DOI:
10.1039/C7SC05420C
(Edge Article)
Chem. Sci., 2018,
9, 3215-3220
Rapid access to diverse, trifluoromethyl-substituted alkenes using complementary strategies†
Received
21st December 2017
, Accepted 21st February 2018
First published on 22nd February 2018
Abstract
Two synergistic approaches to the facile assembly of complex α-trifluoromethyl alkenes are described. Using α-trifluoromethyl-β-silyl alcohols as masked trifluoromethyl alkenes, cross-coupling or related functionalization processes at distal electrophilic sites can be executed without inducing Peterson elimination. Subsequent Lewis acidic activation affords functionalized α-trifluoromethyl alkenes. Likewise, the development of a novel α-trifluoromethylvinyl trifluoroborate reagent complements this approach and allows a one-step cross-coupling of (hetero)aryl halides to access a broad array of complex α-trifluoromethyl alkenes.
Introduction
The ability of fluorine to modulate the pharmacological properties of biologically active molecules and to improve metabolic stability has spurred the development of an array of synthetic methods for both incorporation and modification of fluorinated motifs within complex molecular settings.1 Of interest are α-trifluoromethyl alkenes, for they are known peptide isosteres,2 are medicinally relevant moieties in pharmacologically active compounds,3 and are synthetic intermediates to other fluorinated species4 (Scheme 1). For instance, cyclopropanation yields trifluoromethyl-substituted cyclopropanes, which are established tert-butyl isosteres,5a and radical defluorinative alkylation gives 1,1-gem-difluoroalkenes, which are proposed ketone mimics.5b–e Although several routes for preparing α-trifluoromethyl alkenes are reported, they are frequently lacking in functional group compatibility or ease of operation. Current approaches for the synthesis of this motif fall under three categories: (1) Wittig-type methylenation of trifluoromethyl ketones (TFMKs) under strongly basic conditions;6 (2) Suzuki, Negishi, and Kumada cross-couplings that employ an arylboron species and highly volatile bromo-3,3,3-trifluoroprop-1-ene;7 (3) Peterson-type olefination of TFMKs that utilizes a Grignard reaction in its synthetic sequence, thus limiting protic group tolerance.8
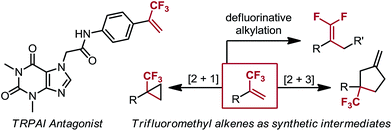 |
| | Scheme 1 α-Trifluoromethyl alkenes in a pharmaceutically active compound and as synthetic intermediates. | |
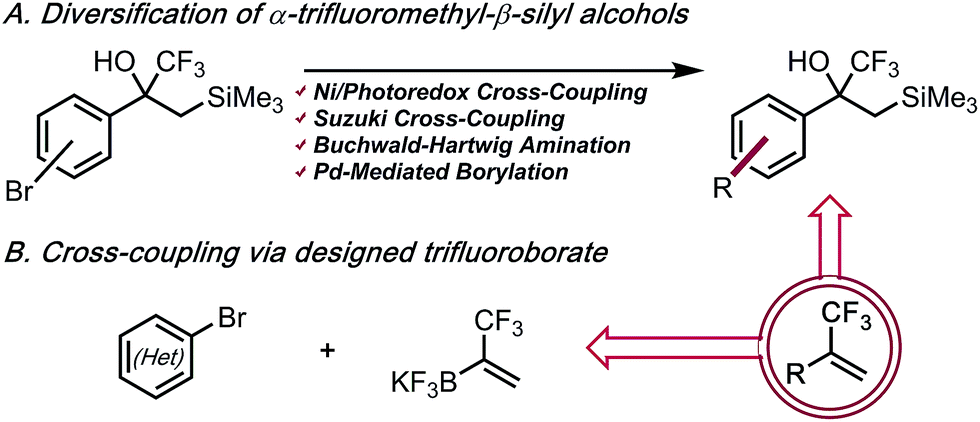
As part of a program to access functionalized fluorinated motifs, we became interested in efficient routes to α-trifluoromethyl alkenes that were both structurally complex and functionally diverse. Two complementary strategies were envisioned: (1) employment of cores with α-trifluoromethyl-β-silyl alcohols as “masked” trifluoromethyl alkenes, thus allowing molecular complexity to be installed without incurring undesired side reactions of the alkene; (2) utilization of a solid, bench-stable organotrifluoroborate reagent for rapid, late-stage introduction of the 3,3,3-trifluoro-1-propenyl motif (Scheme 2). This reagent eliminates the use of a volatile compound with limited bench-stability in favor of a crystalline, bench-stable solid that can be readily synthesized on a multigram scale and provides access to an umpolung bond disconnection. Additionally, the use of aryl bromides rather than arylboron species offers improvement on the availability and cost of starting materials.
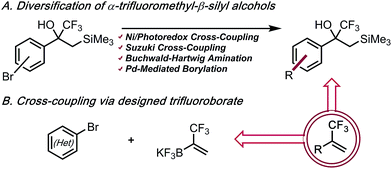 |
| | Scheme 2 α-Trifluoromethyl alkenes via two complementary approaches. | |
Results and discussion
Functionalization of masked α-CF3 alkenes
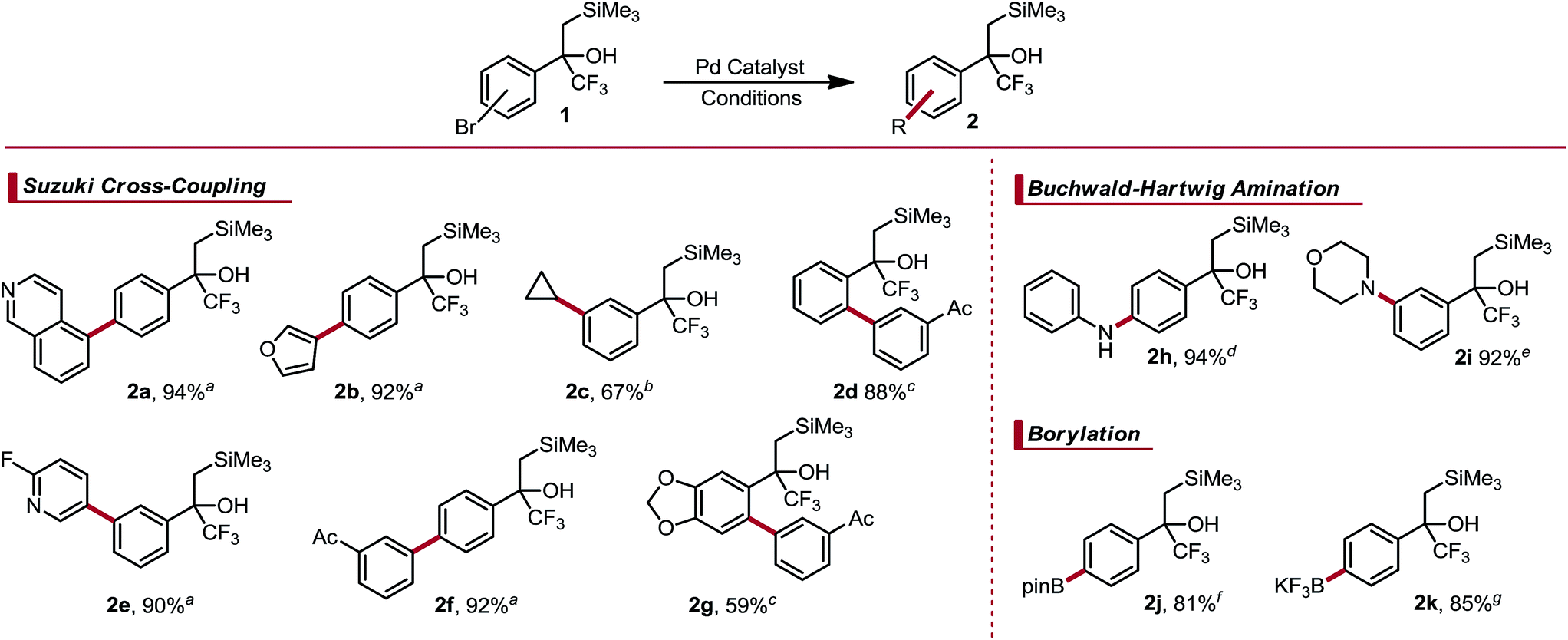
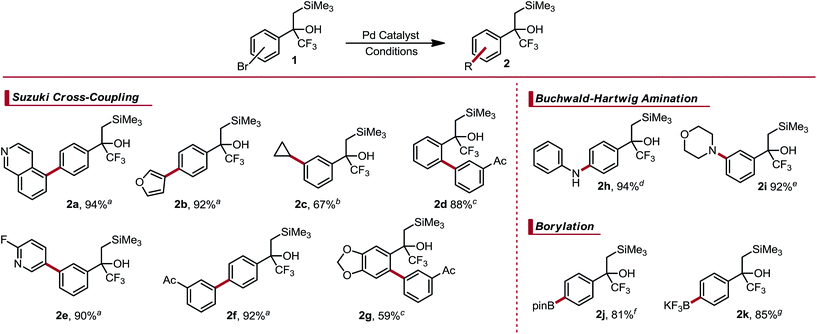
α-Trifluoromethyl-β-silyl alcohols were reported recently by Leadbeater and co-workers as viable precursors to α-trifluoromethyl alkenes.8 The scope of this approach was hampered by the lengthy synthetic sequence used to prepare each example and the general intolerance of their approach toward Lewis or Brønsted acidic functional groups. However, α-trifluoromethyl-β-silyl alcohols were noted to be surprisingly stable despite the known propensity of tertiary β-silyl alcohols to undergo spontaneous elimination under both weakly acidic or basic conditions.9 The origin of this stability is likely derived from electronic deactivation and steric shielding by the α-trifluoromethyl group. We imagined that these α-trifluoromethyl-β-silyl alcohols could serve as masked CF3 alkenes, which would enable rapid diversification to assemble species that would not only be incompatible with the original sequence, but also give rise to α-trifluoromethyl alkenes that would be otherwise challenging, if not impossible, to prepare. Indeed, we envisioned that we could further elaborate these substructures by executing Csp2–Csp3 photoredox cross-coupling,10 Suzuki cross-coupling,11 Buchwald–Hartwig amination,12 and palladium-mediated borylation.13 After functionalization, the relatively mild nature of dehydrative elimination would furnish structurally complex α-trifluoromethyl alkenes. The amenability of α-trifluoromethyl-β-silyl alcohols toward Suzuki–Miyaura coupling was first investigated. Three regioisomeric, brominated cores were prepared and subjected to cross-coupling with aryl trifluoroborates. Good reactivity without concomitant olefination was observed. The para- and meta-substituted aryl bromides coupled in high yield with aryl- and heteroaryl trifluoroborates under relatively standard conditions (Table 1).14 Csp2–Csp3 Cross-coupling using potassium cyclopropyltrifluoroborate also proceeded well using standard conditions.15a The steric bulk of the α-trifluoromethyl-β-silyl moiety initially impeded coupling of the ortho-substituted aryl bromide core. Using high throughput experimentation (HTE),15b suitable conditions for ortho-coupling were quickly identified (see ESI† for details on HTE studies). Electron rich, sterically bulky phosphine ligands were the most efficacious, with 1,2,3,4,5-pentaphenyl-1′-(di-tert-butylphosphino)ferrocene (QPhos) proving optimal. Using these conditions, cross-coupling proceeded smoothly to afford the ortho-arylated structure 2d. These conditions could also be extended to a dioxolane core 2g.
Table 1 Palladium-catalyzed cross-couplings of α-trifluoromethyl-β-silyl alcohols
Reaction conditions: Pd(OAc)2 (1 mol%), RuPhos (3 mol%), organotrifluoroborate (1.1 equiv.), Na2CO3 (2 equiv.), EtOH (0.18 M), 85 °C, 24 h.
Reaction conditions: XPhos Pd G2 (3 mol%), organotrifluoroborate (1.2 equiv.), K2CO3 (3 equiv.), 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 CPME/H2O (0.25 M), 100 °C, 24 h.
Reaction conditions: Pd(OAc)2 (5 mol%), QPhos (12 mol%), organotrifluoroborate (1.1 equiv.), K2CO3 (2 equiv.), 2:1 dioxane/H2O (0.25 M), 85 °C, 24 h.
Reaction conditions: XPhos Pd G2 (2 mol%), aniline (3 equiv.), Cs2CO3 (1.4 equiv.), PhMe (0.5 M), 100 °C, 24 h.
Reaction conditions: XPhos Pd G2 (2 mol%), morpholine (3 equiv.), Cs2CO3 (2.5 equiv.), 5:1 PhMe/tBuOH (0.42 M), 80 °C, 12.5 h.
Reaction conditions: XPhos Pd G2 (2 mol%), B2pin2 (3 equiv.), KOAc (3 equiv.), dioxane (0.5 M), 110 °C, 2 h.
Reaction conditions: XPhos Pd G2 (0.5 mol%), XPhos (1 mol%), BBA (3 equiv.), KOAc (3 equiv.), EtOH (0.1 M), 80 °C then KHF2 (6.75 equiv.), MeOH (0.1 M). :1 CPME/H2O (0.25 M), 100 °C, 24 h.
Reaction conditions: Pd(OAc)2 (5 mol%), QPhos (12 mol%), organotrifluoroborate (1.1 equiv.), K2CO3 (2 equiv.), 2:1 dioxane/H2O (0.25 M), 85 °C, 24 h.
Reaction conditions: XPhos Pd G2 (2 mol%), aniline (3 equiv.), Cs2CO3 (1.4 equiv.), PhMe (0.5 M), 100 °C, 24 h.
Reaction conditions: XPhos Pd G2 (2 mol%), morpholine (3 equiv.), Cs2CO3 (2.5 equiv.), 5:1 PhMe/tBuOH (0.42 M), 80 °C, 12.5 h.
Reaction conditions: XPhos Pd G2 (2 mol%), B2pin2 (3 equiv.), KOAc (3 equiv.), dioxane (0.5 M), 110 °C, 2 h.
Reaction conditions: XPhos Pd G2 (0.5 mol%), XPhos (1 mol%), BBA (3 equiv.), KOAc (3 equiv.), EtOH (0.1 M), 80 °C then KHF2 (6.75 equiv.), MeOH (0.1 M).
|
|
Amination of these aryl bromides proceeded smoothly with both aniline and morpholine, providing the corresponding products (2h and 2i) in high yield. The latter emphasizes the utility of these α-trifluoromethyl-β-silyl alcohols as masked trifluoromethyl alkenes, given the propensity of trifluoromethyl alkenes to undergo defluorinative amination under basic conditions.16
Although numerous arylboron species are commercially available, there is a paucity when compared to commercially available aryl halides. One could envision borylating trifluoroalkenyl-substituted aryl bromides, but this approach is complicated by the facile defluorinative borylation of trifluoromethyl alkenes, resulting in 1,1-gem-difluoroalkenes.17 Consequently, we were interested in the borylation of these masked trifluoromethyl alkenes to generate arylboron species capable of serving as points for further diversification. Palladium-catalyzed borylation using either bis(pinacolato)diboron18 or bisboronic acid (BBA)19 occurred efficiently and in good yield to provide access to arylboronic ester 2j and aryltrifluoroborate 2k (Table 1). Notably, treatment of the intermediate boronic acid obtained from the BBA approach with excess KHF2 induced no observable Peterson elimination. This is a testament to the stability of the masking motif, given that both fluoride-induced and weakly Lewis acid-induced elimination pathways are known with β-silyl alcohols.9b,20 Arylboron species 2j and 2k were effective cross-coupling substrates, reacting in good yield with 5-bromoindole and 7-bromo-3-fluoroquinoline to give the products 2l and 2m, respectively (Scheme 3).
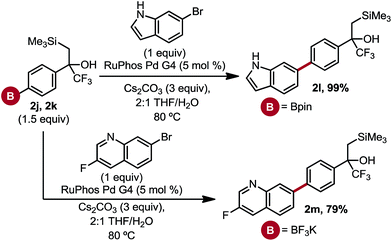 |
| | Scheme 3 Suzuki cross-coupling of borylated α-trifluoromethyl-β-hydroxy alcohols. All yields are isolated yields after purification. For details, see the ESI.† | |
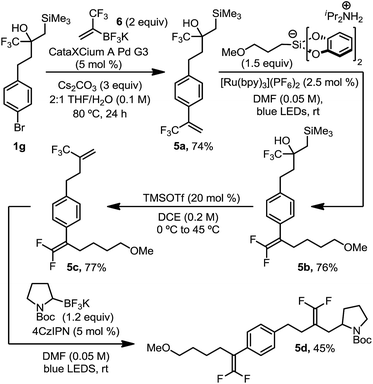 |
| | Scheme 4 Orthogonal introduction and reaction of α-trifluoromethyl alkenes. All yields are isolated yields after purification. For details, see the ESI.† | |
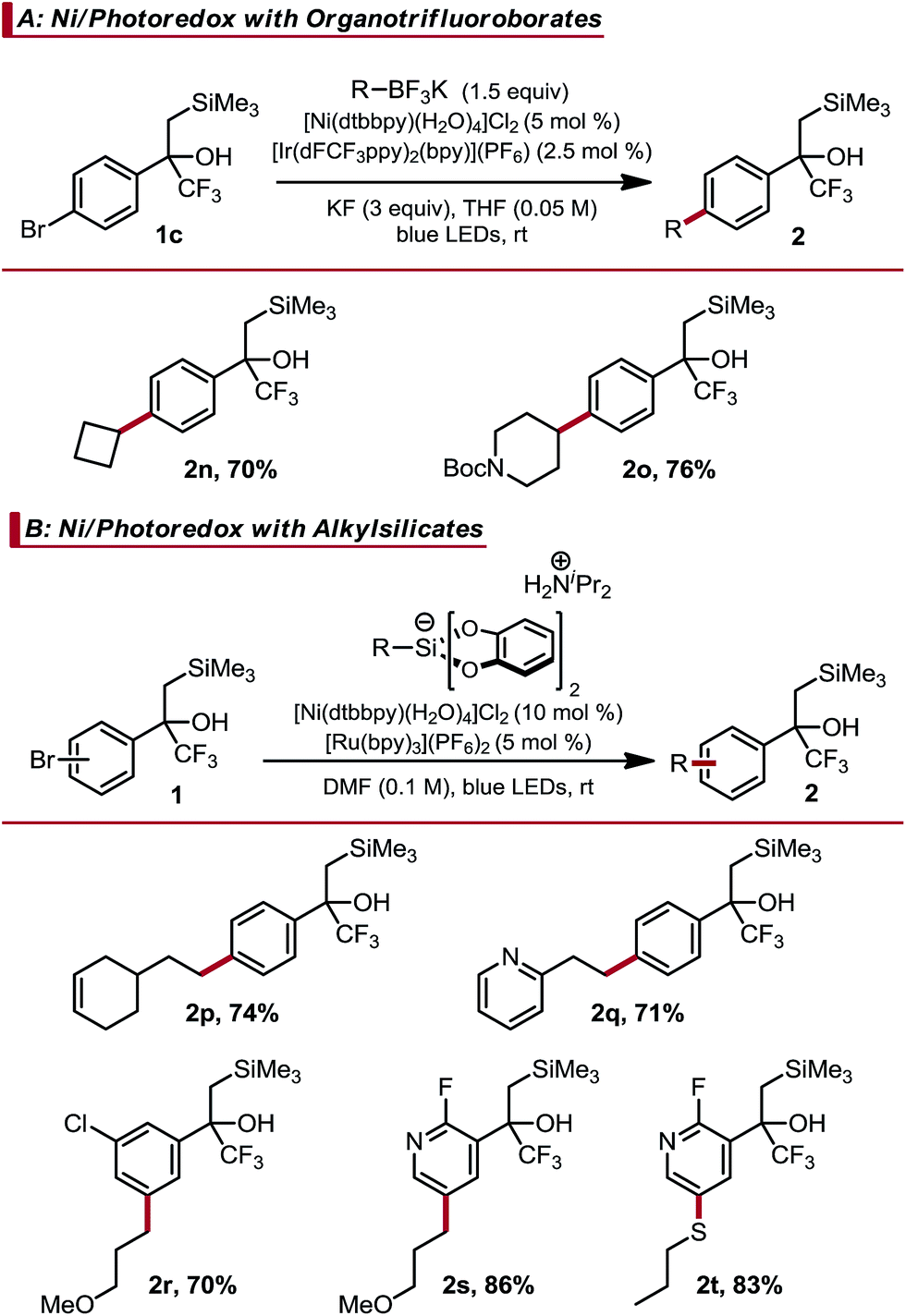
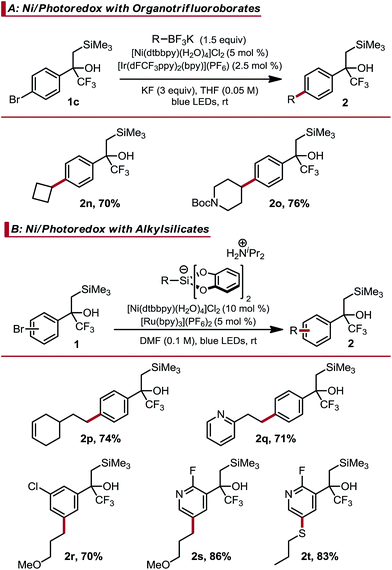
Given the difficulty observed when attempting to prepare Csp2–Csp3 linkages using two-electron manifolds,21 Ni/photoredox dual catalytic processes were next examined. This approach is convenient for Csp2–Csp3 cross-coupling within complex molecular settings because it can be executed under mild conditions and displays broad functional group tolerance.10 A key feature of these methods is the use of carbon-centered radicals to enable room temperature transmetalation. As such, certain moieties that are prone to radical chemistry will engage in off-cycle reactions. For example, trifluoromethyl alkenes are known to undergo facile radical defluorinative alkylation, likely precluding them from this form of catalysis.5b–d Indeed, we conducted competition experiments and found alkylation of the alkene to be competitive with Ni-catalyzed cross-coupling (see ESI† for experimental details). Thus, this reaction class is ideal for demonstrating α-trifluoromethyl-β-silyl alcohols as masked trifluoromethyl alkenes. Dual catalytic cross-coupling was initially investigated using secondary organotrifluoroborates and these brominated, masked alkenes.22 Selection of the appropriate additive was crucial in achieving good reactivity. Carbonates and other basic additives, which are typically employed in Csp2–Csp3 couplings involving organotrifluoroborates, provided only partial conversion. Hypothesizing that the basic additive may deprotonate the relatively acidic α-trifluoromethyl alcohol present in the aryl bromide, we examined alternative additives to sequester the BF3 generated upon oxidative fragmentation of the organotrifluoroborate. KF was identified as the most effective additive, enabling the Csp2–Csp3 cross-coupled products to be obtained in good yield (Table 2(A)). Success here is again remarkable because: (1) BF3 can catalyze Peterson elimination23 and (2) KF can be used despite the high fluorophilicity of silicon. Other radical precursors could also be utilized. Alkyl bis(catecholato)silicates coupled in good yield with brominated, masked trifluoromethyl alkenes, although higher catalyst loadings were required to achieve complete conversion in a timely manner (Table 2(B)).24 Simple aryl masked trifluoromethyl alkenes, in addition to more complex meta-chloro (2r) and pyridyl (2s and 2t) examples, cross-coupled in good yield. Ni/photoredox catalyzed thioarylation was also possible (2t).25
Table 2 Ni/Photoredox dual catalytic cross-coupling of masked trifluoromethyl alkenesa
|
All yields are isolated yields after purification. For details, see the ESI.
|
|
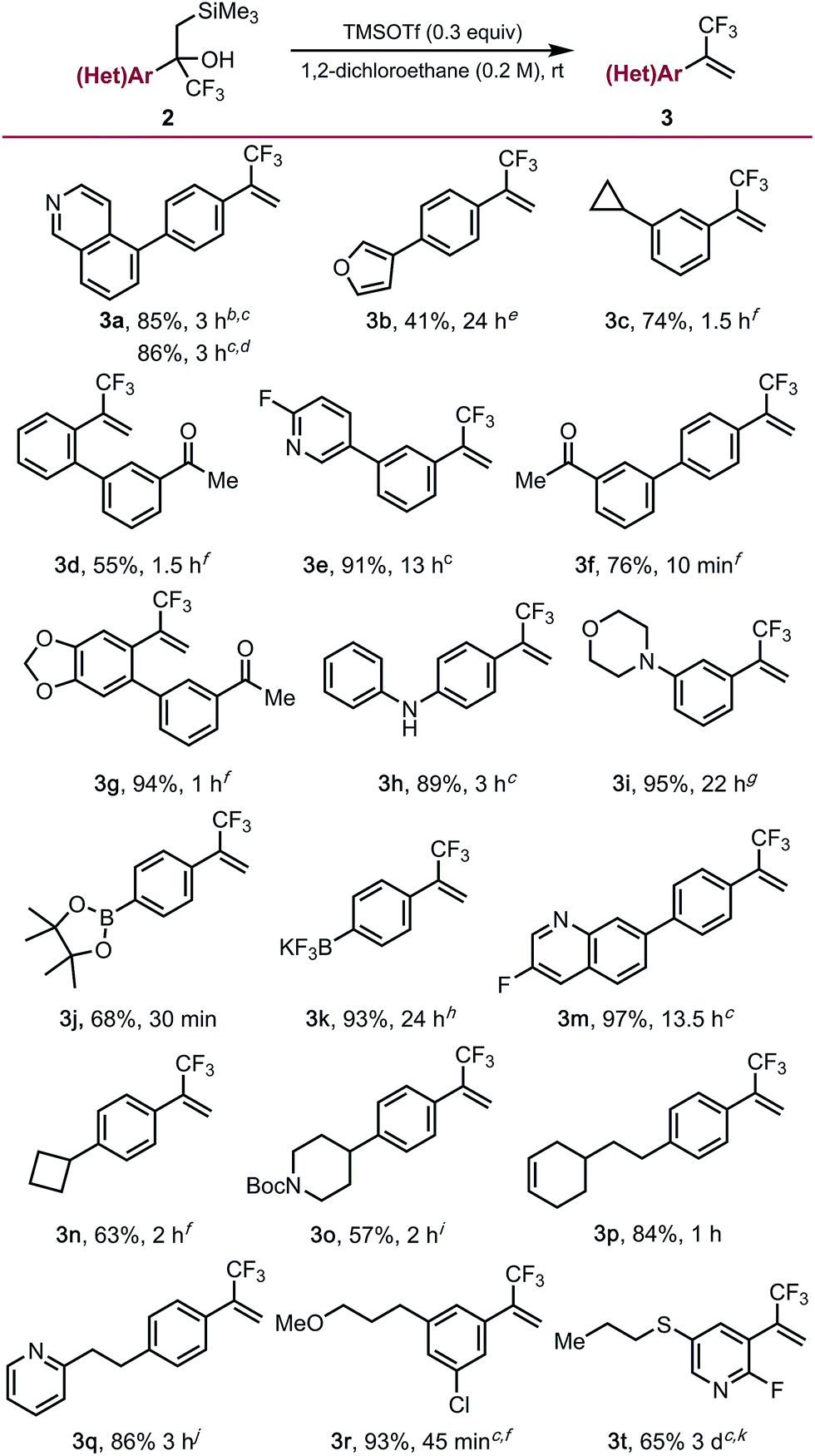
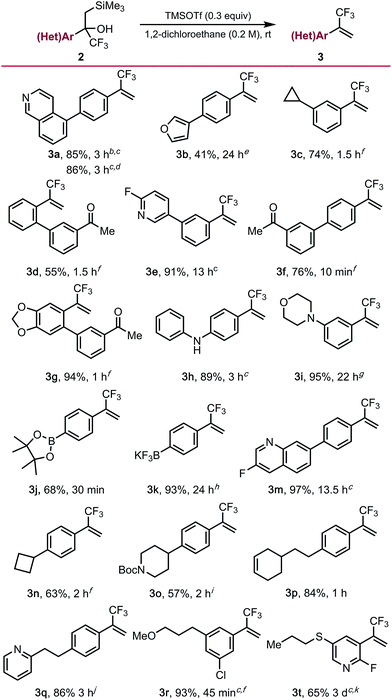
To demonstrate the full potential of this strategy for diversification, unmasking of the functionalized α-trifluoromethyl-β-silyl alcohols using trimethylsilyl trifluoro-methanesulfonate (TMSOTf) was explored. Although it proceeded smoothly for most substrates, a major challenge was conducting the Peterson elimination in the presence of Lewis basic sites. We found that elimination can be accomplished for some of the more recalcitrant substrates if a super-stoichiometric amount (1.3 equiv.) of TMSOTf are employed (e.g., isoquinoline 3a [Table 3]). Alternatively, the addition of an equivalent of HCl to protonate the basic site (e.g., the nitrogen of 2a) enables successful elimination using sub-stoichiometric amounts (0.6 equiv.) of TMSOTf. Substrates containing less basic amines (3e, 3h, 3m) readily underwent elimination using catalytic amounts of TMSOTf, supporting the hypothesis of Lewis base-mediated deactivation of TMSOTf. Substrates containing ketone (3d, 3f, 3g), alkene (3p), strained rings (3c, 3n), alkyl ether (3r), morpholine (3i), and acid sensitive furan (3b) functional groups underwent elimination in moderate to good yields. Unfortunately, while unmasking of free indole substrate 2l was successful, undesired side reactions were observed on the indole moiety. Elimination proceeded smoothly in the presence of an N-Boc piperidine (3o), although reprotection of the free amine was required upon reaction completion. Unmasking of α-trifluoromethyl-β-silyl alcohol 2t required additional TMSOTf and extended reaction times due to deactivation of the alcohol toward elimination by the electron-deficient 2-fluoropyridine core. Related alcohol 2s was resistant toward elimination, only undergoing partial conversion to the trifluoromethyl alkene even under rather forcing conditions. Interestingly, boronic ester 2j and potassium organotrifluoroborate 2k were also compatible with the Peterson elimination conditions. However, in the case of the latter, exposure of 3k to KHF2 upon reaction completion was required to return all the material to the trifluoroborate salt due to concomitant silyl-mediated defluorination.
Table 3 α-Trifluoromethyl alkenes via Peterson elimination of cross-coupled productsa
|
Elimination to α-trifluoromethyl alkenes. General reaction conditions: α-trifluoromethyl-β-silyl alcohol (1.0 equiv.), 1,2-dichloroethane (0.2 M), TMSOTf (0.3 equiv.), rt. All yields are isolated yields after purification. Some substrates required additional TMSOTf or increased reaction temperature, for additional details see the ESI.
Using 1.3 equiv. TMSOTf.
Conducted at 90 °C.
Conducted using 1.0 equiv. of HCl and 0.6 equiv. of TMSOTf.
Using 0.1 equiv. TMSOTf.
Using 0.2 equiv. TMSOTf.
Using 0.9 equiv. TMSOTf.
Using 2.0 equiv. TMSOTf.
Using 2.2 equiv. TMSOTf.
Using 1.2 equiv. TMSOTf.
Using 6.0 equiv. TMSOTf.
|
|
Direct access to α-trifluoromethyl alkenes using potassium trifluoromethylvinyl trifluoroborate
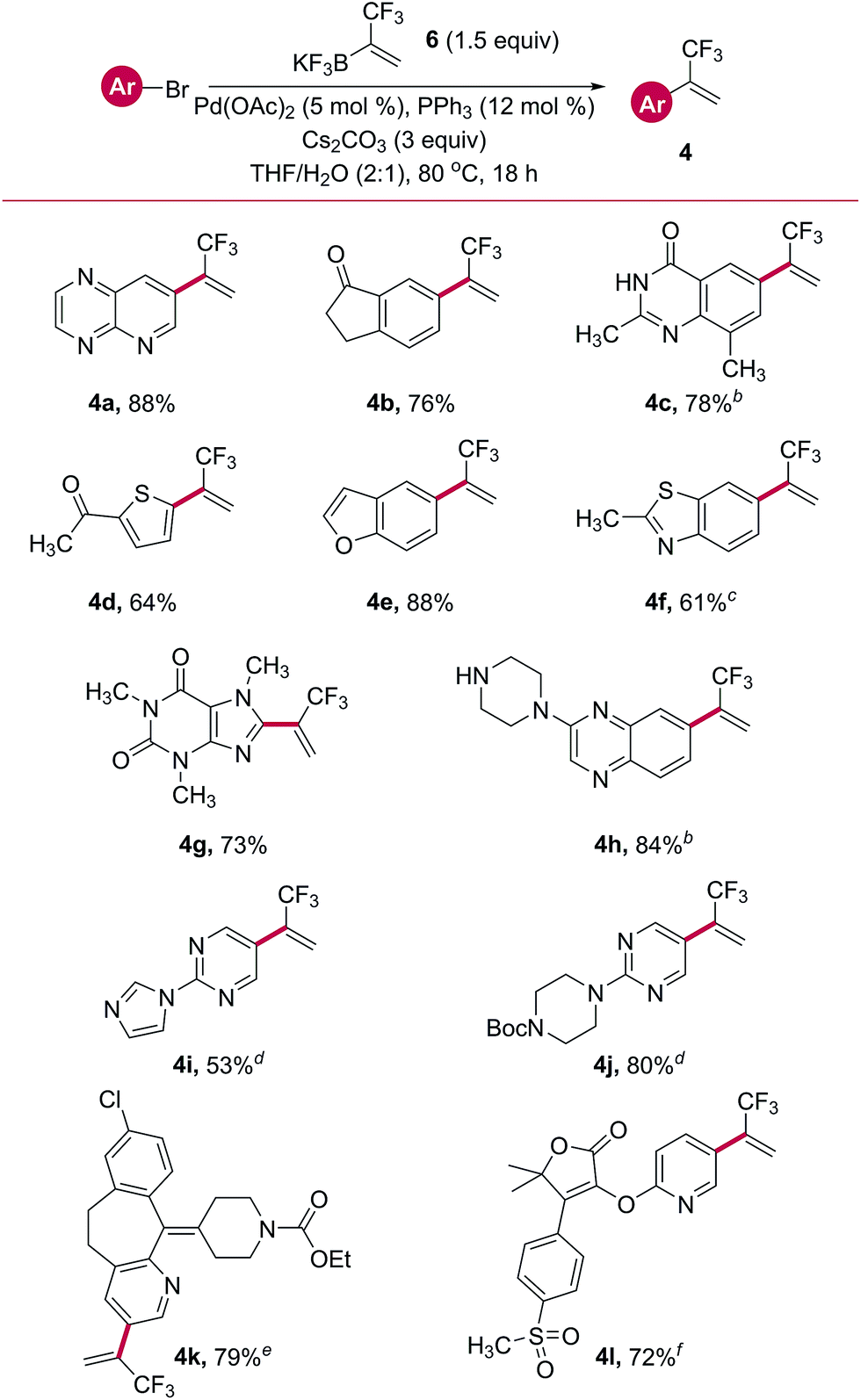
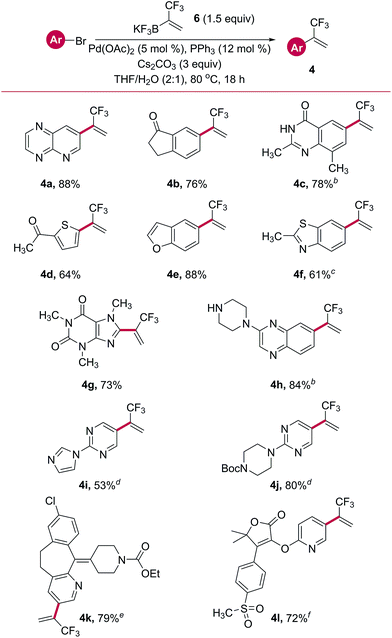
Direct, one-step Suzuki cross-coupling of potassium trifluoromethylvinyl trifluoroborate 6 with a host of aryl and heteroaryl bromides was next pursued. Initial success was found when using Pd(OAc)2 with PPh3 as a ligand (Table 4). Several aryl- and heteroaryl bromides containing a wide variety of functional groups were successfully cross-coupled, including a pyrido[2,3-b]pyrazine (4a), indanone (4b), thiophene (4d), benzofuran (4e), and a caffeine derivative (4g). Other substrates required variation of the phosphine ligand for palladium to impart good reactivity. A survey of commercially available palladium pre-complexes was sufficient. Both a quinazolinone (4c) and a benzopyrazine containing a free secondary amine (4h) were successfully cross-coupled using the PCy3 Pd G4 pre-complex. Similarly, SPhos Pd G4 performed well in the cross-coupling with a bromo-benzothiazole (4f). Two examples, a benzocycloheptapyridine (4k, a derivatized form of Claritin®) and a pyridylfuranone (4l), demonstrated the amenability of this reaction to complex, highly functionalized, drug-like structures.
Table 4 Palladium cross-couplings with organotrifluoroboratea
|
Unless otherwise noted, reactions were performed using bromide (1.0 equiv., 1 mmol), 6 (1.5 equiv.), Pd(OAc)2 (5 mol%), PPh3 (12 mol%), and Cs2CO3 (3 equiv.) in THF/H2O (2:1, 0.11 M) at 80 °C for 18 h.
Performed with PCy3 Pd G4 in place of Pd source and ligand.
Performed with SPhos Pd G4 in place of Pd source and ligand.
Performed with XPhos Pd G4 in place of Pd source and ligand.
Performed with Pd G4 dimer (2.5 mol%) in place of Pd source.
Performed with Catacxium A Pd G3 in place of Pd source and ligand.
|
|
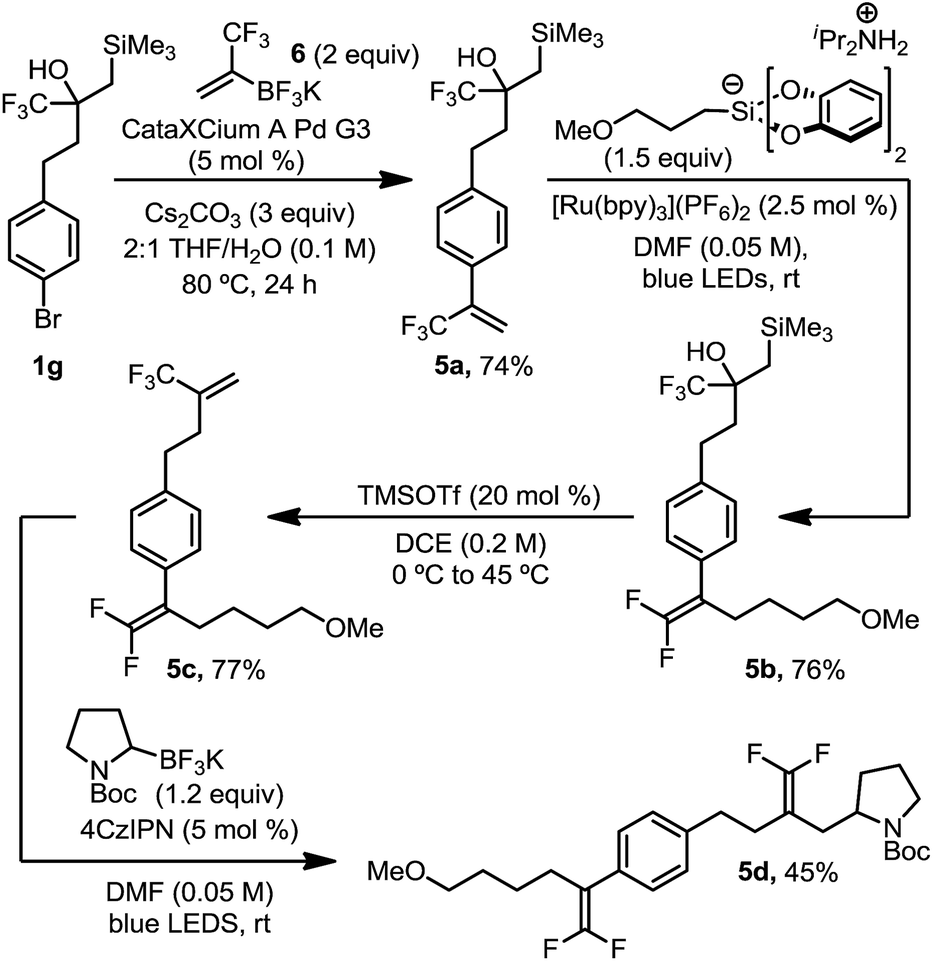
This organotrifluoroborate reagent, 6, provided an opportunity to demonstrate the complementarity and orthogonality of these two distinct approaches (Scheme 4). As such, we executed a synthesis in which two trifluoromethyl alkenes were sequentially introduced and functionalized. Suzuki cross coupling of potassium trifluoromethylvinyl trifluoroborate 6 with brominated, masked trifluoromethyl alkene 1g afforded α-trifluoromethyl styrene 5a in good yield. Radical defluorinative alkylation of the trifluoromethyl alkene using diisopropylammonium bis(catecholato) (3-methoxypropyl) silicate provided gem-difluoroalkene 5b, which subsequently underwent alcohol elimination to reveal an aliphatic trifluoromethyl alkene 5c in good yield. Trifluoromethyl alkene 5c then proceeded through a second selective radical defluorinative alkylation using N-Boc-pyrrolidine trifluoroborate to afford differentiated gem-difluoroalkene 5d. Synthesis of such a structure by any other means would prove challenging, if not impossible, using state-of-the-art approaches.
Conclusions
In conclusion, two complementary approaches for the synthesis of highly functionalized trifluoromethyl alkenes have been realized. First, the viability of α-trifluoromethyl-β-silyl alcohols as masked trifluoromethyl alkenes was validated, enabling rapid diversification through several cross-coupling approaches. The elaborated products readily underwent elimination to generate their corresponding trifluoromethyl alkenes. Second, the one-step (trifluoromethyl)vinylation of functional group-rich aryl- and heteroaryl bromides was achieved using potassium trifluoromethylvinyl trifluoroborate. Taken together, these two complementary approaches provide the means to access an array of structurally diverse α-trifluoromethylated arenes rapidly that can be further leveraged to produce novel, fluorinated species.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
The authors are grateful for the financial support provided by NIGMS (R01 GM 113878). J. P. P. and C. B. K. are grateful for NIH NRSA postdoctoral fellowships (F32 GM125241 and F32 GM117634). R. J. W. is grateful for an NSF GRFP fellowship. We sincerely thank Dr Simon Berritt of the University of Pennsylvania (UPenn) for his assistance and use of the Penn Merck High Throughput Experimentation Center with its funding provided by NIH S10 OD011980. We thank Dr Charles W. Ross, III (UPenn) for his assistance in obtaining HRMS data, Dr Patrick Carroll (UPenn) for his assistance in obtaining x-ray crystal structure data (CCDC 1811877), and the late Dr George Furst (UPenn) for his assistance in obtaining NMR data. We thank Merck Research Laboratories and GlaxoSmithKline for the generous donation of aryl halides, Johnson-Matthey for donation of precious metal salts, and Frontier Scientific for the donation of boronic acids.
Notes and references
-
(a) N. A. Meanwell, J. Med. Chem., 2011, 54, 2529 CrossRef CAS PubMed;
(b) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC;
(c) K. W. Hagmann, J. Med. Chem., 2008, 51, 4359 CrossRef PubMed;
(d) K. Müeller, C. Faeh and F. Diederich, Science, 2007, 317, 1881 CrossRef PubMed;
(e) T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214 CrossRef CAS PubMed;
(f) C. F. Ni and J. B. Hu, Chem. Soc. Rev., 2016, 45, 5441 RSC.
-
(a) J. Xiao, B. Weisblum and P. J. Wipf, J. Am. Chem. Soc., 2005, 127, 5742 CrossRef CAS PubMed;
(b) E. Inokuchi, T. Marumi, A. Nilda, K. Kobayashi, K. Tomita, S. Oishi, H. Shno and N. Fujii, J. Org. Chem., 2008, 73, 3942 CrossRef CAS PubMed;
(c) K. Kobayahi, T. Marum, S. Oishi, H. Ohno and N. Fujii, J. Org. Chem., 2009, 74, 4626 CrossRef PubMed;
(d) S. Fustero, G. Chiva, J. Piera, J. F. Sanz-Cervera, A. Volonterio, M. Zanda and C. Ramirez de Arellano, J. Org. Chem., 2009, 74, 3122 CrossRef CAS PubMed.
-
R. Bakthavatchalam, Compounds useful for treating disorders related to trpa1, WO 2010138879, Dec 2, 2010.
-
(a)
T. Woltering, W. Guba, H. Hilpert, A. Kuglstatter, A. Limberg, U. Obst Sander, E. Pinard and W. Wostl, Preparation of fluorooxazine derivatives for use as BACE1 inhibitors, WO 2014114532, July 31, 2014;
(b)
R. Bakthavatchalam, Compounds useful for treating disorders related to TRPA1, WO 2010138879, Dec. 2, 2010;
(c)
H. Mizuno, Preparation of pyridinyl oxadia-zolones for controlling pests, WO 2009066786, May 28, 2009;
(d)
M. G. Kelly, C. J. Kaub, J. Kincaid, S. Janagani, G. Wu, Z.-L. Wei, K. Sahasrabudhe, M. Duncton, R. B. Upasani, Y. Fang and M. Cox, Preparation of amide derivatives as ion-channel ligands, WO 2007100758, Sep 7, 2007;
(e)
M. G. Kelly, J. Kincaid, M. Duncton, K. Sahasrabudhe, S. Janagani, R. B. Upasani, G. Wu, Y. Fang and Z.-L. Wei, Preparation and biological activity of N-aryl and N-hetaryl substituted benzamides and pyridinecarboxamides as mammalian ion-channel ligands for using as anti-inflammatory and pain relief agents in pharmaceutical compositions, US 20060194801, Aug 31, 2006;
(f)
Y. Zhou, J. Qu, Y. Liu, and Y. Zhao, Preparation method and application of difluoroallyl borate, CN 106366105, Feb 1, 2017;
(g) B. M. Trost and L. Debien, J. Am. Chem. Soc., 2015, 137, 11606 CrossRef CAS PubMed.
-
(a) D. Barnes-Seeman, M. Jain, L. Bell, S. Ferreira, S. Cohen, X.-H. Chen, J. Amin, B. Snodgrass and P. Hatsis, ACS Med. Chem. Lett., 2013, 4, 514 CrossRef CAS PubMed;
(b) S. B. Lang, R. J. Wiles, C. B. Kelly and G. A. Molander, Angew. Chem., Int. Ed., 2017, 56, 15073 CrossRef CAS PubMed;
(c) T. Xiao, L. Li and L. Zhou, J. Org. Chem., 2016, 81, 7908 CrossRef CAS PubMed;
(d) L. Li, T. Xiao, H. Chem and L. Zhou, Chem.–Eur. J., 2017, 23, 2249 CrossRef CAS PubMed;
(e)
K. Uneyama, Hydrogen Bonding in Organofluorine Compounds, in Organofluorine Chemistry, Blackwell Publishing, Oxford, UK, 2006 Search PubMed.
- Representative examples of this procedure:
(a) B. M. Trost and L. Debien, J. Am. Chem. Soc., 2015, 137, 11606 CrossRef PubMed;
(b) T. Kobayashi, T. Eda, O. Tamura and H. Ishibashi, J. Org. Chem., 2002, 67, 3156 CrossRef CAS PubMed.
-
(a) R. Pana, X. Liua and M. Dengb, J. Fluorine Chem., 1999, 2, 167 CrossRef;
(b) B. Jiang and Y. Xu, J. Org. Chem., 1991, 56, 7336 CrossRef CAS;
(c) O. Kobayashi, D. Uraguchi and T. Yamakawa, J. Fluorine Chem., 2009, 130, 591 CrossRef CAS.
- T. A. Hamlin, C. B. Kelly, R. M. Cywar and N. E. Leadbeater, J. Org. Chem., 2014, 79, 1145 CrossRef CAS PubMed.
-
(a) D. J. Peterson, J. Org. Chem., 1968, 33, 780 CrossRef CAS;
(b) L. F. van Staden, D. Gravestock and D. Ager, Chem. Soc. Rev., 2002, 31, 194 Search PubMed.
-
(a) J. C. Tellis, D. N. Primer and G. A. Molander, Science, 2014, 345, 433 CrossRef CAS PubMed;
(b) M. Jouffroy, D. N. Primer and G. A. Molander, J. Am. Chem. Soc., 2016, 138, 475 CrossRef CAS PubMed;
(c) J. C. Tellis, C. B. Kelly, D. N. Primer, M. Jouffroy, N. R. Patel and G. A. Molander, Acc. Chem. Res., 2016, 49, 1429 CrossRef CAS PubMed;
(d) J. Twilton, C. Le, P. Zhang, M. H. Shaw, R. W. Evans and D. W. C. MacMillan, Nat. Rev. Chem., 2017, 1(0052), 1 Search PubMed.
-
(a) G. A. Molander, J. Org. Chem., 2015, 80, 7837 CrossRef CAS PubMed;
(b) R. Martin and S. L. Buchwald, Acc. Chem. Res., 2008, 41, 1461 CrossRef CAS PubMed.
- P. Ruiz-Castillo and S. L. Buchwald, Chem. Rev., 2016, 116, 12564 CrossRef CAS PubMed.
- K. Kubota, J. Iwamoto and H. Ito, Org. Biomol. Chem., 2017, 15, 285 CAS.
-
(a) G. A. Molander and B. Biolatto, J. Org. Chem., 2003, 68, 4302 CrossRef CAS PubMed;
(b) G. A. Molander, B. Canturk and L. E. Kennedy, J. Org. Chem., 2009, 74, 973 CrossRef CAS PubMed.
-
(a) G. A. Molander and P. E. Gormisky, J. Org. Chem., 2008, 73, 7481 CrossRef CAS PubMed;
(b) J. R. Schmink, A. Bellomo and S. Berritt, Aldrichimica Acta, 2013, 46, 71 Search PubMed.
- R. T. Thornbury and F. D. Toste, Angew. Chem., Int. Ed., 2016, 55, 11629 CrossRef CAS PubMed.
- Y. Liu, Y. Zhou, Y. Zhao and J. Qu, Org. Lett., 2017, 19, 946 CrossRef CAS PubMed.
-
(a) T. Ishiyama, M. Murata and N. Miyaura, J. Org. Chem., 1995, 60, 7508 CrossRef CAS;
(b) W. Tang, S. Keshipeddy, Y. Zhang, X. Wei, J. Savoie, N. D. Patel, N. K. Yee and C. H. Senanayake, Org. Lett., 2011, 13, 1366 CrossRef CAS PubMed.
- G. A. Molander, S. L. J. Trice and S. M. Kennedy, J. Org. Chem., 2012, 77, 8678 CrossRef CAS PubMed.
-
Peterson Olefination, in Comprehensive Organic Name Reactions and Reagents, ed. Leo A. Paquette, 2010, vol. 491, p. 2176 Search PubMed.
-
(a)
J. F. Hartwig, Organotransition Metal Chemistry: From Bonding to Catalysis, University Science, Sausalito, CA, 2010 Search PubMed;
(b) A. Rudolph and M. Lautens, Angew. Chem., Int. Ed., 2009, 48, 2656 CrossRef CAS PubMed.
- D. N. Primer, I. Karakaya, J. C. Tellis and G. A. Molander, J. Am. Chem. Soc., 2015, 137, 2195 CrossRef CAS PubMed.
- D. J. Ager, The Peterson Olefination Reaction, Org. React., 2004, 38(1), 1–223 Search PubMed.
-
(a) M. Jouffroy, D. N. Primer and G. A. Molander, J. Am. Chem. Soc., 2016, 138, 475 CrossRef CAS PubMed;
(b) K. Lin, C. B. Kelly, M. Jouffroy and G. A. Molander, Org. Synth., 2017, 94, 1625 Search PubMed.
- M. Jouffroy, C. B. Kelly and G. A. Molander, Org. Lett., 2016, 18, 876 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Rebecca J.
Wiles
,
Rebecca J.
Wiles