
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffects of molecular architecture on morphology and photophysics in conjugated polymers: from single molecules to bulk
Zhongjian
Hu†
 *,
Beiyue
Shao‡
,
Geoffrey T.
Geberth‡
and
David A.
Vanden Bout
*
*,
Beiyue
Shao‡
,
Geoffrey T.
Geberth‡
and
David A.
Vanden Bout
*
Department of Chemistry, University of Texas at Austin, USA. E-mail: dvandenbout@cm.utexas.edu
First published on 4th January 2018
Abstract
Conjugated polymers (CPs) possess a wide range of desirable properties, including accessible energetic bandgaps, synthetic versatility, and mechanical flexibility, which make them attractive for flexible and wearable optoelectronic devices. An accurate and comprehensive understanding about the morphology–photophysics relations in CPs lays the groundwork for their development in these applications. However, due to the complex roles of chemical structure, side-chains, backbone, and intramolecular interactions, CPs can exhibit heterogeneity in both their morphology and optoelectronic properties even at the single chain level. This molecular level heterogeneity together with complicated intermolecular interactions found in bulk CP materials severely obscures the deterministic information about the morphology and photophysics at different hierarchy levels. To counter this complexity and offer a clearer picture for the properties of CP materials, we highlight the approach of probing material systems with specific structural features via single molecule/aggregate spectroscopy (SMS). This review article covers recent advances achieved through such an approach regarding the important morphological and photophysical properties of CPs. After a brief review of the typical characteristics of CPs, we present detailed discussions of structurally well-defined model systems of CPs, from manipulated backbones and side-chains, up to nano-aggregates, studied with SMS to offer deterministic relations between morphology and photophysics from single chains building up to bulk states.
Introduction
Flexible and wearable optoelectronic devices, being light, bendable, low-cost, and even stretchable, represent exciting future technologies in displays, light sources, solar cells, and wearable sensors, to name a few.1–7 The advent of the rapidly blossoming field of flexible optoelectronics has been mostly triggered by the advancements in exploring solution-processable, low-cost, and mechanically flexible semiconducting materials, especially organic semiconductors.3,4 The excellent plasticity and tuneable optical and electrical properties of conjugated polymers (CPs) make them an important class within the realm of organic semiconductors. In most of the CP-based optoelectronic devices, CPs as an active layer are employed in the form of thin films with thicknesses of tens to hundreds of nanometers. Governed jointly by external processing methods (solvents, coating conditions, etc.) and internal molecular structures (backbone, side-chains, etc.), bulk films in most cases are morphologically heterogeneous, possessing domains with varying degrees of order and non-uniform grain boundaries. Such morphological heterogeneity largely originates from the molecular and nanometer scale. Scheme 1A displays a morphological evolution from single CP chains up to a bulk film. Given these facts, optical and electronic information from bulk films is then an average of this large heterogeneity thus limiting the usefulness of conventional analytical tools on bulk films for making connections between structure and function.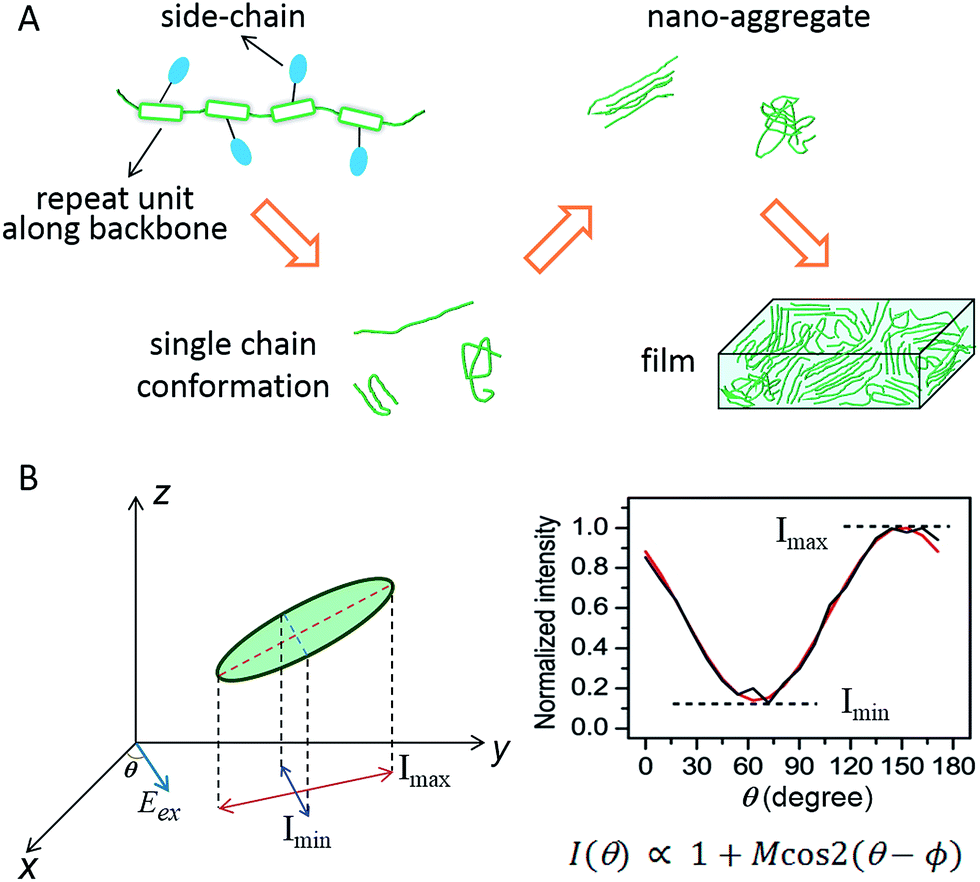 | ||
| Scheme 1 (A) Schematic evolution of morphology from single CP chains to a bulk film. CP chains with varying structural features result in different single chain conformations, which further determine the morphological order in nano-aggregates – the building blocks of bulk films. (B) A schematic representation of the polarization excitation experiments. Eex is the direction of linearly polarized excitation at an angle θ (left). A representative fluorescence intensity trace as a function of excitation polarization angle. The black line is experimental data and the red line is the fitted curve (right). Panel (B) is adapted with permission from ref. 18, Copyright 2012 American Chemical Society. | ||
In order to effectively leverage CP materials in an array of applications, a fundamental understanding and a precise engineering regarding the morphological, optical, and electronic properties at different hierarchy levels from the bottom up are essential. To counter the heterogeneity in bulk CP films, there have been many research efforts dedicated to studying nanoscale and molecular systems, i.e., nano-aggregates and single CP chains, in the past decade. Since the pioneering work from Paul F. Barbara's laboratory in the late 1990s on poly(2-methoxy-5-(2′-ethlyhexyloxy)-p-phenylene vinylene) (MEH-PPV),8 single molecule/aggregate spectroscopy (SMS) has expanded greatly to elucidate the hidden morphological, optical and electronic properties at the nanoscale and single-molecule levels.9–11 For example in order to connect structure and function it is critical to have some measure of the conformation or morphology of individual polymer chains and aggregates. This can be accomplished using fluorescence excitation polarization analysis. A typical wide-field laser optical microscope system for such experiments is illustrated in Scheme 1B. In these experiments, the fluorescence intensity traces are modulated with a rotating linearly polarized excitation light and then fitted with I(θ) ∝ 1 + M![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cos2(θ − ϕ), where θ is the excitation polarization angle and ϕ is the polarization angle at maximum absorption.12–14 The modulation depth, M, represents the anisotropy of the absorption (excitation) tensor projected onto the x–y plane of the laboratory frame and is related to the morphological order of individual molecules or aggregates, with M → 1 signifying an ordered and anisotropic absorber and M → 0 a disordered isotropic absorber. This method can be further expanded to examine the fluorescence anisotropy of the CP emission and to look at anisotropy for both absorption and emission simultaneously.15–17 For more details about the SMS technique in studying CPs in terms of instrumentation, sample preparation, and challenges and limitations, interested readers can refer to review articles.8–11,18–20
cos2(θ − ϕ), where θ is the excitation polarization angle and ϕ is the polarization angle at maximum absorption.12–14 The modulation depth, M, represents the anisotropy of the absorption (excitation) tensor projected onto the x–y plane of the laboratory frame and is related to the morphological order of individual molecules or aggregates, with M → 1 signifying an ordered and anisotropic absorber and M → 0 a disordered isotropic absorber. This method can be further expanded to examine the fluorescence anisotropy of the CP emission and to look at anisotropy for both absorption and emission simultaneously.15–17 For more details about the SMS technique in studying CPs in terms of instrumentation, sample preparation, and challenges and limitations, interested readers can refer to review articles.8–11,18–20
By zooming in on individual CP chains from highly heterogeneous bulk states, single molecules represent the smallest possible scale for studying CPs. Yet, it does not necessarily exclude possible heterogeneities at the single chain level such as different single chain conformations. Recent research endeavours have revealed that CPs can be morphologically and optoelectronically heterogeneous even at the single chain level, given the complex roles of side-chains, backbone, and possible intramolecular interchain interactions.12,21–26 For instance, one needs to be careful of intramolecular interactions due to single chain folding as a result of high molecular weight. In such a case, the molecule becomes a molecular aggregate (involving spatial ‘intramolecular’ interchain interactions) rather than a single isolated non-interacting chain that is under study. Therefore, even working at the molecular level, one still cannot completely break out of molecular heterogeneity.
To uncover deterministic relations between morphology and photophysics in CPs, one needs not only the analysis at the single molecule or nano-aggregate level but, more importantly, structurally well-defined systems. By “structurally well-defined systems”, we mean CPs or model systems with the molecular weight, backbone, side-chains, assembly, etc., under strict control. Meanwhile, a bottom-up approach is also crucial so as to circumvent the complex interactions within a larger scale. By starting with well-defined molecular systems and understanding how factors such as molecular weight, side-chains, and backbone influence the morphological and photophysical behaviour at the molecular level, higher order nanoscale systems – the building blocks of films – can be envisioned by controlled assembly from single CP chains. This then facilitates new insights into the bulk film photophysics. Herein, we will highlight the latest advances regarding deterministic relations between morphology and photophysics from single chains up to bulk states, obtained by combining specific model material systems and single molecule/aggregate spectroscopy. The review concludes with the prospective scientific challenges remaining in the study of photophysics, morphology, and materials design of CPs.
From single molecules to aggregates
Backbone
The backbone, the core structure of a polymer chain, plays a dominant role in determining the single chain conformation and the optoelectronic properties. Recent studies have demonstrated the usage of specifically designed CP systems to probe the effects of backbone structural variation on single chain conformation and corresponding photophysical properties. These structural changes include manipulating defects, conjugation length, rigidity, and chromophore and main chain shape.The impact of different types of defects along the backbone on the conformation and electronic properties has been investigated by the Barbara group using random copolymers of MEH-PPV that intentionally incorporated different chemical defect sites (Fig. 1A).27 As shown by the modulation depth, M, distribution histograms in Fig. 1B, together with molecular dynamics (MD) simulations, the rigid para-terphenyl defects appear to preserve the linear conformation of the backbone, while ortho-terphenyl defects twist the backbone and lead to disordered chain conformations. Saturated defects allow for torsional freedom and lead to a wide range of conformations with high defect inclusion in the polymer chain. The structural variations result in blue-shifted absorption of the polymers with more defects, as conjugation along the polymer chain is interrupted by the defect sites. On the other hand, there is no substantial difference in the emission spectra due to efficient energy transfer to the lowest energy emitting sites, which exist even in highly defected polymers.
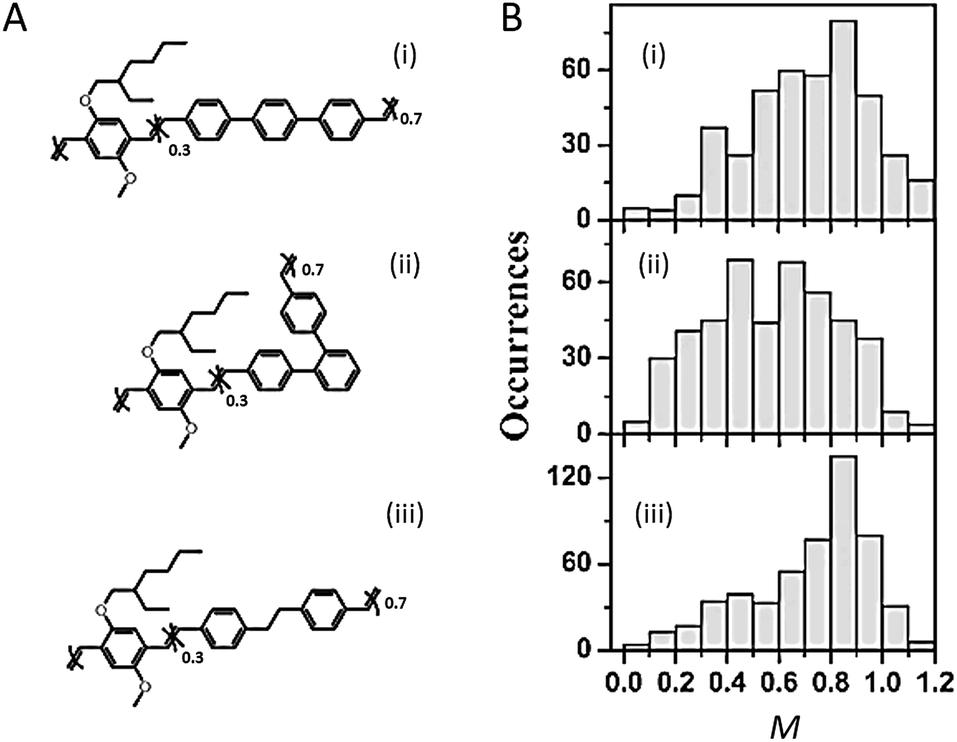 | ||
| Fig. 1 (A) Structures of para-terphenyl-MEH-PPV (i), ortho-terphenyl-MEH-PPV (ii), and saturated MEH-PPV (iii), respectively. (B) Modulation depth, M, distribution histograms of polymers shown in (A). Adapted with permission from ref. 27, Copyright 2011 American Chemical Society. | ||
Hildner et al. studied the impact of conjugation length on fluorescence spectral properties for a ladder-type (p-phenylene) dimer and its corresponding polymer methyl-substituted ladder-type poly(p-phenylene), MeLPPP.28 It was revealed that when going from the dimer to the polymer there is a significant change in the electron–phonon coupling to intramolecular vibration mode. As shown in Fig. 2A, the low-energy skeletal stretch mode at ∼150 cm−1 observed for the single dimer molecule (solid, top) disappears in the spectra of single MeLPPP chains (solid, bottom),28,29 which is attributed to a reduced electron–phonon coupling strength and/or a reduced vibrational energy of the skeletal stretch mode with increasing pi-electron delocalization. Compared to the dimer, the MeLPPP molecules exhibit a broader distribution in zero phonon lines (ZPLs) as a result of an increasing conformational disorder. On the other hand, the linewidths of ZPLs of the dimer and the polymer do not show differences as they are typically determined by interactions with the local environment.
 | ||
| Fig. 2 (A) Low-temperature emission spectrum (solid) of a single dimer (top) and a single MeLPPP molecule (bottom) imbedded in polystyrene. The dashed curves show corresponding ensemble emission spectra. Corresponding structure of the dimer and the MeLPPP polymer are exhibited on the right. Figures are adapted with permission from ref. 28. Copyright 2016 American Chemical Society. (B) Structure of LPPentP (left); fluorescence excitation (black) and emission spectra (grey) of a single LPPentP chain exhibiting single emitting site with multiple absorption sites (right). Adapted with permission from ref. 30, Copyright 2015, John Wiley & Sons, Ltd. | ||
The backbone rigidity strongly affects the morphology of single chains and nanostructures as well as the corresponding photophysics. Basché et al. studied a ladder-type CP poly(ladder-type pentaphenylene) (LPPentP) (Fig. 1C), which is based on poly(para-phenylene) and constituted of stiff chromophores that cannot bend. Purely electronic ZPL have been observed in 66% of single LPPentP chains at low temperature.30 On the contrary, for MEH-PPV, a material with relatively more flexibility, only about 10% of the molecules showed ZPL.31 This lack of ZPL was assigned to linear electron–phonon coupling, which originated in the less rigid backbone of MEH-PPV giving rise to geometrical relaxation following electronic excitation.30 As shown in Fig. 2B, single molecule excitation and emission spectroscopy showed multiple absorption sites with a single emitting site.
This result unmasked the nonemitting donor chromophores, which transfer their excitation energy to a low-energy emitting chromophore, leading to emission from a single emitting site. Gesquiere et al. investigated the effect of backbone rigidity on single chain conformation and crystallinity of nanofibers of polythiophenes.32 By examining PL spectra and emission polarization anisotropy, the authors found that the single polymer chain conformations do not necessarily translate into their respective self-assembled nanofibers,33 as factors such as side-chain interactions, solvent environment, and intermolecular interactions all might contribute to the overall morphology in aggregates. Therefore, care should be taken when correlating the isolated single CP chains' conformation with their morphology in self-assembled nanostructures.
Besides backbone rigidity, the shape of CP backbone also has a profound influence. It has been reported that electron delocalization or conjugation is persistent even with bending and twisting of the individual chromophore segment.34 Bending of the chromophores, however, would make them less stable and more prone to environmental effects such as oxidation, which ultimately reduces the effective conjugation. Such change in chromophore shape (at least for phenylene-vinylenes), rather than the overall conformation of a single chain, greatly influences the spectroscopic properties. A comparison of low-temperature single molecule emission spectral data between PPV oligomers (7-mer and 17-mer) and polymers showed that spectral broadening is intrinsic to single chromophores.35 Bending of the chromophore has a substantial effect on ultrafast electronic dephasing and spectral diffusion, which are two additional potential physical origins (except aggregation) for spectral broadening. The effect of backbone shape on ultrafast structural relaxation and exciton trapping has been studied by Lupton et al. in model systems of linear oligomers and cyclic structures based on carbazole-bridged phenylene–ethynylene–butadiynylene units (Fig. 3A).36Fig. 3B shows that the dimer exhibits the largest red-shift in energy during the first 200 ps, indicating a strong structural relaxation. In contrast, the torsional relaxation is largely inhibited in the structurally rigid macrocycle. Furthermore, the emission linear dichroism data shown in Fig. 3C reveals that exciton delocalization processes are also different: while the exciton acts as a linear transition dipole in linear oligomoers, it localizes randomly on different segments on the cyclic structure and results in linear dichroism values around zero.37 Collectively, these above studies surrounding backbone structure engineering underline the potential for synthetic strategies by manipulating backbone features to tune the chain conformation and the optoelectronic properties.
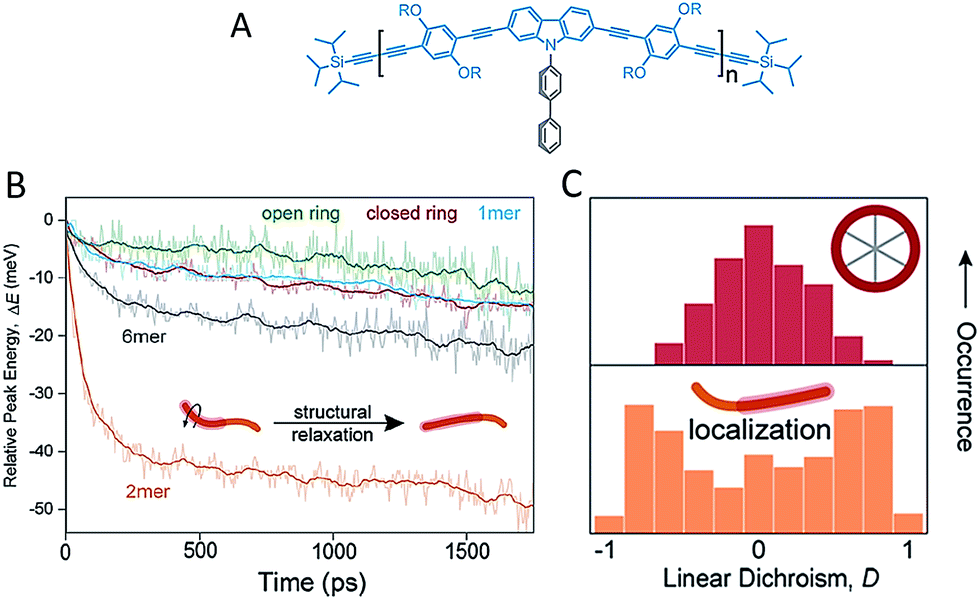 | ||
| Fig. 3 (A) Structure of the repeat unit for the linear oligomers and the ring's rim of the macrocycle. Panel (B) shows the temporal shift of the emission peak plotted as the relative energy change relative to detection time 0. Panel (C) displays the distribution histograms of the linear dichroism (LD) of single molecules of the macrocycle and the dimer. The LD data were estimated from two orthogonal emission polarizations via LD = (Ix − Iy)/(Ix + Iy). Figures are adapted with permission from ref. 36, Copyright 2015 American Chemical Society. | ||
Side-chains
While the backbone of a CP provides the basis for the chain conformation and photophysics, side-chains function far more than just as solubilising groups. Significant experimental evidence has shown that side-chains can drastically alter not only the morphology, but also the photophysics, introducing an extra means of ‘tuning’ the materials' properties. Factors such as the arrangement, size, side-chain density, and interactions between side-chains or with the surrounding matrix can alter the morphology and photophysics from the single molecule level.The impact of regioregularity of side-chains on the morphology and physics of single chains has been interrogated in the prototypical poly(3-hexylthiophenes) (P3HTs) by our group and the Chen group (Fig. 4A). SMS studies on P3HTs with high molecular weight (Mn above 30 kDa) revealed higher values of the modulation depth (mean M value of 0.65) in regioregular-P3HT (rr-P3HT), indicating more ordered single chain conformation than the rra-P3HT case (mean M value of 0.43) (Fig. 4B).12,23,38 It should be noted that with these high Mn, intramolecular chain folding occurs. Therefore, the disordered single chain conformation in regiorandom-P3HT (rra-P3HT) could be due to disordered intramolecular interchain packing and/or disordered unpacked chain segments. SMS data reported later on for P3HTs with Mn of 10 kDa, at which no chain folding would be expected,22,39 showed that the mean M values of rr-P3HT and rra-P3HT are close, with M values of 0.70 and 0.62, respectively.12 This observation suggests that the conformation of the short rra-P3HT chains is not severely disrupted by the random side-chain arrangement. It also indicates that the random side-chain arrangement in the rra-P3HT has a more dramatic influence on the interchain packing, which is intramolecular in the case of high Mnrra-P3HT. Theoretical calculations of the dihedral angle between adjacent thiophene rings from MD simulated structures of rra-P3HT revealed that the head-to-head (H–H) side-chain coupling twists the single chain conformation characterized by a dihedral angle of ∼55 degrees (Fig. 4C), and therefore leads to unfavorable packing between chains and a shorter conjugation length along the chains.13 Furthermore, to check if a longer distance of H–H coupling (i.e., reduced side-chain density) could mediate the twisted backbone conformation observed in rra-P3HT, we studied the regiorandom poly(3-hexyl-2,5-thienylene vinylene) (rra-P3HTV), which has an increased H–H coupling distance due to the presence of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds between thiophene rings.13 SMS study revealed that rra-P3HTV single chains exhibit highly ordered single chain conformation. This result, in turn, substantiates that the twisted conformation in rra-P3HT is induced by close H–H arrangement, which can be largely eliminated by the longer distance of H–H coupling in rra-P3HTV (Fig. 4C).
C bonds between thiophene rings.13 SMS study revealed that rra-P3HTV single chains exhibit highly ordered single chain conformation. This result, in turn, substantiates that the twisted conformation in rra-P3HT is induced by close H–H arrangement, which can be largely eliminated by the longer distance of H–H coupling in rra-P3HTV (Fig. 4C).
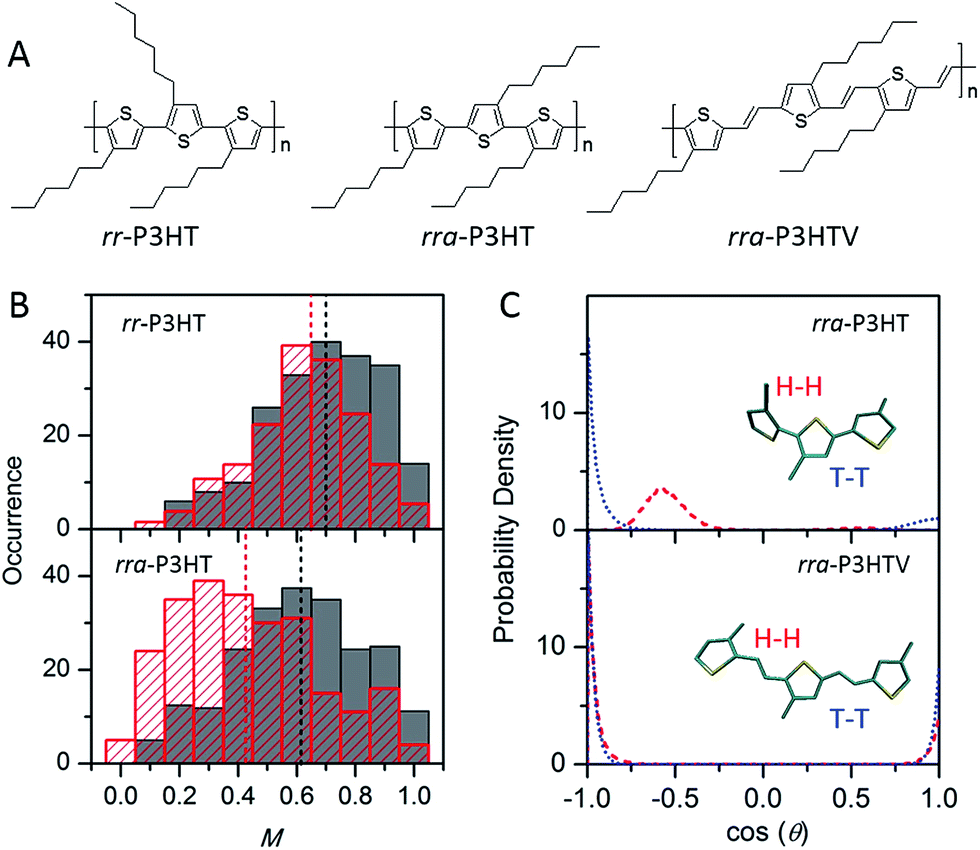 | ||
| Fig. 4 (A) Structures of rr-P3HT, rra-P3HT, and rra-P3HTV. (B) M distribution histograms of rr-P3HT (top) and rra-P3HT (bottom) with different Mn (grey for 10 kDa, red for 146 kDa). Figure is adapted with permission from ref. 12, Copyright 2014 American Chemical Society. (C) Probability density distribution of the dihedral angle between neighbouring thiophene rings for the H–H (red) and T–T (blue) couplings in rra-P3HT (top) and rra-P3HTV (bottom). Figure is adapted with permission from ref. 13, Copyright 2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Experimental evidence has also shown that bulky side-chains usually prevent single polymer chains from forming a closely packed or an ordered conformation.40–42 Vacha et al. have reported that the grafting of long polystyrene (PS) side-chains on polythiophene significantly suppresses photoblinking and high anisotropy as observed for polythiophene with short side-chains. This was attributed to inhibited exciton localization to one or a few segments in polythiophenes with PS branch side-chains as a result of hampering the collapse into ordered chain conformations.40,41 Side-chain structural modifications can also be utilized to inhibit interactions at the single molecule and aggregate level through means such as backbone protection with self-threading side-chains as reported by Scheblykin et al.42 At the single chain level, these bulky substituents increase the effective conjugation length by preventing defect or kink formation, but much more distinctly they inhibit aggregation and packing. This prevents interchain energy transfer while allowing intrachain transfer along the backbone.
Recent experiments, both at the bulk and single molecule level have utilized hydrogen-bonding (H-bonding) and other dynamic bonding interactions for device and materials design applications. In bulk films dynamic H-bonding has been used as a form of non-covalent cross-linking in order to enhance material elasticity for flexible devices and facilitate a means of ‘healing’ cracks in the devices due to mechanical strain.2,43 At the single molecule level, we have taken advantage of H-bonding in order to selectively fold polymeric systems into desired configurations.44Fig. 5A shows structures of a polymer with no H-bonding (control polymer) and two H-bonding capable polymers containing carboxylic acid and urea groups, respectively. Compared to the control polymer, the two H-bonding capable polymer chains exhibited relatively higher average M values, indicative of more ordered folded conformations (Fig. 5B). The urea-containing polymer in particular (Fig. 5A-3), which was designed to display ‘face-on’ stacking of chromophores, has the largest mean M value (0.8) of the three polymers. In addition, this polymer also exhibits ∼0.06 eV red shift in the emission relative to the other two polymers. While these data do not definitively prove that these polymers fold as designed, it is strong evidence to support the idea that side-chains alone can be used to force polymers to fold in certain ways.
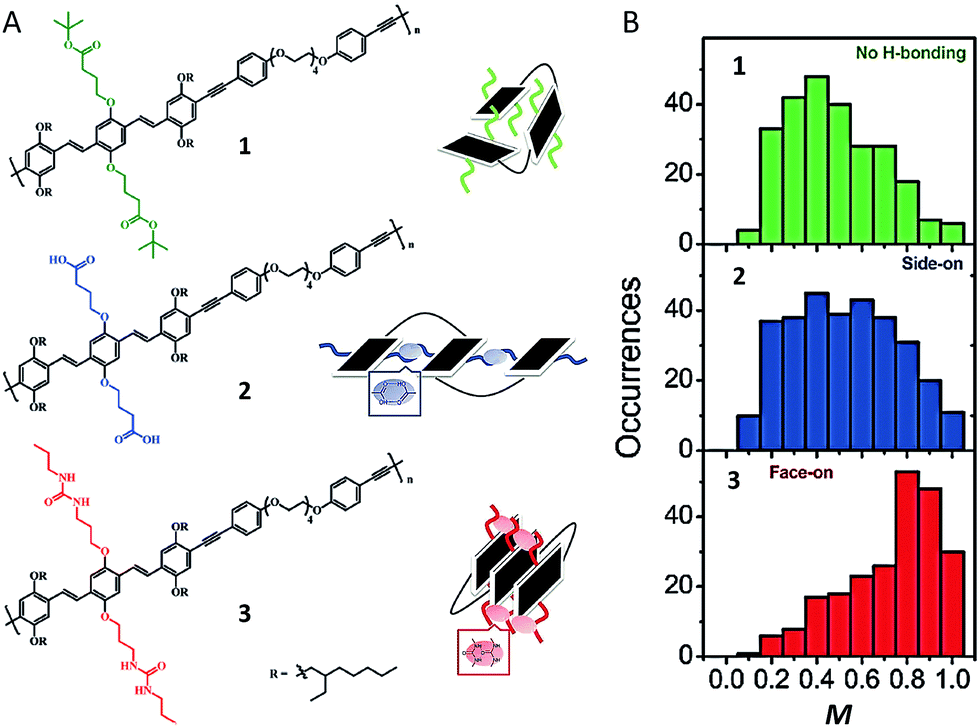 | ||
| Fig. 5 (A) Structures of polymers with no H-bonding (1) and H-bonding capable polymers containing carboxylic acids (2) and urea (3) groups. A corresponding schematic drawing of proposed folding is given next to each structure. (B) M distribution histograms of polymers. Figures are adapted with permission from ref. 44, Copyright 2017 The Royal Society of Chemistry. | ||
Furthermore, the modification of side-chains in CPs can also affect interactions with matrices, thus altering phase separation. Vacha et al. recently utilized this approach to isolate side-chain engineered polyfluorene molecules in vertical cylinders of a phase-separated block copolymer film. With the successes in this material's engineering, the authors have demonstrated single-molecule electroluminescence and accessed specific photophysics in polyfluorene.45 This work offers a unique approach to engineering single CP chains in surrounding media and device configurations.
Oligomers
To interrogate the morphological and photophysical interactions between conjugated segments (i.e., chromophores) in CPs, CPs themselves, even at single chain level in some cases, are not necessarily excellent models due to the possible and random existence of bends, kinks, twists or chemical defects along single chains. In comparison, shorter conjugated oligomers (COs) have specific advantages in structural control, synthetic repeativity, and purification.46 By eliminating the inherent heterogeneity in long CP chains, COs have been used as excellent model systems for accurately probing the morphological and physical behaviour between chromophores in CPs.For the prototypical MEH-PPV, the bimodal distribution of fluorescence emission (i.e., red and blue emission) has been observed in both single molecule and bulk studies. The origin of the red emission, originally ascribed to low energy sites due to intramolecular interchain aggregation,47 has been questioned and elucidated by intrachain chromophores with a longer conjugation length.48,49 By studying PPV oligomers with SMS and theoretical calculations, Vacha et al. revealed that distinct spectral forms can be due to variation in conjugation length caused by torsional defects.50 In addition, spectral jumps were also observed between different spectral forms possibly due to torsional flips of a single phenylene ring, suggesting the dynamic nature of exciton delocalization even in short PPV oligomers. Kim et al. studied the effect of chain length on conformation and exciton dynamics in oligothiophenes.51 The authors observed that with the increase in chain length, from 4-mer to 6-mer to 10-mer, the conformational order and the exciton delocalization reduces due to the bending of chains. COs have also been used to fabricate aggregates to probe morphology and spectroscopic information at nanoscale by the Peteanu group.52,53
Linked conjugated oligomers (LCOs), in which COs are joined through covalent linkers, not only inherit the aforementioned specific advantages of COs but also offer possible controllable interactions between conjugated segments. The latter offers a unique route in studying interactions between chromophores either along a single chain or spatially between different chains. The H-bonding capable polymers demonstrated in Fig. 4 also serve as a good example here. The dynamic nature of energy transfer between chromophores was elegantly examined by Lupton et al. using LCOs with two oligomers in either an oblique-angle or parallel geometry (Fig. 6A).54 The Förster resonance energy transfer (FRET) dynamics was examined by probing the emission polarization and the intensity as a function of time. Fig. 6A shows the data obtained for the open dimer. Interestingly, strong fluctuations of emission polarization were detected when the two oligomer chromophores were active as shown in the fluctuation of linear dichroism in Fig. 6A. This suggests that the FRET pathways switch reversibly between the two oligomers, that is, each oligomer temporarily takes the role of FRET donor or acceptor prior to photodegradation occurring in one, possibly due to temporal stabilization of a certain chromophore. This study clearly demonstrates that the excitation energy in CPs should be pictured as a dynamic process where a random three-dimensional FRET pathway is present. The morphology dependent energy transfer process in MEH-PPV was explored by Ghiggino et al. using a pendant LCO of PPV which has a saturated backbone spacer between the pendant PPV oligomers.55 Despite the nominally same lengths of the pendant PPV oligomers, energy transfer to single emitting sites was detected for most LCO molecules using defocused wide-field microscopy. This result indicates that the specific morphology of each individual chromophore, as well as its interaction with the surrounding matrix, play significant roles in determining their electronic landscape and the energy transfer processes.
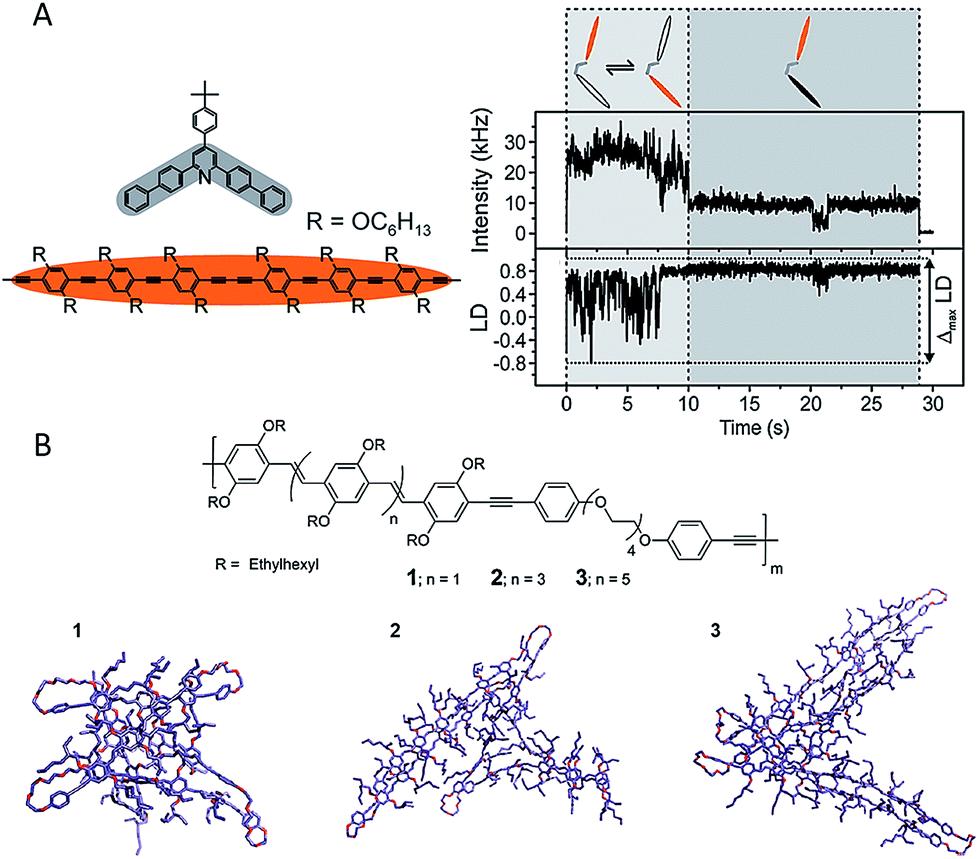 | ||
| Fig. 6 (A) Left: two oligomer chromophores (orange) were organized using one clamping unit (gray) to give an open dimer. Right top: fluorescence intensity transient of an open dimer. Right bottom: the linear dichroism (LD) transient estimated from two orthogonal emission polarizations. Both chromophore units were active within the first 10 s (light-gray shaded area) with strong fluctuations of the LD, as indicated schematically in the cartoon. A bleaching event took place after 10 s, leaving only one chromophore active (dark-gray shaded area), with correspondingly reduced LD fluctuations. Figures are adapted with permission from ref. 54, Copyright 2015 American Chemical Society. (B) MD simulated conformations for PPV based LCOs with different length of oligomers (from left to right: trimer, pentamer, and heptamer). Figures are adapted with permission from ref. 57, Copyright 2013 American Chemical Society. | ||
In order to study how to control the folding properties of polymeric systems, our group designed LCOs in which conjugated bis(2-ethylhexyl)-p-phenylene vinylene (BEH-PPV) oligomers are joined by different morphological directing groups, i.e., flexible, bent, and rigid linkers.56 The LCOs with rigid linkers produce ordered single chain conformations wherein the oligomers are aligned, while by contrast, the LCOs with bent linkers exhibit distorted conformations. Interestingly, the LCOs with flexible linkers exhibit a dependence of single chain conformation on the length of the conjugated oligomer segments (Fig. 6B).57 By increasing the length of oligomers, more ordered conformations of the LCO single chains are observed due to the stronger interactions between longer conjugated units as demonstrated by the structures from MD simulations shown in Fig. 6B. The mean M value increases from 0.39 for the LCO of trimer to 0.75 for the heptamer. Furthermore, recently our group demonstrated that by modifying the side-chain interaction via hydrogen-bonding, the LCO of trimers with flexible linkers can be tuned from disordered to ordered conformations (Fig. 5).44 Recently, Barnes et al. have designed LCOs with different linker lengths and examined how the length affects the folding of LCOs.58 In this study, the relatively short flexible linkers could act more like a simple pivot or defect in CPs. The authors discovered that the longer flexible linker leads to chromophore interaction as long as the chromophore movement is allowed. In contrast, the use of short linkers can constrain the chain folding. These systematic studies have provided not only a clear picture of the factors affecting the folding properties in polymeric systems but also a new route for synthetic consideration to achieve desired morphologies and functionalities in a more controllable way.
Nano-aggregates
For CP films applied in various optoelectronic applications, the morphology is usually dominated by nanoscale domains which are connected with relatively amorphous domain boundaries, as shown schematically in Scheme 1. Therefore, the nanoscale domains not only are building blocks for bulk films but act as a bridge between the gap of single CP chains and bulk states. Isolated and easily handled nano-aggregates can be viewed as model systems of the domains in bulk films. The knowledge about the morphology and physics in the nano-aggregates is important to understand, predict, and control the film phenomena.59–66 Barbara et al. studied the size-dependence of spectroscopic properties of MEH-PPV nanoparticles and found that bulk-like properties can be achieved with MEH-PPV nanoparticles larger than 10 nm.67 Frechet et al. found that with enhanced regioregularity of side-chains the crystallinity of P3HT nanoparticles increases and the PL spectrum red shifts correspondingly. Barnes et al. revealed that the crystallinity of P3HT nanoparticles increases with the size, which could be responsible for a fast PL depolarization in large crystalline nanoparticles.68 In a recent study, our group fabricated P3HT aggregates with a triblock polymer (P3HT–PtBA–P3HT, here PtBA represents a flexible linker of poly(tert-butyl acrylate)) in a series of solvents of which the dielectric constant are tuned ranging from 2 to 20. These experiments produced compelling evidence of the charge-transfer state as a major reason for the drop of P3HT emission quantum yield in aggregated state. It should be noted that the CP nano-aggregates reported in these initial works were usually prepared with a simple reprecipitation method. Typically, in this method CPs in a good solvent are quickly mixed with a poor solvent, leading to a rapid agglomeration of CP chains. Hence, the morphology of nano-aggregates formed in this way is not precisely defined.To get a clearer picture about the relations between nanoscale morphology and photophysics, control in the morphology of nano-aggregate was exerted via fabrication techniques. Grey et al. assembled P3HT nanofibers slowly from a hot toluene solution and discovered photophysical behaviour of J-aggregates (i.e., stronger 0–0 transition relative to 0–1 side-band) (Fig. 7A and B).69 The formation of J-type nanofibers can be ascribed to enhanced intrachain order over the interchain counterpart. In comparison, mixed H-type nanofibers (i.e., with dampened 0–0 emission) can be formed via rapid growth in anisole. Meanwhile, Barnes et al. demonstrated a molecular weight dependence of the H- and J-type character in P3HT nanofibers, where small chains tend to pack to H-type aggregates while high molecular weight chains tend to have J-type character due to enhanced chain extension as a result of folding.70 Grey et al. further studied the influence of J- and H-type character in the nanofibers on the exciton interconversion and interactions between triplet and singlet using excitation intensity modulation spectroscopy.71 This method involves fluorescence intensity modulation by repetitive cycles of a sequence of variable-intensity excitation pulses on single nanostructures.72 The fluorescence intensity drop (modulation depth) at the beginning of each pulse is ascribed to singlets quenching by triplets. It was found that for the assembled nanofibers with J-type character, the modulation depth is much higher than the H-type nanofibers (Fig. 7C). This interesting observation has been elucidated by the formation of triplets from delocalized and long-lived polarons in the J-type nanofibers, evidenced by the fluorescence modulation depth and its sensitivity to oxygen. Furthermore, the discoveries referenced above demonstrate the control of self-assembled nanostructures in understanding and manipulating the exciton coupling and spin state photophysics.
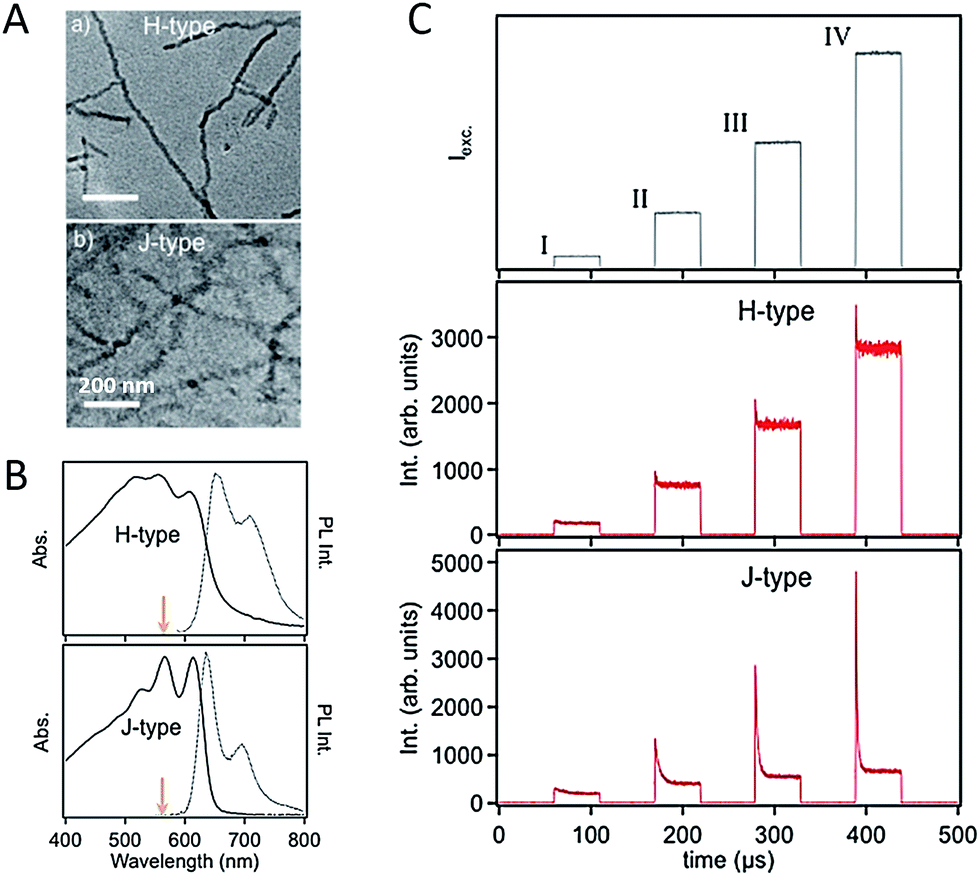 | ||
| Fig. 7 (A) TEM images of H- (top) and J-type (bottom) P3HT nanofibers. (B) Corresponding absorption and PL spectra for the H- and J-type NFs. (C) Top: a cycle containing four excitation windows, each with a different excitation intensity, is repeatedly applied to a selected single NF; middle and bottom: time-averaged fluorescence transient (synchronously time-averaged over many cycles) of a single H-type and J-type NFs, respectively, acquired while modulating the excitation laser intensity as shown in the top panel. Figures are adapted with permission from ref. 71, Copyright 2014 American Chemical Society. | ||
A more precise control in the morphology and size of the nano-aggregates was realized using a so-called solvent vapour annealing (SVA) technique developed by Vogelsang et al.,73–75 which is schematically illustrated in Scheme 2. In this method a concentrated single-molecule sample is treated with a solvent vapor of a mixture of good and poor solvents, where the good solvent can dissolve both the single CP chains and the poly(methyl methacrylate) (PMMA) matrix while the poor solvent can only dissolve the matrix. By tuning the ratio of the good and poor solvent and the concentration of the initial single molecule sample, the size of the formed aggregates can be controlled. During SVA, the diffusion of CP molecules inside the swollen PMMA matrix triggers the formation of CP aggregates. These authors assembled highly ordered MEH-PPV aggregates consisting of 25–45 chains and observed ultralong distances of energy transfer from 20–30 nm up to 60 nm.73
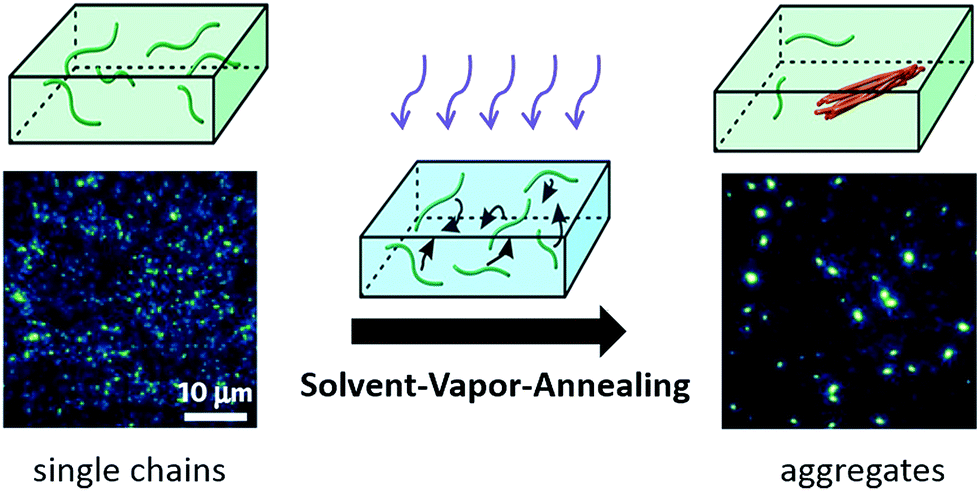 | ||
| Scheme 2 Solvent-vapor-annealing method to assemble single polymer chains into nano-aggregates in a controllable way. Figure is adapted with permission from ref. 75, Copyright 2015 National Academy of Sciences. | ||
The deterministic effect of side-chain arrangement and size on the interchain interaction was systematically investigated by our group using SVA assembled aggregates of rr-P3HT, rra-P3HT and a polythiophene derivative with bulky side-chains poly(3-(2′-methoxy-5′-octylphenyl)thiophene) (POMeOPT) (Fig. 8A).12 It was found that the ordered side-chains in rr-P3HT strongly favour the ordered interchain morphology, as indicated by an average M value of 0.80 for aggregates with ∼100 chains (Fig. 8B). With this size, the aggregates exhibit emission intensity blinking as a single molecule, indicating an efficient exciton migration in these ordered rr-P3HT aggregates (Fig. 8C). Exciton diffusion up to a length scale of 30–40 nm was approximately estimated from the blinking data in bigger sized aggregates. In stark contrast, the random distribution of side-chains in rra-P3HT drastically disrupts the interchain morphology even between only a few chains as demonstrated in Fig. 8B, which correspondingly inhibits efficient exciton coupling and migration between the chains (Fig. 8C). With POMeOPT aggregates, we discovered that the interchain exciton coupling is strongly prohibited (except for relatively short chain aggregates) evidenced by a chain-by-chain photo-degradation behaviour displayed in Fig. 8C. The long interchain packing distance (∼7 Å) and the slightly disordered packing account for a part, but definitely not all, of the reduction in the interchain excitonic coupling in POMeOPT. The competition between intrachain and interchain coupling mechanisms has further been revealed by comparing the POMeOPT aggregates with different polymer chain size in terms of their blinking behaviour and spectral characteristics. Very recently, Vogelsang et al. also demonstrated successful control over interchain coupling (i.e., H-type, J-type, and suppressed electronic coupling) in isolated CP aggregates by tuning the side-chain structure of CPs and the SVA process.76
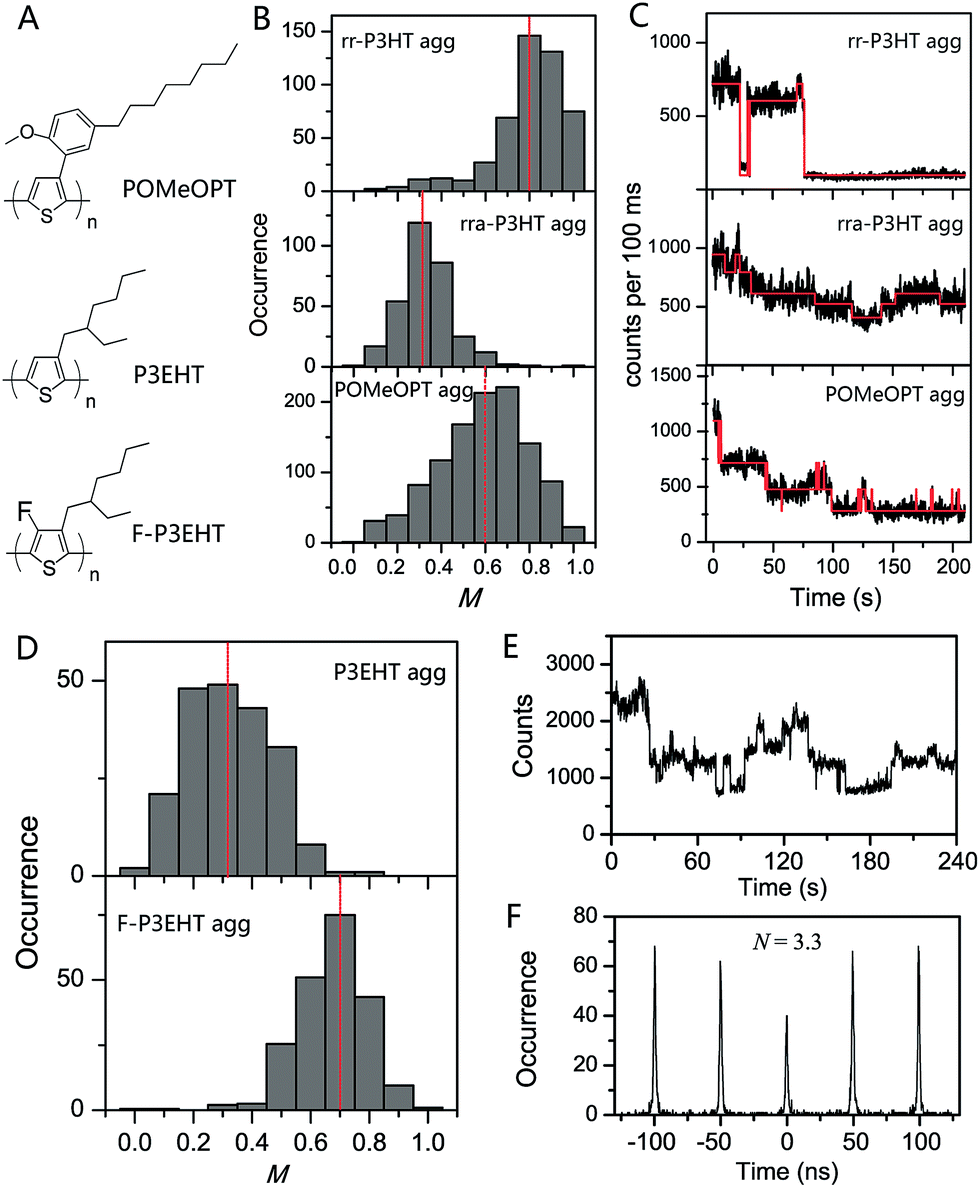 | ||
| Fig. 8 (A) Structures of POMeOPT, P3EHT, and F-P3EHT. (B) M distribution histograms of 100-chain rr-P3HT aggregates, 6-chain rra-P3HT aggregates, and 4-chain POMeOPT aggregates. The fluorescence time traces at high excitation power are shown in panel (C) correspondingly. Panel (D) displays the M distribution histograms of 5-chain P3EHT aggregates and 4-chain F-P3EHT aggregates. Panel (E) and (F) show the fluorescence time trace and photon correlation result of a typical 4-chain F-P3EHT aggregate. Figures are adapted with permission from ref. 12 and 77, Copyright 2014 American Chemical Society and Copyright 2017 National Academy of Sciences. | ||
Backbone fluorination has proven to be an important strategy in developing high-performance conjugated polymers for photovoltaic applications. The fluorination effect of the polythiophene backbone on the electronic and optical properties was recently interrogated by our group using two polymers, i.e., regioregular poly(3-ethylhexylthiophene) (rr-P3EHT) and poly(3-ethylhexyl-4-fluorothiophene) (rr-F-P3EHT) (Fig. 8A).77 Despite the high regioregularity, the rr-P3EHT single chains exhibit a broad distribution of conformations due to the large size of the side-chains. This, in turn, results in relatively disordered interchain morphology in SVA assembled aggregates with several polymer chains (Fig. 8D). On the other hand, for rr-F-P3EHT, a more extended and ordered single chain conformation was confirmed with both M measurement and MD simulation. Thus, ordered single F-P3EHT chains generated highly ordered interchain packing in F-P3EHT aggregates as seen in the M distribution histogram in Fig. 8D. Surprisingly, for the typical 4-chain F-P3EHT aggregates, about 4-step photodegradation behaviour was observed (Fig. 8E) and ∼3.5 emitters were calculated in the photon correlation measurement (Fig. 8F). Therefore, despite the ordered interchain morphology and the close interchain packing (∼3.8 Å), the exciton coupling in F-P3EHT aggregates is hampered. This is attributed to the dominant and competitive coupling along F-P3EHT chains due to the fluorine-induced extended backbone. Meanwhile, it was also found that the highly ordered packing in F-P3EHT surprisingly leads to a reduction in the energetic heterogeneity of the individual chains as the intrachain coupling is increased, based on the aggregate spectral data. This provides an unusual result that the ensemble of chains in the aggregate has a narrower distribution of energies than the corresponding distribution of individual polymer chains. This study highlights the intricate and subtle interplay between interchain and intrachain exciton coupling as a function of interchain ordering and single chain conformation.
Conclusions and outlook
Morphology and photophysics play significant roles in determining the functionality and performance of CP materials in a broad range of optoelectronic applications. The combination of the SMS technique and structurally well-defined model systems offers the ability to explore the information washed out by heterogeneities in ensemble measurements. With manipulations in side-chains, backbone, oligomer model systems, and controlled assembly of CP aggregates, recent research efforts have revealed new insights into the morphological dependence of exciton coupling, exciton trapping, energy transfer, charge transfer, excitation interactions, etc., at different hierarchy levels. Taken together, these results create a clearer picture of how the morphology and photophysics of CPs evolve from individual chains up to bulk states. Meanwhile, these investigations serve to motivate researchers to develop material systems with the desired morphology and functions that are appropriate for specific applications. We believe that the continuous efforts in SMS on CPs will keep contributing to revealing complicated phenomena in CPs and pushing forward the materials and device engineering for advancements in organic semiconductor electronics.The majority of SMS works so far have been focused on prototypical CP systems such as PPV and P3HT. Investigations on material systems with better performance and/or new functionality, such as low bandgap CPs, n-type CPs, and conjugated metallopolymers, are still lacking although these CP systems have been in the field for years. Many fundamental questions regarding in-depth molecular morphology and photophysics in these complex systems are still open. For these complicated CP systems, simplified model systems such as oligomers and copolymers could play more important roles to sort out research targets from the heterogeneous pool.
From the technique point of view, the vast majority of the current SMS approaches are dominantly based on fluorescence. Momentum is needed to push forward the advancement of the technique, for instance, by developing new characterization techniques in single molecule spectroscopy and by integrating SMS with other characterization or manipulating techniques. The Orrit group recently demonstrated exciting simultaneous measurement of both absorption and PL emission of single MEH-PPV molecules using near-critical xenon in photothermal microscopy.78 Such a demonstration has enriched the tool kit of SMS. Further revelation of single molecule absorption will certainly benefit our understanding of photophysical behavior of single molecules. Additionally, the coupling of SMS with other characterization techniques is expected to be very useful in the study of morphology, optical and electronic properties of CPs. For instance, scanning tunneling spectroscopy, with a capability of probing local single chain or aggregate morphology and electronic properties, can be combined with the SMS to explore more direct property relationships. Atomic force microscopy can also work together with SMS, as demonstrated by the Vacha group,79,80 to investigate the optical and electronic properties as a function of mechanical stimuli of single chain or aggregate, which will offer useful information for developing flexible optoelectronics of CPs.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
G. G. would like to acknowledge the help, support, and mentorship of Dr Robert Orwoll. D. A. V. B. acknowledges support from the National Science Foundation (Grant CHE-1310222). We would also like to acknowledge Dr Hsien-Yi Hsu for helpful discussions and support editing the manuscript.References
- J. Xu, S. Wang, G.-J. N. Wang, C. Zhu, S. Luo, L. Jin, X. Gu, S. Chen, V. R. Feig, J. W. F. To, S. Rondeau-Gagné, J. Park, B. C. Schroeder, C. Lu, J. Y. Oh, Y. Wang, Y.-H. Kim, H. Yan, R. Sinclair, D. Zhou, G. Xue, B. Murmann, C. Linder, W. Cai, J. B.-H. Tok, J. W. Chung and Z. Bao, Science, 2017, 355, 59–64 CrossRef CAS PubMed.
- J. Y. Oh, S. Rondeau-Gagné, Y.-C. Chiu, A. Chortos, F. Lissel, G.-J. N. Wang, B. C. Schroeder, T. Kurosawa, J. Lopez, T. Katsumata, J. Xu, C. Zhu, X. Gu, W.-G. Bae, Y. Kim, L. Jin, J. W. Chung, J. B. H. Tok and Z. Bao, Nature, 2016, 539, 411–415 CrossRef CAS PubMed.
- S. Logothetidis, Handbook of flexible organic electronics: Materials, manufacturing and applications, Elsevier, 2014 Search PubMed.
- T. Someya, M. Kaltenbrunner and T. Yokota, MRS Bull., 2015, 40, 1130–1137 CrossRef.
- C. Liao, M. Zhang, M. Y. Yao, T. Hua, L. Li and F. Yan, Adv. Mater., 2015, 27, 7493–7527 CrossRef CAS PubMed.
- S. Casalini, C. A. Bortolotti, F. Leonardi and F. Biscarini, Chem. Soc. Rev., 2017, 46, 40–71 RSC.
- R.-P. Xu, Y.-Q. Li and J.-X. Tang, J. Mater. Chem. C, 2016, 4, 9116–9142 RSC.
- P. F. Barbara, A. J. Gesquiere, S. J. Park and Y. J. Lee, Acc. Chem. Res., 2005, 38, 602–610 CrossRef CAS PubMed.
- M. Orrit, T. Ha and V. Sandoghdar, Chem. Soc. Rev., 2014, 43, 973–976 RSC.
- J. M. Lupton, Adv. Mater., 2010, 22, 1689–1721 CrossRef CAS PubMed.
- M. Vacha and S. Habuchi, NPG Asia Mater., 2010, 2, 134–1142 CrossRef.
- Z. Hu, T. Adachi, R. Haws, B. Shuang, R. J. Ono, C. W. Bielawski, C. F. Landes, P. J. Rossky and D. A. Vanden Bout, J. Am. Chem. Soc., 2014, 136, 16023–16031 CrossRef CAS PubMed.
- Z. Hu, T. Adachi, Y.-G. Lee, R. T. Haws, B. Hanson, R. J. Ono, C. W. Bielawski, V. Ganesan, P. J. Rossky and D. A. Vanden Bout, ChemPhysChem, 2013, 14, 4143–4148 CrossRef CAS PubMed.
- T. Adachi, J. Brazard, P. Chokshi, J. C. Bolinger, V. Ganesan and P. F. Barbara, J. Phys. Chem. C, 2010, 114, 20896 CAS.
- M. C. Traub, G. Lakhwani, J. C. Bolinger, D. Vanden Bout and P. F. Barbara, J. Phys. Chem. B, 2011, 115, 9941–9947 CrossRef CAS PubMed.
- D. Thomsson, H. Lin and I. G. Scheblykin, ChemPhysChem, 2010, 11, 897–904 CrossRef CAS PubMed.
- O. Mirzov, R. Bloem, P. R. Hania, D. Thomsson, H. Lin and I. G. Scheblykin, Small, 2009, 5, 1877–1888 CrossRef CAS PubMed.
- J. C. Bolinger, M. C. Traub, J. Brazard, T. Adachi, P. F. Barbara and D. A. Vanden Bout, Acc. Chem. Res., 2012, 45, 1992–2001 CrossRef CAS PubMed.
- D. Woll, E. Braeken, A. Deres, F. C. De Schryver, H. Uji-i and J. Hofkens, Chem. Soc. Rev., 2009, 38, 313–328 RSC.
- H. Kobayashi, S. Onda, S. Furumaki, S. Habuchi and M. Vacha, Chem. Phys. Lett., 2012, 528, 1–6 CrossRef CAS.
- A. Thiessen, J. Vogelsang, T. Adachi, F. Steiner, D. Vanden Bout and J. M. Lupton, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E3550–E3556 CrossRef CAS PubMed.
- Z. Hu, J. Liu, L. Simón-Bower, L. Zhai and A. J. Gesquiere, J. Phys. Chem. B, 2013, 117, 4461–4467 CrossRef CAS PubMed.
- T. Adachi, J. Brazard, R. J. Ono, B. Hanson, M. C. Traub, Z. Q. Wu, Z. C. Li, J. C. Bolinger, V. Ganesan, C. W. Bielawski, D. A. V. Bout and P. F. Barbara, J. Phys. Chem. Lett., 2011, 2, 1400–1404 CrossRef CAS.
- L. Simine and P. J. Rossky, J. Phys. Chem. Lett., 2017, 8, 1752–1756 CrossRef CAS PubMed.
- D. Raithel, S. Baderschneider, T. B. de Queiroz, R. Lohwasser, J. Köhler, M. Thelakkat, S. Kümmel and R. Hildner, Macromolecules, 2016, 49, 9553–9560 CrossRef CAS.
- F. Paquin, H. Yamagata, N. J. Hestand, M. Sakowicz, N. Bérubé, M. Côté, L. X. Reynolds, S. A. Haque, N. Stingelin, F. C. Spano and C. Silva, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 88, 155202 CrossRef.
- G. Bounos, S. Ghosh, A. K. Lee, K. N. Plunkett, K. H. DuBay, J. C. Bolinger, R. Zhang, R. A. Friesner, C. Nuckolls, D. R. Reichman and P. F. Barbara, J. Am. Chem. Soc., 2011, 133, 10155–10160 CrossRef CAS PubMed.
- S. Baderschneider, U. Scherf, J. Köhler and R. Hildner, J. Phys. Chem. A, 2016, 120, 233–240 CrossRef CAS PubMed.
- R. Hildner, U. Lemmer, U. Scherf, M. van Heel and J. Köhler, Adv. Mater., 2007, 19, 1978–1982 CrossRef CAS.
- M. F. Zickler, F. A. Feist, J. Jacob, K. Müllen and T. Basché, Macromol. Rapid Commun., 2015, 36, 1096–1102 CrossRef CAS PubMed.
- F. A. Feist, M. F. Zickler and T. Basche, ChemPhysChem, 2011, 12, 1499–1508 CrossRef CAS PubMed.
- Z. J. Hu, J. H. Liu, L. Simon-Bower, L. Zhai and A. J. Gesquiere, J. Phys. Chem. B, 2013, 117, 4461–4467 CrossRef CAS PubMed.
- J. H. Liu, I. A. Mikhaylov, J. H. Zou, I. Osaka, A. E. Masunov, R. D. McCullough and L. Zhai, Polymer, 2011, 52, 2302–2309 CrossRef CAS.
- J. M. Lupton, ChemPhysChem, 2012, 13, 901–907 CrossRef CAS PubMed.
- K. Becker, E. Da Como, J. Feldmann, F. Scheliga, E. T. Csanyi, S. Tretiak and J. M. Lupton, J. Phys. Chem. B, 2008, 112, 4859–4864 CrossRef CAS PubMed.
- A. Thiessen, D. Würsch, S.-S. Jester, A. V. Aggarwal, A. Idelson, S. Bange, J. Vogelsang, S. Höger and J. M. Lupton, J. Phys. Chem. B, 2015, 119, 9949–9958 CrossRef CAS PubMed.
- A. V. Aggarwal, A. Thiessen, A. Idelson, D. Kalle, D. Würsch, T. Stangl, F. Steiner, S.-S. Jester, J. Vogelsang, S. Höger and J. M. Lupton, Nat. Chem., 2013, 5, 964–970 CrossRef CAS PubMed.
- P.-Y. Chen, A. Rassamesard, H.-L. Chen and S.-A. Chen, Macromolecules, 2013, 46, 5657–5663 CrossRef CAS.
- J. Liu, I. A. Mikhailov, J. Zou, I. Osaka, A. E. Masunov, R. D. McCullough and L. Zhai, Polymer, 2011, 52, 2302–2309 CrossRef CAS.
- T. Sugimoto, Y. Ebihara, K. Ogino and M. Vacha, ChemPhysChem, 2007, 8, 1623–1628 CrossRef CAS PubMed.
- T. Sugimoto, S. Habuchi, K. Ogino and M. Vacha, J. Phys. Chem. B, 2009, 113, 12220–12226 CrossRef CAS PubMed.
- D. Sahoo, K. Sugiyasu, Y. Tian, M. Takeuchi and I. G. Scheblykin, Chem. Mater., 2014, 26, 4867–4875 CrossRef CAS.
- F.-J. Kahle, C. Saller, A. Köhler and P. Strohriegl, Adv. Energy Mater., 2017, 7, 1700306 CrossRef.
- B. Shao, X. Zhu, K. N. Plunkett and D. A. Vanden Bout, Polym. Chem., 2017, 8, 1188–1195 RSC.
- Y. Honmou, S. Hirata, H. Komiyama, J. Hiyoshi, S. Kawauchi, T. Iyoda and M. Vacha, Nat. Commun., 2014, 5, 4666 CAS.
- S. S. Zade, N. Zamoshchik and M. Bendikov, Acc. Chem. Res., 2011, 44, 14–24 CrossRef CAS PubMed.
- J. Yu, D. H. Hu and P. F. Barbara, Science, 2000, 289, 1327–1330 CrossRef CAS PubMed.
- A. Köhler, S. T. Hoffmann and H. Bässler, J. Am. Chem. Soc., 2012, 134, 11594–11601 CrossRef PubMed.
- F. A. Feist, G. Tommaseo and T. Basche, J. Phys. Chem. C, 2009, 113, 11484–11490 CAS.
- H. Kobayashi, K. Tsuchiya, K. Ogino and M. Vacha, Phys. Chem. Chem. Phys., 2012, 14, 10114–10118 RSC.
- T. W. Kim, W. Kim, K. H. Park, P. Kim, J. W. Cho, H. Shimizu, M. Iyoda and D. Kim, J. Phys. Chem. Lett., 2016, 7, 452–458 CrossRef CAS PubMed.
- J. Hong, S. Jeon, J. J. Kim, D. Devi, K. Chacon-Madrid, W. Lee, S. M. Koo, J. Wildeman, M. Y. Sfeir, L. A. Peteanu, J. Wen and J. Ma, J. Phys. Chem. A, 2014, 118, 10464–10473 CrossRef CAS PubMed.
- W. Y. So, J. Hong, J. J. Kim, G. A. Sherwood, K. Chacon-Madrid, J. H. Werner, A. P. Shreve, L. A. Peteanu and J. Wildeman, J. Phys. Chem. B, 2012, 116, 10504–10513 CrossRef CAS PubMed.
- T. Stangl, S. Bange, D. Schmitz, D. Wursch, S. Hoger, J. Vogelsang and J. M. Lupton, J. Am. Chem. Soc., 2013, 135, 78–81 CrossRef CAS PubMed.
- E. N. Hooley, A. J. Tilley, J. M. White, K. P. Ghiggino and T. D. Bell, Phys. Chem. Chem. Phys., 2014, 16, 7108–7114 RSC.
- X. Zhu, B. Shao, D. A. Vanden Bout and K. N. Plunkett, Macromolecules, 2016, 49, 3838–3844 CrossRef CAS.
- M. C. Traub, K. H. DuBay, S. E. Ingle, X. Zhu, K. N. Plunkett, D. R. Reichman and D. A. Vanden Bout, J. Phys. Chem. Lett., 2013, 4, 2520–2524 CrossRef CAS.
- J. M. Lucas, J. A. Labastide, L. Wei, J. S. Tinkham, M. D. Barnes and P. M. Lahti, J. Phys. Chem. A, 2015, 119, 8010–8020 CrossRef CAS PubMed.
- Y. Jiang and J. McNeill, Chem. Rev., 2017, 117, 838–859 CrossRef CAS PubMed.
- F. Panzer, M. Sommer, H. Bässler, M. Thelakkat and A. Köhler, Macromolecules, 2015, 48, 1543–1553 CrossRef CAS.
- F. Panzer, H. Bässler, R. Lohwasser, M. Thelakkat and A. Köhler, J. Phys. Chem. Lett., 2014, 5, 2742–2747 CrossRef CAS PubMed.
- D. Tuncel and H. V. Demir, Nanoscale, 2010, 2, 484–494 RSC.
- J. Pecher and S. Mecking, Chem. Rev., 2010, 110, 6260–6279 CrossRef CAS PubMed.
- D. Tenery and A. J. Gesquiere, ChemPhysChem, 2009, 10, 2449–2457 CrossRef CAS PubMed.
- C. F. Wu, H. S. Peng, Y. F. Jiang and J. McNeill, J. Phys. Chem. B, 2006, 110, 14148–14154 CrossRef CAS PubMed.
- Z. Hu and A. J. Gesquiere, Chem. Phys. Lett., 2009, 476, 51–55 CrossRef CAS.
- J. K. Grey, D. Y. Kim, B. C. Norris, W. L. Miller and P. F. Barbara, J. Phys. Chem. B, 2006, 110, 25568–25572 CrossRef CAS PubMed.
- J. A. Labastide, M. Baghgar, I. Dujovne, B. H. Venkatraman, D. C. Ramsdell, D. Venkataraman and M. D. Barnes, J. Phys. Chem. Lett., 2011, 2, 2089–2093 CrossRef CAS.
- E. T. Niles, J. D. Roehling, H. Yamagata, A. J. Wise, F. C. Spano, A. J. Moule and J. K. Grey, J. Phys. Chem. Lett., 2012, 3, 259–263 CrossRef CAS.
- M. Baghgar, J. A. Labastide, F. Bokel, R. C. Hayward and M. D. Barnes, J. Phys. Chem. C, 2014, 118, 2229–2235 CAS.
- A. K. Thomas, J. A. Garcia, J. Ulibarri-Sanchez, J. Gao and J. K. Grey, ACS Nano, 2014, 8, 10559–10568 CrossRef CAS PubMed.
- A. J. Gesquiere, Y. J. Lee, J. Yu and P. F. Barbara, J. Phys. Chem. B, 2005, 109, 12366–12371 CrossRef CAS PubMed.
- J. Vogelsang, T. Adachi, J. Brazard, D. A. V. Bout and P. F. Barbara, Nat. Mater., 2011, 10, 942–946 CrossRef CAS PubMed.
- J. Vogelsang and J. M. Lupton, J. Phys. Chem. Lett., 2012, 3, 1503–1513 CrossRef CAS PubMed.
- T. Stangl, P. Wilhelm, K. Remmerssen, S. Höger, J. Vogelsang and J. M. Lupton, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E5560–E5566 CrossRef CAS PubMed.
- T. Eder, T. Stangl, M. Gmelch, K. Remmerssen, D. Laux, S. Höger, J. M. Lupton and J. Vogelsang, Nat. Commun., 2017, 8, 1641 CrossRef PubMed.
- Z. Hu, R. T. Haws, Z. Fei, P. Boufflet, M. Heeney, P. J. Rossky and D. A. Vanden Bout, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 5113–5118 CrossRef CAS PubMed.
- L. Hou, S. Adhikari, Y. Tian, I. G. Scheblykin and M. Orrit, Nano Lett., 2017, 17, 1575–1581 CrossRef CAS PubMed.
- H. Kobayashi, S. Hirata and M. Vacha, J. Phys. Chem. Lett., 2013, 4, 2591–2596 CrossRef CAS.
- M. Vacha, D. K. Sharma and S. Hirata, J. Photochem. Photobiol., C, 2017 DOI:10.1016/j.jphotochemrev.2017.11.003.
Footnotes |
| † Present address: Center for Integrated Nanotechnologies, Materials Physics and Applications Division, Los Alamos National Laboratory, Email: E-mail: zjhu05@gmail.com |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2018 |