
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe price of flexibility – a case study on septanoses as pyranose mimetics†
Christoph P.
Sager‡
 a,
Brigitte
Fiege‡
a,
Pascal
Zihlmann‡
a,
Raghu
Vannam‡
b,
Said
Rabbani
a,
Roman P.
Jakob
c,
Roland C.
Preston
a,
Adam
Zalewski
a,
Timm
Maier
c,
Mark W.
Peczuh
*b and
Beat
Ernst
*a
a,
Brigitte
Fiege‡
a,
Pascal
Zihlmann‡
a,
Raghu
Vannam‡
b,
Said
Rabbani
a,
Roman P.
Jakob
c,
Roland C.
Preston
a,
Adam
Zalewski
a,
Timm
Maier
c,
Mark W.
Peczuh
*b and
Beat
Ernst
*a
aUniversity of Basel, Institute of Molecular Pharmacy, Pharmacenter of the University of Basel, Klingelbergstrasse 50, 4056, Basel, Switzerland. E-mail: beat.ernst@unibas.ch
bDepartment of Chemistry, University of Connecticut, 55 N. Eagleville Road U3060, Storrs, CT, 06279 USA. E-mail: mark.peczuh@uconn.edu
cUniversity of Basel, Biozentrum: Focal Area Structural Biology, Klingelbergstrasse 70, 4056 Basel, Switzerland
First published on 8th November 2017
Abstract
Seven-membered ring mimetics of mannose were studied as ligands for the mannose-specific bacterial lectin FimH, which plays an essential role in the first step of urinary tract infections (UTI). A competitive binding assay and isothermal titration calorimetry (ITC) experiments indicated an approximately ten-fold lower affinity for the seven-membered ring mannose mimetic 2-O-n-heptyl-1,6-anhydro-D-glycero-D-galactitol (7) compared to n-heptyl α-D-mannopyranoside (2), resulting exclusively from a loss of conformational entropy. Investigations by solution NMR, X-ray crystallography, and molecular modeling revealed that 7 establishes a superimposable H-bond network compared to mannoside 2, but at the price of a high entropic penalty due to the loss of its pronounced conformational flexibility. These results underscore the importance of having access to the complete thermodynamic profile of a molecular interaction to “rescue” ligands from entropic penalties with an otherwise perfect fit to the protein binding site.
Introduction
The flexibility of a molecule defines its intrinsic conformational entropy. Rigid molecules adopt a highly populated conformational ground state with no or only a few alternative conformations. In contrast, flexible molecules populate multiple conformers with low barriers for interconversion between them. Flexibility has implications on intermolecular interactions in different manners (Fig. 1).1 Emil Fischer's lock-and-key model (A) assumes two rigid partners and therefore requires precise complementary shapes to allow tight binding.2–6 However, in most cases, either the ligand (B)7–12 or the receptor (C)13,14 exhibits flexibility while its binding partner is rather rigid. In these cases, complex formation requires an induced fit5 or a conformational selection mechanism.15–18 Finally, considerable reorganizational energy is required for interactions in which both ligand and receptor are flexible (D).19 A detailed knowledge of the thermodynamic profile allows for categorization of a binding event with respect to these four scenarios and affects the design of ligands. The enthalpic and entropic contributions to these intermolecular associations, fundamental for characterizing the thermodynamic profile of an association, can be determined by isothermal titration calorimetry (ITC).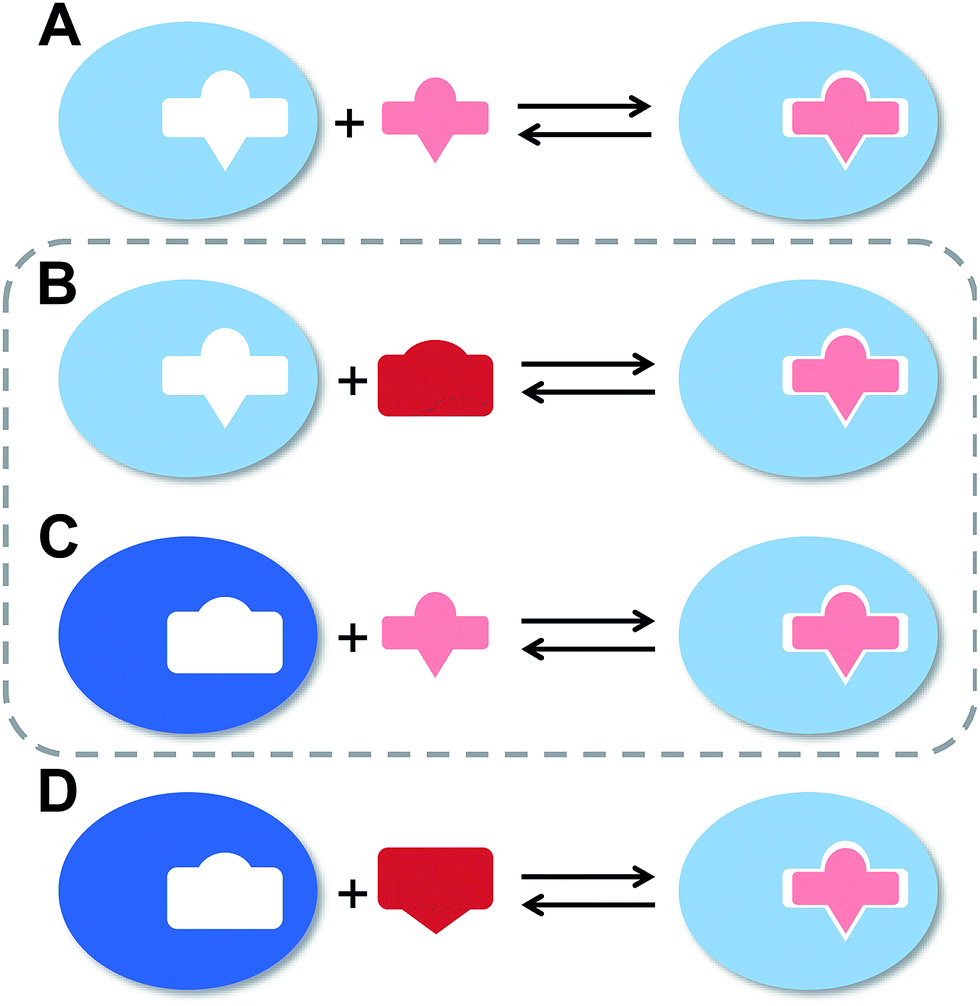 | ||
| Fig. 1 The four types of receptor–ligand interactions. (A) Rigid receptor, rigid ligand. (B) Rigid receptor, flexible ligand. (C) Flexible receptor, rigid ligand. (D) Flexible receptor, flexible ligand. | ||
In the present study, an example of a receptor–ligand interaction according to type B (Fig. 1) was studied in detail. The receptor is the lectin FimH, located at the tip of bacterial pili. It mediates the attachment of uropathogenic E. coli (UPEC) to urothelial cells of the mammalian host and therefore represents the first step of the process leading to urinary tract infection (UTI).20–22 This adhesion impedes the clearance of bacteria from the urinary tract and is therefore essential for the infection of urothelial cells.23–25 Since the physiological ligand of FimH is the highly mannosylated glycoprotein uroplakin 1A located on urothelial cells,20–22 a broad variety of mannose-based FimH antagonists have been developed for blocking this interaction and thereby the bacterial infection of bladder cells.26–34 Their successful application in a UTI mouse model clearly supports their therapeutic potential for treatment and prevention of UTI.26,31,35
To date, only the aglycone of mannosidic FimH antagonists has been successfully varied,26–37 whereas the replacement of mannose by other hexoses, e.g.D-glucose or D-galactose, led to a severe reduction or even loss of affinity.38 We were interested to know whether septanose derivatives, i.e. seven-membered ring homologs of pyranoses,39 could be utilized as ring-expanded FimH antagonists. Our interest was motivated by previous studies where the jackbean lectin concanavalin A (ConA) bound methyl septanosides in its carbohydrate binding pocket, albeit with reduced affinity relative to the pyranose ligand.40,41 ConA naturally recognizes the α1-3, α1-6-dimannosylmannoside42 as well as methyl α-pyranosides of mannose and glucose.43,44 In contrast to the natural selectivity for α-configured saccharides, methyl β-septanosides were bound by ConA whereas α-septanosides were not.
Here we report results from an investigation of septanoses binding to the isolated lectin domain of FimH (FimHLD), which is locked in the high-affinity state.45,46 Flexibility was fundamentally important for binding of the best seven-membered ring ligand, 2-O-n-heptyl-1,6-anhydro-D-glycero-D-galactitol (7) (informally referred to as 2-O-n-heptyl-1-deoxy-septanose), as FimHLD selected one conformation among those on its shallow conformational energy surface.
Results
Synthesis of septanose ligands
The design of the septanose ligands was guided by the structure of the mannose-binding pocket of FimH. It consists of a deep pocket lined with polar amino acid residues forming an extended H-bond network with the natural ligand. The entrance to the mannose-binding pocket, the so-called “tyrosine gate”, is furnished with two tyrosines and an isoleucine and interacts with aliphatic or aromatic aglycones of FimH antagonists.38 For mimicking mannose-based FimH antagonists, methyl β-D-glycero-D-galacto-septanosides 3 and 4 along with the related 1,6-anhydro-D-glycero-D-galactitols 5–7 were synthesized (3 and 4 will be referred to as methyl septanosides, and 5–7 as 1-deoxy-septanoses). For all these seven-membered ring analogs of α-D-mannose, the configuration at C3 to C6 (septanose numbering) is identical to the configuration at C2 to C5 in D-mannose. In addition, the n-heptyl substituent on the C2 hydroxyl group of septanose 4 and 7 was expected to mimic the n-heptyl aglycone at the anomeric center (C1) of α-D-mannopyranoside 2 (Scheme 1). | ||
| Scheme 1 Syntheses of FimH antagonists with a septanose core as replacement for α-D-mannopyranoside 1 and 2. Methyl β-septanoside 3 and 1,2-di-deoxy-septanose 5 have been previously described.40 | ||
For the syntheses of 3 & 4 and 6 & 7, acetonide protected oxepine 8 was utilized as a key starting material.47 The previously described diastereoselective epoxidation and methanolysis of 8 provided methyl β-D-glycero-D-galacto-septanoside 9 (84%).47,48 Alkylation of its C2 hydroxyl group gave 2-O-n-heptyl septanoside 10 in 73% yield. By removal of the acetonide group on 9 and 10, methyl septanosides 3 and 4 were obtained in 93% and 82% yields, respectively. The preparation of 6 and 7 relied on the regio- and diastereoselective hydroboration–oxidation of 8 to give 11 (65%).49 Deprotection of 1-deoxy-septanose 11 then yielded 6 (90%). Alternatively, 7 was obtained by alkylation of 11 at C2 to form compound 12 followed by deprotection (77% over two steps).
Competitive binding assay
Compounds 1–7, along with other analogs (see ESI†), were evaluated for their binding to FimHLD. Compounds were first screened in a competitive binding assay,36 where a polymeric oligomannose–biotin conjugate coupled via streptavidin to horseradish peroxidase is added simultaneously with a serial dilution of the test ligand to wells coated with FimHLD. After incubation, the peroxidase substrate 3,3′,5,5′-tetramethylbenzidine is added and the binding is quantified by colorimetric detection. For the compounds 1–7, the competitive binding assay revealed IC50 values ranging from 64 nM to ∼400 μM (Table 1). The addition of an n-heptyl substituent at C2 of 3 (→ 4) and 6 (→ 7) led to significantly improved inhibition. Septanoside 4 exhibits an 11-fold increase in affinity over 3, and 1-deoxy-septanose 7 a 307-fold improvement over 6. Thus, the n-heptyl substituent in 4 and 7 likely mimics the aglycone of the corresponding pyranoside 2.| Compound | IC50a [μM] | K D [μM] |
|
|
|
N |
|---|---|---|---|---|---|---|
| a IC50 values were measured in a competitive binding assay.36 b K D values and thermodynamic parameters were determined by ITC experiments at a temperature of 25 °C. | ||||||
| 1 (methyl α-D-mannoside) | 5.6 ± 0.5 | n.d. | ||||
| 2 (n-heptyl α-D-mannoside) | 0.064 ± 0.02 | 0.029 | −43.0 | −50.3 | 7.3 | 1.00 |
| 3 | 31.6 ± 4.2 | n.d. | ||||
| 4 | 2.83 ± 0.4 | 2.20 | −32.3 | −27.9 | −4.4 | 0.98 |
| 5 | 86.8 ± 7.9 | n.d. | ||||
| 6 | 421 ± 30.5 | n.d. | ||||
| 7 | 1.37 ± 0.3 | 0.26 | −37.5 | −49.4 | 11.8 | 1.00 |



ITC experiments with compounds 2, 4 and 7
Isothermal titration calorimetry (ITC) provided a deeper insight into the nature of the intermolecular interaction of FimHLD with 2-n-heptyl-septanoside 4 and 2-n-heptyl-1-deoxy-septanose 7 in comparison with n-heptyl α-D-mannopyranoside (2). All three test compounds contain an n-heptyl unit – either as aglycone (→ 2) or as substituent in the 2-position (→ 4 and 7, respectively). The n-heptyl group is essential to establish the important hydrophobic contacts within the tyrosine gate. The KD values obtained from ITC experiments were in relative agreement with the IC50 values from the competitive binding assay, i.e. in both assays 2 was the strongest binder followed by 7 and then 4 (Table 1).ITC data (Table 1 and Fig. 2A) revealed that the almost 100-fold loss in affinity of compound 4 compared to compound 2 is due to a diminished enthalpic contribution 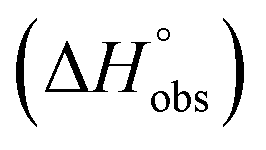 that is partly compensated by a more favorable entropy term
that is partly compensated by a more favorable entropy term  . The less beneficial enthalpy term for septanoside 4 likely results from an increased desolvation penalty due to its increased polar surface area (108.6 Å for 4 compared to 99.4 Å for 2)50 as well as from looser interactions with the FimHLD. As a consequence of a looser fit, the flexibility of septanoside 4 is less compromised, explaining the substantially improved entropy term by 11.7 kJ mol−1 (Table 1).
. The less beneficial enthalpy term for septanoside 4 likely results from an increased desolvation penalty due to its increased polar surface area (108.6 Å for 4 compared to 99.4 Å for 2)50 as well as from looser interactions with the FimHLD. As a consequence of a looser fit, the flexibility of septanoside 4 is less compromised, explaining the substantially improved entropy term by 11.7 kJ mol−1 (Table 1).
 | ||
| Fig. 2 (A) Thermodynamic fingerprints of compounds 2, 4, and 7 binding to FimHLD. (B) Heat capacity (ΔCp) of compounds 2 and 7 binding to FimHLD from ITC experiments performed at three different temperatures (details in ESI†). | ||
In contrast, the binding enthalpies of 2 and 7 are almost identical, so that the affinity loss of 1-deoxy-septanose 7 in comparison to mannoside 2 is exclusively due to a significant entropic penalty ( term). According to eqn (1), the entropy ΔS° consists of desolvation entropy
term). According to eqn (1), the entropy ΔS° consists of desolvation entropy  , conformational entropy
, conformational entropy  , and a term
, and a term 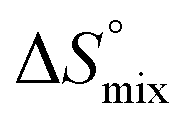 reflecting the loss in translational and rotational degrees of freedom of both binding partners upon complex formation. Because
reflecting the loss in translational and rotational degrees of freedom of both binding partners upon complex formation. Because  for the two antagonists is equal (10 kJ mol−1, eqn (2), where R is the universal gas constant and 55.6 the molarity of water in [M]),51,52 the entropy penalty originates exclusively from a loss of conformational flexibility
for the two antagonists is equal (10 kJ mol−1, eqn (2), where R is the universal gas constant and 55.6 the molarity of water in [M]),51,52 the entropy penalty originates exclusively from a loss of conformational flexibility 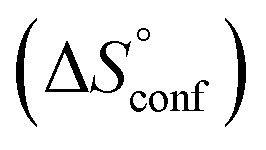 – either caused by the ligand and/or the receptor.
– either caused by the ligand and/or the receptor.
 | (1) |
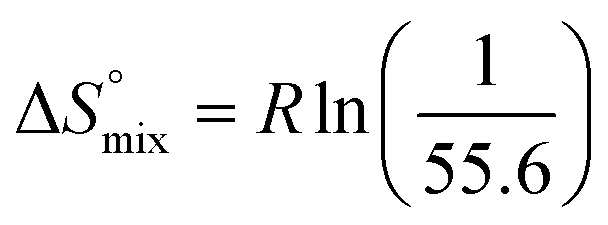 | (2) |
To quantify the contributions of  and
and 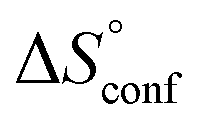 , ITC experiments were conducted at three different temperatures to determine the heat capacity (ΔCp) of mannoside 2 and 1-deoxy-septanose 7 (Fig. 2B and Table 2). ΔCp (eqn (3)), reporting on the entropy contributions from desolvation of ligand and protein, is equal for both complex formations (Fig. 2B). Since at 385 K,
, ITC experiments were conducted at three different temperatures to determine the heat capacity (ΔCp) of mannoside 2 and 1-deoxy-septanose 7 (Fig. 2B and Table 2). ΔCp (eqn (3)), reporting on the entropy contributions from desolvation of ligand and protein, is equal for both complex formations (Fig. 2B). Since at 385 K,  reaches zero, i.e. the hydration shell no longer exists,
reaches zero, i.e. the hydration shell no longer exists, 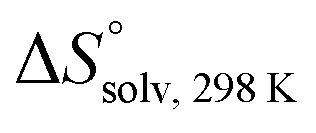 can be calculated according to eqn (4).52–54
can be calculated according to eqn (4).52–54
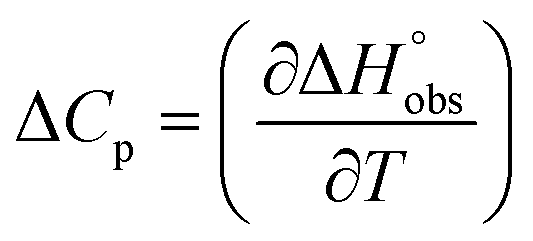 | (3) |
 | (4) |
| Contributor | Mannoside 2 [kJ mol−1] | 1-Deoxy-septanose 7 [kJ mol−1] |
|---|---|---|

|
−69.1 | −68.8 |
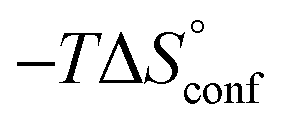
|
66.4 | 70.6 |
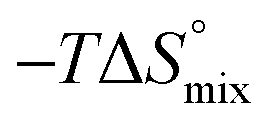
|
10.0 | 10.0 |
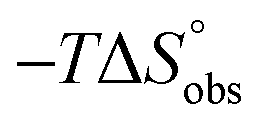
|
7.3 | 11.8 |
The identical ΔCp values (0.902 kJ K−1 ± 0.018 and 0.907 kJ K−1 ± 0.016, respectively) obtained for the interaction of FimHLD with 1-deoxy-septanose 7 and mannoside 2 are a clear indication that both complex formations release comparable numbers of water molecules leading to a comparable gain of desolvation entropy  . Consequently, the entropic penalty for 7 originates from a loss of conformational freedom of either the ligand and/or the protein upon binding (Table 2).
. Consequently, the entropic penalty for 7 originates from a loss of conformational freedom of either the ligand and/or the protein upon binding (Table 2).
To comprehensively interpret the thermodynamics of binding, additional information regarding solution and bound conformations of mannoside 2 and 1-deoxy-septanose 7 are indispensible. The conformational flexibility of septanoses vs. pyranoses as well as the n-heptyl aglycone deserves special attention in this regard. Furthermore, the degree of conformational flexibility of FimHLD has to be addressed. Finally, structural analysis by X-ray crystallography to elucidate the hydrogen bond network established by the two ligands is essential for a final assessment of the two binding processes.
In silico comparison of the solution conformation of mannoside 2 and 1-deoxy-septanose 7
The large conformational entropy penalty for 1-deoxy-septanose 7 in comparison to mannoside 2 could be related to the extent of flexibility of their respective solution conformations. Therefore, the dynamic behavior in solution of each was studied by monitoring two collected variables using metadynamics simulations. The collected variables were defined as the angle between O1–C1–C4 and the dihedral torsion of O1–C1–O5–C5 for mannoside 2 and the corresponding angle O2–C2–C5 and dihedral torsion O2–C2–C1–O6 for compound 7 (for numbering see Fig. 3). Metadynamics simulations revealed an energy landscape for mannoside 2 with one distinct minimum of −154.8 kJ mol−1 of 99° for the angle O1–C1–C4 and 54° for the dihedral angle (indicated by a circle in Fig. 3A), whereas the energy surface for 1-deoxy-septanose 7 was much shallower, showing two minima of −90.4 kJ mol−1 at 99° and 36°, and −89.5 kJ mol−1 at 90° and 90°, respectively (indicated by circles in Fig. 3B). Thus, the metadynamic simulations clearly indicate that 1-deoxy-septanose 7 exhibits a much larger ring flexibility in comparison to mannoside 2. In addition, the minima observed in the metadynamics simulations coincide closely with the bound conformation of the crystal structure. However, compound 7 shows an additional minimum that does not overlap with the bound conformation and therefore adds to the entropic loss.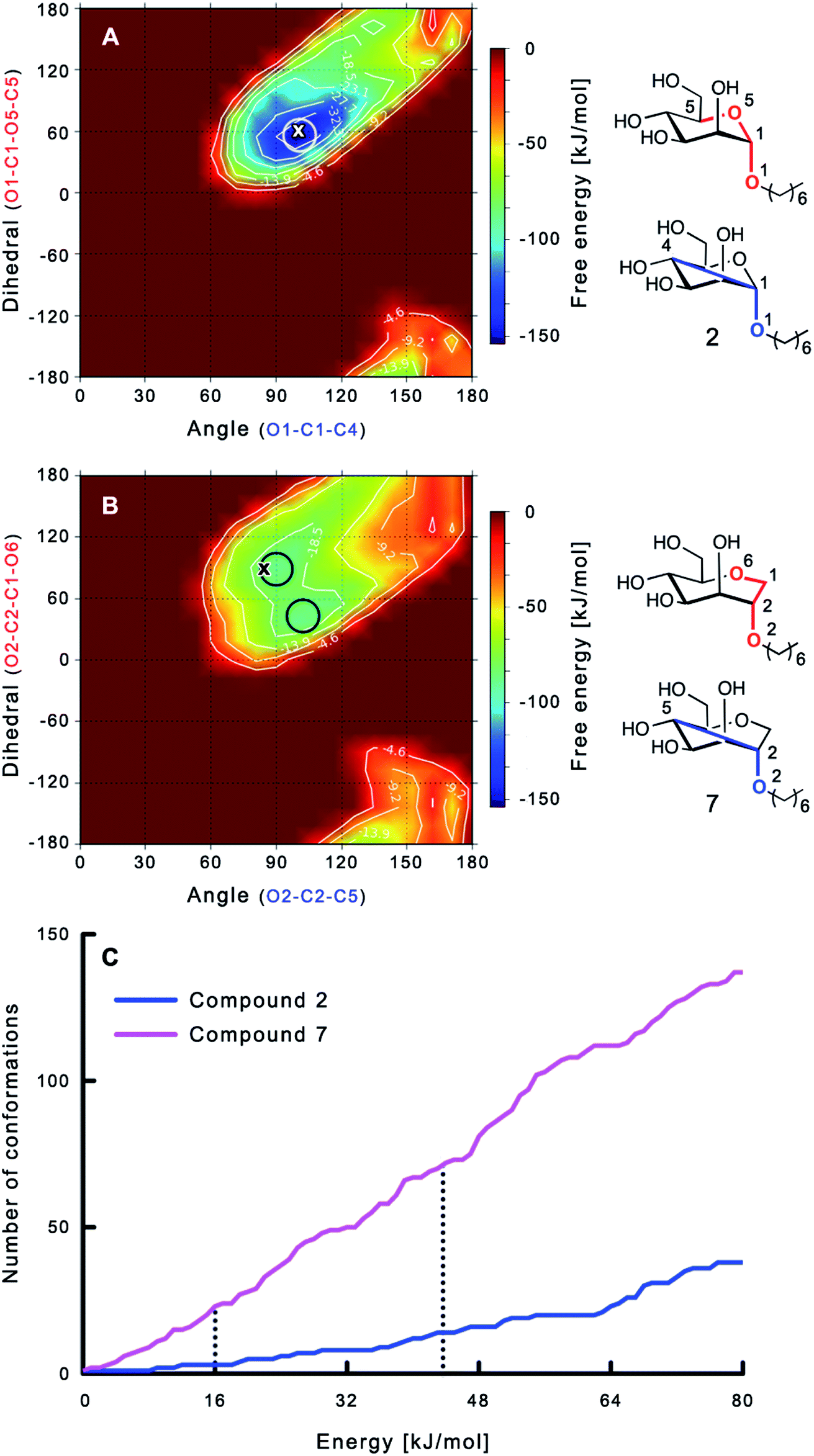 | ||
| Fig. 3 (A, B) Energy surface diagrams of the solution conformation of 2 (A) and 7 (B) obtained from metadynamics analysis. Circles represent the distinct minima observed in metadynamic simulations and crosses indicate the bound conformations in the crystal structures [PDB code 4BUQ (A) & 5CGB (B)]. Energies are given in kJ mol−1 and are color-coded from bordeaux-red (0 kJ mol−1) to dark-blue (−150 kJ mol−1). Vertical axes describe the observed dihedral angle (red), horizontal axes the observed angle (blue), both indicated in the structures to the right. (C) Cumulative frequency distribution analysis of all conformations (within 80 kJ mol−1 from the global minimum). Dashed lines indicate examples that occur at similar energies as denoted in the text. | ||
To further quantify the flexibility issue, all ring conformations were normalized to their respective global minimum. Cumulative frequency analysis revealed that compound 7 exhibits 23 accessible conformations within 16 kJ mol−1 corresponding to the energy range of a hydrogen bond, whereas compound 2 has only access to 3 conformations. When a 44 kJ mol−1 energy range is taken into consideration (equal to the highest barrier in a cyclohexane ring-flip), compound 7 has access to 72 conformations in contrast to the rather rigid mannoside 2 with only 14 accessible conformations (Fig. 3C). In summary, the much higher number of conformations available to septanose derivative 7 results in a higher loss of conformational flexibility upon complexation to FimH and accounts for the difference in entropies of binding to FimHLD for 7 compared to 2.
Ab initio calculations involving interactions of the n-heptyl group
The preference of 1-deoxy-septanose 7 to position its n-heptyl tail in an axial orientation was demonstrated qualitatively by solution NMR experiments in the absence of protein. Karplus analysis of coupling constants 3J2,3 and 3J3,4 (septanose numbering) of 7 obtained from a 1D 1H NMR spectrum at 900 MHz indicated torsion angles in agreement with those expected for the axial conformation (see ESI† for details). To assess whether the tyrosine gate affects the n-heptyl tail in 2 and 7 identically, non-covalent interaction energies were calculated based on the available crystal structures (cf.Fig. 5) with Jaguar.55–57 For the proper quantification of the non-covalent interactions, we performed ab initio DFT calculations using the B3LYP-MM functional in combination with the cc-pVDZ++ basis-set in the gas phase. The calculations led to comparable interaction energies of −18.5 kJ mol−1 and −19.2 kJ mol−1 for the n-heptyl tails of 2 and 7 with the tyrosine-gate. We therefore assume that their contributions to enthalpy as well as entropy are of comparable size.Conformation of FimHLD upon binding of ligand 2 and 7 assessed by NMR CSP experiments
NMR chemical shift perturbation (CSP) experiments of the FimHLD backbone amide resonances were used to assess the conformation of the protein backbone.58 Thus, 1H,15N-HSQC experiments of uniformly 15N-labeled FimHLD (see ESI†) were conducted in the absence and presence of an excess of each ligand. The resonance assignment of FimHLD was available from previous studies.58,59 For both antagonists 2 and 7, the set of protein residues of the mannose binding pocket (e.g. Ala2, Asp54 and Gln133) and a nearby loop (e.g. Thr134, Asn138, Asp141, Gln143) exhibited highly similar CSP effects (Fig. 4; see also Fig. S5 in ESI†). The absence of line broadening effects demonstrates that the protein is rather rigid in complex with both antagonists. Furthermore, the almost identical signals for amino acids remote from the mannose-binding pocket indicate that FimHLD does not undergo global conformational adaptions upon ligand binding. Thus, we can assume that the protein conformation upon binding mannoside 2 and 1-deoxy-septanose 7 is highly similar, consistent with the comparable thermodynamic outcomes of the interactions. | ||
| Fig. 4 NMR chemical shift perturbation experiments of FimHLD with mannoside 2 and 1-deoxy-septanose 7. 1H,15N-HSQC spectra of FimHLD in the absence of antagonist (green) and in the presence of an excess of 2 (blue) and 7 (magenta). Important residues in the binding sites are shown in detail on the panel at the right. | ||
Crystal structure of FimHLD in complex with mannoside 2 and 1-deoxy-septanose 7
The previously reported co-crystal structure of mannoside 2–FimHLD60 (PDB code 4BUQ, Fig. 5A) and the new high-resolution X-ray structure of FimHLD co-crystallized with 7 (PDB code 5CGB; Fig. 5B and Table S3 in ESI†) exhibit a surprisingly high structural similarity with a backbone RMSD of 0.2 Å. Noteworthy, the well-defined, tight FimH mannose-binding pocket installs identical hydrogen bond networks with mannoside 2 and 1-deoxy-septanose 7 (Fig. 5). For both ligands, nine hydrogen bonds with the side-chains of the residues Asn46, Asp54, Gln133, Asn135 and Asp140, and to the backbone of Phe1 and Asp47 are formed (for statistics on diffraction data and structure refinement of ligand 7–FimHLD complex see ESI†).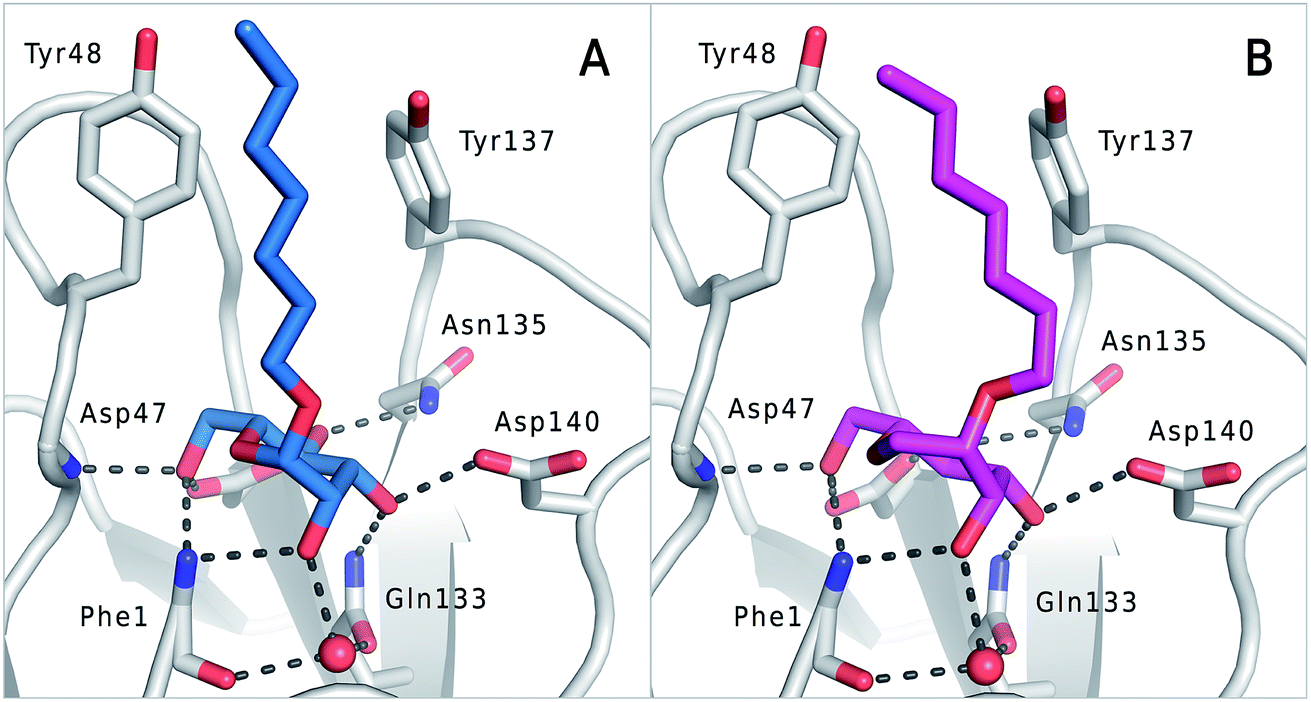 | ||
| Fig. 5 (A) Co-crystal structure of FimHLD with mannoside 2 (PDB code 4BUQ) and (B) co-crystal structure of FimHLD with 1-deoxy-septanose 7 (PDB code 5CGB); the intermolecular hydrogen bond network is depicted by dashed lines. | ||
The dynamic stability of the hydrogen bond networks was assessed by MD simulations starting from the conformations of compound 2 and 7 from the crystal structures in Fig. 5. The simulations were run for the duration of 4.8 ns, followed by an analysis of the hydrogen bond network, where the occupancy of a given hydrogen bond interaction was monitored. The crucial hydrogen bonds with Phe1, Asp54 and Gln133 were retained with comparable occupancies by both compounds 2 and 7 (see Table S3 in ESI†). Therefore, in accordance with ITC experiments, enthalpic contributions from ligand–protein interactions can be predicted to be of similar size.
Discussion
The bacterial lectin FimH mediates the adhesion of uropathogenic E. coli to the bladder epithelium of its host. FimHLD consists of a mannose-binding pocket, which is reached through a hydrophobic cleft equipped with two tyrosines and an isoleucine (tyrosine gate). Its high affinity interaction with mannoside 2 is achieved by an extended hydrogen bond network established between the mannose moiety and the carbohydrate recognition domain of the lectin and by hydrophobic σ–π interactions of the n-heptyl aglycone with the tyrosine gate.38,58,61 Replacement of the mannose moiety by other hexoses, e.g.D-glucose or D-galactose, leads to a substantial reduction or even loss of binding affinity.38Seven-membered ring analogs designed to mimic α-D-mannopyranosides in general exhibit reduced affinities for FimHLD. From a series of septanosides and analogs, methyl septanoside 4 and 1-deoxy-septanose 7 were identified as the best representative binders. The n-heptyl substituent on their 2-position enables σ–π stacking interaction with the tyrosine gate in analogy to mannoside 2.58,61 Nevertheless, 4 (2.2 μM) and 7 (260 nM) showed a 75- and 9-fold loss of inhibitory potency compared to mannoside 2 (29 nM).
To explain the large difference in affinity, a comparison of the thermodynamic fingerprints of their interactions with FimHLD turned out to be instructive. The ITC profile of n-heptyl mannoside (2) served as reference. Binding of 2, like many other mannoside-based antagonists, is largely enthalpy-driven with a major contribution by the nine hydrogen bonds formed between the mannose moiety and the lectin.29,35,38,58 The severe drop in affinity of methyl septanoside 4 compared to mannoside 2 is predominantly due to a substantially smaller enthalpy term (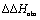 = 22.4 kJ mol−1), accompanied by a pronounced improvement of the entropy term (
= 22.4 kJ mol−1), accompanied by a pronounced improvement of the entropy term ( = 11.7 kJ mol−1). One possible explanation for the severe enthalpy penalty is a steric clash induced by the methoxy group in the 1-position, disordering the essential hydrogen bond network. A less well-defined hydrogen bond network on the other hand would be consistent with an entropy gain of 4 in comparison to mannoside 2 because of the increased conformational flexibility. Interestingly, the almost identical enthalpy terms for mannoside 2 and 1-deoxy-septanose 7 (
= 11.7 kJ mol−1). One possible explanation for the severe enthalpy penalty is a steric clash induced by the methoxy group in the 1-position, disordering the essential hydrogen bond network. A less well-defined hydrogen bond network on the other hand would be consistent with an entropy gain of 4 in comparison to mannoside 2 because of the increased conformational flexibility. Interestingly, the almost identical enthalpy terms for mannoside 2 and 1-deoxy-septanose 7 ( ∼ −50 kJ mol−1) suggest that FimH adopts very similar conformations enabling the formation of all critical hydrogen bonds to both ligands, a hypothesis supported by solution NMR and X-ray co-crystallography.
∼ −50 kJ mol−1) suggest that FimH adopts very similar conformations enabling the formation of all critical hydrogen bonds to both ligands, a hypothesis supported by solution NMR and X-ray co-crystallography.
The origin for the reduced affinity of 7 compared to 2 is a significantly higher entropic penalty. ITC measurements at different temperatures revealed almost identical heat capacities ΔCp for both mannoside 2 and 1-deoxy-septanose 7, indicating comparable desolvation entropies 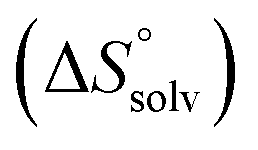 . Since
. Since  reflecting the loss in translational and rotational degrees of freedom upon complex formation can be assumed to be identical, the increased entropic penalty for 7 can be attributed solely to a reduced conformational freedom of the ligand and/or the protein. With NMR chemical shift perturbation experiments it was shown that 1-deoxy-septanose 7 exhibited comparable chemical shift changes and line widths for amino acid residues of the FimH binding pocket as mannoside 2. Thus, both ligands form comparable complexes with FimHLD and therefore exhibit comparable
reflecting the loss in translational and rotational degrees of freedom upon complex formation can be assumed to be identical, the increased entropic penalty for 7 can be attributed solely to a reduced conformational freedom of the ligand and/or the protein. With NMR chemical shift perturbation experiments it was shown that 1-deoxy-septanose 7 exhibited comparable chemical shift changes and line widths for amino acid residues of the FimH binding pocket as mannoside 2. Thus, both ligands form comparable complexes with FimHLD and therefore exhibit comparable  terms. Quantum-mechanical calculations revealed almost identical contributions of the aglycones of 2 and 7 to the interaction energy, suggesting that the loss of conformational entropy is solely due to the higher flexibility of the seven-membered ring of 7 compared to the six-membered ring of 2 in solution. Upon binding to FimHLD, a specific conformation of the septanose ring is required, leading to a significant rigidification of the compound relative to its unbound state. Metadynamics simulations, revealing a flat energy profile for 1-deoxy-septanose 7 with various ring conformations separated by relatively low energetic barriers, support this assumption.
terms. Quantum-mechanical calculations revealed almost identical contributions of the aglycones of 2 and 7 to the interaction energy, suggesting that the loss of conformational entropy is solely due to the higher flexibility of the seven-membered ring of 7 compared to the six-membered ring of 2 in solution. Upon binding to FimHLD, a specific conformation of the septanose ring is required, leading to a significant rigidification of the compound relative to its unbound state. Metadynamics simulations, revealing a flat energy profile for 1-deoxy-septanose 7 with various ring conformations separated by relatively low energetic barriers, support this assumption.
The bound conformation of 7 can therefore be regarded as the result of a conformational selection of the ligand. Whereas conformational selection in terms of protein dynamics has been widely described and quantified by the use of NMR dynamics experiments of the protein,7,15–18 a quantitative description of the impact of ligand flexibility on the thermodynamics of binding is still rare, likely due to the numerous contributing factors, which could be separated and analyzed in this specific example.
Conclusions
ITC results, NMR CSP experiments, computational considerations, and X-ray crystallographic data support the conclusion that 2-n-heptyl 1-deoxy-septanose 7 adopts a conformation enabling the formation of a H-bond network with FimHLD identical to the corresponding mannoside 2. Furthermore, the loss of affinity of 7 is almost exclusively due to a loss in conformational entropy ΔSconf. In silico-analysis and solution NMR data ultimately showed that, although the main solution conformation of the seven-membered ring is similar to the bound conformation, the pronounced flexibility of the ring in solution causes significant entropic penalties upon complex formation.In view of this result, the evaluation of glycomimetics should always include thermodynamic profiling because the quantification of enthalpic and entropic terms can be highly informative for the design of improved antagonists. In the case of FimH antagonists, 1-deoxy-septanose 7 provides an excellent mannopyranoside mimic, i.e. can established an identical H-bond network as the parent compound. However, it suffers from a substantial entropic penalty due to rigidification of the inherent ring flexibility upon complexation. This study excellently exemplifies that flexibility can be an important element in the design of small molecule ligands of target proteins.62,63
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Prof. Stephan Grzesiek at the Biozentrum, University of Basel for the access to 600 and 900 MHz NMR spectrometers. The financial support by the Swiss National Science Foundation (CS & PZ: Swiss National Science Foundation (SNF) grant 200020_129935; RP: SNF grant 200020_146202; TM: SNF grant R'EQUIP 145023) is gratefully acknowledged. B. F. thanks the German Academic Exchange Service (DAAD) for a stipendship. Finally, support for this work was provided to MWP by the National Science Foundation (CAREER CHE-0546311).Notes and references
- S. Moghaddam, Y. Inoue and M. K. Gilson, J. Am. Chem. Soc., 2009, 131, 4012 CrossRef CAS PubMed.
- E. Fischer, Ber. Dtsch. Chem. Ges., 1894, 27, 2985 CrossRef CAS.
- F. W. Lichtenthaler, Angew. Chem., Int. Ed., 1995, 33, 2364 CrossRef.
- R. U. Lemieux and U. Spohr, Adv. Carbohydr. Chem. Biochem., 1994, 50, 1 CrossRef CAS PubMed.
- D. E. Koshland, Angew. Chem., Int. Ed., 1995, 33, 2375 CrossRef.
- P. W. Snyder, J. Mecinovic, D. T. Moustakas, S. W. Thomas III, M. Harder, E. T. Mack, M. R. Lockett, A. Heroux, W. Sherman and G. M. Whitesides, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 17889 CrossRef CAS PubMed.
- C. Diehl, O. Engstrom, T. Delaine, M. Hakansson, S. Genheden, K. Modig, H. Leffler, U. Ryde, U. J. Nilsson and M. Akke, J. Am. Chem. Soc., 2010, 132, 14577 CrossRef CAS PubMed.
- F. Marcelo, Y. He, S. A. Yuzwa, L. Nieto, J. Jiménez-Barbero, M. Sollogoub, D. J. Vocadlo, G. D. Davies and Y. Bleriot, J. Am. Chem. Soc., 2009, 131, 5390 CrossRef CAS PubMed.
- A. R. Patel, G. Ball, L. Hunter and F. Liu, Org. Biomol. Chem., 2013, 11, 3781 CAS.
- D. D. Staas, K. L. Savage, V. L. Sherman, H. L. Shimp, T. A. Lyle, L. O. Tran, C. M. Wiscount, D. R. McMasters, P. E. Sanderson, P. D. Williams, B. J. Lucas Jr, J. A. Krueger, S. D. Lewis, R. B. White, S. Yu, B. K. Wong, C. J. Kochansky, M. R. Anari, Y. Yan and J. P. Vacca, Bioorg. Med. Chem., 2006, 14, 6900 CrossRef CAS PubMed.
- Y. Endo, S. Takehana, M. Ohno, P. E. Driedger, S. Stabel, M. Y. Mizutani, N. Tomioka, A. Itai and K. Shudo, J. Med. Chem., 1998, 41, 1476 CrossRef CAS PubMed.
- Y. Endo, M. Ohno, M. Hirano, A. Itai and K. Shudo, J. Am. Chem. Soc., 1996, 118, 1841 CrossRef CAS.
- K. Gunasekaran and R. Nussinov, J. Mol. Biol., 2007, 365, 257 CrossRef CAS PubMed.
- D. Rauh, G. Klebe and M. T. Stubbs, J. Mol. Biol., 2004, 335, 1325 CrossRef CAS PubMed.
- D. D. Boehr, R. Nussinov and P. E. Wright, Nat. Chem. Biol., 2009, 5, 789 CrossRef CAS PubMed.
- K. Okazaki and S. Takada, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 11182 CrossRef CAS PubMed.
- I. V. Nesmelova, E. Ermakova, V. A. Daragan, M. Pang, M. Menendez, L. Lagartera, D. Solis, L. G. Baum and K. H. Mayo, J. Mol. Biol., 2010, 397, 1209 CrossRef CAS PubMed.
- T. R. Weikl and F. Paul, Protein Sci., 2014, 23, 1508 CrossRef CAS PubMed.
- C. H. Reynolds and M. K. Holloway, ACS Med. Chem. Lett., 2011, 2, 433 CrossRef CAS PubMed.
- X. R. Wu, T. T. Sun and J. J. Medina, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 9630 CrossRef CAS.
- G. Zhou, W. J. Mo, P. Sebbel, G. Min, T. A. Neubert, R. Glockshuber, X. R. Wu, T. T. Sun and X. P. Kong, J. Cell Sci., 2001, 114, 4095 CAS.
- B. Xie, G. Zhou, S. Y. Chan, E. Shapiro, X. P. Kong, X. R. Wu, T. T. Sun and C. E. Costello, J. Biol. Chem., 2006, 281, 14644 CrossRef CAS PubMed.
- A. Ronald, Am. J. Med., 2002, 113, 14 CrossRef.
- D. Abgottspon, G. Rölli, L. Hosch, A. Steinhuber, X. Jiang, O. Schwardt, B. Cutting, M. Smieško, U. Jenal, B. Ernst and A. Trampuz, J. Microbiol. Methods, 2010, 82, 249 CrossRef CAS PubMed.
- M. Scharenberg, D. Abgottspon, E. Cicek, X. Jiang, O. Schwardt, S. Rabbani and B. Ernst, Assay Drug Dev. Technol., 2011, 9, 455 CrossRef CAS PubMed.
- T. Klein, D. Abgottspon, M. Wittwer, S. Rabbani, J. Herold, X. Jiang, S. Kleeb, C. Luthi, M. Scharenberg, J. Bezençon, E. Gubler, L. Pang, M. Smieško, B. Cutting, O. Schwardt and B. Ernst, J. Med. Chem., 2010, 53, 8627 CrossRef CAS PubMed.
- O. Schwardt, S. Rabbani, M. Hartmann, D. Abgottspon, M. Wittwer, S. Kleeb, A. Zalewski, M. Smieško, B. Cutting and B. Ernst, Bioorg. Med. Chem., 2011, 19, 6454 CrossRef CAS PubMed.
- X. Jiang, D. Abgottspon, S. Kleeb, S. Rabbani, M. Scharenberg, M. Wittwer, M. Haug, O. Schwardt and B. Ernst, J. Med. Chem., 2012, 55, 4700 CrossRef CAS PubMed.
- L. Pang, S. Kleeb, K. Lemme, S. Rabbani, M. Scharenberg, A. Zalewski, F. Schadler, O. Schwardt and B. Ernst, ChemMedChem, 2012, 7, 1404 CrossRef CAS PubMed.
- M. Aronson, O. Medalia, L. Schori, D. Mirelman, N. Sharon and I. Ofek, J. Infect. Dis., 1979, 139, 329 CrossRef CAS PubMed.
- C. K. Cusumano, J. S. Pinkner, Z. Han, S. E. Greene, B. A. Ford, J. R. Crowley, J. P. Henderson, J. W. Janetka and S. J. Hultgren, Sci. Transl. Med., 2011, 3, 109ra115 Search PubMed.
- D. Abgottspon and B. Ernst, Chimia, 2012, 66, 166 CrossRef CAS PubMed.
- M. Totsika, M. Kostakioti, T. J. Hannan, M. Upton, S. A. Beatson, J. W. Janetka, S. J. Hultgren and M. A. Schembri, J. Infect. Dis., 2013, 208, 921 CrossRef CAS PubMed.
- J. Bouckaert, Z. Li, C. Xavier, M. Almant, V. Caveliers, T. Lahoutte, S. D. Weeks, J. Kovensky and S. G. Gouin, Chem.–Eur. J., 2013, 19, 7847 CrossRef CAS PubMed.
- S. Kleeb, L. Pang, K. Mayer, D. Eriş, A. Sigl, R. C. Preston, P. Zihlmann, T. Sharpe, R. P. Jakob, D. Abgottspon, A. S. Hutter, M. Scharenberg, X. Jiang, G. Navarra, S. Rabbani, M. Smieško, N. Lüdin, J. Bezençon, O. Schwardt, T. Maier and B. Ernst, J. Med. Chem., 2015, 58, 2221 CrossRef CAS PubMed.
- S. Rabbani, X. Jiang, O. Schwardt and B. Ernst, Anal. Biochem., 2010, 407, 188 CrossRef CAS PubMed.
- Z. Han, J. S. Pinkner, B. Ford, R. Obermann, W. Nolan, S. A. Wildman, D. Hobbs, T. Ellenberger, C. K. Cusumano, S. J. Hultgren and J. W. Janetka, J. Med. Chem., 2010, 53, 4779 CrossRef CAS PubMed.
- J. Bouckaert, J. Berglund, M. Schembri, E. De Genst, L. Cools, M. Wuhrer, C.-S. Hung, J. Pinkner, R. Slättegård, A. Zavialov, D. Choudhury, S. Langermann, S. J. Hultgren, L. Wyns, P. Klemm, S. Oscarson, S. D. Knight and H. De Greve, Mol. Microbiol., 2005, 55, 441 CrossRef CAS PubMed.
- J. Saha and M. W. Peczuh, Adv. Carbohydr. Chem. Biochem., 2011, 66, 121 CrossRef CAS PubMed.
- M. R. Duff Jr, W. S. Fyvie, S. D. Markad, A. E. Frankel, C. V. Kumar, J. A. Gascón and M. W. Peczuh, Org. Biomol. Chem., 2011, 9, 154 Search PubMed.
- S. Castro, M. Duff, N. L. Snyder, M. Morton, C. V. Kumar and M. W. Peczuh, Org. Biomol. Chem., 2005, 3, 3869 CAS.
- D. Gupta, T. K. Dam, S. Oscarson and C. F. Brewer, J. Biol. Chem., 1997, 272, 6388 CrossRef CAS PubMed.
- M. C. Chervenak and E. J. Toone, Biochemistry, 1995, 34, 5685 CrossRef CAS PubMed.
- F. P. Schwarz, K. D. Puri, R. G. Bhat and A. Surolia, J. Biol. Chem., 1993, 268, 7668 CAS.
- D. Eriş, R. C. Preston, M. Scharenberg, F. Hulliger, D. Abgottspon, L. Pang, X. Jiang, O. Schwardt and B. Ernst, ChemBioChem, 2016, 17, 1012 CrossRef PubMed.
- M. M. Sauer, R. P. Jakob, J. Eras, S. Baday, D. Eriş, G. Navarra, S. Bernèche, B. Ernst, T. Maier and R. Glockshuber, Nat. Commun., 2016, 7, 10738 CrossRef CAS PubMed.
- S. D. Markad, S. Xia, N. L. Snyder, B. Surana, M. D. Morton, C. M. Hadad and M. W. Peczuh, J. Org. Chem., 2008, 73, 6341 CrossRef CAS PubMed.
- M. P. DeMatteo, N. L. Snyder, M. Morton, D. M. Baldisseri, C. M. Hadad and M. W. Peczuh, J. Org. Chem., 2005, 70, 24 CrossRef CAS PubMed.
- R. Murali and M. Nagarajan, Carbohydr. Res., 1996, 280, 351 CrossRef CAS.
- P. Ertl, B. Rohde and P. Selzer, J. Med. Chem., 2000, 43, 3714 CrossRef CAS PubMed.
- B. M. Baker and K. P. Murphy, J. Mol. Biol., 1997, 268, 557 CrossRef CAS PubMed.
- K. P. Murphy, D. Xie, K. S. Thompson, L. M. Amzel and E. Freire, Proteins, 1994, 18, 63 CrossRef CAS PubMed.
- K. P. Murphy, P. L. Privalov and S. J. Gill, Science, 1990, 247, 559 CAS.
- R. L. Baldwin, Proc. Natl. Acad. Sci. U. S. A., 1986, 83, 8069 CrossRef CAS.
- Schrödinger Suite 2015-4, Jaguar, Version 9.0, Schrödinger, LLC, New York, NY, 2015 Search PubMed.
- S. T. Schneebeli, A. D. Bochevarov and R. A. Friesner, J. Chem. Theory Comput., 2011, 7, 658 CrossRef CAS PubMed.
- A. D. Bochevarov, E. Harder, T. F. Hughes, J. R. Greenwood, D. A. Braden, D. M. Philipp, D. Rinaldo, M. D. Halls, J. Zhang and R. A. Friesner, Int. J. Quantum Chem., 2013, 113, 2110 CrossRef CAS.
- B. Fiege, S. Rabbani, R. C. Preston, R. P. Jakob, P. Zihlmann, O. Schwardt, X. Jiang, T. Maier and B. Ernst, ChemBioChem, 2015, 16, 1235 CrossRef CAS PubMed.
- S. Vanwetswinkel, A. N. Volkov, Y. G. J. Sterckx, A. Garcia-Pino, L. Buts, W. F. Vranken, J. Bouckaert, R. Roy, L. Wyns and N. A. J. van Nuland, J. Med. Chem., 2014, 57, 1416 CrossRef CAS PubMed.
- G. Roos, A. Wellens, M. Touaibia, N. Yamakawa, P. Geerlings, R. Roy, L. Wyns and J. Bouckaert, ACS Med. Chem. Lett., 2013, 4, 1085 CrossRef CAS PubMed.
- A. Wellens, M. Lahmann, M. Touaibia, J. Vaucher, S. Oscarson, R. Roy, H. Remaut and J. Bouckaert, Biochemistry, 2012, 51, 4790 CrossRef CAS PubMed.
- S. Chung, J. B. Parker, M. Bianchet, L. M. Amzel and J. T. Stivers, Nat. Chem. Biol., 2009, 5, 407 CrossRef CAS PubMed.
- S. A. Moura-Tamames, M. J. Ramos and P. A. Fernandes, J. Mol. Graphics Modell., 2009, 27, 908 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc04289b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |