
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTagging effects on the mid-infrared spectrum of microsolvated protonated methane†
Alexander
Esser
 a,
Harald
Forbert
b and
Dominik
Marx
a
a,
Harald
Forbert
b and
Dominik
Marx
a
aLehrstuhl für Theoretische Chemie, Ruhr-Universität Bochum, 44780 Bochum, Germany
bCenter for Solvation Science ZEMOS, Ruhr-Universität Bochum, 44780 Bochum, Germany
First published on 21st December 2017
Abstract
Although bare protonated methane is by now essentially understood at the level of intramolecular large-amplitude motion, scrambling dynamics and broadband vibrational spectra, the microsolvated species still offer plenty of challenges. One aspect is the effect of the attached solvent molecules on the infrared absorption spectra of microsolvated CH5+ complexes compared to the bare parent molecule. In this study we analyze, based on ab initio molecular dynamics simulations, protonated methane molecules that have been microsolvated with up to three hydrogen molecules, i.e. CH5+·(H2)n. In particular, upon introducing a novel multi-channel maximum entropy methodology described herein, we are able to decompose the infrared spectra of these weakly-bound complexes in the frequency window from 1000 to 4500 cm−1 into additive single mode contributions. Detailed comparisons to the bare CH5+ parent reveal that these perturbed modes encode distinct features that depend on the exact microsolvation pattern. Beyond the specific case, such understanding is relevant to assess tagging artifacts in vibrational spectra of parent molecules based on messenger predissociation action spectroscopy.
I. Introduction
Since its discovery, published in 1952 by Tal'roze and Lyubimova,1 protonated methane (CH5+) has challenged the traditional understanding of what a molecular structure is. Consequently, this molecule has been intensely studied in order to understand its intriguing behavior and, over time, became the prototype of ”fluxional” (or ”floppy”) molecules.2–12 It is the smallest possible carbocation and forms a three-center two-electron bond13–15 where its constituents continuously exchange their sites, resulting in all five hydrogen atoms being dynamically equivalent even in the quantum ground state.14,15 This peculiar phenomenon is dubbed ”hydrogen scrambling” (or just ”scrambling”) and is caused by intramolecular large-amplitude motion on an extremely shallow potential energy surface (PES); see ref. 11 for a comprehensive recent review.Bare protonated methane has been thoroughly studied by theoretical means which goes back to the early days of computational quantum chemistry. This includes pioneering semiempirical work followed by accurate studies of the structures of the ground state and various isomers, their energy differences and the barriers involved in scrambling as well as harmonic vibrational frequencies.13,16–23
The converged static picture23 is that the global minimum of the PES, the e – Cs symmetric structure, is separated from two energetically higher-lying s – Cs and C2v transition states by only about 0.1 and 1 kcal mol−1, respectively. This scenario led early on to the conjecture that the molecule should indeed easily undergo scrambling at room temperature,13 which has been explicitly confirmed two decades later not only at room temperature,24 but even at low temperatures close to the nuclear quantum ground state14 based on ab initio path integral simulations in conjunction with topological bonding analyses;15 these key findings have been confirmed by several other groups using different methods as comprehensively reviewed.11
In addition to a wealth of static quantum chemistry studies, ab initio molecular dynamics (AIMD) simulations25 were used to compute, disentangle and assign the fully anharmonic infrared (IR) spectra of CH5+ and all its H/D isotopologues;9,11,26–28 the overriding impact of isotopic substitution on the properties of the CH5+ isotopologues has been predicted early.29 The decomposition and assignment of these broadband IR spectra in terms of modes was obtained from time-autocorrelation and cross-correlation function approaches based on the underlying ab initio trajectories;27,30,31 see ref. 11 for a comprehensive review of these (ab initio) molecular dynamics based approaches to theoretical spectroscopy. It has been shown that AIMD simulations at a temperature of 300 K, thus relying on classical Newtonian dynamics of the nuclei, emulate the quantum effects of the nuclei quite well and also reproduce the experimental spectrum of CH5+, while at lower temperatures of around 100 K the peaks in the IR spectrum become much sharper as a result of frozen scrambling dynamics.9,26
In addition to these early ab initio path integral and AIMD simulations,5,14,15,24,26,29 diffusion Monte Carlo and vibrational configuration interaction calculations based on parameterized PESs32–39 not only confirmed the previous findings5,14,15,24,26,29 that have been obtained from “on the fly” quantum simulations, but they also contributed significantly to our current knowledge of bare protonated methane. Most notable in this respect are the pioneering IR spectra that have been reported for several H/D isotopologues of protonated methane.35,36 All of the available information taken together leads to the conclusion that bare protonated methane including its isotopologues is now essentially understood in terms of its peculiar intramolecular large-amplitude motion, scrambling dynamics, and broadband IR spectra.11 Yet, largely unexplained remain the available high-resolution vibrational spectra5,12,37,40,41 of bare CH5+, which continue to challenge state-of-the-art theoretical rovibrational spectroscopy.42–46
This favorable situation for bare CH5+ is in stark contrast to our current understanding of microsolvated CH5+ species, which still offer many puzzles concerning the interplay of scrambling dynamics and microsolvation effects. Tagging of protonated methane using hydrogen molecules is possible due to the rather weak but still significant interactions between these so-called tags and the CH5+ core. This approach has been pioneered experimentally by the group of Y. T. Lee47–50 upon introducing an ingenious technique that is now known as messenger predissociation action spectroscopy; it is noted in passing that the accompanying simulations in ref. 48 must be considered with care due to technical shortcomings. Using this flavor of action spectroscopy, Lee and coworkers were able to measure vibrational action spectra of CH5+·(H2)n complexes in the range of n = 1 up to 6, thus excluding the limiting bare case n = 0. The experiment accessed two rather narrow frequency windows from about 2700 to 3200 cm−1 and 4060–4140 cm−1, thus observing the C–H bands of microsolvated CH5+ complexes and the H2 stretching modes, respectively, in the mid-IR regime. These action spectra47–50 remain so far the only existing experimental IR spectra of microsolvated CH5+. We note in passing that the influence of tags on quasi-rigid molecules is generally small,51,52 whereas studying the potentially significant changes of the IR spectra of highly fluxional molecules such as protonated methane is the challenge.
At variance with bare CH5+, theoretical investigations of CH5+·(H2)n species are rather scarce.48,53–58 Moreover, structure optimizations of CH5+·(H2)n lead to a wealth of minimum energy structures of differently tagged species. From the set of optimized structures56 it is clearly seen that the tagging molecules prefer specific sites depending on n, see Fig. 1 where the global minima for n = 1, 2 and 3H2 tags are displayed in panels b, e and h, respectively, compared to the bare CH5+ reference molecule in panel a. Our atom/site labeling scheme (defining the permutations, see below) is the same that has been introduced for bare protonated methane,27 where site 1 is the “outer moiety atom”, site 2 the “inner moiety atom”, site 3 denotes the “in-plane atom” while the sites 4 and 5 are located ”out-of-plane”. Thus, atoms 1 and 2 form the so-called H2 “moiety” while atoms 3, 4 and 5 constitute the CH3 “tripod”, the carbon nucleus of which forms a three-center two-electron bond13–15 together with the two protons of the moiety. The singly tagged CH5+·H2 species has been found to have three close-lying low-energy minimum structures, while the doubly tagged case has three such structures and the n = 3 case features only a single low-energy minimum structure.56
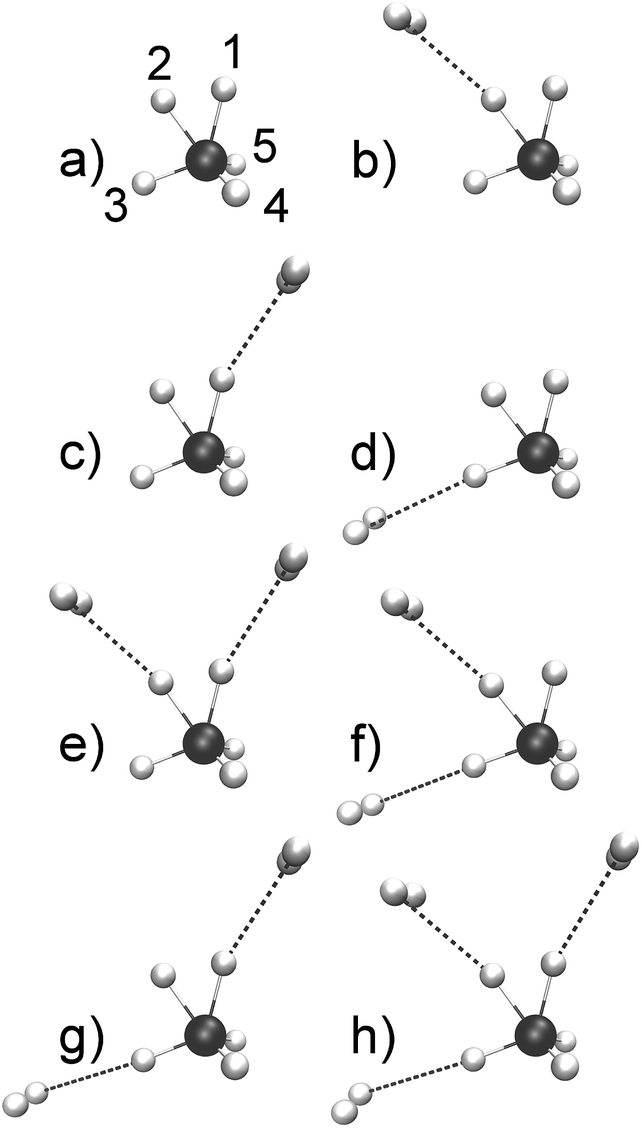 | ||
| Fig. 1 Optimized low-lying minimum energy structures of H2-microsolvated protonated methane based on ref. 56. The structures are sorted according to increasing energy.56 As a reference, the bare protonated methane is shown in its e – Cs ground-state structure in panel a where the site labeling scheme (defining our standard ordering27 for permutations) is also introduced. The singly tagged species has three distinct minimum structures that differ in the location of the tagging molecules as indicated: species b is the energetically lowest-lying minimum, whereas species c is ΔE = 0.35 kcal mol−1 and species d is ΔE = 1.77 kcal mol−1 above the global minimum energy. The doubly tagged species also has three distinct minimum structures where the two tag molecules are located at the positions 1 and 2 (species e being the global minimum), at 2 and 3 (species f with ΔE = 1.19 kcal mol−1), and at 1 and 3 (species g with ΔE = 1.67 kcal mol−1). Finally, the protons 3, 2 and 1 are the preferred sites when three tagging H2 molecules are attached to CH5+ as depicted in panel h. | ||
Analyzing the optimized structures of tagged CH5+ species, such as those depicted in Fig. 1, exclusively provides a static perspective in terms of equilibrium structures. But what about the impact of tagging on large-amplitude motion and ultimately on the scrambling dynamics of the CH5+ core? This question is highly relevant to messenger spectroscopy, where ideally the tags should not (significantly) perturb the microsolvated parent species. In stark contrast, in microsolvating protonated methane, even the weak interactions of the tags with the bare CH5+ species apparently do significantly impact on the vibrational spectra.47–49 This finding, in turn, has been exploited to investigate experimentally the extend to which the vivid scrambling dynamics of CH5+ itself might be affected by perturbations due to the tagging molecules.
Starting with the case of bare CH5+, it is known that so-called “full scrambling” requires two processes as sketched in Fig. 2 (obviously, the following description rests on a quasi-classical picture that involves distinguishable protons whereas quantum effects are discussed in ref. 58). The first one shown in panel a consists of breaking and making of the H2 moiety as a result of (relative) motion of the inner moiety proton toward the in-plane proton via the C2v transition state and reformation of an accordingly displaced moiety, i.e. the two out-of-plane protons are only spectators. The resulting pseudorotational motion involves only three out of five protons, exchanges only one of the protons involved in the e – Cs equilibrium structure, and thus leads to so-called “partial scrambling”.58 The second large-amplitude motion, see Fig. 2b is an internal rotation, where the two moiety atoms are rotating, via the s – Cs transition state, w.r.t. the pseudo C3-axis of the tripod. This is an easy process in view of its very small activation energy, on the order of 0.1 kcal mol−1 (cf.ref. 23) on the PES, that leads to an exchange of the inner and outer moiety protons within the e – Cs structure. When combined with partial scrambling, internal rotation also involves the out-of-plane protons and eventually leads to full scrambling of all five protons. The low-resolution IR spectra resulting from this complex scrambling dynamics have been computed for CH5+ and all its isotopologues and used to assign the experimental broadband spectra.26,27
 | ||
| Fig. 2 Schematic illustration of the pseudorotations and internal rotations in panels (a and b), respectively, that enable hydrogen scrambling in bare CH5+. Note that the atom numbering is merely used here to visualize the scrambling which results in permutations of the atoms, whereas all such permutations are properly taken into account when performing the spectral analysis (see text). | ||
Having understood the IR spectrum of bare CH5+ and its relation to hydrogen scrambling, the question that remains to be answered is about the tagging effect on IR spectra when using messenger vibrational spectroscopy techniques. At the level of only structure, it has been recently demonstrated by low-temperature path integral simulations58 that microsolvation by H2 influences scrambling in distinct ways depending on the number of tagging molecules. The effects of one and three tagging hydrogen molecules were shown to be very similar, while two tagging H2 lead to a distinctly different behavior. Internal rotation (cf.Fig. 2b) is found to be frozen in the case of one and three tagging molecules, while in the n = 2 case internal rotation is still active but partial scrambling is reduced drastically. In contrast, partial scrambling via pseudorotation (cf.Fig. 2a) is still possible for the n = 1 and 3 species of CH5+·(H2)n. Such subtle yet significant perturbation of the scrambling dynamics of the CH5+ core due to tagging raises the question as to the impact of the H2 tags on the IR spectrum of protonated methane depending on their number n.
In this study, we computed the IR spectra of microsolvated protonated methane with up to n = 3 tagging hydrogen molecules in the mid-IR frequency window from 1000 to 4500 cm−1 based on our AIMD simulation approach to theoretical spectroscopy.11 Using classical nuclear dynamics, the scrambling motion is frozen at the chosen simulation temperature, which allows us to precisely address tagging effects on the bending and stretching bands of protonated methane at mid-IR frequencies, whereas the prominent signals due to hydrogen scrambling dynamics are known to be located in the far-IR regime.9,26 At variance with the usual approach relying on Fourier transforms of dipole autocorrelation functions we introduce here a multi-channel signal processing approach that belongs to the family of maximum entropy (ME) methods (sometimes also dubbed MaxEnt). The resulting spectra are smooth and can subsequently be decomposed into so-called mode-specific IR spectra after introducing generalized normal modes. Having these modes at hand, it is possible to dissect the mid-IR spectrum of H2-microsolvated CH5+ and thus to reveal the effect of tagging when using n = 1, 2, or 3 tags.
II. Computational approach
A. Ab initio molecular dynamics simulations
All calculations have been performed using Kohn–Sham density functional theory in the generalized gradient approximation (GGA) within the CPMD program package59 as described in ref. 57 (see also ref. 11 and 58); these available AIMD trajectories underly the present analyses. In order to represent faithfully the relatively weak interactions between the CH5+ core and the tagging H2 molecules as well as their mutual interactions, a dispersion correction60 has been added to the particular GGA functional (LDA + B) used. This efficient “LDA + B + D” electronic structure treatment of CH5+·(H2)n has been carefully validated by benchmarking57 it with respect to coupled cluster and many-body perturbation theory data as published in the comprehensive ESI to ref. 58. The box length was set to 25 a.u. with a plane wave cutoff of 35 Ry and using a 0.34 fs time step for AIMD. In order to prevent the hydrogen molecules from drifting too far away from the CH5+ core, leading eventually to physically meaningful but undesired dissociation events, a soft restraining potential has been applied during the AIMD simulations akin to ref. 58. After sufficient equilibration in the NVT ensemble at 110 K, a total of 10 NVE trajectories per system (with a length of 10 ps each) has been obtained starting from initial conditions that have been sampled from the respective NVT run; note that full scrambling is frozen at 110 K when using classical nuclei as done in these AIMD simulations. All reported data were obtained from averaging the respective properties over all trajectories for each number n = 0, …, 3 of tagging H2 molecules.B. Total IR spectra from single-channel maximum entropy method
Assuming classical nuclear dynamics, the total IR absorption cross section α(ω) is obtained from the well-established method of calculating the Fourier transform (FT) of the autocorrelation function of the total dipole moment of the sample (see e.g.ref. 11 and references given therein). Using the corresponding dipole velocities, α(ω) is given by | (1) |
 | (2) |
Instead of directly evaluating eqn (2) with the time discretized autocorrelation function, a fundamentally different approach is followed by the maximum entropy (ME) method. It is based on the concept of computing the IR spectrum by using a set of model parameters that are determined from the available data (in our case these data are the x, y, z components of the total dipole velocity vector Ṁ as obtained from our trajectories). In the FT approach, the problem arises of how the edges of the available data are treated. The ME method deals with this problem by stating that the best assumption to the data outside the available range is to treat it as unbiased as possible, leading to the equivalent statement of maximizing the information theoretic entropy, being a measure of ignorance in the sense of Shannon and Jaynes, under the constraint that the autocorrelation function agrees with the existing data; we refer to standard literature62–64 for derivation and discussion.
The so-called single-channel ME approach directly yields I(ω) and thus the absorption cross section as
 | (3) |
The model coefficients and residual variance can be determined by using the Burg algorithm,65–67 which takes advantage of a recursive scheme for efficient computation. Using this algorithm, the residual variance is obtained from
| σ(p)2 = σ(p−1)2(1 − (γ(p)p)2) | (4) |
 | (5) |
 | (6) |
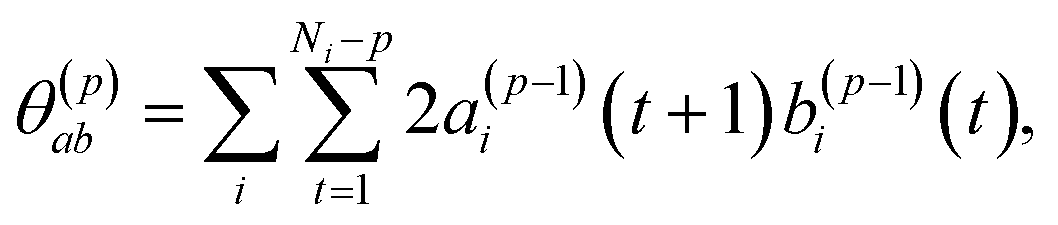 | (7) |
 | (8) |
 | (9) |
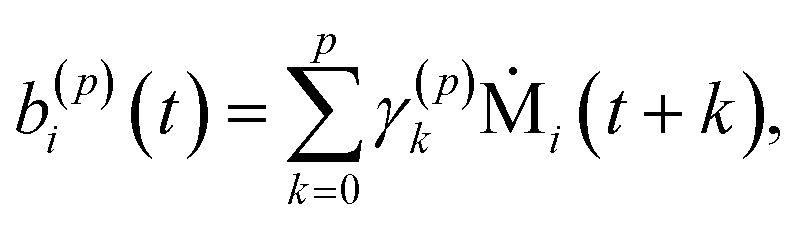 | (10) |
| a(p)i(t) = a(p−1)i(t + 1) − γ(p)pb(p−1)i(t), | (11) |
| b(p)i(t) = b(p−1)i(t) − γ(p)pa(p−1)i(t + 1). | (12) |
| γ(p)k = γ(p−1)k − γ(p)pγ(p−1)p−k, | (13) |
To summarize this concise description, we initialize the algorithm with σ(0)2 as the variance of the data series and a(0)i(t) = b(0)i(t) with the data set, i.e. dipole velocity trajectories {Ṁi(t)}, where the x, y, z components of the vectors Ṁ(t) are treated isotropically as separate scalar trajectories. To obtain the new sets of model parameters, we use the recursive scheme by using eqn (11)–(13), where γ(p)p is determined through eqn (6) with the help of eqn (7) and (8). Each set of model parameters is determined for a specific order p, where an upper bound to the order must be determined. We used the combined information criterion68 to determine the optimal order p in the single-channel ME approach. At the end the model parameters γ(p)k, the reflection coefficient γ(p)p and the residual variance σ(p)2, obtained viaeqn (4), are used in eqn (3) to compute the spectrum.
In stark contrast to using the Fourier approach in conjunction with any smoothing, the maximum entropy spectra are computed here by minimizing the assumptions on the available data series, thus implementing the ‘maximum parsimony’ approach or ‘Occam's razor’ principle. Furthermore, based on the algorithmic selection as implemented by us, the number of parameters is autonomously determined by the algorithm itself and thus without any user interference. The resulting spectrum looks similar to a smoothed Fourier spectrum but does not require any specifications and thus (arbitrary) choices of window functions and window lengths. The reported spectra are thus in a sense ‘optimally smoothed’ spectra that are independent from the specific researcher.
C. Multi-channel maximum entropy method
In order to obtain a consistent positive semi-definite cross-correlation matrix spectrum as required for the subsequent decomposition of the total spectrum into mode-specific IR spectra, αk(ω), according to Sec. II D, a multi-channel ME method is needed where we analyse vector data in contrast to scalar data in the single-channel method. For every center j and every component ζ = x, y, z of center j we assign a separate channel, thus resulting in 3ncenters channels and a 3ncenters by 3ncenters matrix of (cross-)correlation spectra, where ncenters is the number of centers. In each channel we have the product v(i)jζ·qj, where v(i)jζ is the velocity of center j and component ζ in the i-th trajectory and qj is the charge of center j. These channels are then simultaneously fitted to obtain model coefficient matrices Γ(p)k, analogously to the γ(p)k in the single-channel method.We based our particular implementation on the Vieira-Morf algorithm69 as described in ref. 70, where we took advantage of the time reversibility of the molecular dynamics propagation. This leads to the following set of equations
| Σ(p) = Σ(p−1) − Γ(p)pΣ(p−1)Γ(p)†p, | (14) |
| Γ(p)p = (Σ(p−1))1/2(Θaa(p))−1/2Θ(p)ab·(Θ(p)aa)−1/2(Σ(p−1))−1/2, | (15) |
| Γ(p)k = Γ(p−1)k − Γ(p)pΓ(p−1)p−k, | (16) |
| a(p)i(t) = a(p−1)i(t + 1) − Γ(p)pb(p−1)i(t), | (17) |
| b(p)i(t) = b(p−1)i(t) − Γ(p)pa(p−1)i(t + 1). | (18) |
These equations are analogous to the single-channel method, where the Γ(p)k are the model coefficient matrices, Σ(p) the covariance matrix of the residual errors, and a(p)i(t) and b(p)i(t) the forward and backward prediction errors of the different trajectories indexed by i. Note that similarly to the single-channel case,  is the negative unit matrix for all p and the data sets from the molecular dynamics, i.e. the dipole velocity trajectories {Ṁi(t)} enter via
is the negative unit matrix for all p and the data sets from the molecular dynamics, i.e. the dipole velocity trajectories {Ṁi(t)} enter via
| (a(0)i(t))jζ = (b(0)i(t))jζ = v(i)jζ(t)·qj, | (19) |
 | (20) |
Finally,
 | (21) |
 | (22) |
| I(ω) = Ap(ω)Σ(p)A†p(ω), | (23) |
 | (24) |
D. Mode-specific IR spectra and generalized normal modes from multi-channel maximum entropy cross-correlation analysis
In our previous work, mass-weighted velocities of the nuclei have been used to construct the so-called generalized normal coordinates,30,31 building upon a scheme that uses internal coordinates. This approach, yielding only the modes which are encoded in the vibrational density of states, has been extended subsequently71 to yield mode-specific IR spectra, instead of mode-specific power spectra as before, by using Cartesian coordinates in conjunction with weighting the particle velocities by the effective charge qi. In contrast to our previous methods,30,31 the velocities of both nuclei and orbitals are considered here,71 where the latter are described in terms of the time derivatives of the moving positions of the centers of the corresponding maximally localized Wannier functions (see e.g. Sec. 7.2 in ref. 25) that have been determined along the AIMD trajectory from the underlying electronic structure. Including the electronic degrees of freedom in this way71 allows us to assign IR intensities to the individual mode-specific absorptions (instead of just the intensities according to the vibrational density of states when using only the nuclear degrees of freedom). Our previous approach71 has been further improved in the current work (i) by using the ME methodology of a time-reversible algorithm to enforce the proper symmetry of the correlation functions in conjunction with (ii) an automatic order-selection algorithm that ensures the optimal determination of the number of required parameters and thus ‘optimal smoothing’ independently from any user choice.Within our latest decomposition approach,71 the dipole velocity cross-correlation matrix is given by
 | (25) |
 | (26) |
The thereby obtained diagonal elements represent the modes while the remaining off-diagonal elements are a measure of the residual correlations.
Finally, the mode-specific IR absorption cross sections are given by the functions αk,
| αk(ω) = Λk,k(ω)(dk·dk), | (27) |
 | (28) |
 | (29) |
As a result of this systematic spectral decomposition, the total IR absorption cross section can be “re-synthesized” to a good approximation as a sum of mode-specific contributions αk(ω),
 | (30) |
E. Reference structures
In order to compute generalized normal modes as described in the previous section, it is essential to construct reference structures with respect to which the particle displacements and thus these modes can be defined. The methodology described in ref. 30 can only be applied to cases where a single reference structure is sufficient, which is relevant only to quasi-rigid molecules with a well-defined unique equilibrium structure (such as CH4 for instance). In the case of CH5+·(H2)n, trying to construct a single reference structure is obviously not a useful concept in view of the existence of several energetically close-lying isomers as depicted in Fig. 1. It is therefore expected that several such structures, possibly even all of them, are visited during the dynamics of these weakly-bound complexes. For instance, there are three such dynamically accessible structures for both n = 1 and n = 2 according to Fig. 1, being distinct in real space, whereas only a single energetically low-lying structure is relevant for the triply tagged species. Thus, each molecular dynamics trajectory must be split into well-defined segments which are associated to a specific reference structure (constructed as explained below).However, each permutation of the atom labeling due to a successful hydrogen scrambling event within a distinct real space structure formally creates another reference structure when computing and diagonalizing the cross-correlation matrix defined viaeqn (25); cf. the molecular symmetry group concept43 where Longuet-Higgins’ permutation-inversion groups are a special case. This permutation sampling would chop up each trajectory segment that is associated to a specific real space structure into pieces that are associated to a distinct atom labeling pattern, thus creating additional reference structures for mode analysis when using eqn (25). This trivial effect of only changing the permutation state of reference structures can be eliminated by transforming each distinct atom labeling pattern back to our standard ordering27 as defined in Fig. 1a.
In the present case, the starting point of the computation of the reference structures were the respective minimum energy structures of the systems of interest, all of which are shown in Fig. 1. The trajectories were analyzed and split into segments that are close in structure to one of these energetic minimum structures, thus considering not only the global but also all local minima. To this end, all frames were transformed into standard order (which is defined according to the labeling in Fig. 1a and thus includes permutational ordering) and rotated using root-mean-square distance criteria to match as closely as possible any of the available energetic minimum structures. The center of mass motion has been taken out by transformation into the barycenter coordinate system, i.e. the square-root mass-weighted center (all this has been carried out in Cartesian coordinates).
This procedure generates trajectory segments that are associated to the available minimum energy structures. Subsequently, the configurations of these trajectory segments were averaged to create what we call the reference structure of the respective trajectory segment. They thus define structural motifs also in the case of fluxional molecules, where different minima are regularly populated due to large-amplitude motion. This procedure led in all cases to very symmetric reference structures. Moreover, these reference structures turn out to be, apart from slight changes in bond lengths, identical to the corresponding energetic minimum structures and are therefore not shown here. These reference structures and the associated trajectory segments are then used in order to compute the generalized normal modes that describe best the dynamics in these trajectory segments and thus the IR spectra corresponding to the respective structural motif.
For bare protonated methane at 110 K, a single reference structure turned out to be sufficient (after transforming the instantaneous permutation pattern to standard ordering in each trajectory frame) which was essentially the global minimum energy structure, see Fig. 1a. For the singly tagged system, i.e. CH5+·(H2)n with n = 1, two reference structures were required. Analysis of the trajectories showed that roughly 70% of the generated configurations could be assigned to the minimum energy structure shown in Fig. 1b and ≈30% to the one depicted in panel c, whereas the structure in panel d was not observed.
In the case of the doubly tagged n = 2 system, about 97% of the trajectory frames could be assigned to the minimum energy structure shown in panel e, which corresponds to the global minimum. The remaining 3% of the frames were assigned to the structures shown in panel f and panel g. Given its overwhelming population, only the global minimum structure e has been considered in the mode decomposition of the IR spectrum. Finally, the triply tagged system has only one reference structure as displayed in Fig. 1h.
III. Results and discussion
In the following, the total IR spectra α(ω) of the investigated microsolvated systems, i.e. CH5+·(H2)n for n = 1 up to 3, are analyzed and assigned in terms of the additive mode-specific absorption cross sections αk(ω) in the frequency range of 1000–4500 cm−1. As already alluded to in the Introduction, the present analysis focuses specifically on tagging effects on the stretching and bending modes of protonated methane which are located in the mid-IR frequency regime. In addition, the limiting n = 0 case corresponding to the bare CH5+ is analyzed as well using the identical methodology in order to provide a consistent reference to discuss changes of the generalized modes and thus of the total lineshape due to microsolvation as a function of n. Our frequency window includes the bending and stretching modes of protonated methane and its H2-tagged species, which allows us to discuss how H2-tagging affects the bending and stretching modes of bare CH5+. In the subsequent presentation, we will address bare protonated methane first followed by discussing the tagged species. For the latter, we analyze the n = 3 case first since that turned out to be most easily understood, followed by n = 2 and finally by n = 1.A. Bare protonated methane
We first of all compare the mid-IR spectrum of bare CH5+ as obtained using the single-channel maximum entropy method to the one obtained from the Fourier transform at two temperatures, 110 and 300 K, see Fig. 3. It is clearly seen that the spectra computed using the ME and FT methods agree well with each other. In addition to reproducing all features of the FT spectra (without introducing additional modulations) the ME method is seen to reduce the noise level drastically using the same statistics, thus resulting in smoother curves. Although similar results could be obtained by applying an adequate windowing function when computing the FT spectrum, we want to stress that the presented ME spectra are not based on such arbitrarily chosen smoothing procedures, but rather follow from an algorithmic procedure that selects the parameters based on an automatic minimization procedure that results in an ‘optimally smoothed’ spectrum. Furthermore, although the mid-IR spectrum at 110 K shows more structure as expected, it is qualitatively comparable to the one at 300 K. | ||
| Fig. 3 Mid-IR spectra of bare protonated methane at 300 and 110 K in the top and bottom panel, respectively, computed from AIMD simulations with classical nuclei using the single-channel ME method (blue and circles), see Sec. II B, and the usual FT approach (black). The orders for the ME spectra computations were 591 for 300 K and 700 for 110 K. Note that the FT spectra are off-set for the sake of presentation. | ||
In addition to computing the total IR spectra, we also computed the individual modes using our new multi-channel ME approach. The obtained generalized normal modes are shown in detail in the ESI† and agree well with the previously extracted modes.27
In the bare case, five stretching modes above 2000 cm−1 are extracted with our novel procedure, see Fig. 3b and Table 1 for frequencies, in accordance with earlier results27 that were, however, simply based on power spectra. The extracted modes are, in order of decreasing frequency, the asymmetric as well as symmetric stretching modes of the tripod hydrogen atoms (atom 4 and atom 5) abbreviated as ta and ts, the stretching mode of the hydrogen atom at the in-plane position (atom 3) called ti, and the symmetric as well as asymmetric stretching modes of the moiety hydrogen atoms (atom 1 and atom 2) called ms and ma, see Fig. 1a for the atom labels. The tripod stretching modes are of similar intensity and shape while the ms mode is significantly wider and of lower intensity compared to the ma mode. These modes are visualized in the ESI† by means of dynamical animations.
| Mode | CH5+ | CH5+(H2)1–I | CH5+(H2)1–II | CH5+(H2)2 | CH5+(H2)3 |
|---|---|---|---|---|---|
| Hi2 | — | — | — | — | 4374 |
| Hom2 | — | — | 4336 | 4343 | 4343 |
| Him2 | — | 4327 | — | 4334 | 4340 |
| ta | 3309 | 3291 | 3294 | 3294 | 3304 |
| ts | 3218 | 3198 | 3203 | 3200 | 3203 |
| ti | 3080 | 3081 | 3099 | 3098 | 3058 |
| ms | 2706 | 2710 | 2718 | 2673 | 2660 |
| ma | 2429 | 2288 | 2293 | 2242 | 2286 |
| bw | 1500 | 1501 | 1509 | 1513 | 1498 |
| bt | 1477 | 1466 | 1473 | 1472 | 1473 |
| bs | 1430 | 1442 | 1444 | 1446 | 1439 |
| br | 1276 | 1284 | 1309 | 1308 | 1324 |
| bb | 1176 | 1207 | 1161 | 1176 | 1171 |
All bending modes of bare CH5+ are located between 1000 and 1800 cm−1. We extracted five bending modes in this frequency range, see Fig. 3b and Table 1 for frequencies, which also agree with the generalized normal modes that have been obtained earlier27 but using standard FT instead of multi-channel ME techniques. Thus, the following modes were extracted and will be investigated as to their response to microsolvation of CH5+ with n = 1 to 3H2 molecules: wagging bend (bw), twisting bend (bt), scissoring bend (bs), rocking bend (br), and butterfly bend (bb). These modes are also animated in the ESI† for the sake of direct comparison to the corresponding modes in case of the n > 0 species.
At variance with the previous approach,27 however, the current mode spectra are not just projected vibrational density of states, but carry instead the proper IR intensity of each distinct mode, i.e. they are the mode-specific IR absorption cross sections αk(ω) according to eqn (27). Since the remaining cross-correlations according to eqn (28) are small, these mode-specific spectra approximately add up to the corresponding total IR spectrum, α(ω). It is demonstrated in Fig. 4 that, indeed, this approach allows one to re-synthesize the total IR spectrum “mode by mode”, which is shown from bottom to top starting with the highest-frequency mode and progressing to the lowest one according to Table 1.
 | ||
| Fig. 4 Mode-by-mode re-construction of the total IR absorption cross section α(ω) of bare CH5+ at 110 K based on successively adding up all mode-specific IR absorption cross sections αk(ω) according to eqn (27) from bottom to top starting with the highest-frequency modes, see text. The modes were obtained by diagonalization of the cross-correlation matrix (cf.eqn (23)) computed by the multi-channel ME method. The topmost spectrum is the total IR spectrum obtained from the single-channel ME method (where total rotations and all index permutations have been taken out whereas they are included in Fig. 3), the second spectrum from top (thick line) is the re-synthesized total IR spectrum according to eqn (30), whereas the third curve from top (dotted line) quantifies the residual cross-correlations according to eqn (28). Note that there are minor deviations between the single-channel and the total spectrum that result from the sum of all modes obtained by the multi-channel approach. Deviations between the two methods are expected when using finite time-series since the multi-channel method has access to more data in the form of the different channels and thus can better resolve fine structure compared to the single-channel approach (a close-up of the bending region is shown in the ESI†). Moreover, the more detailed multi-channel analysis also leads to a different number of parameters as a result of the ME methodology as determined autonomously by the algorithm given the larger database. | ||
Thus, the decomposition technique introduced here allows one to work out – at a fully quantitative level – which modes contribute to what extent to peaks, shoulders or wings of the total lineshape, which appears to be an interesting theoretical spectroscopic analysis tool beyond the specific case discussed herein.
B. Tagged protonated methane: CH5+(H2)n
The mid-IR spectra of the three microsolvated systems compared to the bare case are shown in Fig. 5. Adding hydrogen tags to bare CH5+ (cf. bottom-most red curve with circles) clearly affects the mid-IR spectrum in systematic ways. Obviously, each added hydrogen tag leads to an additional signal at about 4500 cm−1 that is caused by the respective hydrogen stretching mode. This mode is IR forbidden for an isolated H2 molecule, but it is seen here due to the interaction of the tag with the CH5+ core, thus breaking the symmetry of the attached H2via electronic polarization effects. Besides this striking but trivial change compared to bare protonated methane, already upon adding the first hydrogen tag the asymmetric moiety stretch signal ma, which is at about 2400 cm−1 in the bare case, shows a considerable red-shift by more than 100 cm−1 together with a significant gain in intensity compared to all other bands. Both, frequency shift and intensity increase persist upon tagging CH5+ with more than a single H2 molecule and are most pronounced for the CH5+·(H2)2 species. The other stretching mode of the H2 moiety in CH5+, being the symmetric stretch ms at around 2700 cm−1 in the n = 0 case, is also found to gain systematically more intensity, which is most significant for the CH5+·(H2)2 species, and red-shifts up to 46 cm−1 in the case of n = 3. This is easily understood at the qualitative level since the hydrogen molecules stay attached at the moiety (similar to the structures depicted in Fig. 1) during the dynamics at 110 K and thus perturb mostly the moiety dynamics; hence a pronounced change in the bands caused by the moiety itself is expected.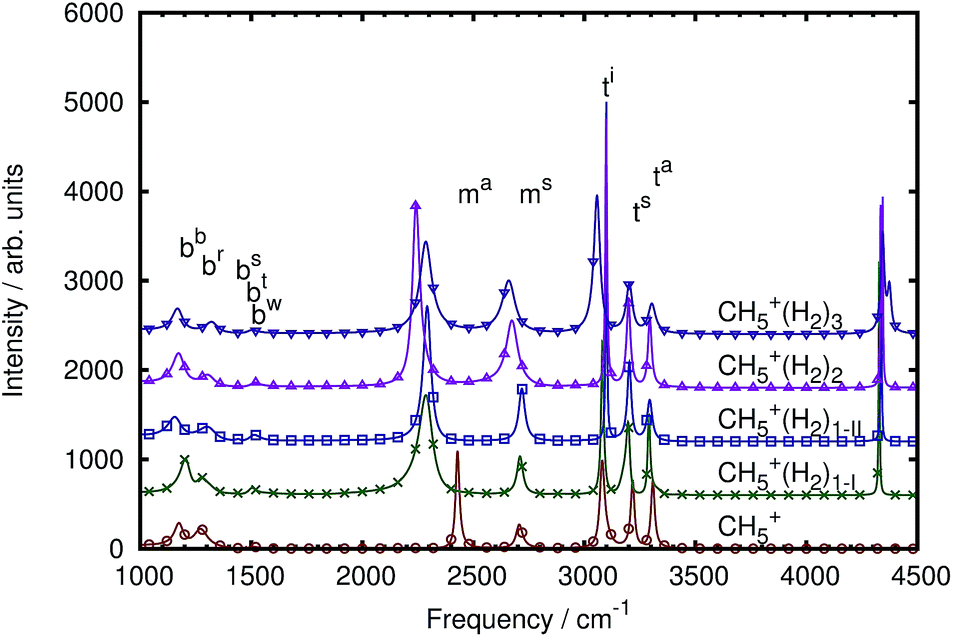 | ||
| Fig. 5 IR spectra of microsolvated protonated methane (with three, two and one H2 molecules attached from top to bottom) compared to bare protonated methane all at 110 K and computed with the multi-channel ME method. See Sec. II C. | ||
The tripod stretches show continuous changes when more tags are added. In the bare system, the three tripod stretches are distinct from each other and this distinction is conserved upon tagging. Only subtle changes within the tripod stretching modes are observed that mainly involve intensities and minor frequency shifts. Since the first tagging hydrogen molecule can be situated at two different positions we present two spectra in the case of using one tag. They show subtle differences in the tripod stretching signals, in particular the intensity of the ta mode shows significantly different intensities depending on the distinct tagging site. Similarly, the ms mode has a higher intensity in the situation where the tag resides at site 2. Further details will be discussed in later sections when focusing on the specific mode absorptions. Adding a second messenger hydrogen molecule leads to a better distinction of the in-plane ti and asymmetric tripod ta stretching modes while the symmetric tripod stretching mode ts is almost unchanged. Finally, adding the third tagging hydrogen molecule results in a very much broadened signal of the in-plane tripod stretching mode ti while the other two resonances, ta and ts, remain as sharp as they were before.
How can these observations be qualitatively understood? The first hydrogen molecule is expected to be attached at the inner moiety hydrogen atom (being atom 2 in Fig. 1a) according to the energetic global minimum structure of CH5+·H2 shown in panel b. Due to the intermolecular interaction a change of the symmetric and asymmetric moiety stretches is expected. Furthermore, a change of the in-plane tripod stretching mode is expected since this atom is now better localized in space, which is reflected in the sharper peak. Since pseudorotation is still possible in this situation, the ti and the ma modes are broader compared to the case with two hydrogen tags. The second hydrogen tag would then be located at the remaining moiety hydrogen atom 1 according to the global minimum structure of the CH5+·(H2)2 species, see panel e in Fig. 1. Thus, internal rotation as sketched in Fig. 2b will be easily possible in view of the symmetric tagging scenario of the moiety. At the same time, the in-plane hydrogen atom 3 will be better localized because atom displacements as illustrated in Fig. 2a, and thus pseudorotation leading to partial scrambling, will be hindered resulting in a sharp resonance of ti. Finally, the third H2 molecule would be added to the in-plane hydrogen atom 3, see panel h in Fig. 1. Thus, once three tags are added, all three in-plane atoms 1, 2, and 3 in CH5+·(H2)3 are interacting equally with H2 molecules, which is a scenario that is equivalent to that in the bare CH5+ limit where all three in-plane atoms are untagged, compare panel h to panel a in Fig. 1. This approximate equivalence in the presence of microsolvation, which is distinctly different from all other tagging patterns that are collected in Fig. 1, allows for easy pseudorotational motion according to the process sketched in Fig. 2a and eventually enables “partial scrambling” in the n = 3 case. This topological argument suggests that partial scrambling should be more easily possible for the n = 3 case compared to both n = 1 and 2, the onset of which in terms of small-amplitude motion leads to a broader signal of the ti and the ma modes.
Addressing finally the perturbations of the bending region, it appears at first sight as if these resonances feature only some mild changes due to tagging. However, it is difficult to discern at the resolution level of Fig. 5 the much more intricate changes of the bending band of CH5+ due to its interaction with the tags. Instead of discussing this here, we refer to Fig. 7 and to the corresponding detailed analysis of the mode-specific IR spectra to be presented together with the mode assignments.
At this stage we conclude, based on visual inspection of the computed total IR spectra, that tagging of protonated methane by a few H2 molecules has a significant effect on all parts of its mid-IR spectrum, from the characteristic triple-peaked tripod stretching region at highest frequencies 3000–3500 cm−1 to the two moiety stretches around 2500 cm−1 down to the bending regime at 1100–1600 cm−1. However, the assessment of spectral changes due to tagging is still qualitative as it is simply based on directly comparing the peaks of the microsolvated cases to those of the bare reference, CH5+. Thus, it remains to be seen if our spectral decomposition and mode assignment techniques are also able to disentangle the IR spectrum of protonated methane once it is microsolvated.
C. Mode assignment of the microsolvated cases
The analysis was carried out as described in the methods section leading to the modes shown in Fig. 6 and 7 for the stretching and bending bands, respectively. The corresponding frequencies are compiled in Table 1 whereas the atomic displacements that generate these localized modes are visualized in the ESI† in terms of dynamical animations. | ||
| Fig. 6 Mode decomposition and assignment of microsolvated protonated methane with labeled stretching resonances as follows. Hi2: H2 stretch at in-plane site, Him2: H2 stretch at inner moiety site, Hom2: H2 stretch at outer moiety site, ta: antisymmetric out-of-plane stretch in tripod, ts: symmetric out-of-plane stretch in tripod, ti: in-plane stretch in tripod, ms: symmetric stretch in moiety, ma: antisymmetric stretch in moiety; the vertical dotted lines mark the corresponding resonance frequencies of bare CH5+ shown in panel e. The panels a–d show the microsolvated species from top with 3 tags to bottom with 1 tag. The nuclear displacements that underlie these generalized normal modes are visualized in the ESI† by dynamical animations. | ||
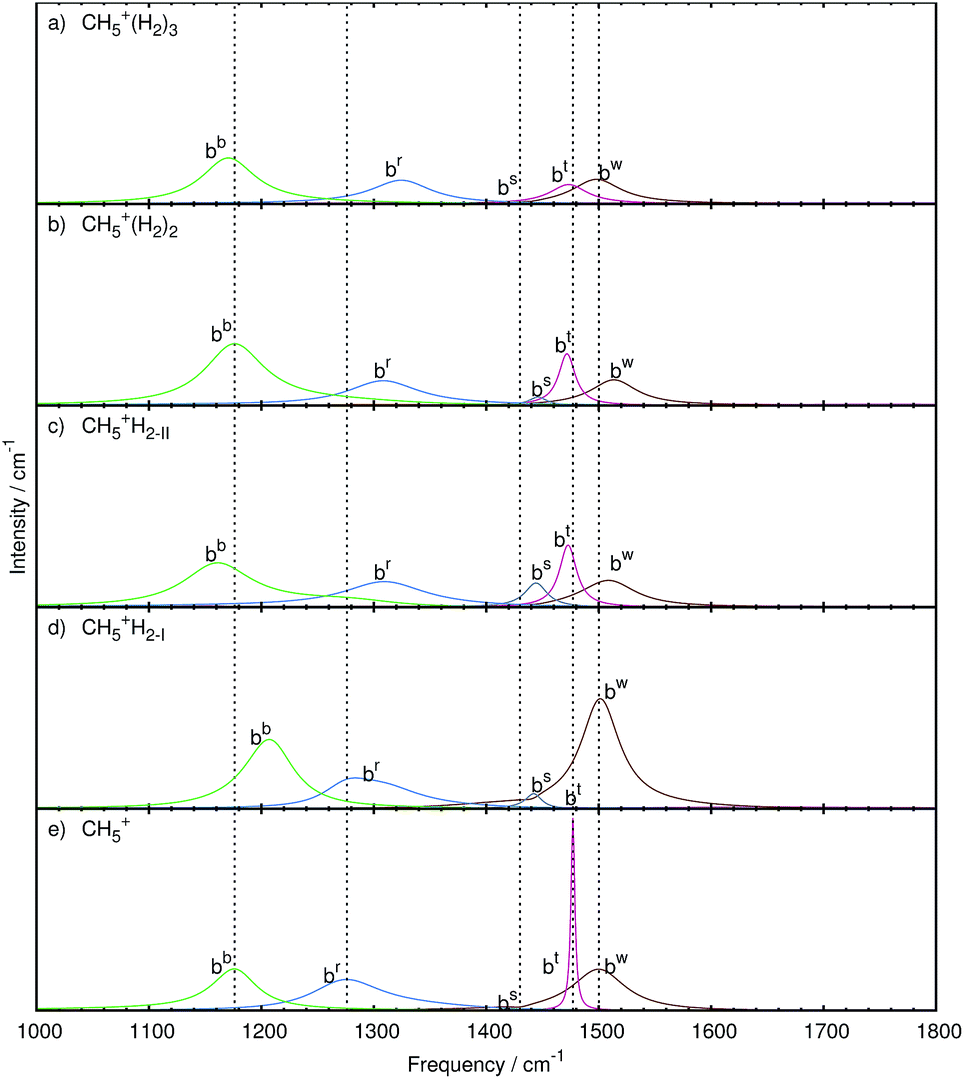 | ||
| Fig. 7 Mode decomposition and assignment of microsolvated protonated methane with labeled bending resonances as follows. bw: wagging bend bt: twisting bend bs: scissoring bend br: rocking bend bb: butterfly bend; the vertical dotted lines mark the corresponding resonance frequencies of bare CH5+ shown in panel e. The panels a–d show the microsolvated species from top with 3 tags to bottom with 1 tag. Note that some mode-specific spectra have been scaled to allow for a better visualization using scaling factors as follows. bw: 10, bt: 100, bs: 10, br: 1, and bb: 1. The nuclear displacements that underly these generalized normal modes are visualized in the ESI† by dynamical animations. | ||
In the following, the stretching and bending modes will be discussed separately starting with the former ones. Within each mode class we proceed from the simplest case, which is the one with three messenger species attached being closest to bare CH5+, to the most difficult one. The latter is the situation when a single H2 molecule interacts with bare CH5+ since dynamical site-exchange events become significant. Bare protonated methane, which serves as our reference, has already been reviewed in terms of its mode assignment in Sec. III A to which we now refer.
Three tags. After adding three messenger H2 species to bare CH5+, we obtain three modes in the region of 4500 cm−1 caused by the stretching motion of the three hydrogen molecules. These species, located at the atoms 1, 2 and 3, are clearly distinguished from each other after having disentangled the total spectrum, namely the stretching mode due to the H2 molecules attached to the in-plane position (Hi2) and to the inner (Him2) and outer (Hom2) moiety sites of CH5+, being atoms 1, 2, and 3, respectively, according to the scheme defined in Fig. 1a. The H2 stretching motion from the tag bound to site 2, i.e. Him2, has the lowest quasi-intramolecular frequency, indicating that the interaction between the inner moiety atom and the tag is the strongest.
Next lower in frequency are the tripod stretching modes ta (3304 cm−1), ts (3203 cm−1) and ti (3058 cm−1), which are red-shifted w.r.t. bare CH5+. The intensity of the ti mode is higher w.r.t. the bare reference system and the other two tripod stretches, which reflects the additional interaction of the tripod atoms with the hydrogen tagging molecules. The increased intensity of the ti mode indicates a better defined stretching mode compared to the bare case.
The symmetric and asymmetric stretching modes of the moiety, ms (2660 cm−1) and ma (2286 cm−1), respectively, show a red-shift w.r.t. the bare case (2706 and 2429 cm−1, respectively), which is more pronounced for the asymmetric case. The intensities of both modes match better, compared to the previous cases, due to a significant reduction in the peak height of the ma mode. This is in contrast to the bare and the other microsolvated cases where the ms mode always has a significantly lower intensity. This shows that these modes are now more pronounced indicating that the structure must be better defined overall. We note that for the sake of simplicity and given the mostly rather sharp resonances, we use the peak height of the mode-specific absorption cross section as a proxy for intensity, although the integral of the full peak should be used in principle.
Two tags. The set of stretching modes obtained for CH5+·(H2)2 is shown in Fig. 6b. We obtained the expected two hydrogen stretching modes from the two H2 tags, labeled Hom2 and Him2, and they also show the same trend as before, i.e. the Hom2 mode (at 4343 cm−1) is located at a higher frequency than the Him2 mode (4334 cm−1).
When referring to what we have seen for the CH5+·(H2)3 complex, the removal of the tag located at the in-plane position, i.e. atom 3, results in distinct changes in the C–H stretching modes of the CH5+ core for the n = 2 species. We observe that now the ts and ta modes are similar in intensity to each other, while the ts mode remains similar to the triply tagged case. It is furthermore seen that the ti mode (3098 cm−1) is blue-shifted in contrast to the ta (3294 cm−1) and ts (3200 cm−1) modes that are, as in the n = 3 case, red-shifted w.r.t. the bare reference molecule. This indicates that protonated methane is differently affected upon microsolvating it when using two or three solvent species.
Moreover, also the lineshape of the moiety stretching modes is very different from what is observed in the triply tagged case, whereas it is similar to the situation found for the bare system, i.e. the ms mode is of significantly lower intensity than the ma mode. Although different in their lineshape, the modes of the n = 2 species show the same frequency shifting pattern as those of the triply tagged case, both ms (2673 cm−1) and ma (2242 cm−1) modes being red-shifted.
Since sites 1 and 2 are tagged in the n = 2 case whereas site 3 is not, partial scrambling will be less likely to happen since this process would result in an energetically unfavorable situation given that the resulting structure would be 1.19 kcal mol−1 higher in electronic energy (as provided in the caption of Fig. 1). Assuming partial scrambling to be inactive, we would expect from this observation the ti mode to feature a very narrow and more harmonic type of C–H stretching vibration. Internal rotation, on the other hand, will still be allowed since the structure resulting after this process will be identical to the one before this rotation after a slight readjustment of the two weakly attached H2 molecules. This situation might explain the different behavior of the ti mode shifting as well as the lineshape changes of the moiety stretching modes and the ta mode.
One tag. In the most intricate case of the CH5+·(H2) complex, the single tagging molecule can be attached to sites 1, 2, and 3 as depicted in Fig. 1 in panels b, c, and d, respectively. It turned out that essentially only the two moiety sites 1 and 2 were found to be populated during the AIMD simulations (for about 70 and 30% of the total trajectory length, respectively), whereas the H2 molecule was not observed to be attached to site 3, belonging to the tripod. This finding is consistent with the energetic ordering of the optimized structures,56cf. caption to Fig. 1, where isomer c involving site 1 is only 0.35 kcal mol−1 above the global minimum, whereas attachment of H2 at the tripod site 3 is as much as 1.77 kcal mol−1 higher in energy. Thus, only the two reference structures corresponding to panels b and c, respectively, reflecting directly the two lowest-energy static structures, must be used in the following dynamical vibrational analysis.
By using two reference structures, two sets of modes are obtained as shown in Fig. 6d for reference structure I, where the tag is located at site 2, and in Fig. 6c for reference structure II involving the H2 attachment to site 1. In both cases, a single stretching mode corresponding to the hydrogen molecule is extracted. The Him2 mode in Fig. 6d is located at slightly lower frequency (4327 cm−1) than the Hom2 mode (4336 cm−1) in Fig. 6c. This is consistent with the weaker interaction of the H2 tag when attached to site 1 compared to site 2, which is also indicated by a shorter intermolecular bond length when involving site 2 (1.87 Å) versus site 1 (1.89 Å).
In both reference situations, the modes look very similar at first glance, but subtle differences are still discernable upon more careful inspection. The ta stretching mode observed in Fig. 6c due to reference structure II shows a long tail on its red-wing that overlaps with the blue wing of the ts mode. In contrast, in reference structure I shown in Fig. 6d all three tripod stretching modes are well separated. The red-shift of the ta (3291 and 3294 cm−1) and ts (3198 and 3203 cm−1) modes is comparable to each other. Comparing the ti mode in both sets reveals distinct differences regarding shifting and lineshape. In reference structure I, the ti mode is with 3081 cm−1 essentially unshifted whereas in the case of reference structure II this mode is blue-shifted (3099 cm−1), which is similar to what has been observed in case of the doubly tagged case (3098 cm−1).
Also the moiety stretching modes again look quite similar to each other as they both show a similar ratio as the bare system, where the ms mode has a considerably lower intensity than the ma mode. Note, however, that the ma mode is subject to a significantly increased linewidth and that the ms mode shows only a minor intensity increase compared to the bare system in panel e of Fig. 6. Both moiety stretches are red-shifted, where the ma mode shows a more pronounced red-shift than the ms mode. Additionally, the ma mode shows the smallest red-shift in reference structure II (2293 cm−1), whereas this mode is stronger red-shifted in the doubly tagged case (2242 cm−1).
Qualitative trends. To sum up the discussion of the stretching modes which determine the overall shape of the high frequency part of the IR spectrum of CH5+·(H2)n, we conclude that tagging affects the associated peaks in distinct ways as follows. First of all, the ts mode is relatively uninfluenced by adding the first and second tagging molecule to the bare species, while the ta mode changes readily upon microsolvation. Upon adding the third tagging H2 molecule, both the ta and ts mode show the same behavior of reduced intensity and red-shifting w.r.t. bare CH5+. The ti mode, on the other hand, displays a distinctly different behavior as a function of the number of tagging molecules: one finds essentially no shift once the bare system is tagged at its so-called inner moiety site or a red-shift if it is triply tagged, whereas a blue-shift is observed if it is microsolvated by a single hydrogen molecule which is located at the outer moiety site, or alternatively in the case of adding two tags. Focusing on the moiety stretching modes, it becomes clear that the ma mode shows the most significant change upon microsolvation in form of a strong red-shift. This can be understood when considering the intricate three-center electronic bonding situation in (bare) protonated methane13–15 as summarized in the Introduction. The tripod stretching modes involve exclusively the usual two-center two-electron bonding for each of the three C–H covalent bonds involved. These bonds are rather strong15 and, therefore, are not that much affected as a result of non-covalent interactions with the tagging hydrogen molecule (at the in-plane location). In stark contrast, when considering the moiety stretching modes, it is a three-center two-electron bond that binds the two hydrogens involved to the carbon site. This bonding type is clearly weaker than the two-center one15 and, therefore, these softer C–H bonds are more strongly influenced when forming non-covalent bonds with the tag, which explains a more pronounced frequency red shift when they are tagged. The impact on the ms mode is different for the singly tagged species as a blue shift is observed, which is explained by the asymmetric nature of the tagging in this limiting case, which hinders the full displacement of the tagged hydrogen moiety atom compared to the bare case. The moiety atoms are again symmetrically tagged when adding more than one hydrogen molecule, hence the expected red-shift of the modes is observed as in the ma case.
These features herald at the level of mid-IR spectra the purely structural changes upon microsolvation that have been obtained recently from time-independent ab initio path integral simulations at low temperature,58 namely that the quantum localization of CH5+ due to microsolvation is very similar in the case of adding either one and three hydrogen molecules, while the case of two H2 is distinctly different. This suggests, already at this stage, that high-resolution messenger vibrational spectroscopy should be a suitable tool47–50 to uncover even subtle modifications of hydrogen scrambling due to microsolvation of protonated methane.
Three tags. In the case of attaching n = 3H2 tags to protonated methane, the bw and bt modes show a strongly decreased intensity together with a red-shift (to 1498 and 1473 cm−1, respectively) w.r.t. the parent system, CH5+. Furthermore, these resonances are changed from relatively sharp to broad signals, which clearly reflects the significant impact of the tagging molecules on the intramolecular bending dynamics of the microsolvated CH5+ core molecule.
The bs mode is blue-shifted (to 1439 cm−1) with a similarly low intensity as in the bare system. Comparison with the other microsolvated cases studied shows that both in the bare and the n = 3 case the bs mode is of lowest intensity while in the CH5+·(H2)2 species it gains intensity and turns out to be strongest in the n = 1 limit. The br mode, on the other hand, is blue-shifted (by 48 cm−1) but comparable in lineshape to the bare system. The bb mode is red-shifted (to 1171 cm−1) and mainly conserved in shape with an increase in intensity.
Two tags. Upon removing the tagging molecule located at atom 3, the bt mode of the doubly tagged species has a similar lineshape as the one in the singly tagged species in reference structure II including a red-shift (1472 cm−1). The lineshape changed from that of a rather broad feature in the triply tagged case to a narrow signal for n = 2. No change in the lineshape is observed for the bw mode, which shifts to the blue (1513 cm−1) compared to bare system. Also the br mode has a similar structure but shows a blue-shift (1308 cm−1). While the bs mode is of low intensity in the bare and triply tagged systems, it is found to gain intensity in the n = 2 case.
One tag. As in the case of the stretching band, two sets of bending modes are obtained depending on where the single H2 molecules is attached at the moiety, i.e. at site 2 (yielding reference structure I) or at site 1 (reference structure II).
The bw mode shown in Fig. 7c for reference structure II is broad and of low intensity, which is strikingly different from the one observed for reference structure I in Fig. 7d, where it is found to become an extremely strong signal that is even more intense than in bare protonated methane. The bt mode of reference structure II is similar to the one obtained from the doubly tagged case with a similar shift (1472 cm−1) and lineshape, which underlines the similarities between those two tagging cases akin to what has been found when analyzing the stretching bands. On the other hand, the bt mode obtained from reference structure I is very low in intensity. For both tagging sites, the bs mode has gained intensity w.r.t. the bare case. While the br mode is essentially similar in all tagging cases in terms of intensity, it varies only in the magnitude of the blue-shift compared to the corresponding CH5+ resonance. The shift is least pronounced in the case of reference structure I.
IV. Conclusions and outlook
The effect of microsolvation by one, two and three H2 molecules on the bending and stretching bands in the mid-infrared absorption spectrum of tagged protonated methane, CH5+·(H2)n, has been investigated systematically in comparison to bare CH5+ serving as our internal reference. The spectra in the frequency window from 1000 to 4500 cm−1 have been computed based on ab initio molecular dynamics trajectories obtained at 110 K with classical nuclei using an advanced maximum entropy methodology. The maximum entropy spectra are computed here by strictly minimizing the assumptions on the available data series, thus implementing the ‘maximum parsimony’ approach or ‘Occam's razor’ principle at the level of the algorithm. The reported spectra are thus in a sense ‘optimally smoothed’ spectra that are independent from the user, in contrast to standard Fourier smoothing procedures that rely on specifications and thus (arbitrary) choices of window functions and window lengths. Moreover, given the same trajectory data, this method is shown to provide smoother spectra than those obtained via Fourier transforming the dipole autocorrelation function, while preserving the correct intensities. This technique is able to process multi-channel data which enables us to extract generalized normal coordinates as well as the associated mode-specific IR absorption coefficients from molecular dynamics simulations. The full spectra can be “re-synthesized” to a good approximation simply as a sum of these mode-specific partial spectra, which allows one to quantify their contributions to the total IR lineshape in a mode-by-mode fashion.The computed total mid-IR spectra of CH5+·(H2)n feature subtle but significant changes w.r.t. bare CH5+, which depend crucially on the number of tagging molecules, n. The case with three tags turns out to be most easily understood since only a single tagging motif, where all three H2 molecules remain attached to specific proton sites, is found to contribute to the dynamics. In stark contrast, there exist three microsolvation motifs in the n = 2 and n = 1 cases as defined by optimized minimum energy structures. For CH5+·(H2)2, however, only the lowest-energy structure is found to contribute to the dynamics to an overwhelming extent. Most difficult in terms of both spectral decomposition and assignment is the n = 1 species, where the two lowest-energy structures are significantly populated. Moreover, they are found to dynamically interchange along the trajectories and produce distinct spectral fingerprints.
The impact of microsolvation on the mid-IR spectrum of bare CH5+ is complex and affects the peak positions and intensities of the well separated stretching and bending regions in characteristic ways. The stretching frequency regime is shaped by three and two modes at higher and lower frequency that can be associated to stretching dynamics involving mainly the tripod and moiety atoms, respectively, of the microsolvated CH5+ core molecule. Also the bending band consists of five modes with vastly different intensities in this case. Quite interestingly, the two microsolvation motifs that are dynamically visited in the n = 1 case lead to pronounced differences in the bending modes in terms of their intensities and frequency shifts.
An important qualitative finding based on our systematic decomposition of the bending and stretching spectra of microsolvated protonated methane is that the pronounced frequency shift of the asymmetric moiety stretching resonance serves as a unique mid-IR marker to distinguish the tagged species from bare protonated methane. The strong frequency response of this moiety stretch due to even weak non-covalent intermolecular interactions with tagging species has been traced back to the three-center two-electron bonding nature which binds two hydrogens to the carbon site, in stark contrast to the three standard two-center two-electron C–H bonds that form the tripod. This observation is particularly useful since the two moiety stretching signals are, even at higher temperatures, well separated features in the mid-IR spectrum of protonated methane.
We close by stressing that multi-channel maximum entropy analysis of (ab initio) molecular dynamics trajectories is a powerful tool that can be applied broadly to microsolvated molecules in order to understand their IR spectra. In particular, the approach allows one to quantify and assign the distinct influence of the microsolvating species on the vibrational dynamics of the parent molecule.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We gratefully thank Alex Witt and Sergei Ivanov for supplying the ab initio trajectories of the microsolvated species that have been used for the present analyses. This work was supported by the Cluster of Excellence RESOLV (EXC 1069) funded by Deutsche Forschungsgemeinschaft and computational resources were provided by HPC@ZEMOS, HPC-RESOLV, RV-NRW, and BOVILAB@RUB.References
- V. L. Tal'roze and A. K. Lyubimova, Dokl. Akad. Nauk SSSR, 1952, 86, 909 Search PubMed.
- R. J. Saykally, Science, 1988, 239, 157 CAS.
- T. Oka, Philos. Trans. R. Soc. London, Ser. A, 1988, 324, 81 CrossRef CAS.
- G. E. Scuseria, Nature, 1993, 366, 512 CrossRef.
- D. Marx and M. Parrinello, Science, 1999, 284, 59 CrossRef CAS.
- P. R. Schreiner, Angew. Chem., Int. Ed., 2000, 39, 3239 CrossRef CAS PubMed.
- D. Gerlich, Phys. Chem. Chem. Phys., 2005, 7, 1583 RSC.
- S. Borman, Chem. Eng. News, 2005, 83, 45 Search PubMed.
- P. Kumar and D. Marx, Phys. Chem. Chem. Phys., 2006, 8, 573 RSC.
- A. B. McCoy, Int. Rev. Phys. Chem., 2006, 25, 77 CrossRef CAS.
- S. D. Ivanov, A. Witt and D. Marx, Phys. Chem. Chem. Phys., 2013, 15, 10270 RSC.
- T. Oka, Science, 2015, 347, 1313 CrossRef CAS PubMed.
- V. Dyczmons and W. Kutzelnigg, Theor. Chim. Acta, 1974, 33, 239 CrossRef CAS.
- D. Marx and M. Parrinello, Nature, 1995, 375, 216 CrossRef CAS.
- D. Marx and A. Savin, Angew. Chem., Int. Ed. Engl., 1997, 36, 2077 CrossRef CAS.
- A. Gamba, G. Morosi and M. Simonetta, Chem. Phys. Lett., 1969, 3, 20 CrossRef CAS.
- G. A. Olah, G. Klopman and R. H. Schlosberg, J. Am. Chem. Soc., 1969, 91, 3261 CrossRef CAS.
- S. Ehrenson, Chem. Phys. Lett., 1969, 3, 585 CrossRef CAS.
- V. Dyczmons, V. Staemmler and W. Kutzelnigg, Chem. Phys. Lett., 1970, 5, 361 CrossRef CAS.
- P. C. Hariharan, W. A. Lathan and J. A. Pople, Chem. Phys. Lett., 1972, 14, 385 CrossRef CAS.
- P. R. Schreiner, S.-J. Kim, H. F. Schaefer III and P. v. R. Schleyer, J. Chem. Phys., 1993, 99, 3716 CrossRef CAS.
- M. Kolbuszewski and P. R. Bunker, J. Chem. Phys., 1996, 105, 3649 CrossRef.
- H. Müller, J. Noga, W. Klopper and W. Kutzelnigg, J. Chem. Phys., 1997, 106, 1863 CrossRef.
- D. Marx and M. Parrinello, Z. Phys. D, 1997, 41, 253 CrossRef CAS.
- D. Marx and J. Hutter, Ab Initio Molecular Dynamics, Cambridge University Press, Cambridge, 2009 Search PubMed.
- O. Asvany, P. Kumar, P. B. Redlich, I. Hegemann, S. Schlemmer and D. Marx, Science, 2005, 309, 1219 CrossRef CAS PubMed.
- S. D. Ivanov, O. Asvany, A. Witt, E. Hugo, G. Mathias, B. Redlich, D. Marx and S. Schlemmer, Nat. Chem., 2010, 2, 298 CrossRef CAS PubMed.
- A. Witt, S. D. Ivanov, G. Mathias and D. Marx, J. Phys. Chem. Lett., 2011, 2, 1377 CrossRef CAS.
- D. Marx and M. Parrinello, Science, 1999, 286, 1051 CrossRef.
- G. Mathias and M. D. Baer, J. Chem. Theory Comput., 2011, 7, 2028 CrossRef CAS PubMed.
- G. Mathias, S. D. Ivanov, A. Witt, M. D. Baer and D. Marx, J. Chem. Theory Comput., 2012, 8, 224 CrossRef CAS PubMed.
- A. Brown, B. J. Braams, K. Christoffel, Z. Jin and J. M. Bowman, J. Chem. Phys., 2003, 119, 8790 CrossRef CAS.
- A. Brown, A. B. McCoy, B. J. Braams, Z. Jin and J. M. Bowman, J. Chem. Phys., 2004, 121, 4105 CrossRef CAS PubMed.
- A. B. McCoy, B. J. Braams, A. Brown, X. Huang, Z. Jin and J. M. Bowman, J. Phys. Chem. A, 2004, 108, 4991 CrossRef CAS.
- X. Huang, L. M. Johnson, J. M. Bowman and A. B. McCoy, J. Am. Chem. Soc., 2006, 128, 3478 CrossRef CAS PubMed.
- L. M. Johnson and A. B. McCoy, J. Phys. Chem. A, 2006, 110, 8213 CrossRef CAS PubMed.
- X. Huang, A. B. McCoy, J. M. Bowman, L. M. Johnson, C. Savage, F. Dong and D. J. Nesbitt, Science, 2006, 311, 60 CrossRef CAS PubMed.
- X.-G. Wang and T. Carrington Jr, J. Chem. Phys., 2008, 129, 234102 CrossRef PubMed.
- C. E. Hinkle, A. S. Petit and A. B. McCoy, J. Mol. Spectrosc., 2011, 268, 189 CrossRef CAS.
- E. T. White, J. Tang and T. Oka, Science, 1999, 284, 135 CrossRef CAS PubMed.
- O. Asvany, K. M. T. Yamada, S. Brünken, A. Potapov and S. Schlemmer, Science, 2015, 347, 1346 CrossRef CAS PubMed.
- R. Wodraszka and U. Manthe, J. Phys. Chem. Lett., 2015, 6, 4229 CrossRef CAS PubMed.
- H. Schmiedt, S. Schlemmer and P. Jensen, J. Chem. Phys., 2015, 143, 154302 CrossRef PubMed.
- X.-G. Wang and T. Carrington Jr, J. Chem. Phys., 2016, 144, 204304 CrossRef PubMed.
- H. Schmiedt, P. Jensen and S. Schlemmer, Phys. Rev. Lett., 2016, 117, 223002 CrossRef PubMed.
- H. Schmiedt, P. Jensen and S. Schlemmer, Chem. Phys. Lett., 2017, 672, 34 CrossRef CAS.
- D. W. Boo and Y. T. Lee, Chem. Phys. Lett., 1993, 211, 358 CrossRef CAS.
- D. W. Boo, Z. F. Liu, A. G. Suits, J. S. Tse and Y. T. Lee, Science, 1995, 269, 57 CAS.
- D. W. Boo and Y. T. Lee, J. Chem. Phys., 1995, 103, 520 CrossRef CAS.
- D. W. Boo and Y. T. Lee, Int. J. Mass Spectrom., 1996, 159, 209 CrossRef.
- P. J. Kelleher, C. J. Johnson, J. A. Fournier, M. A. Johnson and A. B. McCoy, J. Phys. Chem. A, 2015, 119, 4170 CrossRef CAS PubMed.
- R. Navrtil, J. Jak and J. Roithov, J. Mol. Spectrosc., 2017, 332, 52–58 CrossRef.
- E. Fois, A. Gamba and M. Simonetta, Can. J. Chem., 1985, 63, 1468 CrossRef CAS.
- S. J. Kim, P. R. Schreiner, P. v. R. Schleyer and H. F. Schaefer III, J. Phys. Chem., 1993, 97, 12232 CrossRef CAS.
- S. Roszak and J. Leszczynski, Chem. Phys. Lett., 2000, 323, 278 CrossRef CAS.
- A. Witt, S. D. Ivanov, H. Forbert and D. Marx, J. Phys. Chem. A, 2008, 112, 12510 CrossRef CAS PubMed.
- A. Witt, PhD thesis, Ruhr-Universität Bochum, 2010.
- A. Witt, S. D. Ivanov and D. Marx, Phys. Rev. Lett., 2013, 110, 083003 CrossRef PubMed.
- J. Hutter, et al., CPMD Program Package, http://www.cpmd.org Search PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787 CrossRef CAS PubMed.
- R. Ramirez, T. Lopez-Ciudad, P. Kumar and D. Marx, J. Chem. Phys., 2004, 121, 3973 CrossRef CAS PubMed.
- T. J. Ulrych and T. N. Bishop, Rev. Geophys. Space Phys., 1975, 13, 183 CrossRef.
- R. W. Herring, CRC Report No 1330, Communications Research Centre, Ottawa, Canada, 1980.
- N. Wu, The Maximum Entropy Method, Springer-Verlag, Berlin Heidelberg, 1997 Search PubMed.
- J. P. Burg, Proc. 37th Meet. Soc. Exploration Geophysicists, 1967 Search PubMed.
- J. P. Burg, Paper presented at Advanced Study Institute on Signal Processing, NATO, Enschede, Netherlands, 1968 Search PubMed.
- J. P. Burg, PhD thesis, Stanford University, 1975.
- P. Broersen, IFAC Proc., 1997, 30, 245 CrossRef.
- M. Morf, A. Vieira, D. T. L. Lee and T. Kailath, IEEE Trans. Geosci. Electron., 1978, GE-16, 85 CrossRef.
- P. J. Brockwell, R. Dahlhaus and A. A. Trindade, Statistica Sinica, 2005, 15, 197 Search PubMed.
- J. Sun, G. Niehues, H. Forbert, D. Decka, G. Schwaab, D. Marx and M. Havenith, J. Am. Chem. Soc., 2014, 136, 5031 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc04040g |
| This journal is © The Royal Society of Chemistry 2018 |