
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructure and formation of highly luminescent protein-stabilized gold clusters†
D. M.
Chevrier
 a,
V. D.
Thanthirige‡
b,
Z.
Luo‡
c,
S.
Driscoll‡
a,
P.
Cho
a,
M. A.
MacDonald
a,
Q.
Yao
c,
R.
Guda
b,
J.
Xie
c,
E. R.
Johnson
a,
A.
Chatt
a,
N.
Zheng
d and
P.
Zhang
*a
a,
V. D.
Thanthirige‡
b,
Z.
Luo‡
c,
S.
Driscoll‡
a,
P.
Cho
a,
M. A.
MacDonald
a,
Q.
Yao
c,
R.
Guda
b,
J.
Xie
c,
E. R.
Johnson
a,
A.
Chatt
a,
N.
Zheng
d and
P.
Zhang
*a
aDepartment of Chemistry, Dalhousie University, 6274 Coburg Road, Halifax, NS B3H4J3, Canada. E-mail: peng.zhang@dal.ca
bDepartment of Chemistry, Western Michigan University, Kalamazoo, MI49008, USA
cDepartment of Chemical and Biomolecular Engineering, National University of Singapore, 119260, Singapore
dState Key Laboratory for Physical Chemistry of Solid Surfaces, Collaborative Innovation Center of Chemistry for Energy Materials, Engineering Research Center for Nano-Preparation Technology of Fujian Province, National Engineering Laboratory for Green Chemical Productions of Alcohols-Ethers-Esters, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen, China
First published on 5th February 2018
Abstract
Highly luminescent gold clusters simultaneously synthesized and stabilized by protein molecules represent a remarkable category of nanoscale materials with promising applications in bionanotechnology as sensors. Nevertheless, the atomic structure and luminescence mechanism of these gold clusters are still unknown after several years of developments. Herein, we report findings on the structure, luminescence and biomolecular self-assembly of gold clusters stabilized by the large globular protein, bovine serum albumin. We highlight the surprising identification of interlocked gold-thiolate rings as the main gold structural unit. Importantly, such gold clusters are in a rigidified state within the protein scaffold, offering an explanation for their highly luminescent character. Combined free-standing cluster synthesis (without protecting protein scaffold) with rigidifying and un-rigidifying experiments, were designed to further verify the luminescence mechanism and gold atomic structure within the protein. Finally, the biomolecular self-assembly process of the protein-stabilized gold clusters was elucidated by time-dependent X-ray absorption spectroscopy measurements and density functional theory calculations.
Introduction
Composed of only tens or hundreds of Au atoms, Au clusters or nanoclusters (NCs) are highly stable 1–2 nm diameter particles with distinctive core/surface structures and molecule-like optical properties.1–4 Due to the high surface area and quantum confinement effects of Au NCs, stabilizing ligands often play an important role in directing their structure and properties. Besides small thiol molecules that are commonly used as protecting ligands for Au NCs, large and relatively more complex ligand types such as proteins and other biomolecules have been implemented to stabilize various sizes of Au particles, which further improves their integration into biological- and medical-related applications.5,6 Concurrently with the development of these new bionanomaterials, the biomolecular self-assembly process that controls the formation of Au nanostructures has intrigued researchers for years.7–9Protein-stabilized Au NCs are a unique class of bionanomaterials with intense luminescence and highly specific chemical recognition properties suitable for biological imaging and chemical sensing applications.10–16 Conveniently, and rather remarkably, protein molecules can act as both the stabilizing ligand and the structure-directing agent to facilitate the formation of ultra-small Au clusters. This was successfully shown where highly luminescent Au NCs (quantum yield (QY) = ∼6%, over 108 times higher than bulk gold) were stabilized by bovine serum albumin (BSA) using a facile one-pot, protein-directed synthesis.17 This approach reduces the number of steps in Au NC synthesis and avoids the use of harsh chemical reagents, making it greener than general Au NC syntheses.
Despite the promising luminescence property of BSA-stabilized and other protein-stabilized Au NCs,18,19 little is known about the biomolecular self-assembly and atomic structure of Au NCs inside the protein. Complicating this unresolved puzzle are the reported inconsistencies on the relative Au(0)/Au(I) composition measured from X-ray photoelectron spectroscopy (XPS) and the disparity between AunBSA compositions proposed from mass spectrometry (MS).13,20 To further complicate the problem, Au structure inside the BSA protein has been hypothesized to be Au25(SR)18 NCs21,22 even though they do not share similar photoluminescence or optical properties.
Identifying the atomic-level structure and self-assembled formation of Au NCs in the protein molecule would help determine the origin of protein-stabilized Au NC luminescence and lead to better-guided developments in sensing and imaging technologies. Experimental and investigative techniques beyond standard laboratory methods (e.g., FT-IR, XPS, MS) were applied in order to reveal more informative and reliable details on Au NC structure and properties. Synchrotron-based X-ray absorption fine structure (XAFS) spectroscopy was critical in this regard, as it provided the ability to resolve interactions between few-atom Au clusters and stabilizing protein residues from an element-specific perspective. In conjunction with other experimental techniques and in-depth analyses, the atomic structure of Au in BSA was surprisingly found to resemble interlocked gold-thiolate (Au-SR) ring structures, which slowly develop over the course of the protein-directed synthesis. Additional experiments and quantum calculations are presented that consistently support the atomic structure of Au in BSA. The distinctive red luminescence property, as it relates to the newly identified structure, is also examined.
Results and discussion
Structure of protein-stabilized gold clusters
The synthesis of red luminescent BSA-stabilized Au clusters (AuBSA) is schematically shown in Fig. 1. This facile, protein-directed protocol employs amino acid residues of the protein to interact and control the formation of Au clusters. In alkaline solution conditions and with the right concentration of BSA and Au(III) precursors, red luminescent Au clusters are formed (Fig. S1†).17 Our initial investigation utilized extended-XAFS (EXAFS) to determine whether Au structures in AuBSA are related to the core/shell structures of atomically precise NCs, such as Au25(SR)18, which reports on protein-stabilized Au NCs have suggested.13,20 The extended region of the Au XAFS spectrum provides local structural information for Au atoms stabilized by the protein. The scattering features observed in the Fourier transform of the EXAFS spectrum (FT-EXAFS) can be linked to specific bonding environments, either qualitatively compared with reference materials or quantitatively refined with a fitting analysis. Fig. 2A directly compares the FT-EXAFS of luminescent AuBSA with Au25(SR)18 and Au38(SR)24. The spectral regions corresponding to Au–S and Au–Au bonding are highlighted in blue and orange, respectively. The overlap of each reference FT-EXAFS spectrum with AuBSA clearly shows there is no significant Au core structure, as evidenced by the absence of metallic Au–Au scattering in the 2.5–3.0 Å region. Fitting the Au–S region of AuBSA EXAFS (Table S1, Fig. S2†) yielded a coordination number (CN) of 2.1(1) and a bond length of 2.31(2) Å. Thus, most Au atoms are in RS-Au-SR structures (CN = 2 and SR – cysteine from BSA) with no Au core structure, meaning an oxidation state of Au(I) is expected for Au atoms in AuBSA. This is substantiated by inspection of the X-ray absorption near-edge structure (XANES) region of AuBSA with Au(I) and Au(0) reference materials (Fig. S3†). From this initial result, the AuBSA system appears to be better classified as a Au(I)-SR cluster or complex rather than a Au NC with core/shell architecture.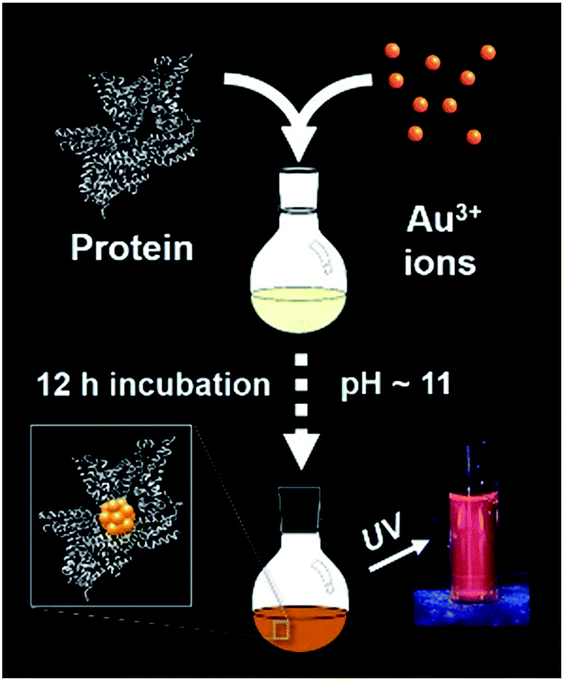 | ||
| Fig. 1 One-pot protocol for the synthesis of protein (bovine serum albumin)-stabilized luminescent Au clusters. | ||
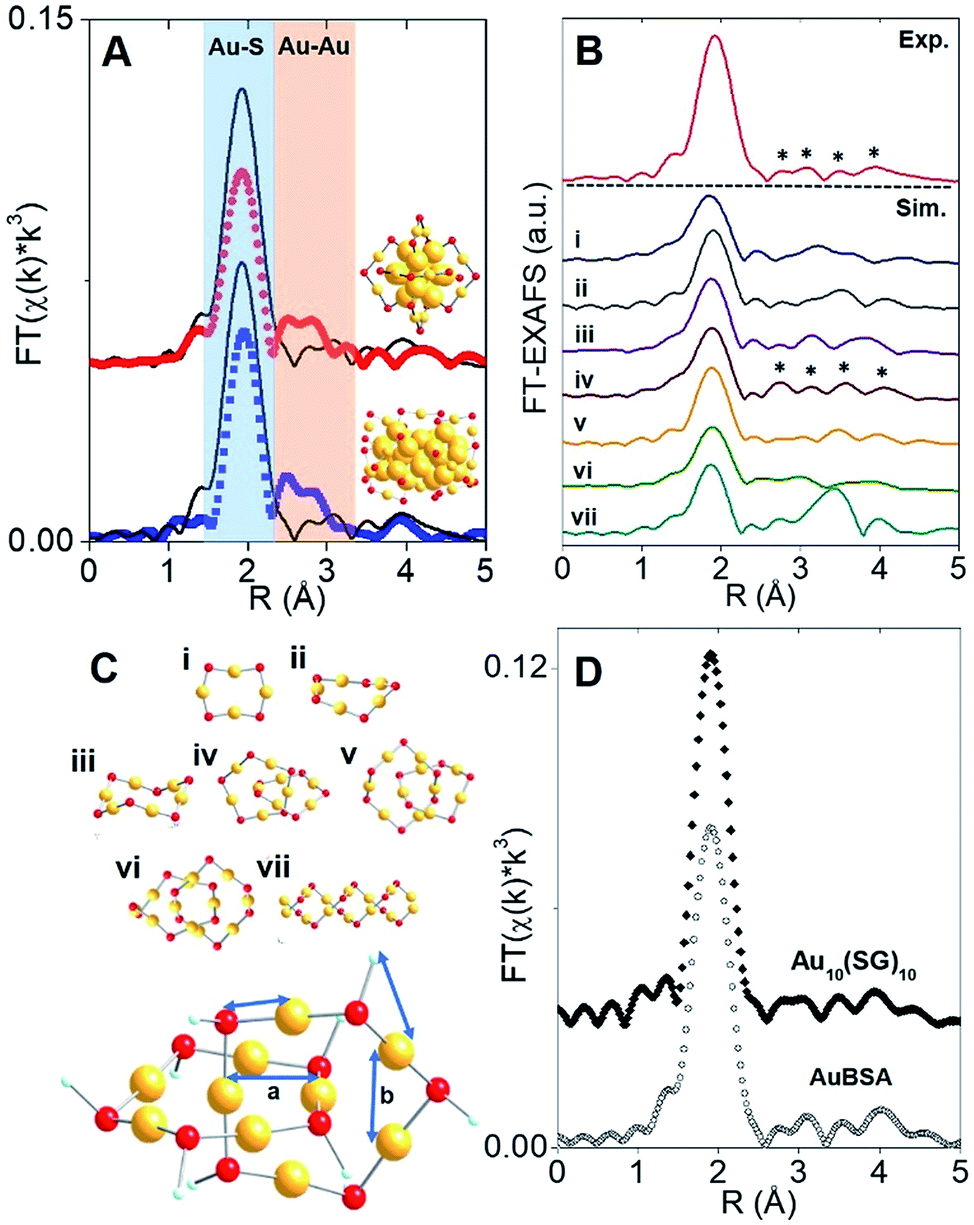 | ||
| Fig. 2 Structural identification of AuBSA clusters. (A) Au L3-edge FT-EXAFS of luminescent AuBSA (black line) with Au25(SR)18 (red dot) and Au38(SR)24 (blue dot) (inset, respective models with Au (yellow) and S (red) atoms). (B) Simulated Au L3-edge FT-EXAFS of Au(I)-SR structures (i) Au4S4 ring, (ii) Au5S5 ring, (iii) Au6S6 ring, (iv) Au10S10 catenane, (v) Au11S11 catenane, (vi) Au12S12 catenane and (vii) Au–S double helix (methyl substituents were omitted from all structures for clarity). (C) Models of Au(I)-SR structures and 4 representative EXAFS scattering paths from the Au10(SR)10 catenane model used for fitting (a – inter-ring and b – intra-ring). (D) Au L3-edge FT-EXAFS of Au10(SG)10 (SG – glutathione) and AuBSA. | ||
Although Au–Au scattering representing Au core structures was not found in the EXAFS fitting of AuBSA, longer-range Au–Au scattering related to aurophilic interactions (i.e., Au(I)–Au(I) interactions of longer distance than a metallic bond) was identified. EXAFS fitting yielded a CN parameter of 0.8(5) and a distance of 3.02(3) Å (Fig. S2, Table S1†) for these Au(I)–Au(I) interactions. This bond distance is indeed much longer than bulk Au–Au bonding (∼2.88 Å) and Au–Au bonding from atomically precise Au NC core structures (∼2.78–2.82 Å) (e.g., Au25(SR)18).23,24 This part of our structural study suggested that there are inter-molecular aurophilic interactions between SR-Au(I)-SR structures inside the BSA protein.
In search of a structural model to obtain more quantitative information from EXAFS fitting (additional scattering paths and corresponding structural parameters), a pre-screening step was implemented by comparing the experimental FT-EXAFS of AuBSA with simulated spectra of all the available Au(I)-SR cluster references, shown in Fig. 2B.25–30 Such references are distinct Au(I)-SR structures known from X-ray crystallography or predicted from theory, and are presented in Fig. 2C (rings (Au4(SR)4, Au5(SR)5, Au6(SR)6), interlocked rings or catenanes (Au10(SR)10, Au11(SR)11, Au12(SR)12) and polymer ([Au(SR)]x)). Screening the simulated EXAFS spectra of several different Au(I)-SR clusters allowed us to closely compare the positions of scattering features that follow the dominant Au–S peak to narrow down the specific conformation of Au(I)-SR structures in AuBSA. The Au10(SR)10 catenane structure (two interlocked Au5(SR)5 rings) had the closest resemblance with 4 similarly positioned scattering features following the Au–S peak. Importantly, the inter-ring Au–Au distance from the crystallographic Au10(SC6H4C(CH3)3)10 structure26 averages 3.05 Å and is the most similar to the aurophilic interaction distance found in AuBSA clusters (3.02 Å), whereas Au11(SR)11 and Au12(SR)12 catenanes have significantly longer average distances of 3.28 and 3.35 Å, respectively.26,30
After identifying the Au10(SR)10 catenane as the best structural match to the Au local structure in AuBSA clusters, EXAFS scattering paths that specifically describe the Au10(SR)10 model (Fig. 2C) were used to fit AuBSA with more local structural detail. In addition to Au–S and intermolecular aurophilic interactions, Au–C scattering (between Au and C adjacent to S) and longer Au–Au scattering (between Au–Au in the same ring) were included. The multi-shell EXAFS fit of these 4 scattering environments for AuBSA is shown in Fig. S4† along with fitting results in Table S2.† Bond distances obtained from the multi-shell EXAFS fit are in line with the average distances from the Au10(SR)10 crystal structure listed in Table 1, reinforcing our structural identification of interlocked Au(I)-SR rings for AuBSA clusters.
We note that this structural finding from EXAFS does not explicitly suggest the composition of AuBSA clusters is Au10BSA, but rather the Au local structural environment is similar to Au10(SR)10 catenanes and not Au25(SR)18 NCs. Other groups have utilized mass spectrometry (MS) to determine that the number of Au atoms to BSA protein appears to range from 9 to 38.17,31–34 Our own MS investigation (MALDI-TOF detection) confirmed that we were obtaining similar results for our own samples, having approximately 20–23 Au atoms per BSA molecule (Fig. S5†). Based on the number of Au atoms per BSA and the dominant Au–S structural environment determined from EXAFS, it is possible that two Au10(SR)10 catenane structures are present in each BSA molecule. The additional 1–3 Au atoms could be attributed to small amount of Au(SR)2 monomers formed in BSA.
Free-standing (i.e., without protein host) Au10(SR)10 stabilized by glutathione (SG) ligands (Au10(SG)10) was examined moving forward to provide additional verification of the interlocked ring structures from an experimental perspective (k-space in Fig. S6 and UV-Vis absorption in Fig. S11†) and for later comparison of luminescence properties. Both AuBSA and Au10(SG)10 have few characteristic absorption features and the comparison is further challenged by the strong absorption from the many amino acid residues in BSA. Thus, the comparison of the Au local structure is more informative at this point. Fig. 2D compares the experimental FT-EXAFS spectrum of Au10(SG)10 with AuBSA, which clearly demonstrated the identical position and pattern of all scattering features previously identified with simulated FT-EXAFS of Au10(SR)10. Detailed EXAFS fitting analysis using the Au10(SR)10 model was performed for Au10(SG)10 and yielded consistent structural parameters to AuBSA (Tables 1 and S2†), reconfirming that interlocked Au(I)-SR ring structures exist in both AuBSA and Au10(SG)10. As a final confirmation, density-functional theory (DFT) was applied35 to optimize the structures of Au5(SR)5 and Au10(SR)10 (where SR is SCH3 for computational efficiency) and the resulting energies support the favourable formation of interlocked ring structures over individual isolated ring structures. Fig. S7† depicts the large stabilization energy of −1.14 eV calculated for interlocking two Au5(SCH3)5 rings to form a Au10(SCH3)10 catenane, offering a valid explanation for the high stability of AuBSA clusters experimentally reported.17 Additionally, the distinct inter-ring and intra-ring Au–Au bonding distributions of optimized Au10(SCH3)10 (Fig. S8 and Table S3† for coordinates) correspond well with the crystal structure and our EXAFS fitting results for AuBSA and Au10(SG)10 (Table 1).
Origin of luminescence
Photoluminescence (PL) reported for Au(I)-SR catenane molecules,30 including Au10(SG)10,36 is weak in comparison to the high emission efficiency of AuBSA clusters. In order to elucidate the origin of the enhanced emission, a property of AuBSA clusters promising for applications in metal-ion sensing and biological imaging, the PL properties were next examined to understand the role of the protecting protein. The Au10(SG)10 system is further utilized to identify physical characteristics that are unique to AuBSA clusters, despite having the same Au cluster structure.AuBSA have PL centered at 650 nm when excited at 365 nm (Fig. S9†). The excitation spectrum shows that two peaks, one at 365 nm and another at 475 nm, are responsible for the observed luminescence. The QY was determined to be 6.8% (photo-physical results are shown in Table S4†). The large stokes shift (285 nm) and long PL decay lifetime (1.065 μs, Table S4†) further suggest the emission from AuBSA clusters is characteristic of phosphorescence, similar to other known Au(I)-SR structures.37,38
Although a virtually-identical Au local structure was determined from our EXAFS analysis, Au10(SG)10 shows a modest difference in emission energy, centered at 620 nm (excitation at 365 nm) instead of 650 nm, but more importantly, a significantly weaker emission intensity (Fig. S9†) with a QY of only 0.48%. The second excitation peak at 475 nm appears as a shoulder instead of a strong peak as seen for AuBSA clusters. This could indicate the larger protecting protein increases the oscillator strength of the electronic transition at this particular energy. The small discrepancy in maximum emission energy between AuBSA and Au10(SG)10 (∼0.09 eV) can be attributed to the difference in ligand type (protein versus tripeptide) having varied electron withdrawing ability.39 If there are inter-cluster interactions between Au(I)-SR structures in AuBSA clusters, this could also further red-shift the emission from orbital splitting.37 With regards to the substantial difference in QY however, the larger encapsulating BSA protein could prevent photo-excited energy loss between Au(I)-SR ring structures and the surroundings. To test the importance of the protecting protein for the enhanced luminescence, the encapsulating protein shell protecting Au(I)-SR clusters was disrupted via enzymatic digestion with trypsin (schematically shown in Fig. 3A). After cleaving BSA molecules with trypsin digestion, the local structure of Au(I)-SR clusters remained mainly intact as shown by Au L3-edge EXAFS (Fig. S10†), but the PL intensity had decreased ∼5 fold (Fig. 3B) and emission maximum red-shifted by 15 nm.
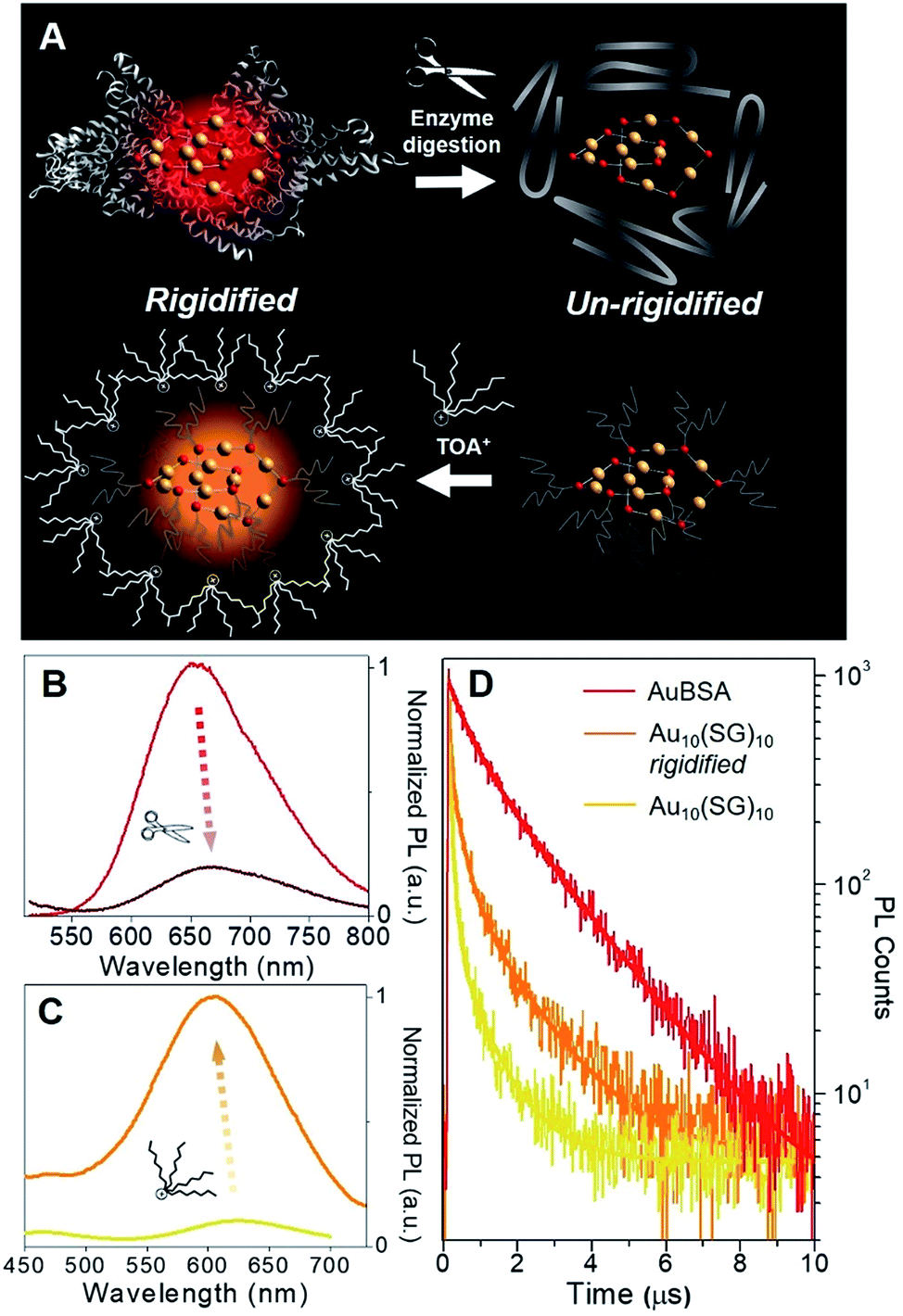 | ||
| Fig. 3 Photoluminescence properties of Au10(SG)10 and AuBSA clusters in rigidified and un-rigidified states. (A) Scheme of un-rigidifying AuBSA clusters with enzyme digestion and rigidifying Au10(SG)10 clusters with TOA+/toluene phase-transfer. (B) Resultant luminescence decrease of AuBSA clusters (un-rigidified, dark red line) and (C) luminescence enhancement of Au10(SG)10 clusters (rigidified, orange line). (D) Photoluminescence decay lifetime traces of AuBSA clusters (red), rigidified Au10(SG)10 clusters (orange) and original Au10(SG)10 clusters (yellow). | ||
The loss of the hierarchical protein structure from enzyme digestion may also affect the structural rigidity of Au(I)-SR clusters, a physical property of thiolate-stabilized Au NCs and other systems that has been recently reported to facilitate efficient emission release.40–42 This is related to protein-stabilized Au clusters since cysteine residues from BSA that bond with Au are fixed to the protein backbone, which could provide the aforementioned structural rigidity of Au(I)-SR clusters within BSA. This would further enable stronger aurophilic interactions between interlocking Au(I)-SR ring structures, inducing ligand to metal–metal charge transfer (LMMCT), an effect known to cause or enhance luminescence for Au(I) oligomers and complexes.38,41,43–45
In order to emulate the emission properties of AuBSA clusters with Au10(SG)10, the protecting and rigidifying environment of the protein was mimicked by performing a water/toluene phase-transfer experiment41,46 with Au10(SG)10 (Fig. S11,† experimental details found in Materials and methods). Tetraoctylammonium (TOA+) ions act as phase-transfer agents, which bring Au10(SG)10 clusters into the organic-phase via electrostatic interaction between ammonium (NR4+) and carboxylic end groups (COO−) of SG ligands. The aliphatic octyl chains from TOA+ protect and isolate Au10(SG)10 clusters from the solvent environment and restrict molecular vibrations or rotations of the SG ligands (Fig. 3A, bottom), thereby creating a “rigidified” state.
The luminescence of rigidified Au10(SG)10 was enhanced by > 10 fold (QY = 5.0%) with a blue-shift of 15 nm (620 nm → 605 nm, see Fig. 3C). The 15 nm blue-shift observed here is in line with the 15 nm red-shift seen above in the opposite direction for AuBSA (Fig. 3B), further verifying a similar rigidifying effect on the Au(I)-SR clusters. The average PL decay lifetime for rigidified Au10(SG)10 had also nearly doubled (from 0.120 to 0.210 μs, Fig. 3D and Table S4†). Together, increased PL lifetime and enhanced QY point to a rigidified state created for Au10(SG)10 clusters. Rigidifying Au10(SG)10 and un-rigidifying Au(I)-SR clusters in BSA demonstrate the similar luminescence property that exists in these two systems, further linking the luminescence properties of small thiolate-stabilized Au NCs with the new category of protein-stabilized Au clusters.
Protein-directed self-assembly of gold clusters
Following the identification of interlocked Au(I)-SR ring structures, the biomolecular self-assembly process guided by BSA could then be further understood by monitoring the protein-directed synthesis with a time-dependent XAFS study. Samples at 0, 1, 2, 3, 6, and 12 h were extracted from the synthesis to capture two distinct stages of the protein-directed synthesis, including an ion-exchange involving the Au(III) precursor. Photoluminescence, Au L3-edge XANES and EXAFS spectra were collected for samples at each time point indicated above and are presented in Fig. 4A, B and 5A, respectively (absorbance and k-space of each sample shown in Fig. S12 and S13,† respectively).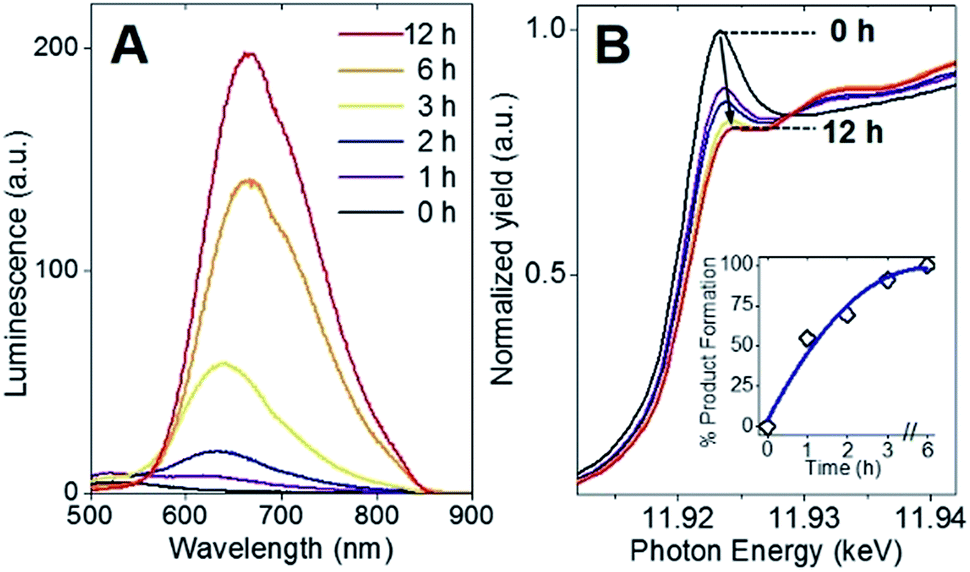 | ||
| Fig. 4 (A) Photoluminescence spectra (excitation λ = 470 nm) and (B) Au L3-edge XANES (inset, white-line integration analysis for Au(III) to Au(I) formation) following the course of the protein-directed assembly of Au clusters. | ||
 | ||
| Fig. 5 (A) Au L3-edge FT-EXAFS with fitted spectra and reference materials. (B) Relative DFT energies of AuCl3 (set to 0 eV), Au(OH)3, and intermediate products of the precursor ion-exchange process. (C) Coordination and Debye–Waller (Au–Au shell only) plot for time-dependent EXAFS fitting results. | ||
Photoluminescence increased steadily over the course of the protein-directed synthesis until 12 h. Even after 36 h, the photoluminescence intensity did not increase further (Fig. S14†). Au L3-edge XANES spectra in Fig. 4B show the white-line intensity (i.e., first feature that follows the absorption edge, and the intensity of this feature reflects the valence electron occupancy, which is mainly the 5d level for Au at the L3-edge) decreased over the course of the synthesis, which signified a reduction of Au(III) precursor to Au(I)-SR clusters. A white-line valence state integration analysis (inset, Fig. 4B) reveals no further reduction of Au(I) after 6 h into the synthesis. S K-edge XANES spectra were also collected for each sample to detect structural changes to the protein through cleavage of disulfide bonds (Fig. S15†). Near-edge features decrease in intensity and change in spectral appearance (relative intensity of two main features at 2472 and 2473 eV) over the course of the protein-directed synthesis. From pristine BSA (mainly disulfide) to time-dependent AuBSA samples, the S K-edge XANES indicate disulfide bonds are disrupted over 12 h to accommodate bonds with Au atoms, resulting in a final XANES spectrum at 12 h that resembles cysteine and Au(I)-SR references.
Using Au–X EXAFS scattering paths (where X is Cl, O, S or Au), quantitative structural parameters were determined by fitting the time-dependent Au L3-edge EXAFS spectra (Fig. 5A and Table S5†). A time plot of Au–X CN parameters is shown in Fig. 5C to convey a two-stage process (0–3 h and 3–12 h). The first scattering path at ∼1.7 Å (spectra not phase corrected) for the 0 h spectrum in Fig. 5A was determined from EXAFS fitting to be Au–O bonding at 2.02(1) Å and surprisingly not Au–Cl bonding from the Au(III) precursor (HAuCl4). A comparison with HAuCl4 (starting material) and Au(OH)3 reference compounds is shown above the 0 h sample in Fig. 5A to further demonstrate the similarity to the latter reference. EXAFS fitting of Au–O scattering for Au(OH)3 gave a bond length of 2.005(4) Å (Fig. S16†), similar to the 0 h sample. DFT calculations support the transformation of gold chloride to gold hydroxide in the presence of excess OH− ions (Tables S6–S8†). The formation energy was calculated for each step of the ion-exchange reaction with each intermediate chlorohydroxoaurate complex, shown in Fig. 5B. It was found that the formation of Au(OH)3 from AuCl3 has a total formation energy of −5.65 eV with the energies of the ion-exchange steps ranging from −1.82 to −1.96 eV. With excess NaOH present, elevated temperatures and vigorous mixing in the protein-directed synthesis of AuBSA clusters, the initial formation of Au(OH)3 is highly favourable.
After 1 h into the synthesis, Au–S bonding appears (feature at ∼2 Å in Fig. 5A) with an EXAFS fitted distance of 2.31(1) Å. At this point in the synthesis, Au atoms begin to form strong covalent bonds with cysteine residues in BSA, which were originally bonded to other cysteine residues via disulfide bonds. The nature of the Au–S bonding does not change from 3 h until the end of the synthesis, with an average bond distance of 2.32(1) Å and a CN of 2.1(1). Notably, appreciable luminescence is first observed around 3 h (Fig. 4A), which is the point where SR-Au(I)-SR structures have mainly formed with no remaining Au–O interactions. The complete ligand-exchange (hydroxide to cysteine) and formation of SR-Au(I)-SR structures from 0–3 h can be regarded as Stage I of the protein-directed synthesis.
Stage II of the protein-directed synthesis is the optimization or final assembly of SR-Au(I)-SR structures. Au–Au interactions were resolvable from EXAFS spectra for samples 3, 6, and 12 h, with a consistent bond distance around 3.02 Å and a CN parameter decreasing from ca. 2 to 1 (Table S5†), which is suggestive of inter-ring Au–Au interactions early on in Stage II. From 3 to 12 h, the Debye–Waller factor (σ2, accounts for thermal and positional disorder of neighbouring atoms in a particular scattering shell) and associated error value decreases for inter-ring aurophilic interactions (plotted in Fig. 5C, right y-axis), indicating that Au(I)-SR clusters are becoming more ordered inside of the protein as incubation and mixing progresses. Since the change in Au local structure is minimal from 3 to 12 h, the increase in luminescence during this stage could be due to the structural optimization of BSA to accommodate the formed Au(I)-SR interlocking ring structures within the protein. No further change in Au local structure was observed even after 36 h of incubation and mixing (Fig. S17 and Table S5†).
Conclusions
This work has elucidated the structure of highly luminescent gold nanoclusters stabilized by bovine serum albumin. Using a multi-step procedure that included screening of gold-thiolate model structures, experimental and simulated X-ray absorption spectroscopy, analogous free-standing gold nanocluster synthesis and rigidifying/un-rigidifying experiments, the structure of the bioinorganic nanoclusters was revealed along with the structural origin of their luminescence property and biomolecular self-assembly mechanism. These findings accentuate the remarkable capability of a protein nano-reactor for producing bioinorganic clusters with intriguing structures and luminescence properties, and should be of importance for future preparation and application of luminescent protein-gold bionanomaterials. The investigative approach demonstrated in this work could also be extended to other biomolecule-metal materials where a large biological system interferes with characterization of the encapsulated metal component.Materials and Methods
Chemicals
HAuCl4·3H2O (Alfa Aesar, 99.99% metal basis), bovine serum albumin (BSA) (Sigma Aldrich, ≥96% purity), NaOH (ACP Chemicals, ≥97.0% purity), tetraoctylammonium bromide (TOABr, 98%), toluene (ACS grade), nanopure water (Barnstead millipore system, 18.2 MΩ cm), dialysis tubing (Fisherbrand, 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–14000 MWCO).
000–14000 MWCO).
Protein-directed synthesis of Au clusters
AuBSA were synthesized following a procedure from Xie et al.17 A 5 mL aqueous solution of bovine serum albumin (50 mg mL−1) was incubated at 37 °C with vigorous mixing. Once BSA dissolved (5 min), a 5 mL aqueous solution of HAuCl4·3H2O (10 mM) was added to the reaction vessel containing BSA. 500 μL of a 1 M NaOH solution was added to the reaction 2 min later, raising the pH of solution to ∼11–12. The reaction proceeded with incubation at 37 °C and stirring for at least 12 h. Nanopure water (18.2 MΩ cm) was used to make up all solutions for the synthesis. For the time-dependent study, samples were taken at 0, 1, 2, 3, 6, 12 and 36 h from the reaction, immediately frozen and then lyophilized for further studies (0 h sample was taken as soon as NaOH was added to the reaction). Luminescence was preserved after the lyophilisation (Fig. S1†). Samples measured for multi-shell EXAFS fitting were dialyzed against nanopure water (18.2 MΩ cm) for 24 h with fresh nanopure water changed every 8 h. For destabilizing AuBSA via enzymatic digestion, 0.5 mL of a freshly prepared 0.1 mg mL−1 trypsin solution was added to 1 mL of the purified AuBSA. This solution was mixed and incubated for 1 day.Au(I)-SC12H25 polymer synthesis (Au(I) reference)
This synthesis closely follows a protocol by Cha et al.47 Briefly, a 0.1 M tetrahydrofuran (THF) solution of n-dodecanethiol was added to a 0.02 M THF solution of HAuCl4·3H2O of equal volume. The mixture was stirred for 1 day and a white precipitate was formed. This was collected as the product, which was washed and dried under vacuum before XAFS measurement.Au10(SG)10 synthesis
In a typical synthesis, GSH (0.1 M, 0.20 mL) was mixed with 4.30 mL of nanopure water, followed by the addition of HAuCl4·3H2O (0.02 M, 0.50 mL) under gentle stirring (500 rpm) at 25 °C for 5 min. A precipitate was formed, which was then dissolved by adjusting pH to ∼7 with NaOH (0.5 M). The solution was aged for 2 h at 40 °C, and the resultant solution of oligomeric Au(I)-thiolate complexes was lyophilized without purification for further characterizations.44Au10(SG)10 rigidification studies
In a typical procedure, 10 mg of Au10(SG)10 dissolved in 10 mL of nanopure water and 10 mg of TOABr dissolved in 5 mL of toluene were added together into a 20 mL scintillation vial. The pH of aqueous solution was then adjusted to pH 9.0 by adding NaOH to ensure carboxyl groups of glutathione were in the anionic form. The electrostatic interaction between the carboxylate anions of the glutathione ligand and the hydrophobic TOA cations in the toluene phase is strong, therefore the TOA+-paired Au10(SG)10 clusters can be readily transferred to the toluene phase (referred to as the “rigidified” state) by stirring the two solutions. The toluene phase was then separated and washed with copious amounts of water to wash away all the water-soluble impurities.Absorption and photoluminescence of AuBSA
UV-Vis and fluorescence spectroscopy measurements for the time-dependent study another supporting measurements besides the rigidifying experiments were collected with the Cary 100 Bio and the Cary Eclipse spectrophotometers, respectively. Solutions were measured using a quartz cuvette sample holder.X-ray absorption fine structure (XAFS)
Au L3-edge XAFS data was collected from the CLS@APS (Sector 20-BM) beamline at the Advanced Photon Source (operating at 7.0 GeV) in Argonne National Labs, Chicago, IL, USA. Each powdered AuBSA sample was measured in transmission mode simultaneously with a gold foil reference at the Au L3-edge (11.919 keV). Au L3-edge EXAFS spectra for Au25(SR)18 and Au38(SR)24 NCs (both were measured at room temperature in powdered form) were borrowed from previous studies.48,49 The amplitude reduction factor used for EXAFS fitting was determined using a Au(I)-SR polymer material. The Au–S coordination number was fixed at 2 to obtain a value of 0.93 which was used for all Au L3-edge EXAFS fitting. All measurements were collected at room temperature and ambient pressure. EXAFS data was transformed and normalized into k- and R-space using the WinXAS program with conventional procedures described in previous work.50 A k-range of 3.0–12.0 Å−1 was used for all FT-EXAFS spectra. Self-consistent multiple-scattering calculations were performed using the FEFF8.2 program to obtain scattering amplitudes and phase-shift functions used for fitting various scattering paths (Au–O, Au–Cl, Au–S, and Au–Au) in the EXAFS data.51 Reported uncertainties for EXAFS fitting results were computed from off-diagonal elements of the correlation matrix, which were weighted by the square root of the reduced chi-squared value obtained from each simulated fit. The amount of experimental noise was also taken into consideration for each Fourier transformed R-space spectrum from 15–25 Å.52 S K-edge XANES data were collected from the SXRMB beamline at the Canadian Light Source (operating at 2.9 GeV). Each powdered AuBSA sample and reference material was measured in fluorescence mode under room temperature and vacuum conditions. All samples were mounted onto a copper sample using carbon tape.EXAFS simulations
Simulated EXAFS spectra of Au(I)-SR clusters were calculated using the FEFF8.2 program with coordinates from crystal structure files or DFT-optimized structures. The L3-edge EXAFS spectrum for each Au site was calculated using a self-consistent field radius set at 4.5 Å to encompass enough atoms in the cluster for full multiple scattering calculations. A Debye–Waller factor of 0.0036 Å2 was used for Au sites based on the Au–S EXAFS fitting result in Table S1,† similar to a value chosen by Yamazoe et al.53 The averaged EXAFS spectrum was Fourier-transformed using an identical k-space region to AuBSA (3.0–12.0 Å−1) and k1-weighting for the pre-screening process.Steady-state and time-resolved photoluminescence (PL) studies of Au10(SG)10 and AuBSA
Steady-state PL and temperature-dependent PL measurements were carried out with an Edinburgh spectrofluorimeter (F900S). The PL quantum yields were measured using Rhodamine B in ethanol as a standard.54 For temperature-dependent PL measurements, the fluorimeter was coupled with Optistat DN cryostat (Oxford instruments) and ITC temperature controller. The measurements were carried out from 77 to 303 K. The vacuum in the cryostat was maintained with a leybold turbo molecular pump. Spectra were taken at different temperatures after a wait period of 10 minutes. The error in temperature setting was ±0.5 K. The Au10(SG)10 sample was dissolved in a 65:35 glycerol/water mixture for temperature-dependent PL measurements. All the samples were purged with N2 to get rid of dissolved oxygen to omit problems in solvent freezing temperatures as well as the effect of oxygen on phosphorescence lifetimes. Optical absorption measurements before and after temperature-dependent PL measurements have shown no change suggesting the samples did not change during the measurements. Time-resolved PL decay lifetime measurements were measured using a time-correlated single photon counting technique after excitation at 373 nm with a diode laser excitation and the measurements were carried out with an Edinburgh F900S spectrofluorimeter. The PL lifetimes were measured at their respective PL maxima with an emission slit width of 10 nm. Cooled Hamamatsu R-921P PMT was used as the detector.
DFT calculations
Geometry optimization was performed on all species using the LC-ωPBE functional55–57 and the XDM58,59 dispersion correction, with the Couty-Hall modified LANL2DZ basis set for Au,60 6-31G* for C and H, and 6-31+G* for S. Single-point energy calculations on the optimized geometries were carried out using the same LC-ωPBE-XDM method with the aug-cc-pVDZ-PP61 basis set for Au and aug-cc-pVDZ62,63 for C, H, and S. The XDM damping parameters were a1 = 0.8134, a2 = 1.3736 for the geometry optimizations and a1 = 1.1800, a2 = 0.4179 for the single-point energy calculations, as in previous work that demonstrated the excellent performance of LC-ωPBE-XDM for modeling of aurophilic effects in gold complexes.35 All calculations were performed using the Gaussian 09 software package,64 along with the postg program (freely available at http://schooner.chem.dal.ca) for the dispersion energies. The EMSL Basis Set library was used to obtain all basis set and ECP input parameters.65EXAFS fitting methodologies
Characterization with XAFS has been useful for investigating site-specific structural properties of Au NCs.24,48,66–68 The X-ray absorption perspective has several advantages in this study by probing specific absorption edges to observe changes in the oxidation state and structural environment during the course of the BSA-directed synthesis. More specifically, X-ray absorption near-edge structure (XANES) is used to examine the interaction between excited core electrons and unoccupied valence levels, offering information on the electronic structure of the absorbing atom. Extended X-ray absorption fine structure (EXAFS) deconvolutes and refines the post-edge XAFS oscillations to determine coordination numbers, bond lengths, and Debye–Waller factor structural parameters. As a result, the combination of XANES and EXAFS techniques can provide invaluable information on the Au local structure within a complex system such as a protein molecule. For time-dependent samples of AuBSA taken at 0, 1, 2, 3, 6, 12 and 36 h, two or three scattering paths were fitted to the experimental EXAFS spectrum. For these EXAFS fits, the E0 shift values were correlated for each shell with all other parameters running free. This was done to help meet the Nyquist criterion for number of dependent and independent parameters when conducting EXAFS fitting.69 The Au10(SR)10 catenane model was used to generate simulated scattering paths to fit the AuBSA sample and the Au10(SG)10 sample with more Au scattering environments. Unlike the two-/three-shell fit, coordination numbers for each scattering path were fixed to their ideal values according to the Au10(SR)10 catenane model. Bond lengths (R) and Debye–Waller (σ2) parameters ran free for the fit with all E0 shift values correlated to help satisfy the Nyquist criterion. A total of 4 single scattering paths were used (identified in Fig. 1D) and fitted over a R-range of 1.5–4.0 Å.Contributions
DMC conducted the sample synthesis and structural determination of protein-gold samples, the synchrotron X-ray experiments and analysis, the organization of all results and wrote the manuscript. PZ supervised DMC in carrying out the research work and contributed to the writing of the manuscript. QY conducted the MALDI-TOF measurement. VDT and RG performed the steady-state and time-resolved photoluminescence measurements and Au10(SG)10 rigidification studies. ZL prepared the Au10(SG)10 sample and was supervised by JX. SD and ERJ conducted all DFT calculations. PC and MAM contributed to the early stage EXAFS work of protein-gold nanocluster samples. NZ contributed to the design of the paper. AC co-supervised DMC's work. All authors helped to revise the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
D. M. C. was supported by the NSERC CGS-Alexander Graham Bell scholarship during this work. P. Z., A. C. and E. R. J. acknowledge NSERC Discovery Grants for funding. CLS@APS facilities (Sector 20-BM) at the Advanced Photon Source (APS) are supported by the U.S. Department of Energy (DOE), NSERC Canada, the University of Washington, the Canadian Light Source (CLS), and the APS. Use of the APS is supported by the DOE under Contract No. DE-AC02-06CH11357. The CLS is financially supported by NSERC Canada, CIHR, NRC, and the University of Saskatchewan. J. X. acknowledges Ministry of Education, Singapore for funding support (R-279-000-481-112). V. D. T. and G. R. acknowledge the support of ACS-PRF #53999-ND5 and Western Michigan University startup. Technical support at CLS@APS (Sector 20, APS) facilities from Dr Robert Gordon and Dr Zou Finfrock is acknowledged and greatly appreciated. Technical support at SXRMB facilities (CLS) from Dr Yongfeng Hu and Dr Qunfeng Xiao is also acknowledged and greatly appreciated.References
- P. D. Jadzinsky, G. Calero, C. J. Ackerson, D. A. Bushnell and R. D. Kornberg, Science, 2007, 318, 430–433 CrossRef CAS PubMed.
- Z. Y. Li, N. P. Young, M. Di Vece, S. Palomba, R. E. Palmer, A. L. Bleloch, B. C. Curley, R. L. Johnston, J. Jiang and J. Yuan, Nature, 2008, 451, 46–48 CrossRef CAS PubMed.
- S. Chen, R. Ingram, M. Hostetler, J. Pietron, R. Murray, T. Schaaff, J. Khoury, M. Alvarez and R. Whetten, Science, 1998, 280, 2098–2101 CrossRef CAS PubMed.
- M. Azubel, J. Koivisto, S. Malola, D. Bushnell, G. L. Hura, A. L. Koh, H. Tsunoyama, T. Tsukuda, M. Pettersson, H. Hakkinen and R. D. Kornberg, Science, 2014, 345, 909–912 CrossRef CAS PubMed.
- M. B. Dickerson, K. H. Sandhage and R. R. Naik, Chem. Rev., 2008, 108, 4935–4978 CrossRef CAS PubMed.
- S. S. Shankar, A. Rai, B. Ankamwar, A. Singh, A. Ahmad and M. Sastry, Nat. Mater., 2004, 3, 482–488 CrossRef CAS PubMed.
- M. Sarikaya, C. Tamerler, A. K. Jen, K. Schulten and F. Baneyx, Nat. Mater., 2003, 2, 577–585 CrossRef CAS PubMed.
- W. J. Crookes-Goodson, J. M. Slocik and R. R. Naik, Chem. Soc. Rev., 2008, 37, 2403–2412 RSC.
- H. Wei, Z. Wang, J. Zhang, S. House, Y. G. Gao, L. Yang, H. Robinson, L. H. Tan, H. Xing, C. Hou, I. M. Robertson, J. Zuo and Y. Lu, Nat. Nanotechnol., 2011, 6, 93–97 CrossRef CAS PubMed.
- C.-L. Liu, H.-T. Wu, Y.-H. Hsiao, C.-W. Lai, C.-W. Shih, Y.-K. Peng, K.-C. Tang, H.-W. Chang, Y.-C. Chien, J.-K. Hsiao, J.-T. Cheng and P.-T. Chou, Angew. Chem., Int. Ed., 2011, 50, 7056–7060 CrossRef CAS PubMed.
- X. Le Guével, N. Daum and M. Schneider, Nanotechnology, 2011, 22, 275103 CrossRef PubMed.
- L. Shang, S. Dong and G. U. Nienhaus, Nano Today, 2011, 6, 401–418 CrossRef CAS.
- D. M. Chevrier, A. Chatt and P. Zhang, J. Nanophotonics, 2012, 6, 64504 CrossRef.
- J. Xie, Y. Zheng and J. Y. Ying, Chem. Commun., 2010, 46, 961–963 RSC.
- Y. Wang, Y. Wang, F. Zhou, P. Kim and Y. Xia, Small, 2012, 8, 3769–3773 CrossRef CAS PubMed.
- L. Shang, S. Brandholt, F. Stockmar, V. Trouillet, M. Bruns and G. U. Nienhaus, Small, 2012, 8, 661–665 CrossRef CAS PubMed.
- J. Xie, Y. Zheng and J. Y. Ying, J. Am. Chem. Soc., 2009, 131, 888–889 CrossRef CAS PubMed.
- P. L. Xavier, K. Chaudhari, P. K. Verma, S. K. Pal and T. Pradeep, Nanoscale, 2010, 2, 2769–2776 RSC.
- H. Wei, Z. Wang, L. Yang, S. Tian, C. Hou and Y. Lu, Analyst, 2010, 135, 1406–1410 RSC.
- P. L. Xavier, K. Chaudhari, A. Baksi and T. Pradeep, Nano Rev., 2012, 3, 1–16 Search PubMed.
- M. Zhu, C. M. Aikens, F. J. Hollander, G. C. Schatz and R. Jin, J. Am. Chem. Soc., 2008, 130, 5883–5885 CrossRef CAS PubMed.
- M. W. Heaven, A. Dass, P. S. White, K. M. Holt and R. W. Murray, J. Am. Chem. Soc., 2008, 130, 3754–3755 CrossRef CAS PubMed.
- P. Zhang, J. Phys. Chem. C, 2014, 118, 25291–25299 CAS.
- T. Yao, Z. Sun, Y. Li, Z. Pan, H. Wei, Y. Xie, M. Nomura, Y. Niwa, W. Yan, Z. Wu, Y. Jiang, Q. Liu and S. Wei, J. Am. Chem. Soc., 2010, 132, 7696–7701 CrossRef CAS PubMed.
- C. A. Simpson, C. L. Farrow, P. Tian, S. J. L. Billinge, B. J. Huffman, K. M. Harkness and D. E. Cliffel, Inorg. Chem., 2010, 49, 10858–10866 CrossRef CAS PubMed.
- M. R. Wiseman, P. A. Marsh, P. T. Bishop, B. J. Brisdon and M. F. Mahon, J. Am. Chem. Soc., 2000, 122, 12598–12599 CrossRef CAS.
- H. Grönbeck, M. Walter and H. Häkkinen, J. Am. Chem. Soc., 2006, 128, 10268–10275 CrossRef PubMed.
- D. J. LeBlanc and C. J. L. Lock, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1997, 53, 1765–1768 Search PubMed.
- R. Bau, J. Am. Chem. Soc., 1998, 7863, 9380–9381 CrossRef.
- S. S.-Y. Chui, R. Chen and C.-M. Che, Angew. Chem., Int. Ed., 2006, 45, 1621–1624 CrossRef CAS PubMed.
- X. Le Guevel, B. Hotzer, G. Jung, K. Hollemeyer, V. Trouillet and M. Schneider, J. Phys. Chem. C, 2011, 115, 10955–10963 CAS.
- M. A. Habeeb Muhammed, P. K. Verma, S. K. Pal, A. Retnakumari, M. Koyakutty, S. Nair and T. Pradeep, Chem.–Eur. J., 2010, 16, 10103–10112 CrossRef CAS PubMed.
- K. Chaudhari, P. L. Xavier and T. Pradeep, ACS Nano, 2011, 5, 8816–8827 CrossRef CAS PubMed.
- N. Fernández-Iglesias and J. Bettmer, Nanoscale, 2014, 6, 716–721 RSC.
- A. Otero-De-La-Roza, J. D. Mallory and E. R. Johnson, J. Chem. Phys., 2014, 140, 18A504 CrossRef PubMed.
- Y. Negishi, K. Nobusada and T. Tsukuda, J. Am. Chem. Soc., 2005, 127, 5262–5270 CrossRef PubMed.
- J. M. Forward, D. Bohmann, J. P. Fackler and R. J. Staples, Inorg. Chem., 1996, 34, 6330–6336 CrossRef.
- S. Cha, J. Kim, K. Kim and J. Lee, Chem. Mater., 2007, 19, 6297–6303 CrossRef CAS.
- Z. Wu and R. Jin, Nano Lett., 2010, 10, 2568–2573 CrossRef CAS PubMed.
- Y. Yu, Z. Luo, D. M. D. M. D. M. Chevrier, D. T. D. T. Leong, P. Zhang, D. Jiang and J. Xie, J. Am. Chem. Soc., 2014, 136, 1246–1249 CrossRef CAS PubMed.
- K. Pyo, V. D. Thanthirige, K. Kwak, P. Pandurangan, G. Ramakrishna and D. Lee, J. Am. Chem. Soc., 2015, 137, 8244–8250 CrossRef CAS PubMed.
- N. Goswami, Q. Yao, Z. Luo, J. Li, T. Chen and J. Xie, J. Phys. Chem. Lett., 2016, 7, 962–975 CrossRef CAS PubMed.
- V. W.-W. Yam, C.-L. Chan, C.-K. Li and K. M.-C. Wong, Coord. Chem. Rev., 2001, 216–217, 173–194 CrossRef CAS.
- Z. Luo, X. Yuan, Y. Yu, Q. Zhang, D. T. Leong, J. Y. Lee and J. Xie, J. Am. Chem. Soc., 2012, 134, 16662–16670 CrossRef CAS PubMed.
- X. Dou, X. Yuan, Y. Yu, Z. Luo, Q. Yao, D. T. Leong and J. Xie, Nanoscale, 2014, 6, 157–161 RSC.
- Y. Negishi and T. Tsukuda, Chem. Phys. Lett., 2004, 383, 161–165 CrossRef CAS.
- S. Cha, J. Kim, K. Kim and J. Lee, Chem. Mater., 2007, 19, 6297–6303 CrossRef CAS.
- M. A. Macdonald, D. M. Chevrier, P. Zhang, H. Qian, R. Jin, D. M. D. M. Chevrier, P. Zhang, H. Qian and R. Jin, J. Phys. Chem. C, 2011, 115, 15282–15287 CAS.
- M. A. Macdonald, P. Zhang, N. Chen, H. Qian and R. Jin, J. Phys. Chem. C, 2011, 115, 65–69 CAS.
- F. Liu, D. Wechsler and P. Zhang, Chem. Phys. Lett., 2008, 461, 254–259 CrossRef CAS.
- A. L. Ankudinov, B. Ravel, J. J. Rehr and S. D. Conradson, Phys. Rev. B, 1998, 58, 7565–7576 CrossRef CAS.
- M. Newville, B. I. Boyanov and D. E. Sayers, J. Synchrotron Radiat., 1999, 6, 264–265 CrossRef CAS PubMed.
- S. Yamazoe, S. Takano, W. Kurashige, T. Yokoyama, K. Nitta, Y. Negishi and T. Tsukuda, Nat. Commun., 2016, 7, 10414 CrossRef CAS PubMed.
- F. L. Arbeloa, P. R. Ojeda and I. L. Arbeloa, J. Lumin., 1989, 44, 105–122 CrossRef.
- O. A. Vydrov and G. E. Scuseria, J. Chem. Phys., 2006, 125, 234109 CrossRef PubMed.
- O. A. Vydrov, J. Heyd, A. Krukau and G. E. Scuseria, J. Chem. Phys., 2006, 125, 74106 CrossRef PubMed.
- O. A. Vydrov, G. E. Scuseria and J. P. Perdew, J. Chem. Phys., 2007, 126, 154109 CrossRef PubMed.
- A. D. Becke and E. R. Johnson, J. Chem. Phys., 2007, 127, 154108 CrossRef PubMed.
- A. Otero-de-la Rozza and E. R. Johnson, J. Chem. Phys., 2013, 138, 204109 CrossRef PubMed.
- M. Couty and M. B. Hall, J. Comput. Chem., 1996, 17, 1359–1370 CrossRef CAS PubMed.
- K. A. Peterson and C. Puzzarini, Theor. Chem. Acc., 2005, 114, 283–296 CrossRef CAS.
- J. T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef.
- D. E. Woon and J. T. H. Dunning, J. Chem. Phys., 1993, 98, 1358–1371 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revis. E.01, Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
- K. L. Schuchardt, B. T. Didier, T. Elsethagen, L. Sun, V. Gurumoorthi, J. Chase, J. Li and T. L. Windus, J. Chem. Inf. Model., 2007, 47, 1045–1052 CrossRef CAS PubMed.
- L. D. Menard, H. Xu, S.-P. Gao, R. D. Twesten, A. S. Harper, Y. Song, G. Wang, A. D. Douglas, J. C. Yang, A. I. Frenkel, R. W. Murray and R. G. Nuzzo, J. Phys. Chem. B, 2006, 110, 14564–14573 CrossRef CAS PubMed.
- O. Guliamov, A. I. Frenkel, L. D. Menard, R. G. Nuzzo and L. Kronik, J. Am. Chem. Soc., 2007, 129, 10978–10979 CrossRef CAS PubMed.
- T. Shibata, H. Tostmann, B. Bunker, A. Henglein, D. Meisel, S. Cheong and M. Boyanov, J. Synchrotron Radiat., 2001, 8, 545–547 CrossRef CAS PubMed.
- E. A. Stern, Phys. Rev. B, 1993, 48, 9825–9827 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc05086k |
| ‡ Authors have made equal contributions. |
| This journal is © The Royal Society of Chemistry 2018 |