
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An unusual stereoretentive 1,3-quaternary carbon shift resulting in an enantioselective RhII-catalyzed formal [4+1]-cycloaddition between diazo compounds and vinyl ketenes†
Kevin X.
Rodriguez
,
Tara C.
Pilato
and
Brandon L.
Ashfeld
 *
*
Department of Chemistry and Biochemistry, University of Notre Dame, Notre Dame, Indiana 46556, USA. E-mail: bashfeld@nd.edu
First published on 19th February 2018
Abstract
Enantioselective quaternary carbon construction in the assembly of cyclopentenones employing a RhII-catalyzed, formal [4+1]-cycloaddition is described. A Rh2(S-TCPTTL)4-catalyzed cyclopropanation of a vinyl ketene with a disubstituted diazo compound initiates a stereoretentive, accelerated ring expansion to provide the cycloadduct in good to excellent yields and enantioselectivity.
Introduction
While the evolution of new enantioselective [4+1]-cycloadditions has advanced considerably in recent years,1 enabling the stereoselective polyfunctionalization of a disubstituted C1 subunit to yield quaternary stereogenic carbons remains a significant challenge. The majority of asymmetric [4+1]-cycloadditions rely heavily on the polarization of a compatible 1,3-diene component to provide optically pure heterocycles. For example, in 2007 Fu disclosed an enantioselective CuI-catalyzed [4+1]-cycloaddition of 2,3-dihydrofurans employing enones and diazoesters to control absolute stereochemistry at a tertiary center.2 More recently, Shi employed a phosphine-mediated, Morita–Baylis–Hillman-like [4+1]-annulation of electron-deficient alkylidene oxindoles to provide dihydrofurans in high selectivity.3 Similarly, the chiral Lewis acid-catalyzed addition of sulfur and nitrogen ylides to electrophilic Michael acceptors (e.g., ortho-quinone methides, aza-dienes, etc.) has been shown to effectively construct the corresponding 5-membered heterocycle through an overall [4+1]-cycloannulation with good to excellent levels of enantioselectivity.4,5 However, carbocycle generation requires a complementary 1,3-diene addition that is complicated by a competitive cyclopropanation.2,3,5,6 Although a subsequent rearrangement of the resulting vinyl cyclopropane (VCP) yields the formal [4+1]-cycloadduct, the high activation energy and commonly accepted diradical mechanism renders absolute stereocontrol inherently difficult.7 Hudlicky and others have shown that optically active cyclopropanes undergo stereoselective migrations, but the static (non-migrating) cyclopropane carbon directs a diastereochemical outcome (Fig. 1a).8 However, cyclopropanation of a 2-substituted-1,3-diene places a non-stereogenic methylene at the static position. To the best of our knowledge, chirality transfer in VCP-rearrangements that rely solely on the configurational stability of a migrating quaternary center has not been established (Fig. 1b).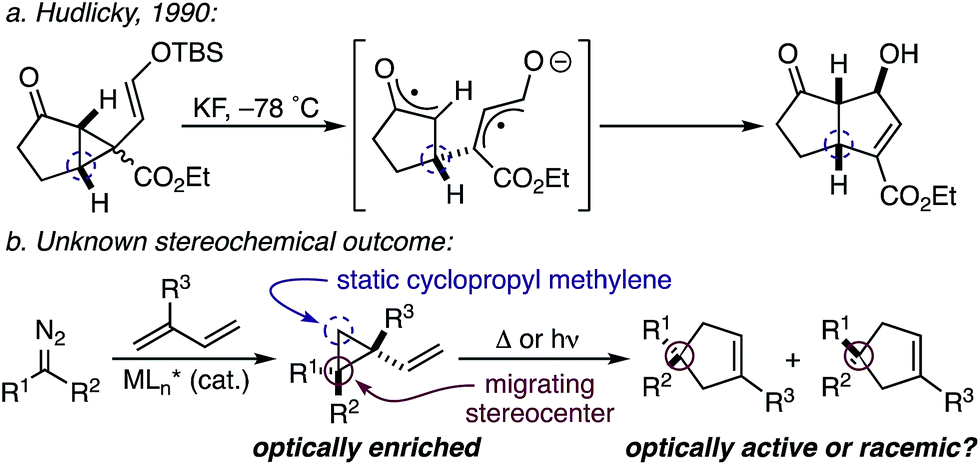 | ||
| Fig. 1 Origin of stereoinduction in vinyl cyclopropane rearrangements. | ||
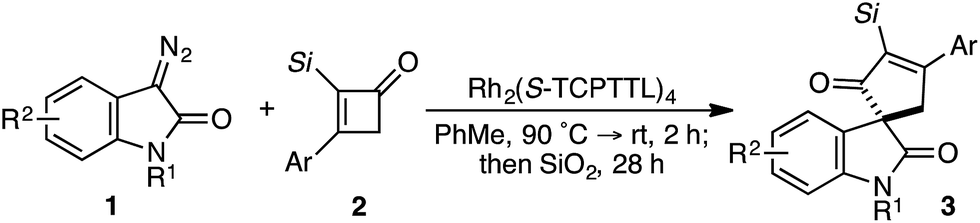
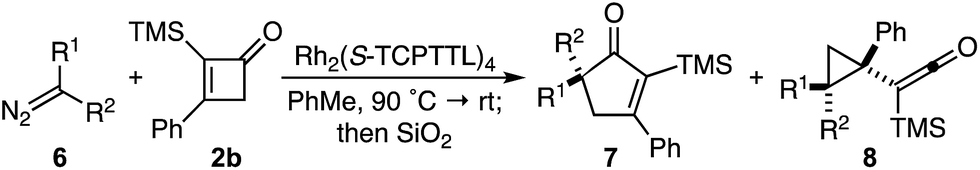
Based on our previous studies employing vinyl ketenes diazo compounds in [4+1]-cycloadditions,9 we speculated that improved orbital alignment between the ketene's orthogonal π-system and that of the migrating C–C bond would lead to greater enantiocontrol throughout the rearrangement.10 Motivated by the therapeutic potential of biologically active oxindole natural products, and synthetic challenge that the C3-spirooxindole stereocenter presents, we strategically chose diazooxindoles as our C1-synthon (Fig. 2a).11 While diastereoselective strategies are known,11a,11c,12 few asymmetric approaches to assemble this quaternary center exist.1b,13 Herein, we describe the first enantioselective, formal [4+1]-cycloaddition between a diazo compound 1 and the vinyl ketene generated in situ from cyclobutenone 2 to provide cycloadduct 3 involving a stereoselective quaternary carbon migration from cyclopropyl ketene 4 (Fig. 2b).7a,14
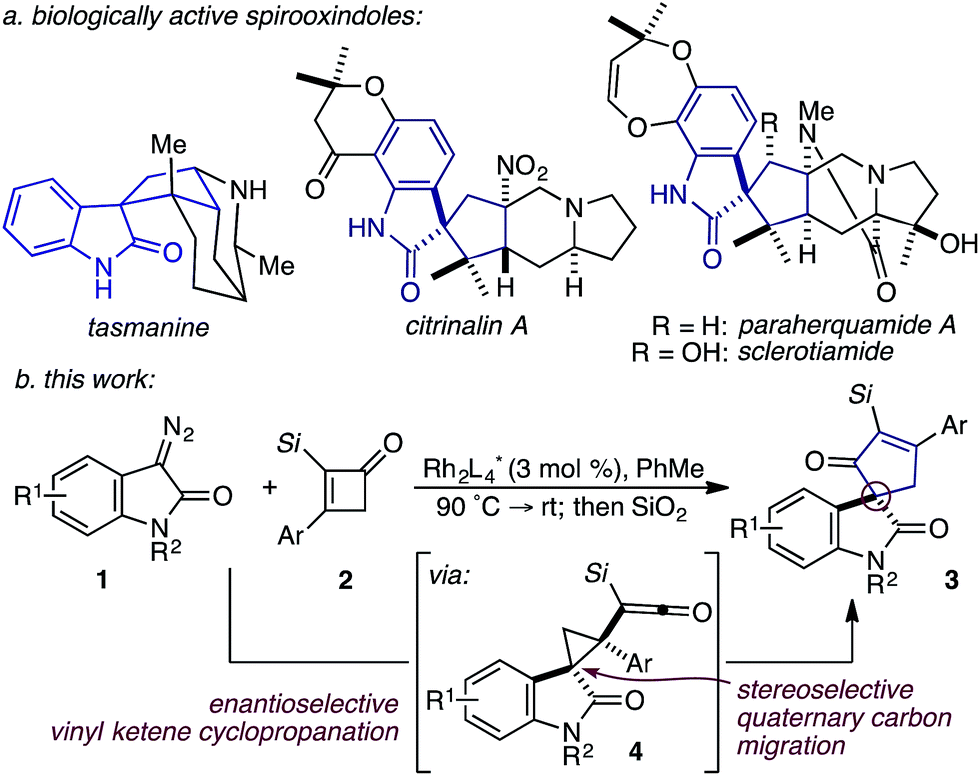 | ||
| Fig. 2 (a) Representative spirooxindole alkaloids; (b) RhII-catalyzed, enantioselective formal [4+1]-cycloaddition. | ||
Despite the preponderance of chiral RhII cyclopropanation catalysts,15 a number of critical issues threatened to derail our efforts prior to undertaking this study. First, examples of diazooxindoles reacting as C1 synthons in asymmetric cycloannulations with high levels of enantioselectivity are rare.1d,16 Additionally, the combination of a Lewis basic ketene carbonyl oxygen and electrophilic metallocarbene could complicate the initial cyclopropanation event.17 Likewise, we were cognizant that the inherent reactivity of vinyl ketenes to undergo dimerization and other side reactions may lead to unproductive or non-stereoselective pathways.17a,17c–f Based on Danheiser's seminal work with vinyl ketenes,18 we opted to generate these relatively underutilized, formal 1,4-dipoles in situ from the corresponding α-silyl cyclobutenones.19
Results and discussion
In spite of these potential complications, we began by examining the enantioselective construction of spirooxindole 3a from diazooxindole 1a and cyclobutenone 2a (Table 1).18,19 While Davies' Rh2(R-DOSP)4 catalyst provided 3a in 81% yield and 14% ee (entry 1), the adamantyl-derived Rh2(R-PTAD)4 improved selectivity to 52% ee, albeit with a modest loss in yield (entry 2).20 Employing either Rh2(S-IBAZ)4 or Rh2(R-BTPCP)4 resulted in diminished levels of enantioselectivity (entries 3 and 4),21 but the tetrachloropthalimide-derived carboxylate Rh2(S-TCPTTL)4 gave 3a in 95% yield and 72% ee (entry 5).22 Tetrafluoropthalimide Rh2(S-TFPTTL)4 and naphthaloyl-tert-leucine tetracarboxylate Rh2(S-NTTL)4 failed to improve selectivity (entries 6 and 7).23|
|
|||||
|---|---|---|---|---|---|
|
|
|||||
| Entry | 5 | Time (h) | Temp (°C) | Yield (%) | ee (%) |
| a Conditions: slow addition of 1a (0.12 mmol) over 1 h to 2a (0.10 mmol) and 5 (3 mol%) in PhMe (0.1 M). See ESI for detailed experimental procedures. b Addition of SiO2 (10 mmol) after 2 h. | |||||
| 1 | 5a | 3 | 100 | 81 | 14 |
| 2 | 5b | 3 | 100 | 60 | 52 |
| 3 | 5c | 3 | 100 | 68 | 8 |
| 4 | 5d | 3 | 90 | 87 | 28 |
| 5 | 5e | 3 | 90 | 95 | 72 |
| 6 | 5f | 3 | 90 | 91 | 62 |
| 7 | 5g | 3 | 90 | 81 | 40 |
| 8 | 5e | 48 | 25 | 32 | 88 |
| 9 | 5e | 30 | 25 | 90 | 90 |
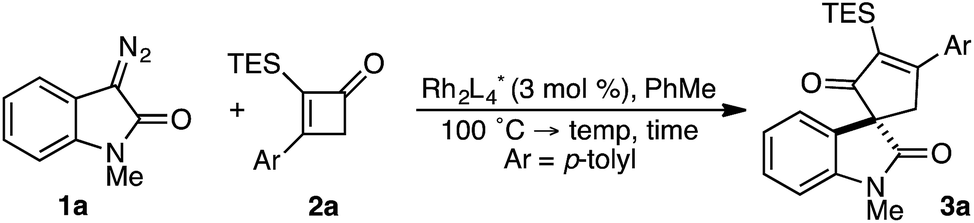
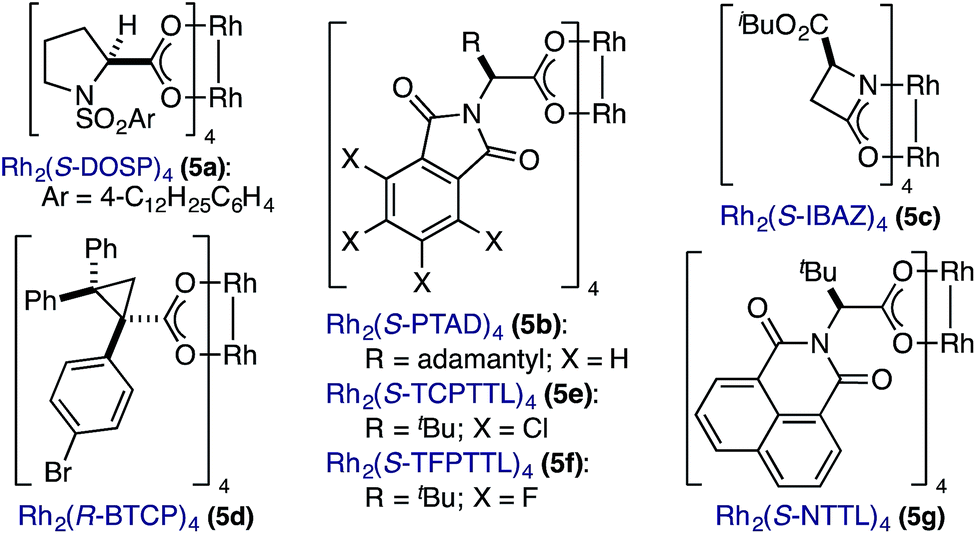
Lowering the reaction temperature following vinyl ketene formation improved selectivity to 88% ee, but the longer reaction times had a negative impact on the yield of 3a (entry 8). While vinyl ketene cyclopropanation occurred rapidly at 25 °C, a sluggish ring expansion resulted in substantial amounts of undesired side products. Attempts to isolate the cyclopropyl ketene intermediate led to the serendipitous discovery that conversion to the cyclopentenone was accelerated upon exposure to silica gel.8a,24 Thus, introduction of SiO2 following cyclopropanation yielded cycloadduct 3a in 90% yield and 90% ee (entry 9).25 Although speculative at this stage, it would appear that the mild Lewis acidic environment created by the addition of SiO2 facilitates the ring expansion event without negatively impacting the chiral integrity of the migrating oxindole C3-stereocenter. A survey of various Lewis acids (e.g., BF3·OEt2, MgI2, Yb(OTf)3) failed to provide a marked improvement in the yield of cycloadduct 3a over SiO2. With an optimized set of conditions in hand, we next assessed the extent of chirality transfer in the cyclopropyl ketene rearrangement.
Careful monitoring of the formal [4+1]-cycloaddition between 1a and the vinyl ketene from 2b in the presence of Rh2(TCPTTL)4 enabled us to track the stereochemical progression en route to cycloadduct 3b (Scheme 1). After 2 h at room temperature, cyclopropyl ketene 4a was observed as a single diastereomer in 95% ee. Subsequent addition of SiO2 converted 4a to cycloadduct 3b in 73% yield and 90% ee. The modest loss of 5% optical enrichment would indicate a well-defined step-wise or competing step-wise and concerted cyclopropyl ketene rearrangements. In a separate experiment, cooling of the reaction mixture (ca. −20 °C) after 2 h led to selective crystallization of 4a, and X-ray crystal diffraction revealed a syn relationship of the oxindole arene and ketene across the cyclopropane and an R configuration of the C3-oxindole quaternary center. Comparison of the X-ray crystal structure obtained for isolated 3b indicated a net stereoretentive migration of the C3-oxindole.26 Interestingly, the diastereoselectivity observed in the formation of cyclopropane 4a is in stark contrast to the major aryl–aryl syn diastereomers observed in many RhII-catalyzed cyclopropanations of styrene derivatives and donor–acceptor metallocarbenes derived from aryl diazo esters.15a,27 While speculative at this stage, the observed aryl–aryl anti-stereoisomer of 4a may arise due to minimization of eclipsing interactions between the oxindole arene and vinyl ketene p-tolyl group.27b,27c
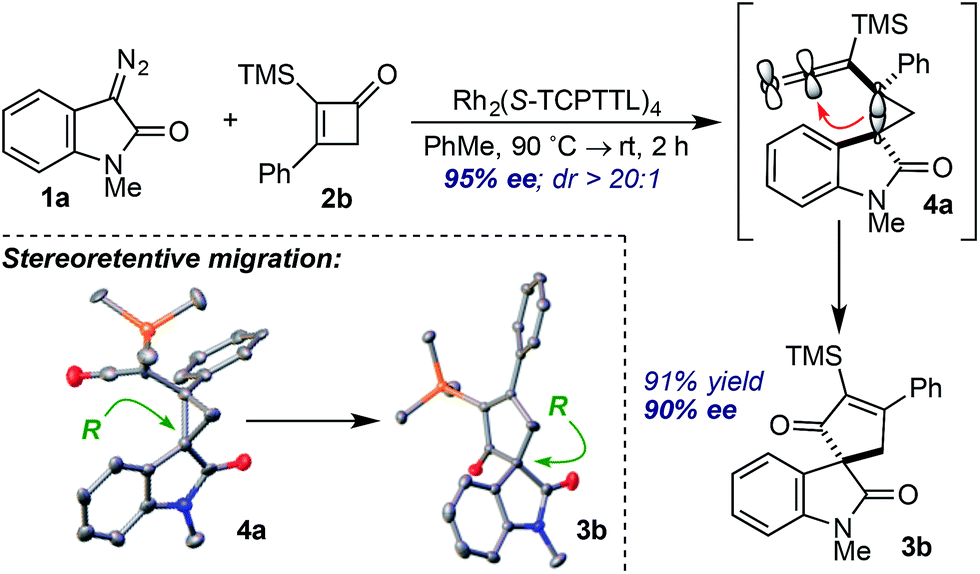 | ||
| Scheme 1 Stereochemical Progression. | ||
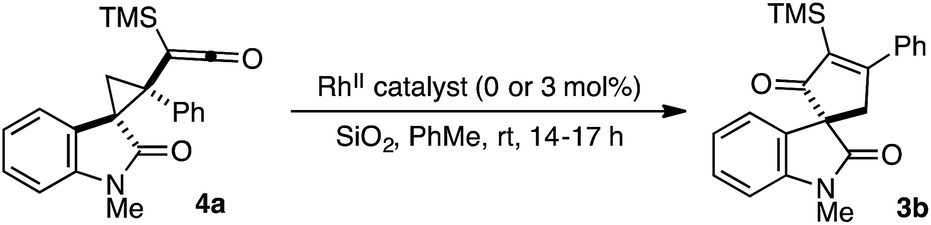
To determine the catalyst influence on the conversion of intermediate 4 to cycloadduct 3, we conducted a series of control experiments to monitor the formation of 3b from isolated cyclopropyl ketene 4a (Table 2). Exposure of racemic 4a to Rh2(TCPTTL)4 under our optimized conditions led to a quantitative yield of racemic 3b (entry 1). Treatment of optically enriched 4a (94% ee) with either Rh2(OAc)4 or no RhII catalyst resulted in comparable yield of 3b with modest loss of optical purity (entries 2 and 3). These results would indicate that the catalyst is not influencing the stereochemical outcome of the cyclopropyl ketene rearrangement.
|
|
||||
|---|---|---|---|---|
| Entry | % ee of 4a | Catalyst | Yield (%) | % ee of 3b |
| 1 | 0 | Rh2(S-TCPTTL)4 | >99 | 0 |
| 2 | 94 | Rh2(OAc)4 | >99 | 84 |
| 3 | 94 | No catalyst | >99 | 86 |
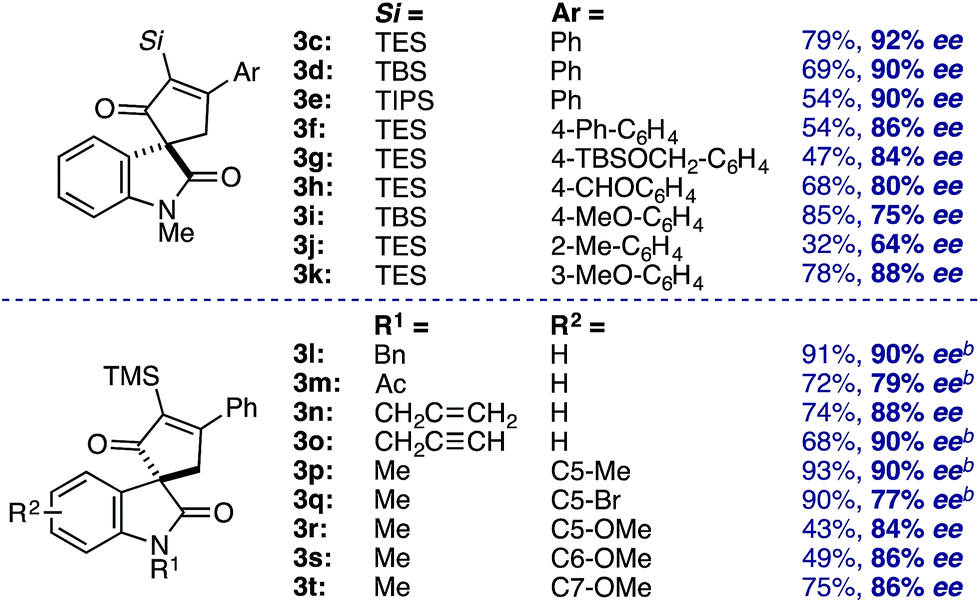
Evaluation of the structural diversity across diazooxindole 1 and cyclobutenone 2 in the Rh2(TCPTTL)4-catalyzed [4+1]-cycloaddition provided the corresponding enantioselectivities (Table 3). While variations of the α-silyl group on 2 did not significantly affect selectivity, yields decreased with increasing size of the vinyl silane (3c–e). Additionally, ortho-substituents on the phenyl ring resulted in a decrease in both yield and enantioselectivity (3j). Various N-alkyl, acyl, benzyl, allyl, and propargyl diazooxindoles gave the corresponding cyclopentenones 3l–p in 79–90% ee. Notably, N-allyl cyclopropanation in 3n was not observed. Oxindole arene substitution did not adversely affect the cycloaddition, resulting in stereoselective quaternary carbon assembly in 77–90% ee (3q–u). It is worth noting that, modest improvements in selectivity were observed by performing the reaction at 4 °C over 48 h for cycloadducts 3m, 3n, and 3p–r. Subsequent recrystallization of 3b led to further optical enrichment (≥98% ee), and the absolute stereochemistry of all substrates was assigned by analogy.
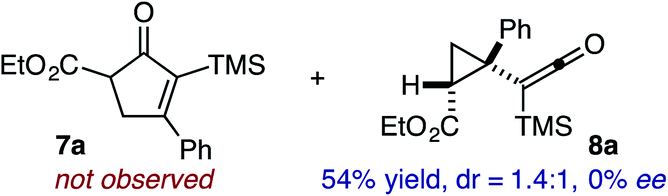
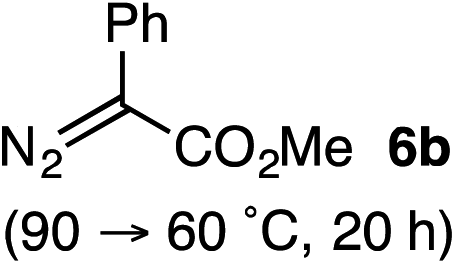
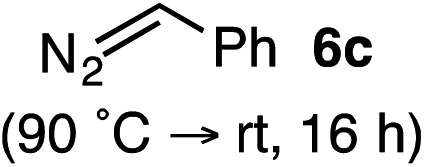
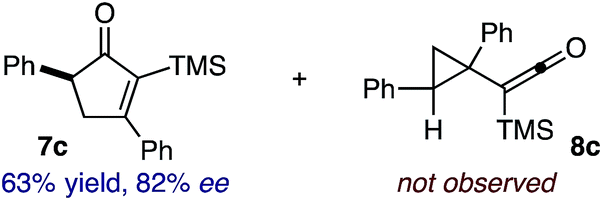
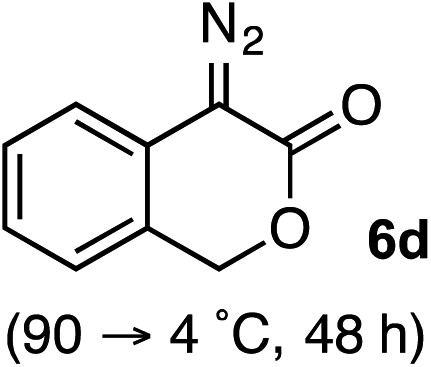
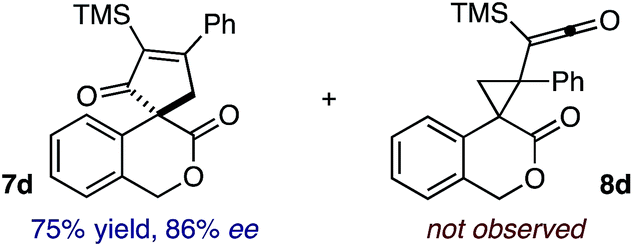
Based on the crown configuration of Rh2(TCPTTL)4, Charette and co-workers proposed a π–π stacking interaction between the carbene donor aryl ring and the pthalimido groups of the catalyst wall as key to the observe stereoinduction in cyclopropanations.26 Speculating that a non-aryl diazo compound would result in reduced enantioinduction, we subjected ethyl diazoacetate (6a) to our standard conditions, but failed to observe formation of cyclopentenone 7a even after prolonged reaction times (Table 4). The stalled migration led to cyclopropane 8a in a 1.4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of diastereomers and 0% ee. However, employing phenyl diazoester 6b resulted in a 1:2.3 ratio of cycloadduct 7b and ketene 8b after 20 h at 60 °C. While adduct 7b was obtained in 80% ee, we isolated cyclopropane 8b as a single diastereomer in 56% ee. Curiously, neither prolonged reaction times (≥48 h) nor the addition of other Lewis acids (i.e., MgI2) improved the conversion of either 8a or 8b to the corresponding cyclopentenones. Although elevated temperatures failed to provide cycloadduct 7a or improve the yield of cyclopropane 8a from diazoester 6a, conducting the formal [4+1]-cycloaddition of 6b above 60 °C led to diminished levels of optical enrichment for 7b while not affecting the amount of each product obtained. In contrast, phenyl diazomethane (6c) underwent rapid conversion to cycloadduct 7c in 63% yield and 82% ee from presumptive cyclopropyl ketene 8c. Likewise, diazochromanone 6d yielded spirocycle 7d in 75% yield and 86% ee and no cyclopropane 8d. These composite results would indicate that the presence of an α-aryl substituent is key to enantioselectivity and enabling a facile cyclopropyl ketene rearrangement en route to the formal [4+1]-cycloadduct.28
:1 ratio of diastereomers and 0% ee. However, employing phenyl diazoester 6b resulted in a 1:2.3 ratio of cycloadduct 7b and ketene 8b after 20 h at 60 °C. While adduct 7b was obtained in 80% ee, we isolated cyclopropane 8b as a single diastereomer in 56% ee. Curiously, neither prolonged reaction times (≥48 h) nor the addition of other Lewis acids (i.e., MgI2) improved the conversion of either 8a or 8b to the corresponding cyclopentenones. Although elevated temperatures failed to provide cycloadduct 7a or improve the yield of cyclopropane 8a from diazoester 6a, conducting the formal [4+1]-cycloaddition of 6b above 60 °C led to diminished levels of optical enrichment for 7b while not affecting the amount of each product obtained. In contrast, phenyl diazomethane (6c) underwent rapid conversion to cycloadduct 7c in 63% yield and 82% ee from presumptive cyclopropyl ketene 8c. Likewise, diazochromanone 6d yielded spirocycle 7d in 75% yield and 86% ee and no cyclopropane 8d. These composite results would indicate that the presence of an α-aryl substituent is key to enantioselectivity and enabling a facile cyclopropyl ketene rearrangement en route to the formal [4+1]-cycloadduct.28
|
|
|
|---|---|
| Diazo 6 | Distribution of 3- vs. 5-membered cycloadducts |
| a Conditions: see ESI for detailed experimental procedures. | |
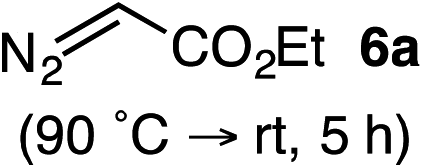
|
|
|

|
|
|
|
|
Subsequent efforts toward functionalizing the spirooxindole cycloadducts focused on evaluating the stability of the quaternary spirocenter. Catalytic hydrogenation of optically enriched spirooxindole 3b (>99% ee via recrystallization) provided spirocyclopentane 9 in 85%, 6:1 diastereoselectivity favoring catalyst approach from the C2-oxindole face, and >99% ee (Scheme 2). Curiously, protodesilylation of 3b at room temperature using TBAF proceeded in quantitative yield, but gave enone 10 in 72% ee. However, conducting the reaction at −20 °C effectively removed the α-TMS group to provide enone 10 in comparable yield and without loss of optical purity. While stereochemical stability of the C3-quaternary center is a reasonable expectation, the combination of our results and those representative examples in the literature would indicate that the 1,3-dicarbonyl arrangement of the spirocycle in 3 can lead to α-epimerization under selected conditions29,30
 | ||
| Scheme 2 Stereochemical progression. | ||
Conclusions
In summary, we have developed an asymmetric RhII-catalyzed formal [4+1]-cycloaddition between diazo compounds and in situ generated vinyl ketenes that leads to the direct stereoselective assembly of quaternary stereogenic carbons. The reaction produces cyclopentenones in good to excellent yields with up to 92% ee. A detailed mechanistic study of the cyclopropyl ketene rearrangement, origin of the observed chirality transfer, and applications toward target-directed total synthesis are currently under investigation and will be reported in due course.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Foundation (CHE 1056242 and 1665440) and Walther Cancer Foundation Advancing Basic Cancer Research Program. K. X. R. was supported by a Walther Cancer Foundation ENSCCII Training Grant. We thank Dr Allen G. Oliver (University of Notre Dame) for assistance with the X-ray crystallography, and are grateful to a referee for suggesting that we perform the protodesilylation of 3b at lower temperatures. This paper is dedicated to Professor Stephen F. Martin in recognition for his receipt of the 2017 Ernest Guenther Award in the Chemistry of Natural Products.Notes and references
- (a) T. Kaur, P. Wadhwa, S. Bagchi and A. Sharma, Chem. Commun., 2016, 52, 6958–6976 RSC; (b) J.-R. Chen, X.-Q. Hu, L.-Q. Lu and W.-J. Xiao, Chem. Rev., 2015, 115, 5301–5365 CrossRef CAS PubMed; (c) S. Kramer and G. C. Fu, J. Am. Chem. Soc., 2015, 137, 3803–3806 CrossRef CAS PubMed; (d) Z.-Y. Cao, Y.-H. Wang, X.-P. Zeng and J. Zhou, Tetrahedron Lett., 2014, 55, 2571–2584 CrossRef CAS.
- S. Son and G. C. Fu, J. Am. Chem. Soc., 2007, 129, 1046–1047 CrossRef CAS PubMed.
- F.-L. Hu, Y. Wei and M. Shi, Chem. Commun., 2014, 50, 8912–8914 RSC.
- (a) Q.-Q. Yang and W.-J. Xiao, Eur. J. Org. Chem., 2017, 2017, 233–236 CrossRef CAS; (b) X.-L. Lian, A. Adili, B. Liu, Z.-L. Tao and Z.-Y. Han, Org. Biomol. Chem., 2017, 15, 3670–3673 RSC; (c) X.-L. Jiang, S.-J. Liu, Y.-Q. Gu, G.-J. Mei and F. Shi, Adv. Synth. Catal., 2017, 359, 3341–3346 CrossRef CAS.
- P.-F. Zheng, Q. Ouyang, S.-L. Niu, L. Shuai, Y. Yuan, K. Jiang, T.-Y. Liu and Y.-C. Chen, J. Am. Chem. Soc., 2015, 137, 9390–9399 CrossRef CAS PubMed.
- (a) N. R. O'Connor, J. L. Wood and B. M. Stoltz, Isr. J. Chem., 2016, 56, 431–444 CrossRef; (b) Y. Liu, S.-J. Han, W.-B. Liu and B. M. Stoltz, Acc. Chem. Res., 2015, 48, 740–751 CrossRef CAS PubMed; (c) X. Han, W. Yao, T. Wang, Y. R. Tan, Z. Yan, J. Kwiatkowski and Y. Lu, Angew. Chem., Int. Ed., 2014, 53, 5643–5647 CrossRef CAS PubMed.
- (a) T. Hudlicky and J. W. Reed, Angew. Chem., Int. Ed., 2010, 49, 4864–4876 CrossRef CAS PubMed; (b) E. J. Corey and A. G. Myers, J. Am. Chem. Soc., 1985, 107, 5574–5576 CrossRef CAS; (c) B. M. Trost and M. J. Bogdanowicz, J. Am. Chem. Soc., 1973, 95, 5311–5321 CrossRef CAS.
- (a) R. W. Coscia and T. H. Lambert, J. Am. Chem. Soc., 2009, 131, 2496–2498 CrossRef CAS PubMed; (b) T. Hudlicky, N. E. Heard and A. Fleming, J. Org. Chem., 1990, 55, 2570–2572 CrossRef CAS; (c) R. L. Danheiser, J. J. Bronson and K. Okano, J. Am. Chem. Soc., 1985, 107, 4579–4581 CrossRef CAS; (d) R. P. Short, J. M. Revol, B. C. Ranu and T. Hudlicky, J. Org. Chem., 1983, 48, 4453–4461 CrossRef CAS; (e) R. L. Danheiser, C. Martinez-Davila, R. J. Auchus and J. T. Kadonaga, J. Am. Chem. Soc., 1981, 103, 2443–2446 CrossRef CAS; (f) T. Hudlicky, F. F. Koszyk, T. M. Kutchan and J. P. Sheth, J. Org. Chem., 1980, 45, 5020–5027 CrossRef CAS; (g) T. Hudlicky, T. M. Kutchan, S. R. Wilson and D. T. Mao, J. Am. Chem. Soc., 1980, 102, 6351–6353 CrossRef CAS.
- (a) K. X. Rodriguez, N. Kaltwasser, T. A. Toni and B. L. Ashfeld, Org. Lett., 2017, 19, 2482–2485 CrossRef CAS PubMed; (b) J. L. Meloche and B. L. Ashfeld, Angew. Chem., Int. Ed., 2017, 56, 6604–6608 CrossRef CAS PubMed.
- (a) A. D. Allen and T. T. Tidwell, Chem. Rev., 2013, 113, 7287–7342 CrossRef CAS PubMed; (b) L. Gong, M. A. McAllister and T. T. Tidwell, J. Am. Chem. Soc., 1991, 113, 6021–6028 CrossRef CAS.
- (a) E. V. Mercado-Marin, P. Garcia-Reynaga, S. Romminger, E. F. Pimenta, D. K. Romney, M. W. Lodewyk, D. E. Williams, R. J. Andersen, S. J. Miller, D. J. Tantillo, R. G. S. Berlinck and R. Sarpong, Nature, 2014, 509, 318–324 CrossRef CAS PubMed; (b) A. D. Huters, E. D. Styduhar and N. K. Garg, Angew. Chem., Int. Ed., 2012, 51, 3758–3765 CrossRef CAS PubMed; (c) K. A. Miller and R. M. Williams, Chem. Soc. Rev., 2009, 38, 3160–3174 RSC; (d) A. C. Whyte, J. B. Gloer, D. T. Wicklow and P. F. Dowd, J. Nat. Prod., 1996, 59, 1093–1095 CrossRef CAS PubMed; (e) R. Güller and H.-J. Borschberg, Helv. Chim. Acta, 1993, 76, 1847–1862 CrossRef.
- (a) S. E. Reisman, J. M. Ready, A. Hasuoka, C. J. Smith and J. L. Wood, J. Am. Chem. Soc., 2006, 128, 1448–1449 CrossRef CAS PubMed; (b) R. M. Williams, J. Cao, H. Tsujishima and R. J. Cox, J. Am. Chem. Soc., 2003, 125, 12172–12178 CrossRef CAS PubMed.
- (a) B. M. Trost, D. A. Bringley, T. Zhang and N. Cramer, J. Am. Chem. Soc., 2013, 135, 16720–16735 CrossRef CAS PubMed; (b) B. M. Trost, N. Cramer and S. M. Silverman, J. Am. Chem. Soc., 2007, 129, 12396–12397 CrossRef CAS PubMed.
- (a) R. Sarpong, J. T. Su and B. M. Stoltz, J. Am. Chem. Soc., 2003, 125, 13624–13625 CrossRef CAS PubMed; (b) J. E. Baldwin, Chem. Rev., 2003, 103, 1197–1212 CrossRef CAS PubMed; (c) T. Hudlicky and M. Natchus, J. Org. Chem., 1992, 57, 4740–4746 CrossRef CAS.
- (a) K. M. Chepiga, C. Qin, J. S. Alford, S. Chennamadhavuni, T. M. Gregg, J. P. Olson and H. M. L. Davies, Tetrahedron, 2013, 69, 5765–5771 CrossRef CAS PubMed; (b) J. Hansen and H. M. L. Davies, Coord. Chem. Rev., 2008, 252, 545–555 CrossRef CAS PubMed; (c) H. M. L. Davies and E. G. Antoulinakis, in Organic Reactions, John Wiley & Sons, Inc., 2004, DOI:10.1002/0471264180.or057.01; (d) H. Lebel, J.-F. Marcoux, C. Molinaro and A. B. Charette, Chem. Rev., 2003, 103, 977–1050 CrossRef CAS PubMed.
- (a) Z.-Y. Cao, X. Wang, C. Tan, X.-L. Zhao, J. Zhou and K. Ding, J. Am. Chem. Soc., 2013, 135, 8197–8200 CrossRef CAS PubMed; (b) Z.-Y. Cao, F. Zhou, Y.-H. Yu and J. Zhou, Org. Lett., 2013, 15, 42–45 CrossRef CAS PubMed; (c) A. Awata and T. Arai, Synlett, 2013, 24, 29–32 CAS.
- (a) C. Werlé, R. Goddard, P. Philipps, C. Farès and A. Fürstner, J. Am. Chem. Soc., 2016, 138, 3797–3805 CrossRef PubMed; (b) C. Werlé, R. Goddard and A. Fürstner, Angew. Chem., Int. Ed., 2015, 54, 15452–15456 Search PubMed; (c) H. Wang, D. M. Guptill, A. Varela-Alvarez, D. G. Musaev and H. M. L. Davies, Chem. Sci., 2013, 4, 2844–2850 RSC; (d) H. M. L. Davies and D. Morton, Chem. Soc. Rev., 2011, 40, 1857–1869 RSC; (e) H. M. L. Davies and J. R. Manning, Nature, 2008, 451, 417–424 CrossRef CAS PubMed; (f) M. P. Doyle and D. C. Forbes, Chem. Rev., 1998, 98, 911–936 CrossRef CAS PubMed.
- (a) K. Benda, T. Knoth, R. L. Danheiser and E. Schaumann, Tetrahedron Lett., 2011, 52, 46–48 CrossRef CAS PubMed; (b) W. F. Austin, Y. Zhang and R. L. Danheiser, Org. Lett., 2005, 7, 3905–3908 CrossRef CAS PubMed; (c) R. L. Danheiser and H. Sard, J. Org. Chem., 1980, 45, 4810–4812 CrossRef CAS; (d) J. H. Rigby and Z. Wang, Org. Lett., 2003, 5, 263–264 CrossRef CAS PubMed; (e) C. P. Davie and R. L. Danheiser, Angew. Chem., Int. Ed., 2005, 44, 5867–5870 CrossRef CAS PubMed; (f) J. L. Loebach, D. M. Bennett and R. L. Danheiser, J. Org. Chem., 1998, 63, 8380–8389 CrossRef CAS; (g) J. L. Loebach, D. M. Bennett and R. L. Danheiser, J. Am. Chem. Soc., 1998, 120, 9690–9691 CrossRef CAS.
- (a) P.-h. Chen and G. Dong, Chem.–Eur. J., 2016, 22, 18290–18315 CrossRef CAS PubMed; (b) P.-h. Chen, J. Sieber, C. H. Senanayake and G. Dong, Chem. Sci., 2015, 6, 5440–5445 RSC.
- J. R. Denton and H. M. L. Davies, Org. Lett., 2009, 11, 787–790 CrossRef CAS PubMed.
- (a) V. N. G. Lindsay, D. Fiset, P. J. Gritsch, S. Azzi and A. B. Charette, J. Am. Chem. Soc., 2013, 135, 1463–1470 CrossRef CAS PubMed; (b) C. Qin, V. Boyarskikh, J. H. Hansen, K. I. Hardcastle, D. G. Musaev and H. M. L. Davies, J. Am. Chem. Soc., 2011, 133, 19198–19204 CrossRef CAS PubMed.
- M. Yamawaki, H. Tsutsui, S. Kitagaki, M. Anada and S. Hashimoto, Tetrahedron Lett., 2002, 43, 9561–9564 CrossRef CAS.
- (a) A. Ghanem, M. G. Gardiner, R. M. Williamson and P. Müller, Chem.–Eur. J., 2010, 16, 3291–3295 CrossRef CAS PubMed; (b) D. Marcoux and A. B. Charette, Angew. Chem., Int. Ed., 2008, 47, 10155–10158 CrossRef CAS PubMed; (c) P. Müller, Y. Allenbach and E. Robert, Tetrahedron: Asymmetry, 2003, 14, 779–785 CrossRef.
- (a) A. Lerchner and E. M. Carreira, Chem.–Eur. J., 2006, 12, 8208–8219 CrossRef CAS PubMed; (b) C. Marti and E. M. Carreira, J. Am. Chem. Soc., 2005, 127, 11505–11515 CrossRef CAS PubMed.
- See the ESI† for full experimental details.
- V. N. G. Lindsay, W. Lin and A. B. Charette, J. Am. Chem. Soc., 2009, 131, 16383–16385 CrossRef CAS PubMed.
- (a) D. T. Boruta, O. Dmitrenko, G. P. A. Yap and J. M. Fox, Chem. Sci., 2012, 3, 1589–1593 RSC; (b) D. T. Nowlan, T. M. Gregg, H. M. L. Davies and D. A. Singleton, J. Am. Chem. Soc., 2003, 125, 15902–15911 CrossRef CAS PubMed; (c) M. P. Doyle, V. Bagheri, T. J. Wandless, N. K. Harn, D. A. Brinker, C. T. Eagle and K. L. Loh, J. Am. Chem. Soc., 1990, 112, 1906–1912 CrossRef CAS.
- V. N. G. Lindsay, C. Nicolas and A. B. Charette, J. Am. Chem. Soc., 2011, 133, 8972–8981 CrossRef CAS PubMed.
- (a) M. S. Morales-Ríos, D. E. González-Juárez, E. Rivera-Becerril, O. R. Suárez-Castillo and P. Joseph-Nathan, Tetrahedron, 2007, 63, 7702–7707 CrossRef; (b) Y. Zhang and J. S. Panek, Org. Lett., 2009, 11, 3366–3369 CrossRef CAS PubMed.
- For representative examples of spirocyclopentyl oxindoles undergoing epimerization at C3, see: (a) A. Madin, C. J. O'Donnell, T. Oh, D. W. Old, L. E. Overman and M. J. Sharp, J. Am. Chem. Soc., 2005, 127, 18054–18065 CrossRef CAS PubMed; (b) S. Atarashi, J.-K. Choi, D.-C. Ha, D. J. Hart, D. Kuzmich, C.-S. Lee, S. Ramesh and S. C. Wu, J. Am. Chem. Soc., 1997, 119, 6226–6241 CrossRef CAS; (c) D. Kuzmich, S. C. Wu, D.-C. Ha, C.-S. Lee, S. Ramesh, S. Atarashi, J.-K. Choi and D. J. Hart, J. Am. Chem. Soc., 1994, 116, 6943–6944 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1557298 and 1557297. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc00020d |
| This journal is © The Royal Society of Chemistry 2018 |