
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dual nickel and Lewis acid catalysis for cross-electrophile coupling: the allylation of aryl halides with allylic alcohols†
Xue-Gong
Jia
,
Peng
Guo
,
Jicheng
Duan
and
Xing-Zhong
Shu
 *
*
State Key Laboratory of Applied Organic Chemistry (SKLAOC), College of Chemistry and Chemical Engineering, Lanzhou University, 222 South Tianshui Road, Lanzhou, 730000, China. E-mail: shuxingzh@lzu.edu.cn
First published on 6th November 2017
Abstract
Controlling the selectivity in cross-electrophile coupling reactions is a significant challenge, particularly when one electrophile is much more reactive. We report a general and practical strategy to address this problem in the reaction between reactive and unreactive electrophiles by a combination of nickel and Lewis acid catalysis. This strategy is used for the coupling of aryl halides with allylic alcohols to form linear allylarenes selectively. The reaction tolerates a wide range of functional groups (e.g. silanes, boronates, anilines, esters, alcohols, and various heterocycles) and works with various allylic alcohols. Complementary to most current routes for the C3 allylation of an unprotected indole, this method provides access to C2 and C4–C7 allylated indoles. Preliminary mechanistic experiments reveal that the reaction might start with an aryl nickel intermediate, which then reacts with Lewis acid activated allylic alcohols in the presence of Mn.
Introduction
Selective cross-electrophile coupling has recently emerged as an increasingly popular approach for constructing C–C bonds.1 The reaction achieves the union of two different bench-stable electrophiles (aryl/alkyl halides, etc.) and avoids using air and/or moisture sensitive organometallic reagents (RMgX, RZnX, , RB(OH)2, etc.).2 Unlike in conventional cross-coupling, where the selectivity is controlled by the oxidative addition of an electrophile and transmetallation of a nucleophile, generally both electrophiles will compete for oxidative addition at the catalyst in cross-electrophile coupling. Thus, the control of the selectivity for the cross-product over the symmetric dimer is a particular challenge, especially when one electrophile is much more reactive than the other. Indeed, such a reaction has been realized very recently3 and significant progress is achieved in nickel catalysis with a metal reductant.4 The substrate, however, remains to be confined to reactive electrophiles (R–I, Br, Cl, OTf, etc.). Given that inert O-electrophiles (alcohol, phenol, ether, etc.) are more readily available and their coupling reactions typically require organometallic reagents,5 the development of a strategy to couple these compounds with electrophiles will be highly attractive but remains extremely challenging.6
, RB(OH)2, etc.).2 Unlike in conventional cross-coupling, where the selectivity is controlled by the oxidative addition of an electrophile and transmetallation of a nucleophile, generally both electrophiles will compete for oxidative addition at the catalyst in cross-electrophile coupling. Thus, the control of the selectivity for the cross-product over the symmetric dimer is a particular challenge, especially when one electrophile is much more reactive than the other. Indeed, such a reaction has been realized very recently3 and significant progress is achieved in nickel catalysis with a metal reductant.4 The substrate, however, remains to be confined to reactive electrophiles (R–I, Br, Cl, OTf, etc.). Given that inert O-electrophiles (alcohol, phenol, ether, etc.) are more readily available and their coupling reactions typically require organometallic reagents,5 the development of a strategy to couple these compounds with electrophiles will be highly attractive but remains extremely challenging.6
The integration of multiple catalytic systems to enhance reactivity and/or selectivity is a concept often employed by biological catalysts7 and is an emerging strategy in molecular catalysis.8 The cooperative effects offer an opportunity to overcome the selectivity challenge in cross-electrophile coupling reactions (Scheme 1). Recently, Weix has reported dual nickel and palladium catalysis, achieving the cross-coupling of two reactive electrophiles (Ar–Br + Ar–OTf).9 MacMillan and Doyle demonstrated that cooperative nickel and photoredox catalysis enabled the coupling between reactive and unreactive electrophiles (aryl/alkyl halides + carboxylic acids).10 This strategy, while powerful, is less effective when a non-radical precursor is used. We considered that the use of a LA (Lewis acid) will lower the activation energy of the inert C–O bond,11 offering an opportunity for the cross-electrophile coupling of unreactive O-electrophiles. We report here a general and practical strategy for cross-electrophile coupling between reactive and unreactive electrophiles by a combination of nickel and LA catalysis (Scheme 1c). The strategy was applied for the allylation of aryl halides with allylic alcohols to afford allylarenes.
 | ||
| Scheme 1 Dual catalysis to address the selectivity challenge in cross-electrophile coupling. | ||
Allylarenes are ubiquitous motifs in various biologically active natural products.12 The precise synthesis of these compounds has been achieved by the coupling reaction of aryl halides with allyl metals,13 the allylation reaction of aryl metals with allylic substrates,14 and the reductive allylation reaction of Ar–Br with allylic acetate.15 These reactions, while powerful, require the pre-activation of at least one reactant. In terms of the atom- and step-economy, the synthesis of allylarenes directly from non-activated precursors will be much more attractive, but remains a particular challenge due to: (1) the selectivity for the allylation of electrophiles (Ar–Br) over nucleophiles (C–OH)16 and (2) the selectivity for the reaction of Ar–Br with C–OH over C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds.17 In this article, we demonstrated such a convenient synthetic route for the selective synthesis of allylarenes from Ar–Br and allylic alcohols.
C bonds.17 In this article, we demonstrated such a convenient synthetic route for the selective synthesis of allylarenes from Ar–Br and allylic alcohols.
Results and discussion
We began our investigation by exploring the reaction of 4-bromotoluene 1aa with cinnamyl alcohol 2a.18 The Mizoroki–Heck reaction product was not observed under reductive conditions. In the absence of a LA, a significant amount of biaryl dimer was obtained, but no cross-product or trace of cross-product 3 were observed (Table S1†). The addition of a catalytic amount of LA significantly improved the selectivity for 3 (Table S2†). Further optimization of the reaction conditions revealed that the use of Ni(dppp)Cl2 (10 mol%), bpy (20 mol%), ZrCl4 (10 mol%), and Mn (3.0 equiv.) in DMA afforded 3 in 85% yield.19With the optimized conditions in hand, we then investigated the reaction scope of aryl bromides. Both the electron-rich and electron-poor aryl bromides gave cross-coupling products in moderate to good yields (Scheme 2, 3–10). Substitution around the aromatic ring was tolerated (3–5). Sterically hindered substitution was tolerated when Ni(diglyme)Br2 was used as a catalyst (7). The reaction was selective for the functionalization of the C–Br bond over the C–Cl and C–F bonds and styrenyl groups, thus enabling later availability for additional transformation (9–13). The functional groups such as tertiary amines, esters, and ketones, as well as a strained ring, were accommodated and remained intact (14–18). Heteroarenes and polyarenes, which are prevalent in pharmaceuticals, can be precisely allylated (18–22). The reaction could be scaled up to gram scale and gave 22 in a 75% yield.
 | ||
| Scheme 2 The scope of aryl bromides. All the data are the averages of the two experiments. 1aa-at (1.5 equiv.) and 2a (1 equiv.) were used. The reactions were for 32–48 h. The yields are the isolated yields. aCatalyst: 10% Ni(diglyme)Br2. b95 h. | ||
While the existing allylation reactions of the allylic alcohols are efficient for allylating nucleophiles,14 this reaction is highly selective for electrophiles. A broad range of nucleophilic aryl bromides were then selectively allylated, leaving the alcohol,20 amine,21 phenol,22 indole23 and silane groups intact (Scheme 3). The utility of this method was illustrated by a regiodivergent synthesis of allylated indoles, which are important structural motifs in many bioactive natural products and drugs.24 Being complementary to conventional methods for the allylation of indoles at the C3 position,23 our approach provides direct access to C2-, C4-, C5-, C6-, and C7-allylated indoles (28–34). Of note, the desired products were obtained in high yields even when an equal amount of the two electrophiles was used.
 | ||
| Scheme 3 The reactions of 2a with nucleophilic aryl bromides. All the data are the averages of the two experiments. The yields are the isolated yields. The reactions were for 32–48 h. aThe conditions in Scheme 2, reaction for 60 h. bThe conditions in Scheme 2, amount of ArBr: 1.0 equiv. cAmount of ArBr: 1.5 equiv. | ||
The reaction proceeded efficiently with a variety of allylic alcohols and in most cases gave the linear (E)-products selectively (Table 1). In addition to the cinnamyl alcohols, unsubstituted and alkyl-substituted allylic alcohols also coupled with a functionalized aryl group (entries 1–5). Although the reaction with α-ethyl substituted alcohol is less selective, the substrate with phenyl substitution gave only the linear (E)-product (entries 6 and 7).
|
|
|||
|---|---|---|---|
| Entry | Allylic alcohol | Product | Yield |
a All the data are the averages of the two experiments. The yields are the isolated yields. The reactions were for 48–50 h. Amount of 1am: 2.0 equiv. 1aa were used for 2j and 2k.
b Catalyst: 10% Ni(dppf)Cl2, 20% 3,4,7,8-tetramethyl-1,10-phenanthroline, 20% AlCl3, 1am (1.5 equiv.).
c [l/b] (linear/branched ratio) = 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, linear product is a 3:1 E/Z isomer.
d [l/b] = 11:1, linear product is a 5:1 E/Z isomer.
e [l/b] = 180:1.
f 15% catalysts were used. :1, linear product is a 3:1 E/Z isomer.
d [l/b] = 11:1, linear product is a 5:1 E/Z isomer.
e [l/b] = 180:1.
f 15% catalysts were used.
|
|||
| 1 |

|

|
80% |
| 2 |

|

|
62%b,c |
| 3 |

|

|
75% |
| 4 |

|

|
73% |
| 5 |

|

|
58% |
| 6 |

|

|
63%b,d |
| 7 |

|
|
76% |
| 8 |

|

|
66%b,e |
| 9 |
|

|
56%f |
| 10 |

|
42 | 60% |


| (1) |
| (2) |
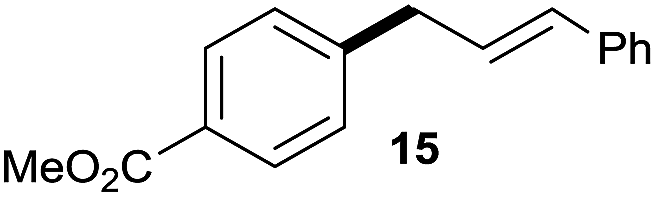
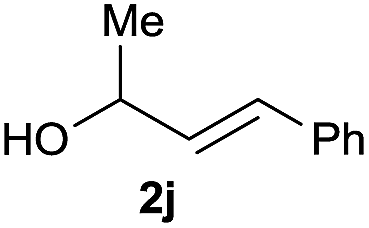
:1 (entry 8). Both nonsymmetrical substrates 2j and 2k gave the same product 42 in useful yields, suggesting that the reaction goes through a π-allyl nickel intermediate.
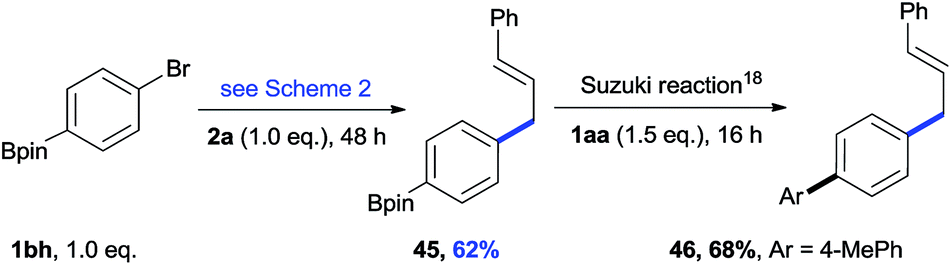
The cross-electrophile coupling reaction complements the Pd-catalyzed reactions well, enabling 1-bromo-4-iodobenzene to be sequentially functionalized with allylic alcohol 2l to the ketone 43 and allylated product 44 (eqn (1)). Our reaction conditions were also compatible with aryl borates and a sequence of C–Br bond allylation and C–B bond coupling is possible for the conversion of substrate 1bh to product 46 (eqn (2)).
The LA was expected to lower the activation energy of the C–OH bond. To confirm this assumption and understand the mechanistic details of this process, we monitored the reaction of 1aa with 2j by GC analysis. In the absence of a LA, no cross-coupling product 42 and side products from 2j were observed in 40 min, but the aryl dimer 51 was steadily increasing (Fig. S1a†). This result indicates that aryl bromide is reactive towards the Ni catalyst, while the allylic alcohol is inert. The use of 15 mol% of ZrCl4 significantly promoted the cross-coupling process, indicating the crucial role of the LA in the activation of allylic alcohols (Fig. S1b†).
Both aryl bromide and allylic alcohol will undergo oxidative addition to Ni(0), generating Ar–NiII(L)Br and allyl-NiII(L)X.16a,b,21b In order to determine which intermediate is formed first, we studied the relative reactivity of 1aa and 2a with in situ generated (bpy)Ni0(cod).25 We would expect, by quenching the reaction, a Ni(II) intermediate would afford Ar–H (47) or allyl-H (48).26Scheme 4 shows that, in every 10 min before the addition of Mn, 4.6% of Ar–H is obtained in each sample but no allyl-H is observed, consistent with Ar–NiII(L)Br forming first. While both cross-product 3 and the Ar–Ar dimer were steadily increasing after the addition of Mn (3 equiv.), allyl-H or allyl–allyl were still not observed (Scheme S1†), suggesting a mechanism in which Ar–NiII(L)Br serves as an intermediate.
 | ||
| Scheme 4 The selectivity of Ar–Br and the allylic alcohol in the initial oxidative addition to Ni(0). A mixture of 1aa/2a (1:1) was added to an in situ generated Ni(0) catalytic system. Samples were collected every 10 min and analysed by GC. Mn was added at 40 min. | ||
To gain more insight into the mechanism of this process, complex Ar–NiII(bpy)Br (49)27 was synthesized from the reaction of (bpy)Ni0(cod) (50) with aryl bromide 1ae. We would expect that, if Ar–NiII(bpy)Br (49) is an intermediate, the initial rate of reaction with complex 49 would be faster than that of the reaction with pre-catalyst 50. Fig. 1 shows that, in 6 min, the use of complex 49 (30 mol%) gave product 7 in almost 30% yield, while only a trace of 7 is formed when pre-catalyst 50 (30 mol%) is used (Fig. 1). This result further indicates that Ar–NiII(L)Br is likely the key intermediate in this reaction.
 | ||
| Fig. 1 The relative reactivity of Ar–NiII(bpy)Br (49) and (bpy)Ni0(cod) (50) capable of catalysing the reaction of 1ae with 2a. Complex 49 or 50 (30 mol%) was added to a solution of 1ae (1.2 or 1.5 equiv.), 2a (1.0 equiv.), bpy (30 mol%), ZrCl4 (10 mol%) and Mn (3.0 equiv.) in DMA. Samples were collected, quenched with H2O, and analysed by GC. | ||
The reduction of Ni(II) complex to Ni(I) by Mn has been extensively reported.4a,b The stoichiometric reactions of 49 with alcohol 2a gave product 7 in 0% yield (without Mn) and 90% yield (with Mn), suggesting that Ni(II) complex 49 might be reduced to the more nucleophilic ArNi(I) first, then react with the allylic alcohols (eqn (3)). The use of electron-poor aryl bromides (e.g.18vs.19 and 32vs.33) generally gave inferior results, which is consistent with this process. While the use of a radical scavenger, such as butylated hydroxytoluene (BHT), hydroquinone, and 1,1-diphenylethylene, did not inhibit the reaction of 1aa and 2a, no product 3 was obtained when 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO, 1.5 equiv.) was employed (Table S8†), consistent with the presence of a single-electron process capable of reducing oxidized Ni catalyst.
 | (3) |
Although a detailed mechanistic picture for this reaction requires further investigation, based on the above results, we tentatively proposed a catalytic cycle, as shown in Scheme 5.28 The oxidative addition of Ar–Br to Ni(0) gives the arylnickel(II) intermediate A. The reduction of A to arylnickel(I) B4a,b followed by the oxidative addition of LA-activated allylic alcohol will give the π-allylnickel(III) intermediate D.29 The reductive elimination of this complex will afford the desired product and recycle the catalyst with Mn. At present, we cannot rule out a radical mechanism as shown in the reaction of aryl halides with alkyl halides.30
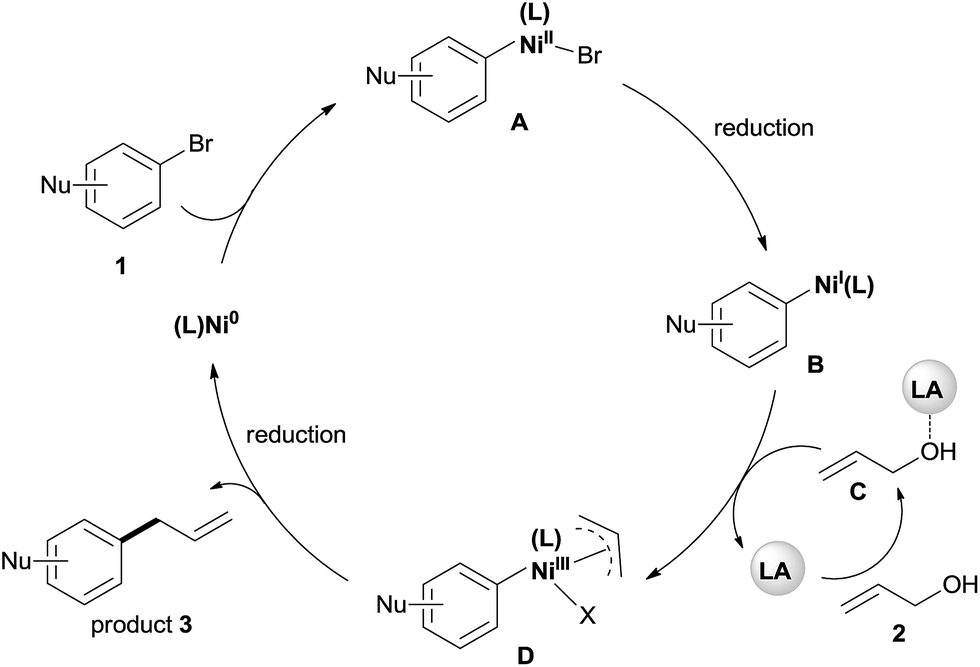 | ||
| Scheme 5 The proposed mechanism for the allylation of aryl bromides with allylic alcohols by dual catalysis. | ||
Conclusions
In summary, we have reported a dual nickel and Lewis acid-catalyzed allylation of aryl halides with allylic alcohols. This approach represents a new strategy for cross-electrophile coupling between reactive and unreactive electrophiles. The reaction tolerated a wide range of functional groups including alcohols, phenols, anilines, silanes, and even borates. In most cases, the reaction gave linear (E)-allylarenes highly selectively. The utility of this method is clearly illustrated by the facile access to C2 and C4–C7 allylated indoles. Preliminary mechanistic studies revealed that the reaction might start with an aryl nickel intermediate, which then reacted with allylic alcohols in the presence of a Lewis acid and reductant. The application of this strategy for the cross-electrophile coupling of other unreactive electrophiles is ongoing in our laboratory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the financial support from the NSFC (21502078, 21772072), FRFCU (lzujbky-2015-k06, lzujbky-2016-ct09), and 1000 Talents Plan Program. We are grateful to Prof. Weiping Tang (UW-Madison) and Prof. Wei Wang (LZU, China) for helpful discussion, and Prof. Yixia Jia (ZJUT, China) for his help in obtaining part of the HRMS data.Notes and references
- Selected reviews: (a) C. E. I. Knappke, S. Grupe, D. Gärtner, M. Corpet, C. Gosmini and A. Jacobi von Wangelin, Chem.–Eur. J., 2014, 20, 682 CrossRef PubMed; (b) T. Moragas, A. Correa and R. Martin, Chem.–Eur. J., 2014, 20, 8242 CrossRef CAS PubMed; (c) D. J. Weix, Acc. Chem. Res., 2015, 48, 1767 CrossRef CAS PubMed; (d) J. Gu, X. Wang, W. Xue and H. Gong, Org. Chem. Front., 2015, 2, 1411 RSC.
- (a) M. R. Netherton and G. C. Fu, Adv. Synth. Catal., 2004, 346, 1525 CrossRef CAS; (b) R. Jana, T. P. Pathak and M. S. Sigman, Chem. Rev., 2011, 111, 1417 CrossRef CAS PubMed; (c) S. L. Zultanski and G. C. Fu, J. Am. Chem. Soc., 2013, 135, 624 CrossRef CAS PubMed; (d) Y. Liang and G. C. Fu, J. Am. Chem. Soc., 2015, 137, 9523 CrossRef CAS PubMed.
- With cobalt catalysis: (a) M. Amatore and C. Gosmini, Angew. Chem., Int. Ed., 2008, 47, 2089 CrossRef CAS PubMed; (b) Y. Cai, A. D. Benischke, P. Knochel and C. Gosmini, Chem.–Eur. J., 2017, 23, 250 CrossRef CAS PubMed, with iron catalysis: (c) W. M. Czaplik, M. Mayer and A. Jacobi von Wangelin, Angew. Chem., Int. Ed., 2009, 48, 607 CrossRef CAS PubMed; (d) C. W. Cheung, F. E. Zhurkin and X. Hu, J. Am. Chem. Soc., 2015, 137, 4932 CrossRef CAS PubMed, with palladium catalysis: (e) A. Krasovskiy, C. Duplais and B. H. Lipshutz, J. Am. Chem. Soc., 2009, 131, 15592 CrossRef CAS PubMed; (f) V. R. Bhonde, B. T. O’Neill and S. L. Buchwald, Angew. Chem., Int. Ed., 2016, 55, 1849 CrossRef CAS PubMed.
- (a) D. A. Everson, R. Shrestha and D. J. Weix, J. Am. Chem. Soc., 2010, 132, 920 CrossRef CAS PubMed; (b) R. Shrestha, S. C. M. Dorn and D. J. Weix, J. Am. Chem. Soc., 2013, 135, 751 CrossRef CAS PubMed; (c) Y. Zhao and D. J. Weix, J. Am. Chem. Soc., 2014, 136, 48 CrossRef CAS PubMed; (d) C. Zhao, X. Jia, X. Wang and H. Gong, J. Am. Chem. Soc., 2014, 136, 17645 CrossRef CAS PubMed; (e) A. H. Cherney and S. E. Reisman, J. Am. Chem. Soc., 2014, 136, 14365 CrossRef CAS PubMed; (f) N. T. Kadunce and S. E. Reisman, J. Am. Chem. Soc., 2015, 137, 10480 CrossRef CAS PubMed; (g) G. A. Molander, K. M. Traister and B. T. O’Neill, J. Org. Chem., 2015, 80, 2907 CrossRef CAS PubMed; (h) X. Wang, S. Wang, W. Xue and H. Gong, J. Am. Chem. Soc., 2015, 137, 11562 CrossRef CAS PubMed; (i) G. Zhang, Y. Xie, Z. Wang, Y. Liu and H. Huang, Chem. Commun., 2015, 51, 1850 RSC; (j) X. Li, Z. Feng, Z.-X. Jiang and X. Zhang, Org. Lett., 2015, 17, 5570 CrossRef CAS PubMed; (k) E. C. Hansen, D. J. Pedro, A. C. Wotal, N. J. Gower, J. D. Nelson, S. Caron and D. J. Weix, Nat. Chem., 2016, 8, 1126 CrossRef CAS PubMed; (l) L. Hu, X. Liu and X. Liao, Angew. Chem., Int. Ed., 2016, 55, 9743 CrossRef CAS PubMed; (m) P. Zhang, C. C. Le and D. W. MacMillan, J. Am. Chem. Soc., 2016, 138, 8084 CrossRef CAS PubMed; (n) J. Liu, Q. Ren, X. Zhang and H. Gong, Angew. Chem., Int. Ed., 2016, 55, 15544 CrossRef CAS PubMed; (o) Z. Duan, W. Li and A. Lei, Org. Lett., 2016, 18, 4012 CrossRef CAS PubMed; (p) A. García-Domínguez, Z. Li and C. Nevado, J. Am. Chem. Soc., 2017, 139, 6835 CrossRef PubMed; (q) S. Ni, W. Zhang, H. Mei, J. Han and Y. Pan, Org. Lett., 2017, 19, 2536 CrossRef CAS PubMed.
- (a) B. M. Rosen, K. W. Quasdorf, D. A. Wilson, N. Zhang, A.-M. Resmerita, N. K. Garg and V. Percec, Chem. Rev., 2011, 111, 1346 CrossRef CAS PubMed; (b) D.-G. Yu, S. Luo, F. Zhao, Z.-J. Shi, in Homogeneous Catalysis for Unreactive Bond Activation, ed. Z.-J. Shi, John Wiley & Sons, Inc., Hoboken, 2014, ch. 5, pp. 347–439 Search PubMed; (c) M. Tobisu and N. Chatani, Acc. Chem. Res., 2015, 48, 1717 CrossRef CAS PubMed.
- For limited reports on reductive coupling between reactive and unreactive electrophiles, see intramolecular versions: (a) E. J. Tollefson, L. W. Erickson and E. R. Jarvo, J. Am. Chem. Soc., 2015, 137, 9760 CrossRef CAS PubMed; (b) L. W. Erickson, E. L. Lucas, E. J. Tollefson and E. R. Jarvo, J. Am. Chem. Soc., 2016, 138, 14006 CrossRef CAS PubMed, elegant examples of intermolecular reactions: (c) M. O. Konev, L. E. Hanna and E. R. Jarvo, Angew. Chem., Int. Ed., 2016, 55, 6730 CrossRef CAS PubMed; (d) A. Correa, T. León and R. Martin, J. Am. Chem. Soc., 2014, 136, 1062 CrossRef CAS PubMed; (e) A. Correa and R. Martin, J. Am. Chem. Soc., 2014, 136, 7253 CrossRef CAS PubMed, reaction with in situ formed Grignard reagent: (f) Z.-C. Cao, Q.-Y. Luo and Z.-J. Shi, Org. Lett., 2016, 18, 5978 CrossRef CAS PubMed.
- (a) A. Warshel, P. K. Sharma, M. Kato, Y. Xiang, H. Liu and M. H. M. Olsson, Chem. Rev., 2006, 106, 3210 CrossRef CAS PubMed; (b) D. Ringe and G. A. Petsko, Science, 2008, 320, 1428 CrossRef CAS PubMed.
- (a) S. Matsunaga and M. Shibasaki, Chem. Commun., 2014, 50, 1044 RSC; (b) J. Park and S. Hong, Chem. Soc. Rev., 2012, 41, 6931 RSC; (c) X.-Z. Shu, M. Zhang, Y. He, H. Frei and F. D. Toste, J. Am. Chem. Soc., 2014, 136, 5844 CrossRef CAS PubMed; (d) X.-Z. Shu, S. C. Nguyen, Y. He, F. Oba, Q. Zhang, C. Canlas, G. A. Somorjai, A. P. Alivisatos and F. D. Toste, J. Am. Chem. Soc., 2015, 137, 7083 CrossRef CAS PubMed.
- L. K. G. Ackerman, M. M. Lovell and D. J. Weix, Nature, 2015, 524, 454 CrossRef CAS PubMed.
- (a) Z. Zuo, D. T. Ahneman, L. Chu, J. A. Terrett, A. G. Doyle and D. W. C. MacMillan, Science, 2014, 345, 437 CrossRef CAS PubMed; (b) Z. Zuo, H. Cong, W. Li, J. Choi, G. C. Fu and D. W. C. MacMillan, J. Am. Chem. Soc., 2016, 138, 1832 CrossRef CAS PubMed; (c) C. P. Johnson, R. T. Smith, S. Allmendinger and D. W. C. MacMillan, Nature, 2016, 535, 322 CrossRef PubMed; coorperativecatalysis with other radical co-generation methods: (d) L. K. G. Ackerman, L. L. Anka-Lufford, M. Naodovic and D. J. Weix, Chem. Sci., 2015, 6, 1115 RSC; (e) Y. Zhao and D. J. Weix, J. Am. Chem. Soc., 2015, 137, 3237 CrossRef CAS PubMed; (f) K. M. Arendt and A. G. Doyle, Angew. Chem., Int. Ed., 2015, 54, 9876 CrossRef CAS PubMed.
- For coorperative metal/Lewis acid catalysis, see a review: (a) C. Wang and Z. Xi, Chem. Soc. Rev., 2007, 36, 1395 RSC, selected examples: (b) Y. Nakao, S. Ebata, A. Yada, T. Hiyama, M. Ikawa and S. Ogoshi, J. Am. Chem. Soc., 2008, 130, 12874 CrossRef CAS PubMed; (c) M. Tobisu, T. Xu, T. Shimasaki and N. Chatani, J. Am. Chem. Soc., 2011, 133, 19505 CrossRef CAS PubMed; (d) K. T. Sylvester, K. Wu and A. G. Doyle, J. Am. Chem. Soc., 2012, 134, 6967 Search PubMed; (e) Y. Minami, H. Yoshiyasu, Y. Nakao and T. Hiyama, Angew. Chem., Int. Ed., 2013, 52, 883 CrossRef CAS PubMed; (f) Y.-X. Li, Q.-Q. Xuan, L. Liu, D. Wang, Y.-J. Chen and C.-J. Li, J. Am. Chem. Soc., 2013, 135, 12536 CrossRef CAS PubMed; (g) Y. Miyazaki, N. Ohta, K. Semba and Y. Nakao, J. Am. Chem. Soc., 2014, 136, 3732 CrossRef CAS PubMed; (h) W. Guan, S. Sakaki, T. Kurahashi and S. Matsubara, ACS Catal., 2015, 5, 1 CrossRef CAS.
- S. Li, Nat. Prod. Rep., 2010, 27, 57 RSC.
- For recent review, see: (a) T. Lindel, N. Marsch and S. K. Adla, Top. Curr. Chem., 2012, 309, 67 CrossRef CAS PubMed, selected examples: (b) A. E. Mattson and K. A. Scheidt, J. Am. Chem. Soc., 2007, 129, 4508 CrossRef CAS PubMed; (c) J. L. Farmer, H. N. Hunter and M. G. Organ, J. Am. Chem. Soc., 2012, 134, 17470 CrossRef CAS PubMed; (d) Y. Yang and S. L. Buchwald, J. Am. Chem. Soc., 2013, 135, 10642 CrossRef CAS PubMed; (e) J.-L. Tao, B. Yang and Z.-X. Wang, J. Org. Chem., 2015, 80, 12627 CrossRef CAS PubMed.
- For recent reviews, see: (a) B. Sundararaju, M. Achard and C. Bruneau, Chem. Soc. Rev., 2012, 41, 4467 RSC; (b) N. A. Butta and W. Zhang, Chem. Soc. Rev., 2015, 44, 7929 RSC, selected examples: (c) T. Hayashi, M. Konishi, K.-I. Yokota and M. Kumada, J. Chem. Soc., Chem. Commun., 1981, 313 RSC; (d) G. W. Kabalka, G. Dong and V. B. enkataiah, Org. Lett., 2003, 5, 893 CrossRef CAS PubMed; (e) M. Kimura, M. Futamata, R. Mukai and Y. Tamaru, J. Am. Chem. Soc., 2005, 127, 4592 CrossRef CAS PubMed; (f) Y. Suzuki, B. Sun, K. Sakata, T. Yoshino, S. Matsunaga and M. Kanai, Angew. Chem., Int. Ed., 2015, 54, 9944 CrossRef CAS PubMed; (g) B. Yang and Z.-X. Wang, J. Org. Chem., 2017, 82, 4542 CrossRef CAS PubMed.
- (a) M. Durandetti, J.-Y. Nedelec and J. Perichon, J. Org. Chem., 1996, 61, 1748 CrossRef CAS PubMed; (b) P. Gomes, C. Gosmini and J. Périchon, Org. Lett., 2003, 5, 1043 CrossRef CAS PubMed; (c) S. Wang, Q. Qian and H. Gong, Org. Lett., 2012, 14, 3352 CrossRef CAS PubMed; (d) L. L. Anka-Lufford, M. R. Prinsell and D. J. Weix, J. Org. Chem., 2012, 77, 9989 CrossRef CAS PubMed.
- An electrophilic π-allyl metallic intermediate is generally formed in allylation reactions, which could be readily trapped with nucleophiles. For reviews, see: ref. 14a and b. Selected references: (a) Y. Kita, R. D. Kavthe, H. Oda and K. Mashima, Angew. Chem., Int. Ed., 2016, 55, 1098 CrossRef CAS PubMed; (b) M. S. Azizi, Y. Edder, A. Karim and M. Sauthier, Eur. J. Org. Chem., 2016, 2016, 3796 CrossRef CAS.
- The reactions of organic halides with allylic alcohols typically give Mizoroki–Heck products rather than allylation products. For a represented review, see: J. Muzart, Tetrahedron, 2005, 61, 4179 CrossRef CAS.
- For detailed conditions, see the Supporting Information.†.
- Although Ni(diglyme)Br2 gave a comparable yield (Table S4 and S6†), the applicability of Ni(dppp)Cl2 for the reactions in Scheme 2 is more general. The role of the phosphine ligand remains unclear at the moment.
- M. Roggen and E. M. Carreira, Angew. Chem., Int. Ed., 2011, 50, 5568 CrossRef CAS PubMed.
- (a) D. Banerjee, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2014, 53, 13049 CrossRef CAS PubMed; (b) Y. Kita, H. Sakaguchi, Y. Hoshimoto, D. Nakauchi, Y. Nakahara, J.-F. Carpentier, S. Ogoshi and K. Mashima, Chem.–Eur. J., 2015, 21, 14571 CrossRef CAS PubMed.
- T. Satoh, M. Ikeda, M. Miura and M. Nomura, J. Org. Chem., 1997, 62, 4877 CrossRef CAS.
- (a) M. Kimura, M. Futamata, R. Mukai and Y. Tamaru, J. Am. Chem. Soc., 2005, 127, 4592 CrossRef CAS PubMed; (b) I. Usui, S. Schmidt, M. Keller and B. Breit, Org. Lett., 2008, 10, 1207 CrossRef CAS PubMed; (c) B. Sundararaju, M. Achard, B. Demerseman, L. Toupet, G. V. M. Sharma and C. Bruneau, Angew. Chem., Int. Ed., 2010, 49, 2782 CrossRef CAS PubMed.
- S. Cacchi and G. Fabrizi, Chem. Rev., 2011, 111, PR215 CrossRef PubMed.
- M. Abla and T. Yamamoto, Bull. Chem. Soc. Jpn., 1999, 72, 1255 CrossRef CAS.
- S. Biswas and D. J. Weix, J. Am. Chem. Soc., 2013, 135, 16192 CrossRef CAS PubMed.
- CCDC 1515176(49) contains the supplementary crystallographic data for this paper.†.
- Alternatively, a mechanism starting with an allyl-Ni(II)X intermediate is still plausible. For related works, see: (a) X. Qian, A. Auffrant, A. Felouat and C. Gosmini, Angew. Chem., Int. Ed., 2011, 50, 10402 CrossRef CAS PubMed; (b) X. Cui, S. Wang, Y. Zhang, W. Deng, Q. Qian and H. Gong, Org. Biomol. Chem., 2013, 11, 3094 RSC , also see: ref. 15d.
- (a) I. Colon and D. R. Kelsey, J. Org. Chem., 1986, 51, 2627 CrossRef CAS; (b) C. Cannes, S. Condon, M. Durandetti, J. Périchon and J.-Y. Nédélec, J. Org. Chem., 2000, 65, 4575 CrossRef CAS PubMed; (c) D. A. Everson, B. A. Jones and D. J. Weix, J. Am. Chem. Soc., 2012, 134, 6146 CrossRef CAS PubMed , also see: ref. 15b.
- D. A. Everson and D. J. Weix, J. Org. Chem., 2014, 79, 4793 CrossRef CAS PubMed , also see: ref. 26.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1515176. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc03140h |
| This journal is © The Royal Society of Chemistry 2018 |