitted for clarity. Selected bond lengths (Å) for 2c: C(1)–C(2) 1.366(3), C(2)–C(3) 1.427(3), C(3)–C(4) 1.366(3), C(4)–C(5) 1.514(3), C(5)–C(6) 1.543(3), C(5)–C(10) 1.524(3), C(6)–C(7) 1.513(3), C(7)–C(8) 1.322(3), C(8)–C(9) 1.468(3), C(9)–C(10) 1.391(3), C(10)–C(11) 1.385(3), C(11)–C(12) 1.382(3), C(12)–C(13) 1.376(3), N(1)–C(9) 1.345(3), N(1)–C(13) 1.352(3), C(1)–B(1) 1.530(3), N(2)–C(4) 1.362(3). | ||
For 3b and 4b, heating their benzene solutions at 90 °C for a few hours can fully convert them to 3c and 4c, respectively, an indication that the activation barriers for the [1,5] H migration of 3b and 4b are lower than those of 1b and 2b. Again, compounds 3c and 4c are stable under air in solution and the solid state, with their two inner H atoms having a cis-geometry. Compounds 1c–4c have a similar yellow colour and are weakly emissive like their 1b–4b isomers. For comparison, the UV-vis and fluorescence spectra of the isolated E-2a, 2b and 2c in THF are shown in Fig. 3. The full spectroscopic and characterization data for 1c–4c are provided in the ESI.† Heating the solution mixture of compounds Z-6a and 6b obtained from the photolysis of E-6a only led to the reversal of 6b back to Z-6a.
One thing that is curious is the cis-configuration of the products 1c–4c, as thermal [1,5] sigmatropic shifts proceed suprafacially and the two inner H atoms should therefore have a trans-geometry. DFT computational analysis indicate that the cis-1c is about 10 kcal mol−1 more stable than trans-1c (see ESI†). To further understand the H migration process, we prepared compound E-4a′ in which the phenyl ring has been deuterated (C6D5). Irradiation of E-4a′ in C6D6 at 365 nm followed by heating at 110 °C led to the quantitative isolation of the cis-4a,10a-DHP 4c′. Analysis and comparison of the 1H NMR spectra of 4c and 4c′ (Fig. 6) established unequivocally that the Ha atom in 4c is completely replaced by a D atom in 4c′, confirming that the migrated H atom is indeed from the phenyl ring and the migratory process is intramolecular. From this finding, we suspect that the cis-product 4c is formed through an internal base (the amino group) promoted configuration inversion of the carbon atom that is bound to Hb. Base induced proton tautomerism in DHPs and derivatives was observed/proposed previously,6 although no detailed spectroscopic information is available.
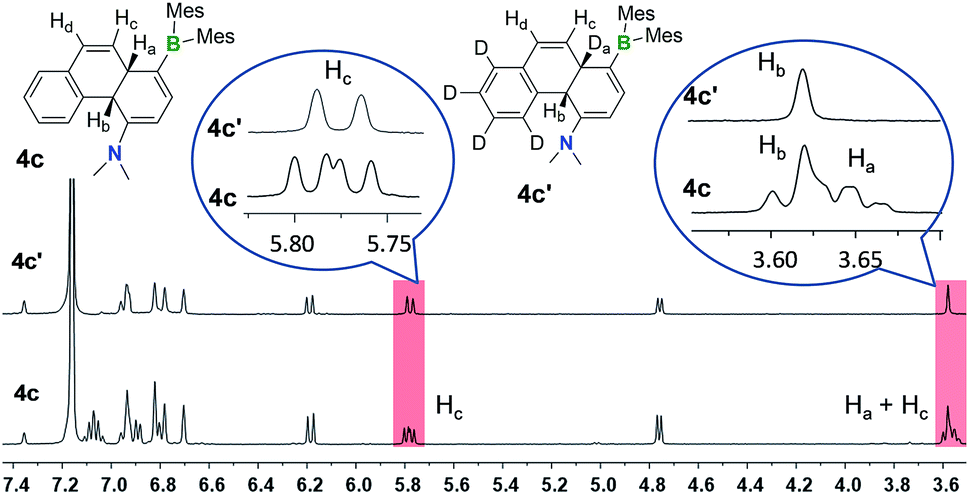 | ||
| Fig. 6 1H NMR spectra of 4c and 4c′ in C6D6 showing the disappearance of peaks and coupling pattern change due to D substitution in 4c′. | ||
To examine the possible involvement of the amino group in the formation of the cis-product 4c, the [1,5] H migration of 4b was repeated in C6D6 (1 mg/0.6 mL of solvent) with the addition of one drop of D2O. Heating at 120 °C for ∼44 hours led to the quantitative isolation of compound 4c′′ (Fig. 7). The 1H NMR spectrum of 4c′′ shows 100% H–D exchange of the H atom ortho to the NMe2 group and ∼50% H–D exchange for the Hb atom (see Fig. S18 in the ESI†). Based on these observations, a plausible mechanism for the 4b to cis-4c conversion is shown in Fig. 8. The 4c-TS in Fig. 8 is likely responsible for the H–D exchange with D2O, where the Hb is in the process of being shuttled to the opposite face of the molecule by the basic N atom.
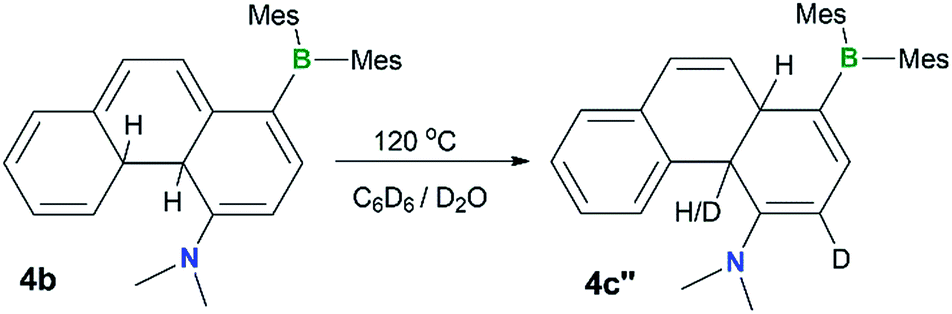 | ||
| Fig. 7 A diagram showing the H–D exchange in the thermal conversion of 4b to 4c′′ in C6D6 in the presence of a drop of D2O. | ||
 | ||
| Fig. 8 Top: proposed mechanism for the transformation of E-4a to cis-4c. Bottom: relative energies of the various species involved in the transformation calculated at the M06-2X/6-31+g(d,p) level of theory, as well as the experimentally determined activation barrier for 4b to 4c isomerization in C6D6. | ||
The kinetics of the [1,5] sigmatropic shift were investigated for 2b, 3b and 4b. The data show unambiguously that the b to c transformation follow first-order kinetics (see ESI Section S6†), consistent with a concerted intramolecular H migration.19 Using the rate constants obtained in benzene at three different temperatures, the activation energies were determined to be 30.0(4), 25(1), and 27.9(5) kcal mol−1 for 2b to 2c, 3b to 3c and 4b to 4c conversion, respectively. The kinetics of the [1,5] H migration is therefore greatly influenced by the nature of the aryl group in b, with the rate constants following the order of MeO-phenyl > phenyl > 2-Me-py. The aromaticity of these aryl groups follows the same order (as calculated by DFT methods, see ESI†). This correlation is reasonable, as the transition state of [1,5] H migration (b to c conversion) involves the aryl ring re-aromatization; thus greater aromaticity likely helps to stabilize the transition state and reduce the activation barrier. DFT computational studies indicate that the energies of cis-1c–4c are about 30 to 33 kcal mol−1 lower than 1b–4b. Thus, the [1,5] H migration is a thermodynamically driven process as illustrated in Fig. 8. Compounds cis-1c–4c are only about 10 kcal mol−1 above E-1a–4a (see Table S65† for the complete DFT data). Again, the donor and the acceptor units are believed to play a key role in the exceptionally high stability of cis-1c–4c and their inactivity toward air under ambient conditions.
Oxidative dehydrogenation of DHPs 1b–4b, 6b and the isolation of phenanthrenes 1d–4d and 6d
Although the DHPs 1b–4b do not react with oxygen under ambient conditions, they do behave like typical DHPs and undergo photo-oxidative dehydrogenation when irradiated at 365 nm under air, producing the fully conjugated phenanthrene derivatives 1d–4d in about 30–40% yields, accompanied by various decomposed and unidentifiable species. Photolysis of E-1a–4a at 365 nm under air at ambient temperature produced a similar mixture of products with 30–37% isolated yields of 1d–4d. Although E-6a only partially converts to 6b when irradiated under nitrogen, it converts to the aromatic compound 6d when irradiated under air, which was isolated in 37% yield. This demonstrates that photocyclization of the simple stilbene derivative compounds E-1a–4a/6a is a viable approach for achieving the amino/boryl functionalised phenanthrene derivatives 1d–4d/6d (Fig. 9), which would not be easy to obtain by other methods. Given the fact that the oxidative dehydrogenation reactions of stilbenes are well established and known to produce phenanthrenes in low to moderate yields when air is used as the oxidant, attempts were not made to further optimize this reaction. E-8a–9a undergo trans to cis isomerisation only when irradiated at 365 nm under air and no dehydrogenated products were observed at all, which is consistent with their inability to undergo photocyclization.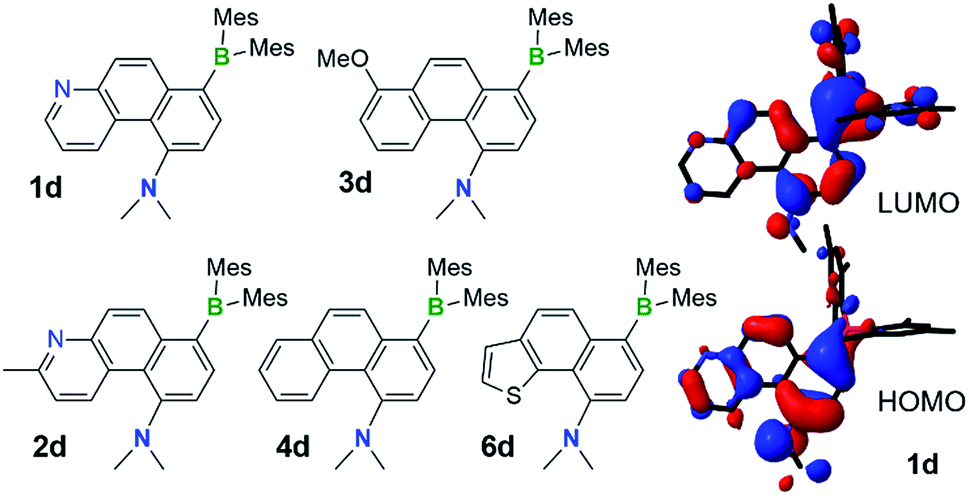 | ||
| Fig. 9 The structures of isolated NMe2 and BMes2 functionalised phenanthrene derivatives 1d–4d and 6d, the HOMO and LUMO diagrams of 1d (DFT at B3LYP/6-31(d) level of theory). | ||
Surprisingly, the DHPs cis-1c–4c did not produce the dehydrogenated products 1d–4d upon irradiation under air. Instead, complete decomposition affording unidentifiable products occurred. These results support that 4a,10a-DHP isomers such as cis-1c–4c are likely not involved in the formation of phenanthrenes and derivatives via the photocyclization of stilbenes. Compounds 1d–4d and 6d were fully characterized by NMR and HRMS analyses, while single-crystal X-ray diffraction analyses were performed on 1d, 2d and 6d. The structures of 2d and 6d are shown in Fig. 10 and that of 1d is provided in the ESI.† In all three structures the amino group is tilted towards the inner side of the three fused aryl rings, with its two methyl groups being above and below the plane of the molecule. The dihedral angle between the NCC plane of the NMe2 and the benzene ring is ∼61° for 1d and 2d, and of ∼71° for 6d, which is clearly necessary in order to minimize steric interactions between the methyl groups and the inner H atom in 1d and 6d. In addition, H-bonding between the N atom and the inner H atom (N⋯H = 2.26 Å, 1d; 2.23 Å, 2d) also favours this tilting of the amino unit. For 6d, a short contact distance is noted (2.79(1) Å) between S and N, which is less than the sum of van der Waals radii of nitrogen and sulphur (3.39 Å). This “chalcogen bonding interaction20” may be responsible for the amino group tilting toward the sulfur atom in 6d. The outer benzene ring and the py ring have a dihedral angle of 11° in 1d and 2d, while the thienyl ring is coplanar with the fused naphthyl ring in 6d. No intermolecular π-stackings are observed in all three crystal lattices.
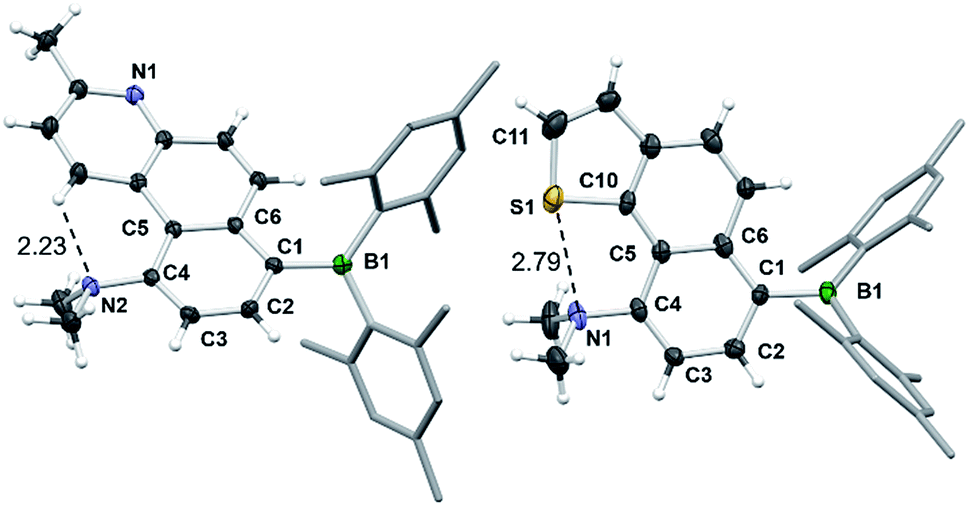 | ||
| Fig. 10 The crystal structures of 2d and 6d with 35% thermal ellipsoids and labels for selected atoms. Selected bond lengths (Å) for 2d/6d: B(1)–C(1) 1.569(4)/1.580(10), C(1)–C(2) 1.383(3)/1.389(10), C(2)–C(3) 1.386(3)/1.420(10), C(3)–C(4) 1.383(3)/1.366(10), C(4)–C(5) 1.493(3)/1.457(10), C(5)–C(6) 1.432(3)/1.460(9), C(4)–N(2)/N(1) 1.419(3)/1.441(8), S(1)–C(10) 1.743(7), S(1)–C(11) 1.722(10). | ||
Compounds 1d–4d and 6d have a light-yellow colour and are brightly fluorescent with λem = ∼480 nm and ΦFL = 0.41–0.54 in THF. The emission spectra of 1d–4d and 6d show significant bathochromic shifts with increasing solvent polarity, which indicates the presence of a polarised excited state (Fig. 11).21 TD-DFT computational results established the involvement of the NMe2 and the BMes2 group in the HOMO and LUMO levels, respectively (Fig. 9), with the vertical excitation to the first excited state primarily involving the HOMO to LUMO transition (>90%) with a very high oscillator strength for 1d–4d and 6d (see ESI†). Nonetheless, in addition to the amino and boryl units, the polycyclic aromatic rings have a great contribution to both HOMO and LUMO levels as shown by the Frontier orbital diagrams of 1d in Fig. 9. One noteworthy feature of compounds 1d–4d and 6d is that they maintain a very high emission quantum efficiency in high polarity solvents such as CH3CN (e.g. 0.53 for 1d, 0.57 for 2d and 0.55 for 4d; see Fig. 11 and ESI†). This may be caused by an increased NMe2 to BMes2 charge transfer contribution to the first excited state in polar solvents, which is known to greatly enhance emission quantum efficiency of the molecule.8,21
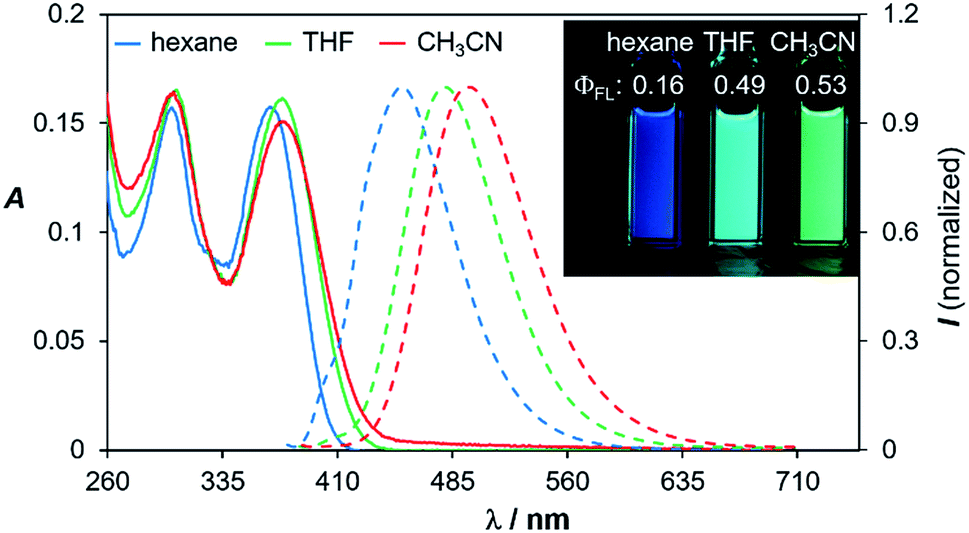 | ||
| Fig. 11 Absorption and fluorescence spectra of compound 1d in three different solvents (1 × 10−5 M). The fluorescence spectra were recorded at λmax of absorption. Inset: photographs showing the emission colours and ΦFL of 1d in the three different solvents. | ||
Conclusions
In summary, a novel series of D–A functionalised stilbenes has been prepared and their photoreactivity established. Following trans–cis isomerisation induced by light, compounds 1a–4a undergo photochemically allowed electrocyclic ring-closure to afford highly persistent and air-stable 1b–4b which represent some of the only examples of fully characterizable trans-4a,4b-DHPs. Based on control experiments, the quinoidal structure imposed by the D–A system is the key to the stability of these DHPs. Upon heating, these species undergo a unique [1,5]-sigmatropic H atom migration and subsequent internal base-induced tautomerization to give cis-4a,10a-DHPs 1c–4c, which represent a new class of stable DHP isomers. Importantly, we have shown that these DHP isomers do not dehydrogenate and are therefore not part of the DHP oxidation pathway. In the presence of air and light, 1b–4b and 6b are converted to their fully conjugated dehydrogenation products 1d–4d and 6d, which are brightly emissive even in high polarity solvents such as CH3CN. The unprecedented stability of the 4a,4b- and 4a,10a-DHP isomers in this study offers rare snapshots into the individual steps involved in the conversion of stilbenes to phenanthrenes, thereby broadening our understanding of this photochemical transformation.Experimental
General procedure
All solvents were freshly distilled over sodium metal and stored under nitrogen prior to use. All starting materials were purchased from Energy Chemical. (Pyridin-2-ylmethyl)triphenylphosphonium chloride,9 2-metho-xybenzyltriphenylphosphonium chloride,10 benzylphosphonium bromide,11 2-thienylmethyltriphenylphosphonium chloride,12 3-thienylmethyltriphenylphosphonium bromide,13 2-dimesityl-borylbenzaldehyde,22 2-bromo-4-(dimethylamino)benzalde-hyde,23 and 5-bromo-2-(dimethylamino)benzaldehyde24 were synthesized according to procedures reported in literature. The starting materials for E-8a and E-9a (BO1′ and BO1m, respectively) were prepared according to procedures outlined in the ESI.† All the reactions were performed under nitrogen and in dry solvents. 1H, 13C and 11B NMR spectra were recorded on a 400 or 700 MHz spectrometer. 1H and 13C chemical shifts are reported in ppm relative to the signals corresponding to the residual non-deuterated solvents (CDCl3: 1H 7.26 ppm, 13C 77.23 ppm; CD2Cl2: 1H 5.32 ppm, 13C 53.84 ppm; C6D6: 1H 7.16 ppm, 13C 128.06 ppm). 11B chemical shifts are reported in ppm relative to the signal of BF3·OEt2 (0.00 ppm). High resolution mass spectrum data were obtained via ESI (Agilent (Q-TOF 6520)) analyser. UV/vis spectra were obtained on an Agilent Cary 300 UV/vis spectrophotometer. Fluorescence spectra were recorded on a Photon Technologies International Quanta Master Model C-60 spectrometer. Fluorescent quantum efficiencies were determined using a Hamamatsu Quantaurus-QY spectrometer (C11347). The purity of all compounds reported were established by 1H NMR and 13C NMR spectra.X-ray crystallographic analyses
The crystal data were collected on a Bruker D8-Venture diffractometer with Mo-target (λ = 0.71073 Å) at 180 K for all compounds except 5a and 6a that were collected at 220 K. Data were processed on a PC with the aid of the Bruker SHELXTL software package25 and corrected for absorption effects. All non-hydrogen atoms were refined anisotropically. The positions of hydrogen atoms were calculated and refined isotropically. The detail of crystal data, collection parameters and results of analyses are provided in the ESI.† The crystal data were deposited to the Cambridge Crystallographic Data Centre with deposition numbers of CCDC 1590110 (1a), 1590111 (3a), 1590113 (4a), 1816881 (5a), 1590195 (6a), 1590116 (7a), 1816882 (9a), 1590114 (1c), 1590115 (2c), 1590112 (1d), 1590109 (2d), 1811865 (6d).‡DFT calculation details
DFT calculations were performed using the Gaussian 09 suite of programs26 at the Center for Advanced Computing at Queen's University. Geometry optimizations and vertical excitations of all compounds were obtained at the B3LYP27/6-31g(d)28 level of theory and the resulting structures were confirmed to be stationary points through vibrational frequency analysis. To obtain more accurate energies, subsequent geometry optimizations were performed with M06-2X29/6-31g(d), followed by single point energy calculations at the M06-2X/6-31+g(d,p) level of theory with implicit solvent effects (C6H6) accounted for using the conducting polarizable continuum model (C-PCM).30Synthesis of compounds E-1a–E-9a
The ylides reagent (0.42 mmol) was dissolved in THF (10 mL) under argon and cooled to 0 °C. Sodium hydride (0.5 mmol, 60% dispersion in mineral oil) was added in small portions and the solution was stirred for 1 hour. The aromatic aldehyde compound (0.5 mmol) was then added and the reaction was warmed to room temperature and stirred for 12 hours. The solution was concentrated in vacuo. The crude product was purified by silica gel chromatography using the appropriate solvents as the eluent.:1 of petroleum ether and ethyl acetate as the eluent). 1H NMR (400 MHz, C6D6): δ 8.47 (d, J = 4.6 Hz, 1H), 7.93 (d, J = 16.3 Hz, 1H), 7.60 (d, J = 8.5 Hz, 1H), 7.45 (d, J = 16.3 Hz, 1H), 7.10–7.04 (m, 2H), 6.78 (d, J = 8.5 Hz, 5H), 6.60–6.53 (m, 1H), 6.35 (d, J = 8.4 Hz, 1H), 2.36 (s, 6H), 2.28 (s, 12H), 2.17 (s, 6H). 13C NMR (101 MHz, CD2Cl2): δ 157.45, 153.25, 149.31, 145.00, 140.63, 139.86, 138.58, 136.03, 135.59, 134.65, 129.03, 128.57, 121.71, 120.12, 111.31, 108.55, 40.19, 23.24, 21.24. 11B NMR (128 MHz, C6D6): δ 70.5. HR-ESIMS (m/z): [M + H]+ calcd for C33H38BN2, 473.3123; found 473.3122.
:1 of petroleum ether and ethyl acetate as the eluent). 1H NMR (400 MHz, C6D6): δ 8.02 (d, J = 16.2 Hz, 1H), 7.60 (d, J = 8.5 Hz, 1H), 7.37 (d, J = 16.2 Hz, 1H), 7.07 (dd, J = 8.5, 5.1 Hz, 2H), 6.81 (s, 4H), 6.66 (d, J = 7.9 Hz, 1H), 6.54 (d, J = 7.5 Hz, 1H), 6.36 (dd, J = 8.5, 2.5 Hz, 1H), 2.41 (s, 3H), 2.39 (s, 6H), 2.29 (s, 12H), 2.18 (s, 6H). 13C NMR (101 MHz, CD2Cl2): δ 157.94, 156.59, 153.21, 144.96, 144.50, 140.69, 139.63, 138.51, 136.32, 135.24, 134.95, 128.97, 128.56, 121.16, 117.47, 111.27, 108.35, 40.21, 24.47, 23.23, 21.24. 11B NMR (225 MHz, C6D6): δ 70.1. HR-ESIMS (m/z): [M + H]+ calcd for C34H40BN2, 487.3279; found 487.3289.
:1 of petroleum ether and CH2Cl2 as the eluent). 1H NMR (400 MHz, C6D6): δ 7.75 (s, 2H), 7.61 (d, J = 8.5 Hz, 1H), 7.18 (d, J = 2.5 Hz, 1H), 7.07–7.02 (m, 1H), 6.94 (dd, J = 7.8, 1.6 Hz, 1H), 6.87 (d, J = 7.7 Hz, 1H), 6.83 (d, J = 7.4 Hz, 4H), 6.49 (d, J = 8.2 Hz, 1H), 6.37 (dd, J = 8.5, 2.6 Hz, 1H), 3.30 (s, 3H), 2.41 (s, 6H), 2.29 (s, 12H), 2.21 (s, 6H). 13C NMR (101 MHz, CD2Cl2): δ 156.93, 153.23, 146.17, 144.66, 140.69, 139.67, 138.51, 134.67, 131.48, 128.49, 127.30, 126.68, 122.62, 120.72, 110.94, 110.77, 108.01, 55.81, 40.21, 23.22, 21.27. 11B NMR (225 MHz, C6D6): δ 70.4. HR-ESIMS (m/z): [M + H]+ calcd for C35H41BNO, 502.3276; found 502.3288.
:1 of petroleum ether and CH2Cl2 as the eluent). 1H NMR (400 MHz, C6D6): δ 7.72 (d, J = 16.1 Hz, 1H), 7.62 (d, J = 8.5 Hz, 1H), 7.11 (d, J = 7.4 Hz, 2H), 7.03 (dd, J = 14.7, 9.3 Hz, 5H), 6.82 (s, 4H), 6.39 (dd, J = 8.5, 2.4 Hz, 1H), 2.47 (s, 6H), 2.29 (s, 12H), 2.19 (s, 6H). 13C NMR (101 MHz, CD2Cl2): δ 153.21, 145.60, 144.65, 140.62, 139.86, 138.61, 138.44, 131.50, 128.60, 128.17, 127.37, 126.83, 110.97, 107.89, 40.22, 23.24, 21.27. 11B NMR (128 MHz, CD2Cl2): δ 72.4. HR-ESIMS (m/z): [M + H]+ calcd for C34H39BN, 472.3170; found 472.3163.
:1 of petroleum ether and CH2Cl2 as the eluent). 1H NMR (400 MHz, C6D6): δ 7.73 (d, J = 16.1 Hz, 1H), 7.63 (d, J = 8.5 Hz, 1H), 7.02 (d, J = 15.7 Hz, 2H), 6.82 (s, 4H), 6.39 (d, J = 8.5 Hz, 1H), 2.46 (s, 6H), 2.29 (s, 12H), 2.19 (s, 6H). 13C NMR (176 MHz, CD2Cl2): δ 152.68, 145.17, 144.18, 140.16, 139.42, 138.17, 137.83, 134.17, 130.99, 128.14, 127.80, 127.72, 127.67, 127.53, 126.59, 126.45, 126.31, 126.12, 125.98, 125.85, 110.61, 107.52, 39.88, 22.82, 20.85. 11B NMR (225 MHz, CD2Cl2): δ 71.3. HR-ESIMS (m/z): [M + H]+ calcd for C34H34D5BN, 477.3484; found 477.3490.
:1 petroleum ether and ethyl acetate as the eluent). 1H NMR (400 MHz, CD2Cl2) δ 8.43 (ddd, J = 4.8, 1.7, 0.8 Hz, 1H), 7.75 (d, J = 7.8 Hz, 1H), 7.53–7.41 (m, 3H), 7.33–7.23 (m, 2H), 7.03 (ddd, J = 7.4, 4.9, 0.9 Hz, 1H), 6.93 (d, J = 16.3 Hz, 1H), 6.85–6.76 (m, 5H), 2.25 (s, 6H), 2.01 (s, 12H). 13C NMR (176 MHz, CDCl3) δ 156.22, 147.81, 147.50, 143.55, 141.09, 140.59, 139.37, 137.25, 135.76, 131.64, 128.64, 128.50, 127.36, 125.63, 121.84, 120.27, 23.30, 21.34. 11B NMR (225 MHz, C6D6) δ 74.68. HR-ESIMS (m/z): [M + H]+ calcd for C31H33BN, 430.2701; found 430.2707.
:1 of petroleum ether and CH2Cl2 as the eluent). 1H NMR (400 MHz, C6D6): δ 7.61 (d, J = 8.5 Hz, 1H), 7.53 (d, J = 16.1 Hz, 1H), 6.99 (dd, J = 9.2, 6.8 Hz, 2H), 6.87–6.76 (m, 5H), 6.72 (t, J = 3.8 Hz, 2H), 6.38 (dd, J = 8.5, 2.4 Hz, 1H), 2.47 (s, 6H), 2.28 (s, 12H), 2.19 (s, 6H). 13C NMR (101 MHz, CD2Cl2): δ 153.18, 145.59, 144.53, 141.23, 140.59, 139.84, 138.56, 128.54, 125.71, 125.63, 122.39, 121.80, 110.86, 107.52, 53, 40.20, 23.23, 21.26. 11B NMR (225 MHz, C6D6): δ 70.9. HR-ESIMS (m/z): [M + H]+ calcd for C32H37BNS, 478.2734; found 478.2751.
:1 of petroleum ether and CH2Cl2 as the eluent). 1H NMR (400 MHz, C6D6): δ 7.59 (d, J = 1.6 Hz, 1H), 7.56 (d, J = 5.6 Hz, 1H), 7.11 (d, J = 15.9 Hz, 1H), 6.93 (d, J = 2.4 Hz, 1H), 6.84 (s, 4H), 6.67 (ddd, J = 8.5, 7.6, 4.3 Hz, 3H), 6.37 (dd, J = 8.5, 2.5 Hz, 1H), 2.44 (s, 6H), 2.30 (s, 12H), 2.20 (s, 6H). 13C NMR (101 MHz, CD2Cl2): δ 153.06, 144.98, 144.33, 143.93, 140.59, 139.40, 138.56, 134.96, 130.86, 128.61, 127.57, 125.50, 124.60, 121.36, 111.02, 107.67, 40.24, 23.22, 21.28. 11B NMR (225 MHz, C6D6): δ 69.06. HR-ESIMS (m/z): [M + H]+ calcd for C32H37BNS, 478.2734; found 478.2742.
:1 petroleum ether and ethyl acetate as the eluent). 1H NMR (400 MHz, C6D6) δ 8.07 (d, J = 15.9 Hz, 1H), 7.83 (d, J = 8.7 Hz, 1H), 7.21 (d, J = 16.1 Hz, 1H), 7.04 (t, J = 7.7 Hz, 1H), 6.95 (s, 1H), 6.78 (s, 4H), 6.68 (d, J = 7.8 Hz, 1H), 6.63 (d, J = 8.6 Hz, 1H), 6.50 (d, J = 7.5 Hz, 1H), 2.41 (s, 3H), 2.39 (s, 6H), 2.27 (s, 12H), 2.13 (s, 6H). 13C NMR (101 MHz, C6D6) δ 157.92, 157.23, 150.18, 144.38, 140.86, 139.07, 135.85, 133.93, 130.58, 129.06, 126.60, 126.00, 120.14, 118.13, 117.56, 115.45, 39.81, 24.60, 23.53, 21.31. 11B NMR (225 MHz, C6D6) δ 76.01. HR-ESIMS (m/z): [M + H]+ calcd for C34H40BN2, 487.3279; found 487.3274.
Photoisomerisation of E-1a–E-9a
Preparative scale reactions
To an oven-dried Schlenk flask was added the stilbene compound (E-1a–E-4a, ∼0.01 mmol). The flask was evacuated and filled with N2 three times. Freshly distilled benzene (4 mL) was injected into the Schlenk flask under nitrogen. The solution was then irradiated with 365 nm UV light at room temperature for 2–7 h until the starting material stilbene was completely consumed according to TLC. After removing the solvent under reduced pressure, compounds 1b–4b were obtained quantitatively as pure products and confirmed by NMR spectroscopy. Compounds 1b–4b cannot be further purified by column chromatography. Compound E-6a partially converts to 6b under 365 nm irradiation, which could not be isolated as a pure compound from the reaction mixture (see Fig. S20–S22 in the ESI†). Compounds E-7a–E-9a only undergoes photoisomerisation to Z-7a–Z-9a (see Fig. S24–S27 in the ESI†). The characterization data provided below are for isolated compounds 1b–4b.Thermal isomerisation of 1b–4b
Preparative scale reactions
To an oven-dried Schlenk flask was added the stilbene compound (E-1a–E-4a, ∼0.03 mmol), which was converted to the corresponding 4a,4b-DHP (1b–4b) in the same manner as described in the photoisomerisation reaction. After the full conversion, the solution was heated at 120 °C for 24 h (1b, 2b), 8 h (3b), 24 h (4b). After being cooled to room temperature, the solvents were then removed under reduced pressure and compounds 1c–4c were obtained nearly quantitatively. These four compounds can be further purified by either column chromatography using petroleum ether and ethyl acetate as the eluent or recrystallization by the slow evaporation of the solvent (benzene or CH2Cl2) under air.Synthesis of compounds 1d–4d and 6d
To a Schlenk flask was added the stilbene compound (E-1a–E-4a and E-6a, ∼0.04 mmol), and THF (40 mL) under the air. The Schlenk flask was then closed and irradiated with 365 nm UV light at room temperature for 2 h. The solution was concentrated in vacuo. The crude product was purified by silica gel chromatography using appropriate solvents as the eluents.:1 of petroleum ether and CH2Cl2 as the eluent). 1H NMR (400 MHz, C6D6): δ 9.56 (d, J = 8.8 Hz, 1H), 8.36 (d, J = 9.3 Hz, 1H), 8.29 (d, J = 9.3 Hz, 1H), 7.72 (d, J = 7.9 Hz, 1H), 7.41 (t, J = 8.2 Hz, 1H), 6.93 (d, J = 7.9 Hz, 1H), 6.79 (s, 4H), 6.63 (d, J = 7.7 Hz, 1H), 3.40 (s, 3H), 2.50 (s, 6H), 2.19 (s, 12H), 2.16 (s, 6H). 13C NMR (176 MHz, C6D6): δ 155.89, 155.78, 144.64, 142.30, 140.86, 139.49, 138.80, 136.36, 132.79, 129.04, 126.94, 125.64, 124.43, 122.88, 121.38, 119.64, 114.87, 105.32, 55.20, 43.82, 23.47, 21.36. 11B NMR (225 MHz, C6D6): δ 72.9. HR-ESIMS (m/z): [M + H]+ calcd for C35H39BNO, 500.3119; found 500.3144.
:1 of petroleum ether and CH2Cl2 as the eluent). 1H NMR (400 MHz, C6D6): δ 9.88 (d, J = 8.7 Hz, 1H), 8.30 (d, J = 9.0 Hz, 1H), 7.70 (d, J = 7.9 Hz, 1H), 7.54 (d, J = 7.9 Hz, 1H), 7.46 (t, J = 7.7 Hz, 1H), 7.33 (t, J = 7.4 Hz, 1H), 7.26 (d, J = 9.0 Hz, 1H), 6.93 (d, J = 7.9 Hz, 1H), 6.81 (s, 4H), 2.46 (s, 6H), 2.19 (s, 12H), 2.18 (s, 6H). 13C NMR (176 MHz, C6D6): δ 155.75, 144.63, 142.54, 140.86, 139.20, 138.90, 136.18, 132.98, 131.66, 129.07, 128.43, 127.58, 127.17, 127.11, 126.00, 125.80, 122.99, 115.01, 43.76, 23.49, 21.37. 11B NMR (225 MHz, C6D6): δ 73.6. HR-ESIMS (m/z): [M + H]+ calcd for C34H37BN, 470.3014; found 470.3036.
:1 of petroleum ether and CH2Cl2 as the eluent). 1H NMR (400 MHz, C6D6): δ 8.26 (d, J = 8.8 Hz, 1H), 7.68 (d, J = 7.6 Hz, 1H), 7.45 (d, J = 8.8 Hz, 1H), 7.22 (s, 1H), 7.11–7.05 (m, 2H), 6.80 (s, 4H), 2.55 (s, 6H), 2.18 (s, 12H), 2.16 (s, 6H). 13C NMR (176 MHz, C6D6): δ 153.95, 145.52, 144.43, 140.89, 139.17, 138.53, 136.30, 134.41, 134.38, 129.10, 128.53, 127.01, 125.36, 123.67, 123.35, 117.44, 45.17, 23.49, 21.35. 11B NMR (225 MHz, C6D6): δ 75.0. HR-ESIMS (m/z): [M + H]+ calcd for C32H35BNS, 476.2578; found 476.2601.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Y. G. Shi, G. F. Hu, T. Peng, N. Wang, P. Chen and S. Wang thank the National Natural Science Foundation of China for financial support (grant no. 21701011, 21501011, 21772012, 21571017) for financial support. S. K. Mellerup, K. Yuan and S. Wang thank the Natural Sciences and Engineering Research Council of Canada (RGPIN1193993-2013) for financial support and the Centre for Advanced Computing at Queen's University for computational facilities. S. K. Mellerup thanks the Canadian government for the Vanier CGS.Notes and references
- For select examples see: (a) R. G. W. Norrish and C. H. Bamford, Nature, 1937, 140, 195 CrossRef CAS; (b) H. E. Zimmerman and A. C. Pratt, J. Am. Chem. Soc., 1970, 92, 6259 CrossRef CAS; (c) Y. Inoue, Chem. Rev., 1992, 92, 741 CrossRef CAS and references therein; (d) G. S. Hammond, J. Saltiel, A. A. Lamola, N. J. Turro, J. S. Bradshaw, D. O. Cowan, R. C. Counsell, V. Vogt and C. Dalton, J. Am. Chem. Soc., 1964, 86, 3197 CrossRef CAS.
- (a) F. R. Stermitz, Org. Photochem., 1967, 1, 247 Search PubMed; (b) M. Scholz, F. Dietz and Z. Mühlstädt, Chem, 1967, 7, 329 CAS; (c) Preparative Organic Photochemistry, ed. A. Schönberg, Springer-Verlag, New York, 1968 Search PubMed; (d) S. T. Reid, Adv. Heterocycl. Chem., 1970, 23, 87 Search PubMed; (e) J. Grimshaw and A. P. de Silva, Chem. Soc. Rev., 1981, 10, 181 RSC; (f) F. B. Mallory, C. S. Wood and J. T. Gordon, J. Am. Chem. Soc., 1964, 86, 3094 CrossRef CAS; (g) D. H. Waldeck, Chem. Rev., 1991, 91, 415 CrossRef CAS; (h) W. H. Laarhoven, Pure Appl. Chem., 1984, 56, 1225 CrossRef CAS; (i) H. Meier, Angew. Chem., Int. Ed., 1992, 31, 1399 CrossRef; (j) M. Irie, Chem. Rev., 2000, 100, 1685 CrossRef CAS PubMed; (k) W. M. Kwok, C. Ma, D. Philips, A. Beeby, T. B. Marder, R. Ll. Thomas, C. Tschuschke, G. Baranovic, P. Matousek, M. Towrie and A. W. Parker, J. Raman Spectrosc., 2003, 34, 886 CrossRef CAS.
- R. B. Woodward and R. Hoffmann, J. Am. Chem. Soc., 1965, 87, 395 CrossRef CAS.
- (a) F. B. Mallory and C. W. Mallory, Org. React., 2005, 30, 1 Search PubMed; (b) J. H. Ho, T. I. Ho and R. S. H. Liu, Org. Lett., 2001, 3, 409 CrossRef CAS PubMed; (c) T. I. Ho, J. Y. Wu and S. L. Wang, Angew. Chem., Int. Ed., 1999, 38, 2558 CrossRef CAS; (d) E. N. Gulakova, D. V. Berdnikova, T. M. Aliyeu, Y. V. Fedorov, I. A. Godovikov and O. A. Fedo-rova, J. Org. Chem., 2014, 79, 5533 CrossRef CAS PubMed; (e) R. Lapouyade, R. Koussini and J. C. Rayez, J. Chem. Soc., Chem. Commun., 1975, 676 RSC; (f) F. B. Mallory and C. W. Mallory, J. Am. Chem. Soc., 1972, 94, 6041 CrossRef CAS; (g) J. Han, T. Wang, Y. Liang, Y. Li, C. Li, R. Wang, S. Feng and Z. Zhang, Org. Lett., 2017, 19, 3552 CrossRef CAS PubMed; (h) T. M. Aliyeu, D. V. Berdnikova, O. A. Fedorova, E. N. Gulakova, C. Stremmel and H. Ihmels, J. Org. Chem., 2016, 81, 9075 CrossRef CAS PubMed; (i) T. Matsushima, S. Kobayashi and S. Watanabe, J. Org. Chem., 2016, 81, 7799 CrossRef CAS PubMed.
- (a) T. D. Doyle, W. R. Benson, D. Banes and N. Filipescu, J. Am. Chem. Soc., 1970, 92, 6371 CrossRef CAS; (b) T. D. Doyle, W. R. Benson and N. Filipescu, J. Am. Chem. Soc., 1976, 98, 3262 CrossRef CAS PubMed.
- J. B. M. Somers, A. Couture, A. Lablanche-Combier and W. H. Laarhoven, J. Am. Chem. Soc., 1985, 107, 1387 CrossRef CAS.
- (a) P. Bortolus, G. Cauzzo and G. Galiazzo, Tetrahedron Lett., 1966, 3717 Search PubMed; (b) P. Bortolus, G. Cauzzo, U. Mazzucato and G. Z. Galiazzo, Z. Phys. Chem., 1969, 63, 29 CrossRef CAS.
- (a) F. Jäkle, Chem. Rev., 2010, 110, 3985 CrossRef PubMed; (b) Y. Ren and F. Jäkle, Dalton Trans., 2016, 45, 13996 RSC; (c) Z. M. Hudson and S. Wang, Acc. Chem. Res., 2009, 42, 1584 CrossRef CAS PubMed; (d) T. W. Hudnall, C. W. Chiu and F. P. Gabbaï, Acc. Chem. Res., 2009, 42, 388 CrossRef CAS PubMed; (e) C. R. Wade, A. E. J. Broomsgrave, S. Aldridge and F. P. Gabbaï, Chem. Rev., 2010, 110, 3958 CrossRef CAS PubMed; (f) D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2009, 49, 46 CrossRef PubMed; (g) E. v. Grotthuss, A. John, T. Kaese and M. Wagner, Asian J. Org. Chem., 2018, 7, 37 CrossRef; (h) L. Ji, S. Griesbeck and T. B. Marder, Chem. Sci., 2017, 8, 846 RSC; (i) C. D. Entwistle and T. B. Marder, Angew. Chem., Int. Ed., 2002, 41, 2927 CrossRef CAS; (j) C. D. Entwistle and T. B. Marder, Chem. Mater., 2004, 16, 4574 CrossRef CAS; (k) S. Y. Li, Z. B. Sun and C. H. Zhao, Inorg. Chem., 2017, 56, 8705 CrossRef CAS PubMed.
- J. M. Janusz, P. A. Young, J. M. Ridgeway, M. W. Scherz, K. Enzweiler, L. I. Wu, L. Gan, J. Chen, D. E. Kellstein and S. A. Green, J. Med. Chem., 1998, 41, 3515 CrossRef CAS PubMed.
- M. Jeran, A. E. Cotman, M. Stephan and B. Mohar, Org. Lett., 2017, 19, 2042 CrossRef CAS PubMed.
- J. A. Smith and K. D. Moeller, Org. Lett., 2013, 15, 5818 CrossRef CAS PubMed.
- Y.-Z. Chen, C.-W. Ni, F.-L. Teng, Y.-S. Ding, T.-H. Lee and J.-H. Ho, Tetrahedron, 2014, 70, 1748 CrossRef CAS.
- J. N. Ngwendson, W. N. Atemnkeng, C. M. Schultze and A. Banerjee, Org. Lett., 2006, 8, 4085 CrossRef CAS PubMed.
- Y. G. Shi, J. Wang, H. Li, G. F. Hu, X. Li, S. K. Mellerup, N. Wang, T. Peng and S. Wang, Chem. Sci., 2018, 9, 1902–1911 RSC.
- (a) L. Ji, R. M. Edkins, A. Lorbach, I. Krummenacher, C. Brueckner, A. Eichhorn, H. Braunschweig, B. Engels, P. J. Low and T. B. Marder, J. Am. Chem. Soc., 2015, 137, 6750 CrossRef CAS PubMed; (b) C. Reus, S. Weidlich, M. Bolte, H.-W. Lerner and M. Wagner, J. Am. Chem. Soc., 2013, 135, 12892 CrossRef CAS PubMed.
- A. A. Bothner-By, Adv. Magn. Opt. Reson., 1965, 1, 195 Search PubMed.
- F. D. Lewis, R. S. Kalgutkar and J. S. Yang, J. Am. Chem. Soc., 2001, 123, 3878 CrossRef CAS PubMed.
- (a) M. V. Sargent and C. J. Timmons, J. Chem. Soc., 1964, 5544 RSC; (b) R. Lapouyade, A. Veyres, N. Hanafi, A. Couture and A. Lablanche-Combier, J. Org. Chem., 1982, 47, 1361 CrossRef CAS; (c) T. I. Ho, J. H. Ho and J. Y. Wu, J. Am. Chem. Soc., 2000, 122, 8575 CrossRef CAS.
- Modern Physical Organic Chemistry, ed. E. V. Anslyn and D. A. Dougherty, Wilstead & Taylor Publishing Services, USA, 2005 Search PubMed.
- (a) P. Metrangolo, H. Neukirch, T. Pilati and G. Resnati, Acc. Chem. Res., 2005, 38, 386 CrossRef CAS PubMed; (b) P. C. Ho, P. Szydlowski, P. J. W. Elder, J. Sinclair, J. Kübel, C. Gendy, L. M. Lee, H. Jenkins, J. F. Britten, D. R. Morim and I. Vargas-Baca, Nat. Commun., 2016, 7, 11299 CrossRef CAS PubMed.
- (a) J. C. Doty, B. Babb, P. J. Grisdale, M. E. Glogowski and J. L. R. Williams, J. Organomet. Chem., 1972, 38, 229 CrossRef CAS; (b) Z. Yuan, C. D. Entwistle, J. C. Collings, D. Albesa-Jov, A. S. Batsanov, J. A. K. Howard, N. J. Taylor, H. M. Kaiser, D. E. Kaufmann, S.-Y. Poon, W.-Y. Wong, C. Jardin, S. Fathallah, A. Boucekkine, J. F. Halet and T. B. Marder, Chem.–Eur. J., 2006, 12, 2758 CrossRef CAS PubMed; (c) D. R. Bai, X.-Y. Liu and S. Wang, Chem.–Eur. J., 2007, 13, 5713 CrossRef CAS PubMed; (d) Z. Yuan, J. C. Collings, N. J. Taylor, T. B. Marder, C. Jardin and J. F. Halet, J. Solid State Chem., 2000, 154, 5 CrossRef CAS; (e) Z. Zhang, R. M. Edkins, J. Nitsch, K. Fucke, A. Steffen, L. E. Longobardi, D. W. Stephan, C. Lambert and T. B. Marder, Chem. Sci., 2015, 6, 308 RSC; (f) Z. Zhang, R. M. Edkins, J. Nitsch, K. Fucke, A. Eichhorn, A. Steffen, Y. Wang and T. B. Marder, Chem.–Eur. J., 2015, 21, 177 CrossRef CAS PubMed.
- B. Neue, R. Fröhlich, B. Wibbeling, A. Fukazawa, A. Wakamiya, S. Yamaguchi and E.-U. Würthwein, J. Org. Chem., 2012, 77, 2176 CrossRef CAS PubMed.
- J. Garcia, J. Sorrentino, E. J. Diller, D. Chapman and Z. R. Woydziak, Synth. Commun., 2016, 5, 475 CrossRef PubMed.
- C. Jimenez, Y. Ellahioui, R. Alvarez, L. Aramburu, A. Riesco, M. Gonzalez, A. Vincente, A. Dahdouh, A. I. Mansour, C. Jimenez, D. Martin, R. G. Sarmiento, M. Medarde, E. Caballero and R. Pelaez, Eur. J. Med. Chem., 2015, 100, 210 CrossRef CAS PubMed.
- SHELXTL, version 6.14, Bruker AXS, Madison, WI, 2000–2003 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09 Revision C.01, 2010 Search PubMed.
- (a) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785 CrossRef CAS; (b) A. D. Becke, J. Chem. Phys., 1993, 98, 1372 CrossRef CAS; (c) A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- (a) R. Ditchfield, W. J. Hehre and J. A. Pople, J. Chem. Phys., 1971, 54, 724 CrossRef CAS; (b) A. D. McLean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215 CrossRef CAS.
- (a) V. Barone and M. J. Cossi, J. Phys. Chem. A, 1998, 102, 1995 CrossRef CAS; (b) R. Cammi, B. Mennucci and J. Tomasi, J. Phys. Chem. A, 1999, 103, 9100 CrossRef CAS; (c) J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1590110, 1590111, 1590113, 1816881, 1590195, 1590116, 1816882, 1590114, 1590115, 1590112, 1590109 and 1811865. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc00560e |
| ‡ These authors have equal contributions to the work reported herein. |
| This journal is © The Royal Society of Chemistry 2018 |