
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTwo-photon fluorescent polysiloxane-based films with thermally responsive self switching properties achieved by a unique reversible spirocyclization mechanism†
Yujing
Zuo
 ,
Tingxin
Yang
,
Yu
Zhang
,
Zhiming
Gou
,
Minggang
Tian
,
Xiuqi
Kong
and
Weiying
Lin
*
,
Tingxin
Yang
,
Yu
Zhang
,
Zhiming
Gou
,
Minggang
Tian
,
Xiuqi
Kong
and
Weiying
Lin
*
Institute of Fluorescent Probes for Biological Imaging, School of Chemistry and Chemical Engineering, School of Materials Science and Engineering, University of Jinan, Shandong 250022, P. R. China. E-mail: weiyinglin2013@163.com
First published on 5th February 2018
Abstract
Responsiveness and reversibility are present in nature, and are ubiquitous in biological systems. The realization of reversibility and responsiveness is of great importance in the development of properties and the design of new materials. However, two-photon fluorescent thermal-responsive materials have not been reported to date. Herein, we engineered thermally responsive polysiloxane materials (Dns-non) that exhibited unique two-photon luminescence, and this is the first report about thermally responsive luminescent materials with two-photon fluorescence. The fluorescence of Dns-non could switch from the “on” to “off” state through a facile heating and cooling process, which could be observed by the naked eye. Monitoring the temperature of the CPU in situ was achieved by easily coating D1-non onto the CPU surface, which verified the potential application in devices of Dns-non. A unique alkaline tuned reversible transition mechanism of rhodamine-B from its spirocyclic to its ring-open state was proposed. Furthermore, Dns-non appeared to be a useful cell adhesive for the culture of cells on the surface. We believe that the constructed thermally responsive silicon films which have promising utilization as a new type of functional fluorescent material, may show broad applications in materials chemistry or bioscience.
Introduction
Responsiveness and reversibility are present in nature, and are ubiquitous in biological systems. In the past decade, the realization of reversibility and responsiveness has been of great importance in the development of properties and the design of new materials.1–3 Fluorescent responsive systems have received more attention for their facile signal detection method, and for the availability of a large number of examples such as sensors or probes.4 Besides this, temperature is an important environmental property, therefore, the development of fluorescence-based thermo-sensitive materials is desirable since such a sensor is effective in monitoring temperature via detecting fluorescence signals.5–8 Previously reported fluorescent temperature responsive systems have been mainly based on twisted intramolecular charge-transfer (TICT) fluorophores,9 cyclodextrin-based phosphorescent compounds,10 spin crossover complexes,11 quantum dots,12 quasi-reversible photodissociation,13 and lanthanide complexes.14 However, these fluorescent thermally responsive materials could only emit single photon fluorescence. Two-photon fluorescence has been applied in luminescent imaging techniques due to its unique advantages, including low photodamage to the samples, weak background fluorescence, and high spatial resolution.15–22 To the best of our knowledge, two-photon fluorescent thermally responsive materials have not been reported to date. Taking into account the merits of both thermally responsive materials and two-photon fluorescence, construction of two-photon fluorescent thermally responsive systems is of great interest and importance. However, there are two vital challenges remaining for designing these systems. First, most reports on thermo-responsive polymers are based on traditional temperature responsive groups such as N-isopropylacrylamides, which are non-fluorescent. In addition, traditional functional fluorophores with two-photon absorption do not show thermally responsive properties. Our initial intention here was to build a thermally responsive system with unique two-photon fluorescence; thus, we envisaged the possibility of exploiting a novel responding mechanism realized by the interactions between the functional groups within the polymer base and the fluorescent dye.Considering that tertiary amines show alkaline properties and have a tendency to react with acid to form a salt, we selected benzoxazine-containing polysiloxanes (P1) which contained the tertiary amine as a key structural element. Furthermore, as a two-photon fluorescent dye, rhodamine-B based fluorescent probes could act as “turn-on” fluorescent switch molecules in response to the targeted cation.23–25 The ion detection mechanism of rhodamine probes is mainly based on the change in structure between its open-cycle and spirocyclic states; thus, we anticipated that the presence of its five-membered ring “opening or closing” states could be critical for thermally responsive properties. Then, we intended to incorporate rhodamine-B into P1 to investigate the novel luminescent responsive properties. Furthermore, rhodamine-B was chosen as the fluorescent moiety, not only for its luminescent performance but, more importantly, for the larger abundance of benzene structures that could make it possible to render physical cross-linking sites on the polysiloxane base.
Herein, rhodamine-B was incorporated into P1, which acted both as a physical cross-linker and luminophore. Transparent films of Dns-non were obtained and they emitted strong photoluminescence under natural light or UV irradiation. Two-photon luminescence of Dns-non was detected. More interestingly, the fluorescence intensity of Dns-non exhibited thermally responsive properties, which could be observed by the naked eye. Furthermore, Dns-non were applied in devices by monitoring the CPU surface temperature in situ. In addition, HeLa cells were cultured on the surfaces of Dns-non to investigate their cytocompatibility and to explore their potential applications in biomedical fields.
Results and discussion
To construct a novel polysiloxane-based benzoxazine system, finding a proper condensation reaction that could bind the benzoxazine functional groups and the polysiloxane base together is of the primary concern. Yagci et al. reported the synthesis of polysiloxane containing benzoxazine moieties in the main chain using hydrosilylation reaction.26 However, a Pt catalyst was applied and the residue of heavy metal catalysts may further limit the application of the products in biology or medical fields.27 We fabricated polysiloxane-based benzoxazine (P1) via facile Mannich-type polycondensation with a high yield using bis[dimethyl(3-aminopropyl)silyl] ether (MMNH2) as the precursor (Scheme 1). An FT-IR spectra of the corresponding compounds provided evidence for the formation of the benzoxazine functionalized polymer P1 (Fig. S1†). The structure of P1 was further confirmed using 1H NMR, 13C NMR, and 29Si NMR analysis (Fig. S2†). In addition, the molecular weight (Mn) of P1 was about 7700 g mol−1, and the PDI was about 1.21 (Table S1†). The increase of the molecular weight also indicates the success of the condensation polymerization. However, with the increase of the molecular weight of P1, the activity of the end group was reduced, which prevented the further growth of the main chain of P1. Mannich condensation is a flexible tool to incorporate a benzoxazine ring into polysiloxane-based materials. | ||
| Scheme 1 (a) Synthesis of P1 by Mannich polycondensation, and (b) illustration of the preparation of Dns-non and their application in electrical devices and bioscience. | ||
Silicon-based films were prepared by an easy “casting” route [Scheme 1(b)]. The films were obtained and named D1-non, D2-non, and D3-non, respectively. Meanwhile, C1-non, C2-non, and C3-non were prepared without any use of rhodamine-B by the same method. Swelling analysis (Fig. S4†) showed that samples were cross-linked by physical cross-linking. Both hydrogen-bond interaction and π–π interaction between different polymer main chains resulted in cross-linking.28 Contact angle measurement results (Fig. S5†) revealed that the surface energy of the films could be easily tuned by merely altering the molar ratio between MMNH2 and BPA. SEM (Scanning Electron Microscopy) images (Fig. S6†) indicated that casting and the thermal treating approach that followed can produce Dns-non with homogenous surfaces.
As shown in Fig. 1(b), Cns-non emitted strong blue photoluminescence under 365 nm UV light. Fig. 1(a) illustrates the fluorescence from C0-non, C1-non, and C2-non excited at 365 nm. Cns-non emitted fluorescence in the spectral region of 460 nm under the excitation at 365 nm. According to the CIE diagram, the luminescence of Cns-non was blue, which was identical to the digital photograph in Fig. 1(b). Unlike carbon, a silicon atom has 5 empty 3d orbitals which can be used as electron acceptors. Luminescence was inferred to be generated by the lone pair electrons of N atoms coordinated with Si. The ligand field further split the 3d orbital of the Si atom, which was degenerated before coordination. Then, the electrons rearranged in the split orbitals resulting in a d–d transition, which could account for the luminescent properties of Cns-non.29,30Dns-non were transparent and presented strong red photoluminescence under natural light with the incorporation of rhodamine-B [Fig. 1(e) and (f)]. We directly stimulated the films using a wavelength of 520 nm. The emission spectra of D1-non were selected for further discussion. As shown in Fig. 1(d), the characteristic emission of rhodamine-B was found to be at a wavelength of 580 nm in D1-non. Based on CIE color coordinates calculated from the emission spectra, the bright red emission of D1-non could be observed by the naked eye when illuminated by natural light.
 | ||
| Fig. 1 (a) Solid-state photoluminescence spectra of Cns-non excited by a wavelength of 365 nm, (b) the photoluminescence image of C1-non excited by a wavelength of 365 nm, (c) the photograph of the CIE chromaticity diagrams for Cns-non, (d) the solid-state photoluminescence spectrum of D1-non excited by a wavelength of 520 nm, (e) the photoluminescence image of D1-non under natural light, (f) the photograph of the CIE chromaticity diagram for D1-non excited by a wavelength of 520 nm, (g) the solid-state photoluminescence spectrum of D1-non excited by a wavelength of 365 nm, (h) the photoluminescence image of D1-non excited by a wavelength of 365 nm, and (i) the photograph of the CIE chromaticity diagram for D1-non excited by a wavelength of 365 nm. | ||
The surface color changed along with the variation of the temperature. We speculated that the UV absorption of Dns-non would change with temperature. The UV absorption of D1-non at different temperatures is shown in Fig. S10.† The absorption of D1-non was mainly derived from rhodamine-B. The absorption decreased with the increasing of the temperature, which fitted the fluorescence results. Rhodamine-B changed to its spirocyclic state at higher temperature and reduced the absorption. As a result, the fluorescence intensity decreased with the increasing of the temperature.
We focused on characterization of the dual color emissive property of D1-non, as dual color emissive materials may offer more potential for stimuli responsive properties. The characteristic peak of rhodamine-B could also be detected at approximately 580 nm, excited by a wavelength of 365 nm [Fig. 1(g)]. However, another emission peak was found at 460 nm, and the emission intensity at 460 nm was 3 times as big as the intensity at 580 nm. Identically, according to the CIE diagram, purple light (a combination of blue and red) could be observed when irradiated at 365 nm, which matched with the color observed by the naked eye. Interestingly, the emission colors of D1-non could be easily tuned from red (0.254, 0.172) to purple (0.592, 0.381) by just controlling the excitation wavelength. Excitation-dependent PL behavior can be useful in multi-color imaging applications.
Dns-non showed fascinating thermally responsive properties when undergoing a heating treatment. As illustrated in Fig. 2(d), after heating to 110 °C for 15 min, the red color of D1-non faded. However, D1-non regained its original color immediately when cooled to room temperature. Apparently, Dns-non presented thermally responsive behavior. Fluorescence spectra of D1-non at different temperatures were produced. As can be seen in Fig. 2(a), after thermal treatment at 110 °C for 15 min, the broad emission peak of D1-non at around 580 nm disappeared. However, remarkably, the cooling down of D1-non induced turn-on emission. Significant changes in emission intensity were observed as the temperature was varied [Fig. 2(b)]. It is obvious that the emission intensity reached the maximum value after cooling for 5 s, but it decreased dramatically at higher temperatures. Meanwhile, blue or red shift of emission peaks was not observed when the temperature was varied. To evaluate the reversibility of the switching operation upon variation of temperature, D1-non was then subjected to temperature cycling between 110 °C and 30 °C.
 | ||
| Fig. 2 (a) Solid-state photoluminescence spectra of D1-non excited by a wavelength of 520 nm measured after heating up and cooling down, respectively, (b) the change of fluorescence of D1-non excited by a wavelength of 520 nm with the time after cooling down, (c) PL intensity upon the cyclic switching of D1-non under alternating conditions of 110 and 30 °C, (d) digital image of D1-non (left: color faded after heating, right: color recovered after cooling down), and (e) digital photographs showing the possibility of using Dns-non as indicators for water temperature using D0-non as the demonstration. | ||
The fact that the luminescence switching operation could be repeated for four consecutive cycles without a fatigue phenomenon [Fig. 2(c)], indicated the fine reversibility of the two state switching process of photoluminescence. The cast films of D0-non to D2-non displayed similar responses to hot water. Taking D0-non as an example, as shown in Fig. 2(e), after heating at 100 °C in a hot water bath, the color of the transparent red film lightened, becoming close to colorless. However, when the temperature of the water dropped, the color was restored. This process could be repeated multiple times with good reproducibility. These results revealed the potential application of Dns-non films as temperature indicators. The pH stability was investigated using D1-non as an example. As shown in Fig. S12,† the fluorescence intensity of D1-non did not change significantly after being treated with solutions of varied pH. The physical cross-linking was efficient. In addition, the tertiary amine structure could act as a buffer, which could further reduce the effect of altering the pH. The results indicated that Dns-non exhibited good pH stability. The effect of common ions on D1-non were examined by treating D1-non with metal ions such as Cu2+, Fe3+, Mg2+, Ba2+, Zn2+, Na+, and K+ in 50% methanol solution at room temperature. The results are shown in Fig. S13.† The addition of Cu2+, Al3+, Mg2+, Ba2+, Na+, Zn2+ and K+ ions led to no noticeable changes in the fluorescence intensity of D1-non, while Fe3+ caused apparent enhancement of the fluorescence intensity. The results indicated that Fe3+ caused rhodamine-B to change to its “ring-opening” state. The application environments of Dns-non do not contain common ions under normal conditions; therefore, the response to several common ions does not affect their applications.
High-performance CPUs built in to PCs have been developed to meet the requirement of higher PC performance. Thinner and more compact designs of CPU have lead to increased heat dissipation, making the CPU temperature rise and causing a shortened life, malfunction and failure of the CPU.31,32In situ indication of the surface temperature of the CPU visually should be taken seriously. Taking into account the temperature responsive property of Dns-non, we anticipated that their application could be expanded to in situ surface thermo-indication of the CPU. As depicted in Fig. 3(a), D1-non was coated onto the upper surface of the CPU. When the computer started running, the color of rhodamine-B faded. As the running time of the computer increased, the surface temperature of the CPU raised gradually, and as a result, the fluorescence intensity of D1-non decreased. However, when the computer was closed, the fluorescence intensity of D1-non recovered as the surface temperature decreased. Furthermore, color saturation of these graphs was selected as a parameter to quantize the fluorescence change [Fig. 3(b)]. The change in color saturation was consistent with the color change observed by the naked eye. Monitoring the surface temperature of the CPU in situ was achieved by easily coating D1-non onto the CPU surface, which verified the potential application in electrical devices of Dns-non.
 | ||
| Fig. 3 (a) Digital images of a D1-non coated CPU with different running times illustrating the color changes, and (b) color saturation of the digital images of D1-non as a function of the computer running time. The error bars represent standard deviation (±S.D.). | ||
The control experiment (Fig. S8†) indicated that the fluorescence of rhodamine-B does not alter after a heating treatment, and the existence of common Si–O–Si chains does not affect the fluorescence of rhodamine-B. Furthermore, as illustrated in Scheme S1,† two kinds of tertiary amine, M1 and M2, were selected as model compounds to get a deeper understanding of the mechanism. We could see from the inserted photograph in Fig. S9(b)† that the red color of Sample 1 faded at an evaluated temperature, and then recovered when cooled down. As shown in Fig. S9(a) and (b),† the fluorescence intensity of Sample 1 decreased after heating. As we expected, the fluorescence intensity increased when cooling down. As shown in Fig. S9(c) and (d),† Sample 2 exhibited a similar phenomenon to that of Sample 1. However, because of the existence of the –COOH group located in M2, the alkalinity of M2 was weaker than that of M1. As a result, the decreasing proportion of the fluorescence intensity of Sample 2 is lower than that of Sample 1. A model control trial further confirmed the tertiary amine regulated reversible transition mechanism. In view of the two-state switching nature of rhodamine-B,33,34 we deduced the mechanism of the fluorescent switch phenomenon of Dns-non. As illustrated in Fig. 4, the existence of a tertiary amine within P1 led to the fluorescent switch behavior of Dns-non. Tertiary amines exert alkaline properties and have a tendency to react with acid to form a salt.23–25 At room temperature, free rhodamine-B rested in its ring-open state. Meanwhile, Dns-non showed characteristic luminescence of rhodamine-B under the irradiation of natural light. When the temperature reached 110 °C, the tertiary amine group captured hydrogen in rhodamine-B at first. Then, the formed COO− attacked the carbon atom adjacent to the benzene ring. As a result, rhodamine-B changed into its spirocyclic state. However, unlike most reported reactions between rhodamine-B and a primary amine without reversibility, the reaction between rhodamine-B and the tertiary amine presented good reversibility within a certain temperature range. When cooling down, N atoms in the polymer main chain attacked the spirocyclic ring, and formed an unstable intermediate. Thereafter, the fluorescence emerged along with the regeneration of rhodamine-B in its “open-cyclic” state. The “switch” process reached an equilibrium. Equilibrium shifted to the “close-cyclic” direction upon heating, whereas it moved to “open-cyclic” when the temperature dropped. This mechanism provided us with a new insight to build novel thermally responsive luminescent materials based on non-responsive groups, which further expands the application of thermally responsive materials.
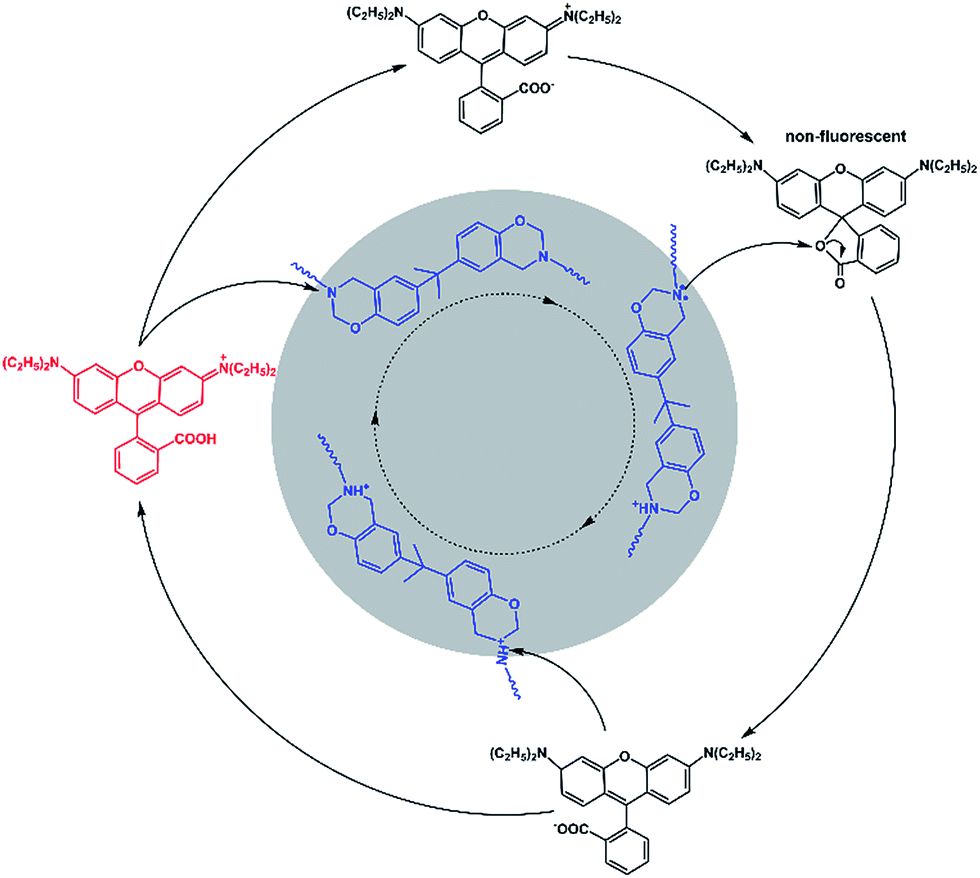 | ||
| Fig. 4 Proposed reaction mechanism for the fluorescence of the Dns-non switch from “turn on” to “off ”circulation. | ||
To investigate the luminescence and the surface properties of Dns-non, fluorescence images of D1-non were captured using a Nikon A1MP confocal microscope. From the bright field image [Fig. 5(a)], phase separation was not found. The results indicated that the homogeneous phase of the film was obtained after rhodamine-B incorporation. D1-non exhibited strong and uniform blue luminescence when excited by a wavelength of 405 nm and captured by the blue channel [Fig. 5(b)]. Meanwhile, from the red channel, bright red luminescence could also be observed when excited by a wavelength of 561 nm [Fig. 5(c)]. The quantified relative fluorescence intensity for blue emission was weaker than for red emission [Fig. 5(d)]. These image results were consistent with the spectroscopic properties of D1-non.
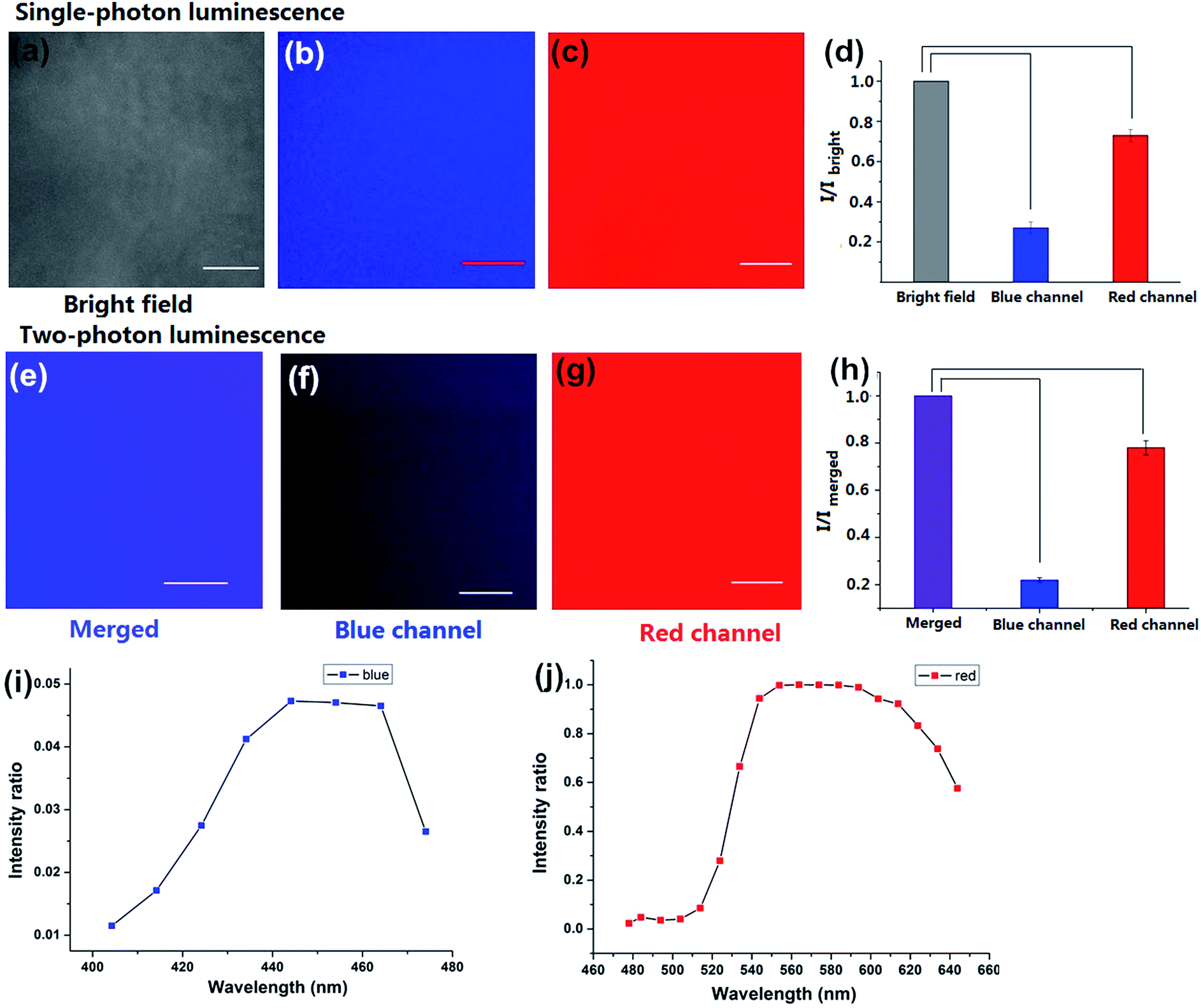 | ||
| Fig. 5 Fluorescence images of D1-non: (a) bright field image, (b) image from the blue channel, (c) image from the red channel, and (d) quantified relative fluorescence intensities for (a)–(c). The error bars represent standard deviation (±S.D.). Two-photon fluorescence images of D1-non: (e) overlay of the blue and red channels, (f) image from the blue channel, (g) image from the red channel, and (h) quantified relative fluorescence intensities for (e)–(g). The error bars represent standard deviation (±S.D.). (i) Two-photon emission spectrum of D1-non in the blue region, and (j) two-photon emission spectrum of D1-non in the red region. | ||
Encouraged by the prominent results of the fluorescence images of D1-non, we decided to examine the fluorescence of D1-non using two-photon fluorescence microscopy. As depicted in Fig. 5(f) and (g), significant luminescence was observed when excited by a wavelength of 760 nm from both the blue channel and the red channel, which demonstrated that D1-non is a suitable material for showing superior two-photon fluorescence properties. Two-photon emission spectra of D1-non are depicted in Fig. 5(i) and (j), where two emission peaks were found at 450 nm and 580 nm, corresponding to blue and red emissions, respectively. The intensity of the red emission derived from rhodamine-B was greater than the blue emission from the polymer, which was identical to the quantified relative fluorescence intensity result [Fig. 5(h)]. Results from the emission spectra further proved that two-photon fluorescence existed in Dns-non. The two-photon absorption cross-sections (δ) of the D1-non solution were determined using a two-photon-induced fluorescence method with the standard fluorescein in 1 mM NaOH aqueous solution as the reference. Fig. S11† shows the two-photon absorption cross-section spectra of D1-non in ethanol solution with a concentration of 10−5 mol L−1. D1-non exhibited the highest cross-section of 232 GM when excited by a wavelength of 720 nm. The two-photon cross-sections of D1-non were higher than those of the reported pure rhodamine-B solutions. The enhanced cross-sections were deduced from the Fluorescence Resonance Energy Transfer (FRET) effect of Pns.
To our best knowledge, this represents the first report on thermally responsive materials that displayed unique two-photon fluorescence properties, which could expand the application of Dns-non into display or fluorescence imaging fields.
Since no 3D fluorescence imaging of silicon luminescent films has been reported previously, we were interested in exploiting the 3D luminescence image of D2-non in this regard. The 3D fluorescence images of D2-non were acquired using 405 and 561 nm excitation and fluorescence emission windows of 435–470 nm and 570–620 nm. As shown in Fig. 6(a) and (c), D2-non demonstrated homogenous strong fluorescence at a depth of up to 19.5 μm in both the blue channel and the red channel. The 3D imaging clearly confirmed the fine luminescence properties of D2-non [Fig. 6(b) and (d)]. Based on the measured uniform and stable surface properties, Dns-non were promising candidates for culturing cells on their upper surfaces. In addition, the unique fluorescence properties of Dns-non may enhance the image quality.
 | ||
| Fig. 6 (a) Planar images of D2-non in the blue channel. Magnification: 40×. (b) The 3D image of (a) at 0–19.5 μm depth. (c) Planar images of D2-non in the red channel. Magnification: 40×. (d) The 3D image of (c) at 0–19.5 μm depth, scale bar: 20 μm. Laser scanning confocal microscope photographs of HeLa cells spreading on D2-non. Magnification: 20×, scale bar: 50 μm: (e) overlay of the green, red, and blue channels, (f) from the green channel, (g) from the red channel, (h) from the blue channel, and (i) from the bright field. (j) Quantified relative fluorescence intensities for (e)–(h). The error bars represent standard deviation (±S.D.). | ||
Polysiloxanes (PDMS) have received extensive attention in the materials and polymer fields because PDMS may offer many superior physical properties.35–37 Bio-applications based on thermally responsive polysiloxane materials are also rare. Due to the importance of polysiloxane-based biomaterials for a range of applications, we investigated the application of Dns-non as cell adhesives. The toxicity of Dns-non towards living HeLa cells was assessed using the standard MTT assays. The MTT assays showed that Dns-non had little cytotoxicity to living cells (Fig. S14†). D1-non was used as the substrate to culture HeLa cells for inspecting the cytocompatibility of the material [Fig. S15†]. As shown in Fig. 6(e), the HeLa cells assembled into mature cytoskeletons, and the immunostaining results provided obvious evidence that the cell could bind to the D1-non substrate. These results made it clear that the physical cross-linking of Dns-non was complete. Films were very stable even when soaked in cell culture medium, showing that neither polymer nor rhodamine-B dissociated in cell culture medium. The cell assay indicated that the formation of adhesion is possible on silicone substrates. Hence Dns-non appeared as useful cell adhesives for the culture of cells on a surface. In addition, the quantified relative fluorescence intensities for the cell images [Fig. 6(j)] showed that both the intensity and contrast ratio of the image from the green channel was fairly weak. However, the merged image [Fig. 6(e)] showed improved contrast ratio and intensity due to the emission from the blue channel and the red channel derived from D1-non itself; thus, the fluorescence of Dns-non increased in external intensity when following cell imaging, thus enhancing the film quality.
Conclusions
In summary, new transparent films of Dns-non were fabricated based on Pns using rhodamine-B as a luminophore. Interestingly, Dns-non exhibited unique two-proton luminescence. Most importantly, the characteristic emission of rhodamine-B in Dns-non vanished after a heating treatment and recovered subsequently when cooling down. A proposed alkaline tuned reversible spirocyclization mechanism was put forward, which provided new perspective for the construction of thermally responsive materials based on conventional groups. Moreover, Dns-non expanded their application to devices when coated onto a CPU surface. Temperature changes of the CPU were easily detected by the naked eye in situ via by observing the color changes of Dns-non. Finally, the good cytocompatibility of Dns-non was demonstrated for HeLa cells which highlighted the potential of such thermally responsive materials for biological applications and cell culture. We expect that the design strategy described herein could be extended to construct more stimuli-responsive materials showing two-photon fluorescence and to explore their application in materials chemistry or bioscience.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by NSFC (21472067, 21672083), Taishan Scholar Foundation (TS201511041), and the startup fund of University of Jinan (309-10004, 1009428).Notes and references
- F. Ercole, T. P. Davis and R. A. Evans, Nervenarzt, 2010, 1, 37–54 CAS.
- Z. Song, K. Wang, C. Gao, S. Wang and W. Zhang, Macromolecules, 2016, 49, 162–171 CrossRef CAS.
- S. Dong, Y. Luo, X. Yan, B. Zheng, X. Ding, Y. Yu, Z. Ma, Q. Zhao and F. Huang, Angew. Chem., 2011, 123, 1945–1949 CrossRef.
- S. Uchiyama, Y. Matsumura, A. P. de Silva and K. Iwai, Anal. Chem., 2003, 75, 5926–5935 CrossRef CAS PubMed.
- Y. M. Lee and J. K. Shim, Polymer, 1997, 38, 1227–1232 CrossRef CAS.
- Y.-C. Chen, R. Xie and L.-Y. Chu, J. Membr. Sci., 2013, 442, 206–215 CrossRef CAS.
- J. Feng, K. Tian, D. Hu, S. Wang, S. Li, Y. Zeng, Y. Li and G. Yang, Angew. Chem., 2011, 50, 8072–8076 CrossRef CAS PubMed.
- J. Zhou and H. Ma, Chem. Sci., 2016, 7, 6309–6315 RSC.
- C. F. Chapman, Y. Liu, G. J. Sonek and B. J. Tromberg, Photochem. Photobiol., 1995, 62, 416–425 CrossRef CAS PubMed.
- R. E. Brewster, M. J. Kidd and M. D. Schuh, Chem. Commun., 2001, 12, 1134–1135 RSC.
- M. Engeser, L. Fabbrizzi, M. Licchelli and D. Sacchi, Chem. Commun., 1999, 18, 1191–1192 RSC.
- Y.-Q. Wang, Y.-Y. Zhang, F. Zhang and W.-Y. Li, J. Mater. Chem., 2011, 21, 6556–6562 RSC.
- A. P. d. Silva, H. Q. N. Gunaratne, K. R. Jayasekera, S. O’Callaghan and K. R. A. S. Sandanayake, Chem. Lett., 1995, 24, 123–124 CrossRef.
- Y. Al-Abed, T. H. Al-Tel, C. Schröder and W. Voelter, Angew. Chem., 1994, 33, 1499–1501 Search PubMed.
- H. Chen, H. Shang, Y. Liu, R. Guo and W. Lin, Adv. Funct. Mater., 2016, 26, 8128–8136 CrossRef CAS.
- D. Kim, H. Moon, S. H. Baik, S. Singha, Y. W. Jun, T. Wang, K. H. Kim, B. S. Park, J. Jung, I. Mook-Jung and K. H. Ahn, J. Am. Chem. Soc., 2015, 137, 6781–6789 CrossRef CAS PubMed.
- M. Pawlicki, H. A. Collins, R. G. Denning and H. L. Anderson, Angew. Chem., 2009, 48, 3244–3266 CrossRef CAS PubMed.
- Y. Gao, G. Feng, T. Jiang, C. Goh, L. Ng, B. Liu, B. Li, L. Yang, J. Hua and H. Tian, Adv. Funct. Mater., 2015, 25, 2857–2866 CrossRef CAS.
- C. S. Lim, G. Masanta, H. J. Kim, J. H. Han, H. M. Kim and B. R. Cho, J. Am. Chem. Soc., 2011, 133, 11132–11135 CrossRef CAS PubMed.
- B. Dong, X. Song, X. Kong, C. Wang, Y. Tang, Y. Liu and W. Lin, Adv. Mater., 2016, 28, 8755–8759 CrossRef CAS PubMed.
- M. Albota, D. Beljonne, J.-L. Brédas, J. E. Ehrlich, J.-Y. Fu, A. A. Heikal, S. E. Hess, T. Kogej, M. D. Levin, S. R. Marder, D. McCord-Maughon, J. W. Perry, H. Röckel, M. Rumi, G. Subramaniam, W. W. Webb, X.-L. Wu and C. Xu, Science, 1998, 281, 1653–1656 CrossRef CAS PubMed.
- J. H. Lee, C. S. Lim, Y. S. Tian, J. H. Han and B. R. Cho, J. Am. Chem. Soc., 2010, 132, 1216–1217 CrossRef CAS PubMed.
- H. N. Kim, M. H. Lee, H. J. Kim, J. S. Kim and J. Yoon, Chem. Soc. Rev., 2008, 37, 1465–1472 RSC.
- T. Nguyen and M. B. Francis, Org. Lett., 2003, 5, 3245–3248 CrossRef CAS PubMed.
- Z. Xu, L. Zhang, R. Guo, T. Xiang, C. Wu, Z. Zheng and F. Yang, Sens. Actuators, B, 2011, 156, 546–552 CrossRef CAS.
- B. Aydogan, D. Sureka, B. Kiskan and Y. Yagci, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5156–5162 CrossRef CAS.
- L. Xue, D. Wang, Z. Yang, Y. Liang, J. Zhang and S. Feng, Eur. Polym. J., 2013, 49, 1050–1056 CrossRef CAS.
- M. Arslan, B. Kiskan and Y. Yagci, Macromolecules, 2015, 48, 1329–1334 CrossRef CAS.
- Y. Zuo, J. Cao and S. Feng, Adv. Funct. Mater., 2015, 25, 2754–2762 CrossRef CAS.
- Y. Zuo, Z. Gou, J. Cao, Z. Yang, H. Lu and S. Feng, Chem.–Eur. J., 2015, 21, 10972–10977 CrossRef CAS PubMed.
- K.-S. Kim, M.-H. Won, J.-W. Kim and B.-J. Back, Appl. Therm. Eng., 2003, 23, 1137–1144 CrossRef.
- S. Gima, T. Nagata, X. Zhang and M. Fujii, Heat Tran. Asian Res., 2005, 34, 147–159 CrossRef.
- X. Chen, S.-W. Nam, M. J. Jou, Y. Kim, S.-J. Kim, S. Park and J. Yoon, Org. Lett., 2008, 10, 5235–5238 CrossRef CAS PubMed.
- Q.-H. Liu, J. Liu, J.-C. Guo, X.-L. Yan, D.-H. Wang, L. Chen, F.-Y. Yan and L.-G. Chen, J. Mater. Chem., 2009, 19, 2018–2025 RSC.
- F. Abbasi, H. Mirzadeh and A. A. Katbab, Polym. Int., 2001, 50, 1279–1287 CrossRef CAS.
- J. Chojnowski and M. Cypryk, in Silicon-Containing Polymers, Springer, 2000, pp. 3–41 Search PubMed.
- H. Yang, M. Xu, L.-X. Guo, H.-F. Ji, J.-Y. Wang, B.-P. Lin, X.-Q. Zhang and Y. Sun, RSC Adv., 2015, 5, 7304–7310 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc05080a |
| This journal is © The Royal Society of Chemistry 2018 |