
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hydrogen peroxide-triggered gene silencing in mammalian cells through boronated antisense oligonucleotides†
Shohei
Mori
a,
Kunihiko
Morihiro‡
*ab,
Takumi
Okuda
a,
Yuuya
Kasahara
ab and
Satoshi
Obika
 *ab
*ab
aGraduate School of Pharmaceutical Sciences, Osaka University, 1-6 Yamadaoka, Suita, Osaka 565-0871, Japan. E-mail: morihiro@bioorg.rcast.u-tokyo.ac.jp; obika@phs.osaka-u.ac.jp
bNational Institutes of Biomedical Innovation, Health and Nutrition (NIBIOHN), 7-6-8 Saito-Asagi, Ibaraki, Osaka 567-0085, Japan
First published on 6th December 2017
Abstract
Hydrogen peroxide (H2O2) is a reactive oxygen species (ROS) involved in various diseases, including neurodegeneration, diabetes, and cancer. Here, we introduce a new approach to use H2O2 to modulate specific gene expression in mammalian cells. H2O2-responsive nucleoside analogues, in which the Watson–Crick faces of the nucleobases are caged by arylboronate moieties, were synthesized. One of these analogues, boronated thymidine (dTB), was incorporated into oligodeoxynucleotides (ODNs) using an automated DNA synthesizer. The hybridization ability of this boronated ODN to complementary RNA was clearly switched in the off-to-on direction upon H2O2 addition. Furthermore, we demonstrated H2O2-triggered gene silencing in mammalian cells using antisense oligonucleotides (ASOs) modified with dTB. Our approach can be used for the regulation of any gene of interest by the sequence design of boronated ASOs and will contribute to the development of targeted disease therapeutics.
Introduction
Nucleic acids are fundamental molecules for biological processes as they are responsible for the storage, regulation, and expression of genetic information. Modulating the biological activities of nucleic acids is of value for therapeutic and diagnostic applications. Antisense oligonucleotides (ASOs) are among the most promising oligonucleotides for the down-regulation of specific genes.1 ASOs hybridize with complementary mRNA to inhibit translation and reduce specific gene expression though the RNAse H degradation process or to modulate mRNA splicing pathways. Several antisense drugs have been approved by the US FDA in recent years.2–5 However, although ASOs are promising tools to control specific gene expression, a considerable drawback of the technology is the inability to regulate the activity of ASOs in a designated manner. If the activities of ASOs could be strictly controlled, as required for therapeutic use, their dosage could be reduced while maintaining their efficiency, and lesion specific knockdown of the target gene would be realized, reducing the off-target effect. Photo-caging technology represents an effective means to the development of the spatial and temporal control of ASO activity.6–16 In particular, nucleobase-caged ASOs have been systematically investigated, thus facilitating the manipulation of biological phenomena by photo-irradiation.17–22 However, the use of other stimuli, such as small molecule-responsive caged ASOs, remain largely unexplored.23 The chemical environment in our body, as well as inside cells, differs in space and over time. The ability of caged nucleic acids to alter their properties under a specific biomarker in a lesion would provide new options for nucleic acid therapeutics.Some diseased cells, such as inflamed cells24 and cancer cells,25 exhibit elevated intrinsic oxidative stress, and the amounts of reactive oxygen species (ROS) are increased. Thus, the development of molecular probes toward ROS is one of the best intriguing research fields.26 Hydrogen peroxide (H2O2) is an ROS that exhibits good reactivity and is present in significant concentrations in vivo, and is thus an ideal candidate as a biomarker of tumours. Compared with normal cells, cancer cells have increased levels of H2O2 of up to 0.5 nmol/104 cells per h.25 There is therefore great interest in developing ASOs that are activated by excess H2O2. We designed a series of boronated nucleosides (dTBpin, dABpin, dCBpin and dGBpin) for the H2O2-triggered activation of ASOs (Fig. 1a). Arylboronic acids and their esters can effectively temporally mask their molecular activity. They are oxidized to phenol derivatives by H2O2 and subsequent 1,6-elimination releases the active molecule (Fig. 1b). Boronated small molecules have been used for the fluorescent detection of H2O2,27 selective gene activation,28 and as cancer therapeutics.29–31 To adapt H2O2-responsive small molecule technology to ASOs, we masked the Watson–Crick face of the nucleobase with pinacol borane. The sterically hindered arylboronate groups will interfere with duplex formation between the ASO and target mRNA. In the presence of H2O2, the boronate group will be swiftly removed, resulting in gene silencing through hybridization of the ASO to the target mRNA (Fig. 1c).
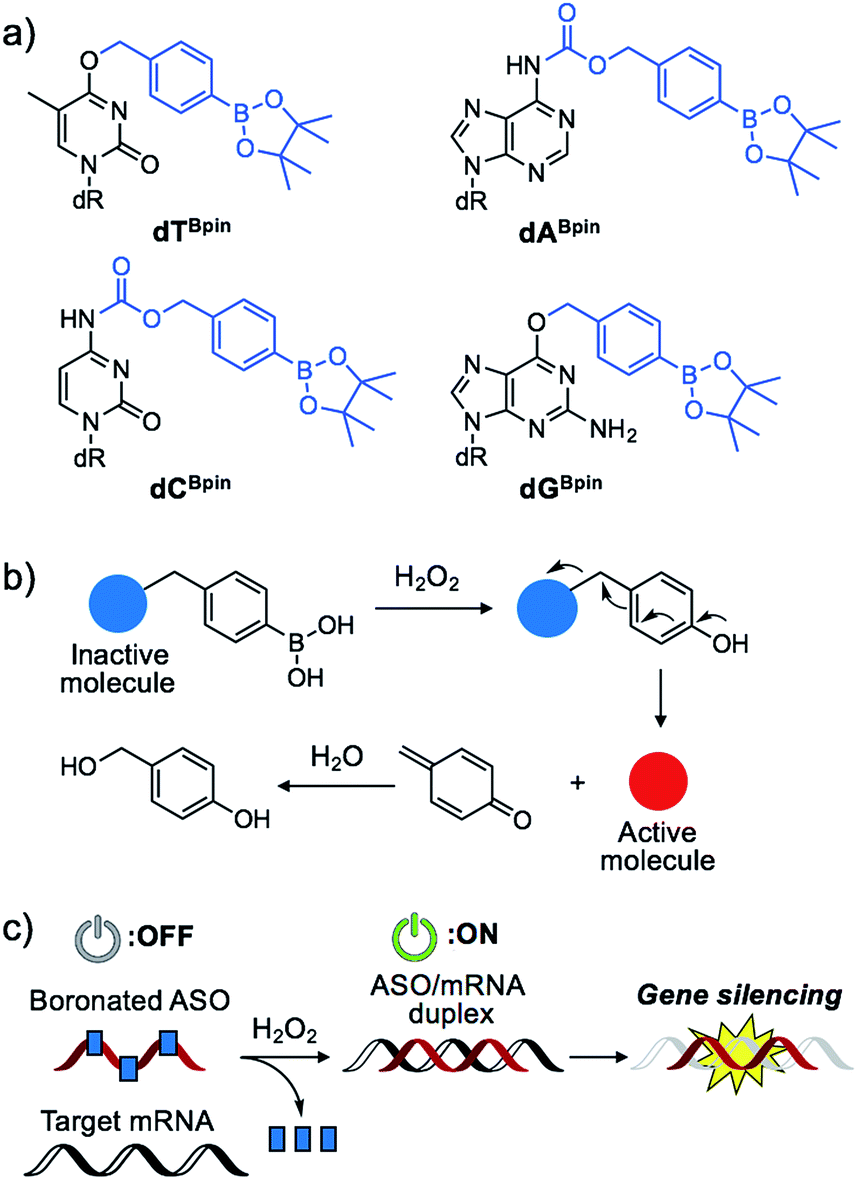 | ||
| Fig. 1 (a) Chemical structure of boronated 2′-deoxyribonucleoside analogues synthesized in this study. (b) Schematic of H2O2-triggered molecule activation. (c) Gene silencing triggered by H2O2 through inactive boronated ASOs. | ||
Results and discussion
Synthesis of boronated nucleosides
The synthesis of boronated nucleosides was started from suitably silylated sugars (Scheme 1). The H2O2 responsive pinacol borane moiety was introduced through a benzylic linker by SNAr reaction of a triazolyl nucleoside derivative32 for dT and by Mitsunobu reaction for dG. The removal of TIPDS by TBAF afforded boronated nucleoside dTBpin (4) and dGBpin (6). In contrast, dABpin and dCBpin have an arylboronate moiety linked via a carbamate spacer on their nucleobase. 4-(Hydroxymethyl) phenylboronic acid pinacol ester 1 was converted to the imidazole derivative in 2 steps. The imidazole was exposed to Meerwein reagent to prepare highly reactive imidazolium salt 2in situ, then reacted with TIPDS-protected dC or dA. TBAF treatment caused degradation of the carbamate moiety, and therefore we used HF-pyridine or TASF for silyl deprotection to afford dABpin (8) and dCBpin (10) (Scheme 1).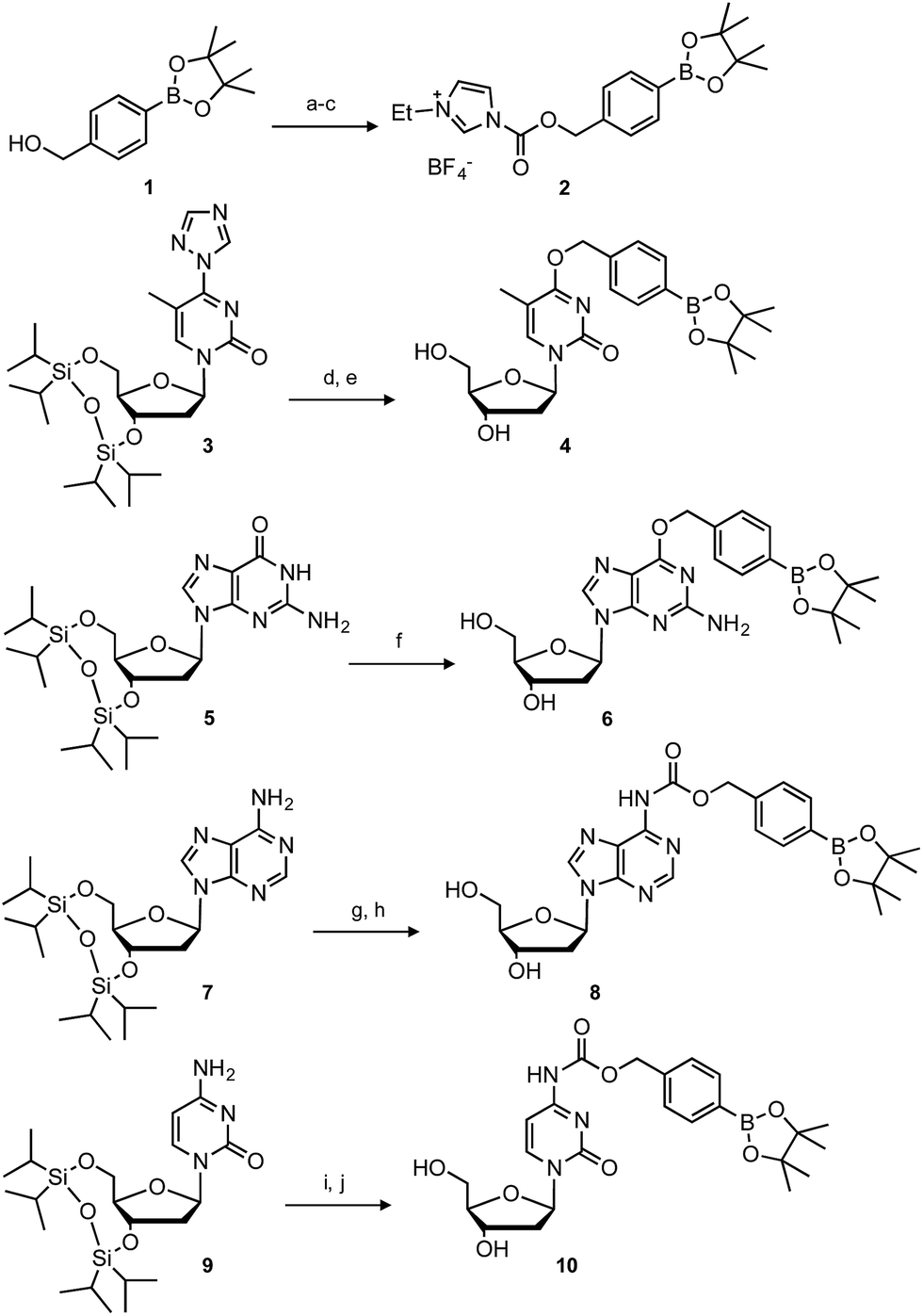 | ||
| Scheme 1 Synthesis of boronated nucleoside analogues. Conditions: (a) triphosgene, Na2CO3, toluene, rt; (b) imidazole, toluene, rt, 87% over 2 steps; (c) Et3OBF4, DCM, rt; (d) 1, DBU, MeCN, rt, 98%; (e) TBAF, THF, 0 °C, 72%; (f) 1, DIAD, PPh3, 1,4-dioxane, rt, then TBAF, 10%; (g) 2, DCM, 0 °C to rt, 91%; (h) TASF, DMF, 0 °C, 62%; (i) 2, DCM, 0 °C to rt, 92%; (j) HF-pyridine, 60 °C, 52%. | ||
H2O2-decaging of boronated nucleosides
With the caged nucleoside monomers in hand, we determined their conversion yields to the corresponding natural nucleosides by H2O2 treatment using HPLC analysis. Exposed to the equimolar equivalent of H2O2 in sodium phosphate buffer (pH = 7.2), virtually all of dTBpin was converted to thymidine within 20 min (96% yield, Fig. 2a). Other boronated nucleosides were also promptly converted to the corresponding nucleosides in 67–88% yields (Fig. 2b and ESI†). The lower yield of dGBpin was due to the generation of a side product, possibly the 8-oxo-dG analogue.33,34 Of the synthesized nucleoside analogues, dTBpin showed the best reaction yield and kinetics for H2O2-decaging. To provide further insights into the decaging mechanism, we examined the inducible reactivity of dTBpin toward other ROS, such as tert-butyl-hydroperoxide (TBHP), hypochlorite (ClO−), hydroxyl radical, tert-buthoxy radical, superoxide (O2−), and nitric oxide. We found that the decaging reaction of dTBpin was highly selective for H2O2 over the other ROS (Fig. 2c). The selective reaction of dTBpin with H2O2 is consistent with the observations of other groups.28–31 We also checked the reactivity of dTBpin toward peroxynitrite (ONOO−), which is reported to be a potential oxidant of arylboronic acid moieties.35 However, in the same reaction conditions, almost no conversion to thymidine was observed (see HPLC chromatograms in ESI†). The half-life of ONOO− in neutral buffer is extremely short36 and an equimolar equivalent of ONOO− may not be sufficient for dTBpin decaging.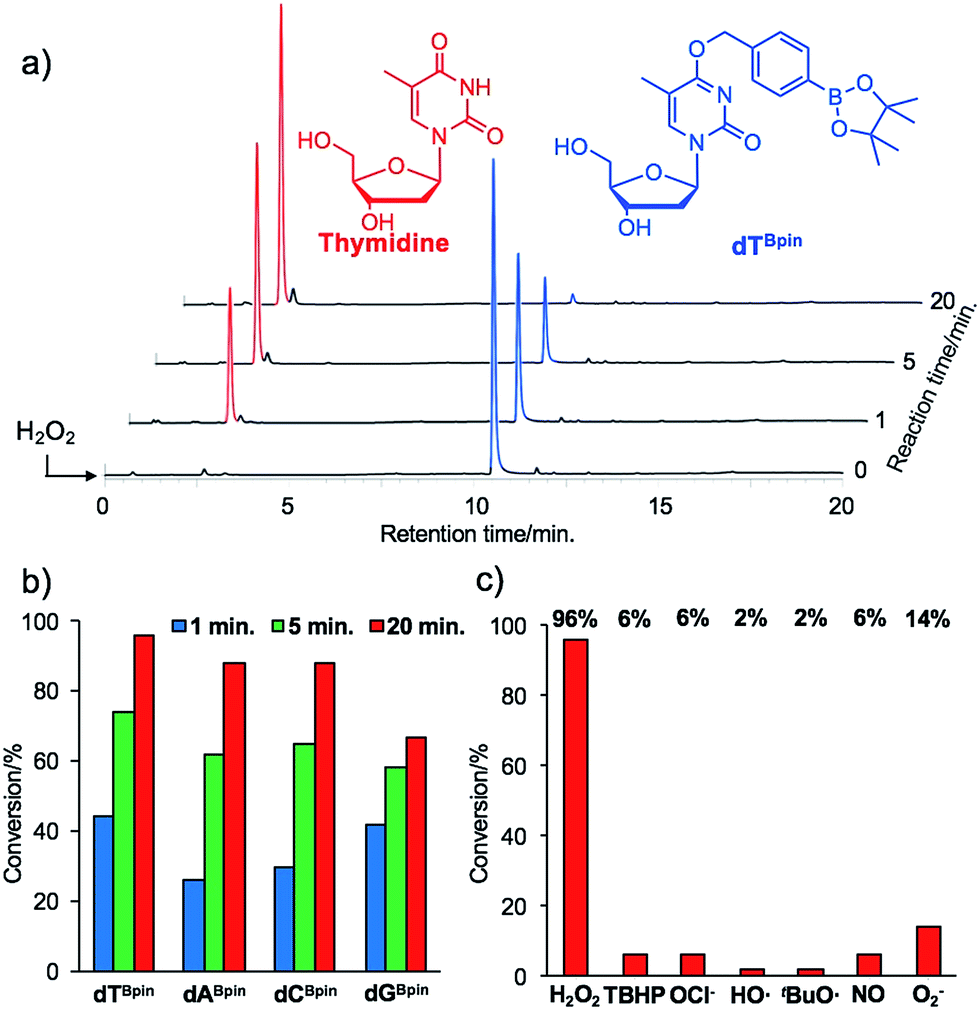 | ||
| Fig. 2 (a) HPLC chromatograms of dTBpin after H2O2 addition at different time points. (b) Conversion rate of each boronated nucleoside analogue to the corresponding natural nucleoside. (c) H2O2-selectivity of dTBpin toward other ROS. | ||
Boronated ODN synthesis
Although the introduction of arylboronate groups into oligodeoxynucleotides (ODNs) is very interesting due to their characteristic function,37,38 the direct incorporation of arylboronic acid moieties using solid phase synthesis is challenging because of their hygroscopicity. Wang and co-workers reported the synthesis of arylboronic acid-modified ODNs using a post-synthetic approach.38 However, this strategy limits the design of boronated ODNs. We predicted that phosphoramidites modified with pinacol borane would be less hygroscopic and could be applied to direct DNA synthesis.We incorporated dTBpin into ODN to take into account both the decaging efficiency and stability of the ODN under DNA synthesis conditions. Fortunately, phosphoramidite 11, prepared from dTBpin nucleoside 4 (see ESI† for the synthesis), could be incorporated into the ODN using standard solid phase synthesis procedures. After cleavage from the solid support with ammonium hydroxide, the protecting groups were removed by exposure to 50 mM K2CO3 in methanol (Fig. 3a). Subsequent HPLC purification afforded highly pure ODN14 in which the pinacol ester group of dTBpin was hydrolysed (see MALDI-TOF and ESI MS analysis in ESI†). ODN14 was observed as the dehydrated form on ESI MS that corresponds to Wang's report.38 To the best of our knowledge, this is the first example of the incorporation of the arylboronic acid group into an ODN without the need to use a post-synthetic approach.
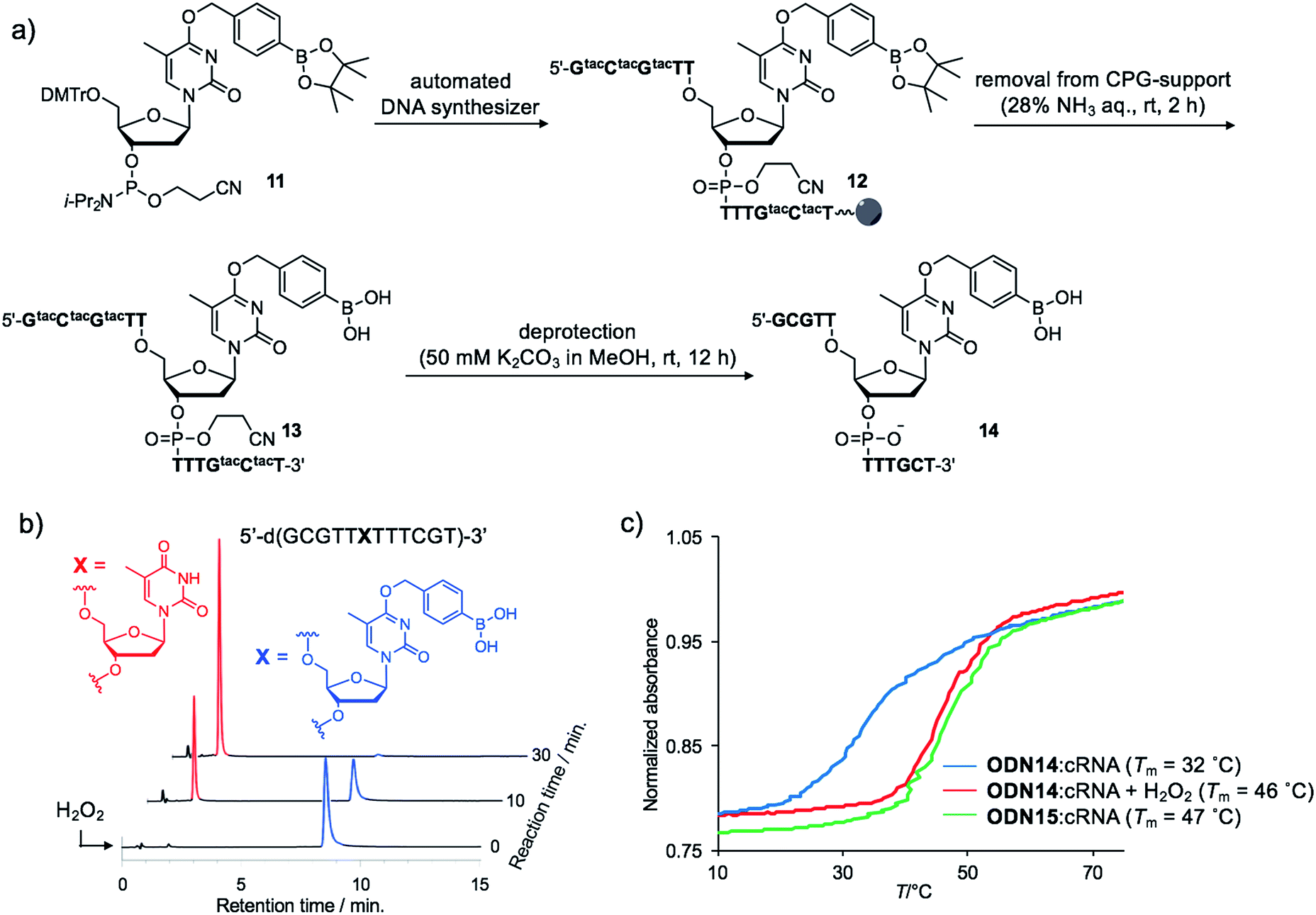 | ||
| Fig. 3 (a) Synthetic scheme of boronated ODN. tac = tert-butylphenoxyacetyl. (b) HPLC chromatograms of ODN14 after H2O2 addition at different time points. (c) Thermal melting curves of the duplex formed between ODN14 and complementary RNA in the presence or absence of H2O2. | ||
H2O2-activation of boronated ODN
Next, we analysed the H2O2 reactivity of dTB-modified ODN14 by HPLC (Fig. 3b). When ODN14 was exposed to an excess amount of H2O2 at rt, the ODN14 peak at 8.8 min completely disappeared and another peak appeared within 30 min in the HPLC profile. The resulting ODN, with a retention time of 2.0 min, was shown by MALDI-TOF MS spectrometry to be ODN14 in which the boronic acid moiety of dTB was absent (see MALDI-TOF MS analysis in SI). The oxidation of dTB by H2O2 remained clean and rapid.The duplex-forming ability of ODN14 toward its complementary RNA (cRNA) strand was measured in the presence and absence of H2O2. Under the H2O2-free condition, the modified duplex ODN14:cRNA (Fig. 3c: blue line) was significantly destabilized compared with the unmodified ODN15:cRNA duplex (Fig. 3c: green line) (ΔTm = 14 °C). The stabilities of the base pairs formed between TBpin and other nucleobases (G, U, and C) were also lower than the T:A base pair (Table S1†). These results indicate that the boronic acid moiety effectively inhibits complementary base pairing, probably due to steric hindrance. The Tm of ODN14:cRNA in the presence of H2O2 (Fig. 3c: red line) was comparable to that of ODN15:cRNA (ΔTm = 1 °C). The mismatch discrimination ability of ODN14 was not decreased by H2O2 addition (Table S1†). These excellent hybridization properties of ODN14 were also seen toward complementary DNA (Table S2†). These results suggest that dTB-modified ODNs enable switching of their hybridization to complementary strand in the off-to-on direction upon H2O2 treatment.
H2O2-triggered gene silencing in mammalian cells
The Tm results prompted us to use the H2O2-responsive caged ODNs in biological systems. We incorporated dTB into the gap region of ASO targeting Mus Scarb1 mRNA (Fig. 4a). Scarb1 has been implicated in several tumours,39 and targeted ASOs may provide a novel cancer therapeutic approach. S0 includes phosphorothioate and locked nucleic acid (LNA) modifications to increase nuclease resistance and affinity toward the target mRNA, respectively. To determine the effect of boronate caging groups on antisense potency, we synthesized a set of three ASOs S1–S3 containing 1–3 dTB units. Hepa-1c1c7 cells, a line derived from mice hepatoma, were treated with the ASOs by simple addition to the medium with CaCl2 (ref. 40) and incubated for 48 h in the absence or presence of H2O2 (10 μM). Scarb1 mRNA levels were then quantified using qRT-PCR (Fig. 4b). Under the H2O2 free condition, the knockdown efficiency of target mRNA by S1 was almost the same as that by S0 (positive control). On the other hand, S2 and S3 showed low antisense efficiency, similar to SA (negative control). This result indicates that at least two dTB units are necessary to mask the gene silencing potency of the ASO. In contrast, when exposed to 10 μM H2O2, the antisense efficiencies of S2 and S3 are comparable to that of S0. The negligible change in gene silencing upon H2O2 treatment without ASO indicates that 10 μM H2O2 does not affect the expression level of Mus Scarb1 mRNA. However, SA showed about 15% lower mRNA expression after H2O2 treatment, suggesting the combination of the modified oligonucleotide and H2O2 may affect the Scarb1 mRNA expression level. Targeting other mRNAs or applying other chemical modifications could help to obtain further insights for this phenomenon. Taken together, these results clearly demonstrate that the ASOs S2 and S3 enable excellent off-to-on switching of gene silencing in mammalian cells triggered by H2O2.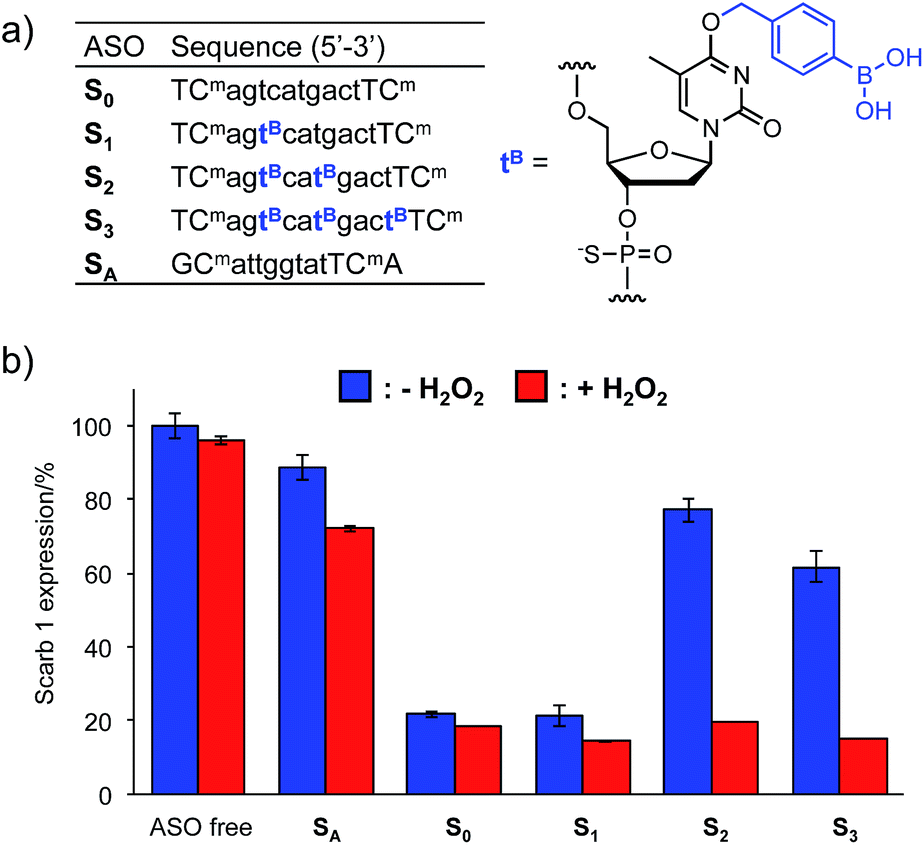 | ||
| Fig. 4 (a) Sequences of ASOs. n = DNA, N = LNA (Cm = LNA-5-Me-cytidine), all internucleosidic linkages are phosphorothioated. (b) Intercellular gene silencing triggered by H2O2 using boronate ASOs. Three independent experiments were averaged and the error bars represent standard deviation. | ||
Conclusions
We designed and synthesized arylboronate-modified nucleosides which are promptly decaged and converted to natural nucleosides by H2O2. The boronated thymidine dTBpin phophoramidite could be directly used in automated DNA synthesis and a TB:A base pair in the duplex could be employed as a temporary mismatch until H2O2 addition. Moreover, dTB-modified ASOs were demonstrated to be H2O2 switches that can control target mRNA expression. H2O2 is known to be involved in various diseases such as cancer, neurodegeneration,41 and diabetes.42 Our results indicate that boronated ASOs are promising tools to control target gene expression in a designated manner and as a prodrug-type nucleic acid therapeutic for selective disease treatment. Furthermore, the design of boronated nucleotides can be easily adapted to other nucleic acid agents such as aptamers, triplex-forming oligonucleotides (TFOs), and small interfering RNAs (siRNAs).Experimental
General
Reagents and solvents were purchased from commercial suppliers and used without purification unless otherwise specified. All experiments involving air and/or moisture sensitive compounds were carried out under an Ar atmosphere. All reactions were monitored with analytical TLC (Merck Kieselgel 60 F254; Merck, Darmstadt, Germany). Flash column chromatography was carried out using EPCLC-W-Prep 2XY (YAMAZEN, Osaka, Japan). Physical data were measured as follows. NMR spectra were recorded on a JNM-ECS-400 spectrometer (JEOL, Tokyo, Japan) using CDCl3 or DMSO-d6 as the solvent with tetramethylsilane as an internal standard. IR spectra were recorded on a FT/IR-4200 spectrophotometer (JASCO, Tokyo, Japan). Optical rotations were recorded on a JASCO P-2200 instrument. FAB mass spectra were measured using a JEOL JIM-700 mass spectrometer. Solid-phase ODN synthesis was performed using an nS-8 II oligonucleotide synthesizer (GeneDesign, Osaka, Japan). MALDI-TOF mass spectra were recorded on an ultrafleXtreme mass spectrometer (Bruker Daltonics, Billerica, MA, USA). ESI mass spectra were recorded on a Xevo G2-XS QTof (Waters, Milford, MA, USA). UV/vis absorption measurements and UV melting experiments were performed using a UV-1650PC UV-vis spectrophotometer equipped with a TMSPC-8 Tm analysis accessory (SHIMADZU, Kyoto, Japan).H2O2-decaging of boronated nucleosides
A reaction solution of boronated nucleoside (1 mM) and H2O2 (1 mM) in a DMSO-containing buffer (10 mM potassium phosphate, pH 7.2, 100 mM NaCl, and DMSO 5% v/v) was incubated at room temperature for the prescribed times and immediately subjected to reversed-phase HPLC analysis using MeCN in 0.1 M triethylammonium acetate buffer (pH 7.0). The conversion rate of boronated nucleoside was determined from the corresponding peak area monitored at 260 nm.Other ROS-decaging of dTBpin
A reaction solution of boronated nucleoside (1 mM) and reactive oxygen species (1 mM) in a DMSO-containing buffer (10 mM potassium phosphate, pH 7.2, 100 mM NaCl, and DMSO 5% v/v) was incubated at room temperature for 12 h and immediately subjected to reversed-phase HPLC analysis using MeCN in 0.1 M triethylammonium acetate buffer (pH 7.0). H2O2, tert-butylhydroperoxide (TBHP), and hypochlorite (NaOCl) were delivered from 30%, 70%, and 10% aqueous solutions respectively. Hydroxyl radical (HO˙) and tert-butoxy radical (tBuO˙) were generated by the reaction of 5 mM (NH4)2Fe(SO4)2, 10 mM EDTA with 1 mM H2O2 or TBHP, respectively. Nitric oxide (NO) was generated from PROLI NONOate. Superoxide (O2−) was produced by xanthine oxidase (4.5 × 10−3 mg/100 μL) in the presence of hypoxanthine (2 mM) and catalase (0.4 mg mL−1). Peroxynitrite (ONOO−) was delivered from NaOH aqueous solution and the concentration of ONOO− was determined using the absorption at 300 nm (ε = 1670 M−1 cm−1). The solution was diluted with phosphate buffer and used immediately. The conversion rate of boronated nucleoside was determined from the corresponding peak area monitored at 260 nm.ODN synthesis
Solid-phase ODN synthesis was performed using commercially available reagents and phosphoramidites. dTBpin phosphoramidite was chemically synthesized as described (Scheme S1†). ODNs were synthesized (with trityl-on) on a 500 Å CPG solid support column (1 μmol scale) using 5-ethylthio-1H-tetrazole (0.25 M in MeCN) as the activator and 0.05 M ((dimethylaminomethylidene)amino)-3H-1,2,4-dithiazaoline-3-thione (DDTT) in pyridine/MeCN (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) for thiolation. The standard synthesis cycle was used for assembly of the reagents and synthesis of the oligonucleotides, except that the coupling time was extended to 5 min for LNA monomers. Cleavage from the solid support and deprotection were accomplished with 28% w/w NH4OH aqueous solution at room temperature for 2 h and potassium carbonate (0.05 M in methanol solution) at room temperature for 12 h, respectively. The crude ODNs were purified on a Sep Pack column (Waters) followed by RP-HPLC on an XBridge™ OST C18 column, 2.5 μm, 10 × 50 mm (Waters) using MeCN in 0.1 M triethylammonium acetate buffer (pH 7.2). The purified ODNs were quantified by UV absorbance at 260 nm and confirmed by MALDI-TOF or ESI mass spectrometry. In ESI MS measurement, boronated ODN and ASO were detected as the dehydrated form, which is consistent with the observation of Wang's group.38
:1, v/v) for thiolation. The standard synthesis cycle was used for assembly of the reagents and synthesis of the oligonucleotides, except that the coupling time was extended to 5 min for LNA monomers. Cleavage from the solid support and deprotection were accomplished with 28% w/w NH4OH aqueous solution at room temperature for 2 h and potassium carbonate (0.05 M in methanol solution) at room temperature for 12 h, respectively. The crude ODNs were purified on a Sep Pack column (Waters) followed by RP-HPLC on an XBridge™ OST C18 column, 2.5 μm, 10 × 50 mm (Waters) using MeCN in 0.1 M triethylammonium acetate buffer (pH 7.2). The purified ODNs were quantified by UV absorbance at 260 nm and confirmed by MALDI-TOF or ESI mass spectrometry. In ESI MS measurement, boronated ODN and ASO were detected as the dehydrated form, which is consistent with the observation of Wang's group.38
H2O2-decaging of boronated ODN
A reaction solution of ODN14 (4 μM) and H2O2 (1 mM) in a buffer (10 mM potassium phosphate, pH 7.2, 100 mM NaCl) was incubated at room temperature for the prescribed times and immediately subjected to reversed-phase HPLC analysis using MeCN in 0.1 M triethylammonium acetate buffer (pH 7.0).UV melting experiments
Melting temperatures (Tm) were determined by measuring the change in absorbance at 260 nm as a function of temperature using a SHIMADZU UV-vis UV-1650PC spectrophotometer equipped with a TMSPC-8 Tm analysis accessory. The melting samples were denatured at 100 °C and annealed slowly to room temperature. Absorbance was recorded in the forward and reverse direction at temperatures between 5 to 90 °C at a rate of 0.5 °C min−1.Cell culture
Hepa-1c1c7 cells were obtained from the European Collection of Authenticated Cell Cultures (ECACC; Salisbury, UK). The cell line was maintained at 37 °C and 5% CO2 in Dulbecco's Modified Eagle's Medium (DMEM; Nacalai Tesque, Kyoto, Japan) supplemented with 10% heat-inactivated foetal bovine serum (FBS) and antibiotics.Evaluation of the in vitro activity of dTB-modified ASOs in the absence or presence of H2O2
Hepa-1c1c7 cells were seeded at 5.0 × 103 cells per well in 96-well plates (Corning, New York, NY, US) containing 10% FBS/DMEM. After 24 h, 100 nM ASO was added and the cells were cultured in medium supplemented with 9 mM CaCl2 in the absence or presence of H2O2 (10 μM). After 48 h, total RNA was isolated using a QIAGEN RNeasy kit (QIAGEN, Valencia, CA, USA). Reduction of target mRNA expression was determined by real time qRT-PCR using a One Step SYBR PrimeScript PLUS RT-PCR kit (Takara Bio Inc., Kusatsu, Japan) and analysed with a StepOnePlus system (Applied Biosystems; Foster City, CA, USA). The primers used in this study were specific for the mouse Scarb1 gene (forward: 5′-TGACAACGACACCGTGTCCT-3′; reverse: 5′-ATGCGACTTGTCAGGCTGG-3′) and for the mouse GAPDH gene (forward: 5′-TGCACCACCAACTGCTTAG-3′; reverse: 5′-GATGCAGGGATGATGTTC-3′). The level of target (Scarb1) gene expression was normalized to that of GAPDH in the absence of H2O2.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by KAKENHI from the Japan Society for the Promotion of Science (JSPS) and the Platform Project for Supporting Drug Discovery and Life Science Research from the Japan Agency for Medical Research and Development (AMED). S. M. is grateful for a Research Fellowship for Young Scientists from JSPS.Notes and references
- T. Yamamoto, M. Nakatani, K. Narukawa and S. Obika, Future Med. Chem., 2011, 3, 339–365 CrossRef CAS PubMed.
- C. M. Perry and J. A. Balfour, Drugs, 1999, 57, 375–380 CrossRef CAS PubMed.
- R. S. Geary, B. F. Baker and S. T. Crooke, Clin. Pharmacokinet., 2015, 54, 133–146 CrossRef CAS PubMed.
- K. R. Q. Lim, R. Maruyama and T. Yokota, Drug Des., Dev. Ther., 2017, 11, 533–545 CrossRef PubMed.
- E. W. Ottesen, Transl. Neurosci., 2017, 8, 1–6 CrossRef PubMed.
- I. A. Shestopalov, S. Sinha and J. K. Chen, Nat. Chem. Biol., 2007, 3, 650–651 CrossRef CAS PubMed.
- X. Ouyang, I. A. Shestopalov, S. Sinha, G. Zheng, C. L. W. Pitt, W. Li, A. J. Olson and J. K. Chen, J. Am. Chem. Soc., 2009, 131, 13255–13269 CrossRef CAS PubMed.
- X. Tang, M. Su, L. Yu, C. Lv, J. Wang and Z. Li, Nucleic Acids Res., 2010, 38, 3848–3855 CrossRef CAS PubMed.
- G. Zheng, L. Cochella, J. Liu, O. Hobert and W. Li, ACS Chem. Biol., 2011, 6, 1332–1338 CrossRef CAS PubMed.
- S. Yamazoe, I. A. Shestopalov, E. Provost, S. D. Leach and J. K. Chen, Angew. Chem., Int. Ed., 2012, 51, 6908–6911 CrossRef CAS PubMed.
- A. Tallafuss, D. Gibson, P. Morcos, Y. Li, S. Seredick, J. Eisen and P. Washbourne, Development, 2012, 139, 1691–1699 CrossRef CAS PubMed.
- J. C. Griepenburg, B. K. Ruble and I. J. Dmochowski, Bioorg. Med. Chem., 2013, 21, 6198–6204 CrossRef CAS PubMed.
- L. Wu, Y. Wang, J. Wu, C. Lv, J. Wang and X. Tang, Nucleic Acids Res., 2013, 41, 677–686 CrossRef CAS PubMed.
- S. Yamazoe, Q. Liu, L. E. McQuade, A. Deiters and J. K. Chen, Angew. Chem., Int. Ed., 2014, 53, 10114–10118 CrossRef CAS PubMed.
- J. C. Griepenburg, T. L. Rapp, P. J. Carroll, J. Eberwine and I. J. Dmochowski, Bioorg. Med. Chem., 2013, 21, 6198–6204 CrossRef CAS PubMed.
- A. Y. Payumo, L. E. McQuade, W. J. Walker, S. Yamazoe and J. K. Chen, Nat. Chem. Biol., 2016, 12, 694–701 CrossRef CAS PubMed.
- D. D. Young, H. Lusic, M. O. Lively, J. A. Yoder and A. Deiters, ChemBioChem, 2008, 9, 2937–2940 CrossRef CAS PubMed.
- A. Deiters, R. A. Garner, H. Lusic, J. M. Govan, M. Dush, N. M. Nascone-Yoder and J. A. Yoder, J. Am. Chem. Soc., 2010, 132, 15644–15650 CrossRef CAS PubMed.
- C. M. Connelly, R. Uprety, J. Hemphill and A. Deiters, Mol. BioSyst., 2012, 8, 2987–2993 RSC.
- J. M. Govan, R. Uperty, M. Thomas, H. Lusic, M. O. Lively and A. Deiters, ACS Chem. Biol., 2013, 8, 2272–2282 CrossRef CAS PubMed.
- F. Schäfer, J. Wagner, A. Knau, S. Dimmeler and A. Heckel, Angew. Chem., Int. Ed., 2013, 52, 13558–13561 CrossRef PubMed.
- J. Hemphill, Q. Liu, R. Uprety, S. Samanta, M. Tsang, R. L. Juliano and A. Deiters, J. Am. Chem. Soc., 2015, 137, 3656–3662 CrossRef CAS PubMed.
- M. Ikeda and M. Kabumoto, Chem. Lett., 2017, 46, 634–640 CrossRef CAS.
- L. Rong, C. Zhang, Q. Lei, M. M. Hu, J. Feng, H. B. Shu, Y. Liu and X. Z. Zhang, Regener. Biomater., 2016, 3, 217–222 CrossRef CAS PubMed.
- T. P. Szatrowski and C. F. Nathan, Cancer Res., 1991, 51, 794–798 CAS.
- X. Chen, F. Wang, J. Y. Hyun, T. Wei, J. Qiang, X. Ren, I. Shin and J. Yoon, Chem. Soc. Rev., 2016, 45, 2976–3016 RSC.
- A. R. Lippert, G. C. Van De Bittner and C. J. Chang, Acc. Chem. Res., 2011, 44, 793–804 CrossRef CAS PubMed.
- J. M. Govan, A. L. Mclver, C. Riggsbee and A. Deiters, Angew. Chem., Int. Ed., 2012, 51, 9066–9070 CrossRef CAS PubMed.
- Y. Kuang, K. Balakrishnan, V. Gandhi and X. Peng, J. Am. Chem. Soc., 2011, 133, 19278–19281 CrossRef CAS PubMed.
- E. J. Kim, S. Bhuniya, H. Lee, H. M. Kim, C. Cheong, S. Maiti, K. S. Hong and J. S. Kim, J. Am. Chem. Soc., 2014, 136, 13888–13894 CrossRef CAS PubMed.
- H. W. Liu, X. X. Hu, K. L. Y. Liu, Q. Rong, L. Zhu, L. Yuan, F. L. Qu, X. B. Zhang and W. Tan, Chem. Sci., 2017, 8, 7689–7695 RSC.
- V. Vendrell-Criado, G. M. Rodriguez-Muñiz, M. C. Cuquerella, V. Lhiaubet-Vallet and M. A. Miranda, Angew. Chem., Int. Ed., 2013, 52, 6476–6479 CrossRef CAS PubMed.
- P. Zhao, L. Zhou, Z. Nie, X. Xu, W. Li, Y. Huang, K. He and S. Yao, Anal. Chem., 2013, 85, 6279–6286 CrossRef CAS PubMed.
- T. Sekiguchi, R. Ito, H. Hayakawa and M. Sekiguchi, J. Biol. Chem., 2013, 288, 8128–8135 CrossRef CAS PubMed.
- J. Zielonka, A. Sikora, M. Hardy, J. Joseph, B. P. Dranka and B. Kalyanaraman, Chem. Res. Toxicol., 2012, 25, 1793–1799 CrossRef CAS PubMed.
- R. Radi, J. Biol. Chem., 2013, 37, 26464–26472 CrossRef PubMed.
- A. E. Hargrove, A. D. Ellington, E. V. Anslyn and J. L. Sessler, Bioconjugate Chem., 2011, 22, 388–396 CrossRef CAS PubMed.
- C. Dai, L. Wang, J. Sheng, H. Peng, A. B. Draganov, Z. Huang and B. Wang, Chem. Commun., 2011, 47, 3598–3600 RSC.
- A. Lacko, M. Nair, S. Paranjape, S. Johnson and W. McConathy, Anticancer Res., 2002, 22, 2045–2049 CAS.
- S. Hori, T. Yamamoto, R. Waki, F. Wada, M. Noda and S. Obika, Nucleic Acids Res., 2015, 43, e128 CrossRef PubMed.
- S. DiMauro and E. A. Schon, Annu. Rev. Neurosci., 2008, 31, 91–123 CrossRef CAS PubMed.
- R. Pop-Busui, A. Sima and M. Stevens, Diabetes/Metab. Res. Rev., 2006, 22, 257–273 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Full experimental details, 1H, 13C and 31P spectra of all new compounds, HPLC charts, ESI-MS spectra of all new ODNs and representative UV melting data. See DOI: 10.1039/c7sc04318j |
| ‡ Present address: Department of Chemistry and Biotechnology, Graduate School of Engineering, The University of Tokyo. |
| This journal is © The Royal Society of Chemistry 2018 |