
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLipid biochemical changes detected in normal appearing white matter of chronic multiple sclerosis by spectral coherent Raman imaging†
K. W. C.
Poon
 *a,
C.
Brideau
a,
R.
Klaver
b,
G. J.
Schenk
b,
J. J.
Geurts
b and
P. K.
Stys
*a
*a,
C.
Brideau
a,
R.
Klaver
b,
G. J.
Schenk
b,
J. J.
Geurts
b and
P. K.
Stys
*a
aHotchkiss Brain Institute, Cumming School of Medicine, University of Calgary, Canada. E-mail: pstys@ucalgary.ca
bDepartment of Anatomy and Neurosciences, Vrije University Medical Center, Amsterdam, The Netherlands. E-mail: J.Geurts@vumc.nl
First published on 2nd January 2018
Abstract
Multiple sclerosis (MS) exhibits demyelination, inflammatory infiltration, axonal degeneration, and gliosis, affecting widespread regions of the central nervous system (CNS). While white matter MS lesions have been well characterized pathologically, evidence indicates that the MS brain may be globally altered, with subtle abnormalities found in grossly normal appearing white matter (NAWM). These subtle changes are difficult to investigate by common methods such as histochemical stains and conventional magnetic resonance imaging. Thus, the prototypical inflammatory lesion likely represents the most obvious manifestation of a more widespread involvement of the CNS. We describe the application of spectral coherent anti-Stokes Raman Scattering (sCARS) microscopy to study such changes in chronic MS tissue particularly in NAWM. Subtle changes in myelin lipid biochemical signatures and intra-molecular disorder of fatty acid acyl chains of otherwise normal-appearing myelin were detected, supporting the notion that the biochemical involvement of the MS brain is far more extensive than conventional methods would suggest.
Introduction
Multiple sclerosis (MS) is a leading cause of non-traumatic neurological disability in young adults1 with a global prevalence of ≈2.3 million.2 The underlying cause remains contentious despite decades of investigation. Patients diagnosed with the various clinical forms of MS (relapsing-remitting, primary progressive and secondary progressive) manifest with signs of neurological dysfunction leading to a decline in sensory, motor, cognitive, bowel/bladder and gait function. Traditionally, MS is considered to be a primary autoimmune inflammatory disease whereby a dysregulated peripheral immune system promotes an attack on the CNS, particularly against the insulating layer of myelin around axons. However, an alternative model proposes that MS may begin as a primary degenerative process, with the autoimmune and inflammatory reactions arising secondarily in response to the degenerating myelin.3–5 The most common phenotype of MS at presentation is relapsing-remitting (RR) disease, wherein patients largely recover during remission. However, approximately 25 years after disease onset the majority of patients will go on to develop a secondary progressive (SP) phase, with accumulation of irreversible disability.4,6–11 The gradual transition from RRMS to SPMS cannot be determined through any definitive clinical, imaging, immunologic, or pathologic criteria.9 A smaller but significant percentage present from the onset with primary progressive MS (PPMS), characterized by a gradual progressive clinical course with no relapses or remissions, and with inflammatory pathology that is less prominent.12 Longitudinal studies have also shown SPMS and PPMS patients to suffer a clinical decline at a similar rate.13 For the more common RRMS, the last two decades have seen a number of immuno-modulatory treatment options. However, the same cannot be said for the progressive MS phenotypes, with the aforementioned anti-inflammatory therapeutics generally shown to be ineffective at slowing accumulation of disability.The MS lesion has been well studied since the report by Charcot in 1868.14 The characteristic pathology includes a combination of focal demyelination, inflammation, gliotic scar formation, and variable degrees of axonal destruction. While these lesions are the most obvious indication of CNS damage, the surrounding regions traditionally labeled as ‘normal-appearing’ white matter (NAWM) have been of particular interest. In the NAWM of MS brains, subtle abnormalities have been detected using advanced magnetic resonance imaging (MRI), including prolonged T1 and T2 relaxation times,15 reduced magnetization transfer ratios (MTR)16 and abnormalities on diffusion tensor imaging.17 MTR and immunohistochemistry correlated experiments show that these abnormalities appear to be dependent on proximity to focal white matter lesions with axonal swelling and increased numbers of microglia/macrophages detected distant from lesions.18 However, a decrease in MTR recordings have also been found preceding lesion formation.19 In patients with a relatively moderate lesion load, signs of progressive brain atrophy point towards more extensive abnormalities in the NAWM20 and new lesions have been found to form in the NAWM even before a breakdown of the blood–brain barrier.21 Taken together these observations suggest pathology that is more widespread than previously appreciated, extending far beyond the classic demyelinating plaque.
The myelin sheath, essential for action potential propagation, is a highly ordered structure composed of ≈70% lipid by dry weight, which exists in a tightly orchestrated balance with major myelin proteins to form the insulating layer around the axon. Historically, myelin lipids have been investigated less extensively than myelin proteins in situ because of their hydrophobicity and a complex variety of different lipid species. However, it has been shown that myelin lipid composition in MS patients differs from that of healthy individuals.6,22 More specifically, decreases in total lipid and phospholipid, especially ethanolamine plasmalogens, cerebrosides, sulfatides and sphingolipids have been reported.6,23–28 In contrast, cholesterol esters, the breakdown products of cholesterol, are increased in MS brain.27–30 Taken together, this suggests that lipid abnormalities might be critically important early events culminating in demyelination. Post-mortem MS brain tissue has been traditionally examined using conventional histological stains such as hematoxylin & eosin, together with myelin-reporting stains such as Luxol Fast Blue (LFB). While these dyes perform well for tissue morphology, cellular infiltration and overt demyelination, they are less sensitive to subtle potentially more widespread biochemical abnormalities. LFB binds to some biological lipids, with no reactivity to cholesterol, cholesterol esters or cerebrosides,31 which is a limitation given that these components are altered in MS. Other lipophilic dyes used to stain myelin (Oil Red O, Sudan Black and Nile Red) exhibit conflicting binding and lack specificity.32,33 Immunohistochemistry may be used to evaluate myelin morphology by targeting major myelin protein complexes such as myelin basic protein (MBP) and proteolipid protein (PLP) and although highly specific, these labels report morphological alterations and loss/gain of protein, but provide no information regarding actual myelin lipid changes. Moreover, such labeling methodologies require extensive tissue processing involving harsh solvents and/or detergent permeabilisation steps that perturb the tissue environment and more crucially, alter the major lipid components of the tissue that may carry important pathological information. A more direct, chemically specific readout of lipid alterations would offer a major advantage in our ability to interrogate subtle biochemical changes in diseased brain.
Label-free imaging techniques such as coherent Raman Scattering, which include variants of coherent anti-Stokes Raman scattering (CARS) and stimulated Raman scattering (SRS)34 have emerged as viable methods to investigate lipid-rich structures such as myelin. These methods are based on Raman spectroscopy, an inelastic scattering technique used to probe the vibrational and rotational modes of molecules and their chemical bonds. With CARS, the wavelengths of two pulsed lasers are tuned to coherently drive a targeted vibrational mode, thereby greatly improving signal intensity and acquisition speeds over spontaneous Raman scattering. As a non-linear imaging modality employing pulsed excitation sources, CARS benefits from excellent axial resolution and diffraction-limited, sub-micron spatial resolution35 without the need for dyes, reporters or chemical processing steps, while additionally providing unique biochemical information regarding the molecular vibrational environment of the sample. Briefly, CARS involves a four wave-mixing process requiring the interaction of a ‘pump’ excitation beam at a frequency ωp and a Stokes beam at frequency ωs. When the frequency difference between ωp and ωs is tuned to match the frequency of a particular Raman active molecular vibration, the resonant oscillators in the sample are coherently driven by the non-linear combination of the excitation fields εp and εs, generating a blue-shifted anti-Stokes signal given by ωas = 2ωp − ωs. The most common way of generating a CARS signal is via a degenerate scheme, whereby the pump excitation field serves as both the pump and the probe simultaneously, whereby the intensity of the anti-Stokes signal is proportional to the square of the number of available vibrational oscillators in a sample. The lipid-rich myelin sheath is well suited for study using CARS by exciting the strong vibrational modes in the high wavenumber CH (carbon–hydrogen) stretching region located between 2800 and 3100 cm−1, well suited for targeting the long acyl chains that comprise myelin lipids. Most studies to date on either peripheral nerves36–40 or the central nervous system41–50 have focused on single frequency imaging for maximum intensity and contrast, whereby a single wavenumber is targeted, typically the strong CH2 symmetric stretch at ∼2850 cm−1. While this method provides label-free images with good signal to noise ratio, it does not provide any additional information regarding the biochemical state of the sample beyond intensity fluctuations suggestive of gain or loss of lipid with respect to the CH2 symmetric stretch and observable morphological variations.51 Conversely, some instruments have prioritized capturing spectral content by using spectrometers and frame grabbers, however, they require multivariate data extraction or mathematical transforms to re-construct images.52,53 Additionally, their forward propagating detection configuration presents challenges in analyzing thick whole mount or in vivo samples. Our spectral adaptation of CARS (sCARS) involves temporally stretching two high peak energy femtosecond pulses via a tuneable, glass-induced54 spectral focusing setup, whose combined spectral bandwidth and resolution are able to coherently drive a large range of vibrational modes in a sample. This allows for the label-free, high-spatial resolution obtainable by standard CARS imaging but with additional spectral selectivity and the generation of spectral data for each pixel of the acquired image (Fig. 1).
 | ||
| Fig. 1 Simplified schematic diagram of sCARS instrumental setup showing fully tuneable dual outputs and an epi-detection system allowing the imaging of various of sample types. | ||
Vibrational spectroscopy to study aspects of MS has largely focused on animal models such as experimental autoimmune encephalomyelitis48,49 or models of artificially-induced demyelination and remyelination.41,55 However, no single model provides a complete picture of MS and few faithfully mimic the progressive phenotype of MS, which is important given that the majority of relapsing-remitting MS patients transition into secondary progressive (SP) disease with time. Many of these models also fail to address the underlying degenerative component that may be an important driver of the disease. To this end, a more complete understanding of MS, especially of the progressive phases, is better obtained through direct analysis of human MS brain samples.
Here we utilise a novel broadband spectral CARS (sCARS) microscope to analyze post-mortem human MS brain tissue. These proof-of-principle results demonstrate patient-specific biochemical lipid changes in myelin in areas surrounding lesions, and importantly, we show subtle but widespread abnormalities of myelin lipids in regions far removed from demyelinating plaques. These observations shed light on the extensive biochemical abnormalities affecting myelin in MS brain that cannot be detected by conventional means, and suggest that such changes may reflect the earliest events eventually leading to inflammatory demyelination.
Materials and methods
Tissue preparation
Human tissue was obtained from The Netherlands Brain Bank (Table S1†). Autopsies were performed with a post-mortem delay of no more than 24 hours. Samples were obtained from four patients with chronic secondary progressive MS. Initial coronal hemispheric slices 10 mm thick were cut from the brain and the corpora callosa were dissected using MRI assisted orientation.56 Samples were fixed in neutral buffered formalin and transferred to 30% sucrose for cryo-protection and subsequent flash freezing. Samples were cryo-sectioned at 100 μm and maintained free-floating until mounting onto positively charged slides (Menzel-Gläser) for staining and imaging. Region-matched control brain samples were also obtained from two controls with non-neurological disease and processed identically.Immunohistochemistry
Immunohistochemical staining was performed as described previously.57 Briefly, sections were incubated with primary antibodies targeting the major myelin protein PLP (proteolipid protein – Serotec, Oxford, UK) and the cell surface receptor HLA-DR (human leukocyte antigen) for activated microglia. Sections were then incubated with Alexa Fluor® 488 labeled goat anti-mouse immunoglobulins (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :400) (Invitrogen, Paisley, UK) or DAB chromogen. All stained sections were cover-slipped and graded by a neuropathologist. White matter lesions, grey matter boundaries and NAWM areas were marked for analysis as shown in Fig. 3. Adjacent unstained sections for sCARS imaging were mounted on glass slides and kept frozen at −20 °C until analysis.
:400) (Invitrogen, Paisley, UK) or DAB chromogen. All stained sections were cover-slipped and graded by a neuropathologist. White matter lesions, grey matter boundaries and NAWM areas were marked for analysis as shown in Fig. 3. Adjacent unstained sections for sCARS imaging were mounted on glass slides and kept frozen at −20 °C until analysis.
 | ||
| Fig. 2 Demonstration of the spectral CARS (sCARS) capabilities of our instrument and broadband spectral vibrational imaging on a mixture of polymer microspheres and biological samples. (a) Polyethylene targeted at 2883 cm−1, (b) polymethyl methacrylate (PMMA) targeted at 2953 cm−1 (c), polystyrene targeted at 3054 cm−1, and (d) a pseudo-coloured spectral image of the entire 2800–3100 cm−1 wavenumber region showing the three distinct polymer types and a green background originating from the Sylgard® 184 silicone elastomer mounting media, which has a sharp peak around 2915 cm−1 (e) sCARS image of mouse adipocyte cells as an example of a randomly ordered lipid structure (f) protein-rich silkworm (Bombyx) silk (g) sCARS image of ex vivo mouse dorsal root myelin as an example of an ordered lipid structure (h) spectra derived from the sCARS image of polymer microspheres and biological samples respectively and the rainbow lookup table used to colorize the images (scale bars = 20 μm). | ||
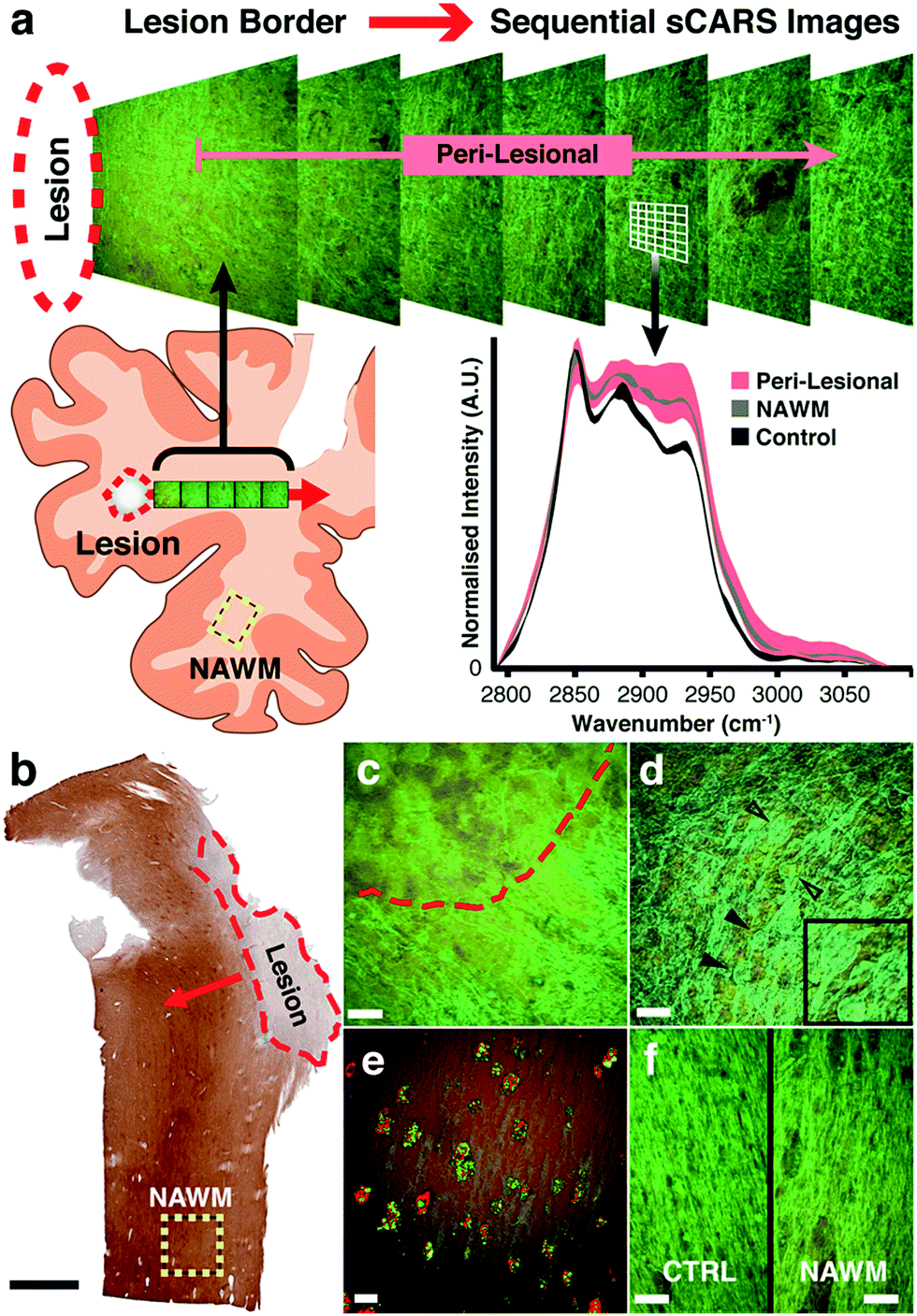 | ||
| Fig. 3 (a) Illustration of the sampling procedure and imaging workflow. Sequential, step-wise pseudo-coloured sCARS images were taken beginning at the border of a demyelinated lesion and moving outward. sCARS spectra were extracted from each pixel of the images for further analysis. As an example, an overlay plot of spectra is shown from the peri-lesional sequence, normal appearing white matter (NAWM) and control areas showing their respective distributions. (b) Example of an MS section immunostained for proteolipid protein (PLP). Red dashed area represents a demyelinated plaque; red arrow shows outward direction of spectral image sampling; yellow dashed square represents area chosen for obtaining ‘normal-appearing’ white matter data. Black scale bar = 2 mm. (c) Pseudo-coloured sCARS spectral image taken at the border or a lesion with gliotic demyelinated tissue above red dashed line, and partially demyelinated tissue below it. (d) Characteristic axonal spheroids (black arrowheads) found throughout a diffuse lesion. Inset: magnification of axonal spheroids. (e) Activated microglia within lesion rim stained for HLA-DR (red) containing digested myelin/lipid residue shown with sCARS (green) (f) side-by-side comparison of pseudo-coloured sCARS images showing the morphological similarities between a region-matched healthy control sample and ‘normal appearing’ white matter. All white scale bars = 20 μm. | ||
sCARS system
The sCARS system is driven by an amplified ytterbium-doped femtosecond fiber laser (Clark-MXR – MI, USA) producing 100 femtosecond pulses at a 1 MHz repetition rate with 10 μJ pulse energies. This laser pumps a custom built dual non-collinear optical parametric amplifiers (Clark-MXR – MI, USA) providing three outputs, two with a tuning range of 650 to 1300 nm and the residual fixed at 1040 nm. For the current experiments, outputs were tuned to 750 nm and 963 nm for pump and Stokes respectively to target the CH wavenumber region. Both pulses were chirped using custom-designed multi-pass S-TIH53 glass blocks (BMV Optical Technologies – Ottawa, Canada). In combination with the high-energy pulses a spectral resolution below 1 cm−1 is theoretically achievable.58 To ensure optimal spectral focusing, the excitation beams have to be equally stretched based on their initial transform limited pulse durations. Therefore pump and Stokes beams travelled through 450 mm and 650 mm of glass respectively, which stretched the pulses to ≈6.9 ps and 2.8 ps (ref. 59) providing a spectral resolution of ≈15 cm−1 to best match the Raman line widths of the CH wavenumber region. This allowed us to scan a 300 wavenumber range (2800–3100 cm−1) by manipulating the temporal overlap and instantaneous frequency interaction between the two pulses through the use of an optical delay line and stepper stage. The beams were coupled into a C1 galvanometer scan head (Nikon – Tokyo, Japan). Pump and Stokes beams passed through a 700 nm long pass dichroic filter (Semrock – NY, USA) and were focused at the sample by a 25 × 1.1NA water immersion objective (Nikon – Tokyo, Japan). Emission was split by a secondary 600 nm short pass dichroic filter (BMV Optical) and collected by a pair of photomultiplier tubes (Hamamatsu City – Hamamatsu, Japan) (CARS > 600 nm and two-photon excitation fluorescence, second harmonic generation < 600 nm). Bandpass filters (Semrock – NY, USA) of 624/40 nm and 535/70 nm respectively precede the detectors for additional signal cleanup. 512 × 512 images were acquired with 2 μs pixel dwell times and a field of view of ≈170 × 170 μm. All hardware interfaces were controlled using LabVIEW (National Instruments – Austin TX, USA) except image scanning, which was controlled using native EZ-C1 software (Nikon – Tokyo, Japan). Calibration was carried out using a mixture of polymer microspheres: 20 μm polyethylene, 11 μm polymethyl methacrylate (Cospheric LLC – Santa Barbara CA, USA), and 15 μm FocalCheck™ polystyrene beads (Molecular Probes – Eugene OR, USA) mounted on a glass slide using Sylgard® 184 silicone elastomer (Dow Corning – Midland MI, USA).Data processing and image analysis
Spectral CARS images were analysed using a combination of custom software written by P. K. Stys and Matlab (Mathworks, 2014a). Briefly, spectral images were obtained in the same fashion as a fluorescence lambda stack by targeting each wavenumber of the CH region sequentially. Through this method, 101 images were obtained containing per-pixel CARS spectral information. These images were then collated into a single image and pseudo-coloured to reflect shape and intensity of the CARS spectrum at each pixel. For analysis, spectra were only normalised. For display, spectra were smoothed using binomial filtering (single-pass), normalised and baseline removed using a cubic spline fit.Results and discussion
Broadband spectral biochemical imaging
To illustrate the capability of our instrument to resolve chemical differences using sCARS microscopy we imaged a mixture of microspheres composed of three different polymers: polyethylene (PE), polymethyl methacrylate (PMMA) and polystyrene (PS). These materials have distinctive and invariant Raman spectra that also serve to calibrate the instrument. By imaging the entire spectral region between 2800–3100 cm−1 we were able to construct a spectrum for each pixel of the image, differentiating each polymer in the mixture by its distinctive vibrational peaks (Fig. 2a–d). By combining a spectral series of images encompassing the entire CH region, an sCARS image can be acquired where each pixel contains a spectrum ranging from 2800–3100 cm−1. sCARS images were color-coded by intensity and spectral content. With more complex samples such as biological tissue, these high-resolution pseudo-colored images allowed simultaneous display of both morphology and chemistry. Several examples of sCARS images from biological samples are shown in Fig. 2e–g. Mouse adipocytes and myelin, while both lipid rich, produce very different sCARS spectra due to different lipid compositions22 and the structural assembly of the lipid molecules. As an example of pure protein imaging, an sCARS image of dewaxed, protein-rich silk fibers from the Bombyx silkworm is shown in Fig. 2f.The respective spectra extracted from each image show the high spectral resolution achieved by our instrument, ranging from the distinct spectra of the polymers (Fig. 2h – left) to the clear differences seen between the various types of lipid-rich structures and proteinaceous sample (Fig. 2h – right). Such spectral specificity allows the pinpointing of subtle molecular changes and lipid packing order in images at diffraction-limited resolution.
sCARS imaging of secondary progressive multiple sclerosis tissue
We then applied our technique to study biochemical lipid changes in human secondary progressive MS brains. Guided by PLP immunostained parallel sections (Fig. 3b and S1†) a series of sCARS images was recorded from both MS and control human brain. For MS tissue, the workflow consisted of sequentially acquiring adjacent images moving outward from the border of demyelinated focal lesions (Fig. 3a). With a field of view of 170 × 170 μm, at least 10 sCARS overlapping images were taken, extending out to >1.53 mm from the lesion for samples A, B and C. Sample D (due to the nature of the lesion outlined below) was sampled 16 times every 500 μm. For control tissue, a triplicate of images were taken alternately from Ctrl1 or Ctrl2. Based on spectrally colour-coded sCARS images, the peri-lesional image sequence exhibited a noticeable color gradient moving outward away from the lesion in some samples, suggesting changes in the lipid vibrational signature. The next step involved extraction of pixel-by-pixel spectra taken from myelin-specific regions while contributions from cell bodies and the extracellular matrix were omitted based on distinct spectral signatures. Classic MS lesions exhibit areas of focal demyelination and concomitant inflammation resulting in variable degrees of axonal damage and astrocytic scarring. While these lesions are histologically obvious, our aim was to investigate biochemical changes in myelin of surrounding peri-lesional and histologically grossly normal areas to detect subtle abnormalities. While the lesions in samples A, B and C were completely demyelinated with distinct borders (Fig. 3c), sample D had many spared myelinated axonal tracts within the lesion that also featured a very diffuse border. Throughout this sample, numerous axonal spheroids of various sizes were also seen (Fig. 3e). As our focus was on myelin lipids, analysis in this particular sample began in the approximate centre of the lesion and continued outwards past the diffuse border. Our instrument was also able to simultaneously collect other non-linear imaging modalities; for example, immunostained microglia along the rim of a lesion were imaged by two-photon excited fluorescence while the ingested myelin lipid debris contained within was simultaneously imaged with sCARS (Fig. 3e). Spectral images were also recorded from an area on the same respective section that morphologically exhibited no overt demyelination or gliosis (yellow dashed square – Fig. 3b), which we termed ‘normal appearing’ white matter (NAWM). For controls, we chose region-matched tissue samples derived from patients with non-neurological causes of death. With standard histological stains, NAWM is morphologically similar to control samples. Pseudo-coloured sCARS images of NAWM and control samples, while also morphologically similar, exhibited a minor colour difference (Fig. 3f) suggesting spectral differences. Spectra obtained from peri-lesional areas for samples A, B and C showed substantial heterogeneity, suggesting significant variation in the underlying myelin lipid biochemistry (Fig. S2†). Conversely, the collection of peri-lesional spectra from sample D were more homogeneous but were very different to controls. The general observation in all samples was one of increased asymmetric CH2 stretch and CH3 chain end symmetric stretch (2880 cm−1 and 2935 cm−1 respectively) in diseased regions (Fig. S2†), compared to non-MS controls. Most intriguing was the finding that all four samples exhibited abnormal spectral signatures in NAWM areas, being distinctly different from region-matched control samples (Fig. S2†).Spectral scatter analysis for classification of pathological changes
For highly ordered lipids in biological membranes such as myelin, the measurement of intensity ratios in the CH stretching modes has been a classical way to observe conformational disorder,23,24 especially after exogenous insult. However, this method only deals with raw intensity changes at single wavenumbers, and does not take into account possible subtle Raman shifts, broad shoulders and overall spectral curve shape. Having access to the entire CH wavenumber region at each pixel (up to 262144 spectra for a 512 × 512 image), myelin lipid spectra were analyzed using a custom algorithm to consider any arbitrary spectral changes occurring in the entire CH wavenumber region. By bracketing relevant myelin lipid spectra between a control spectrum and the most pathologically abnormal myelin spectrum, a score reflecting biochemical changes was calculated. Fig. 4 outlines the data analytical process beginning with the sCARS images of peri-lesional areas, NAWM and non-neurological controls. For analysis, only myelinated axons were considered. Pixels were selected and visually masked (in blue) based on spectral similarity using a reference spectrum and least square difference of spectral similarities in the image with selectable tolerances and intensity thresholds to eliminate dim features of the image (Fig. 4a and b). Next, all relevant spectra were extracted from each image then classified and scored using a set of defined bracketing spectral parameters whereby control = 0 and lesion/pathological = 1 (Fig. 4c). This was accomplished using a non-linear transformation based on a single scalar value that was passed as a fitting coefficient to a standard Levenberg–Marquardt non-linear least squares curve fitting algorithm. The optimized coefficient returned by the fitting represented the “pathological score”. The results were then displayed in a two-dimensional kernel density scatter plot of integrated signal intensity on the y-axis vs. pathological score on the x-axis. Fig. 4d shows a combined kernel density plot from the analysis of sample C with a cluster created from the spectra of 17 overlaid sCARS images (11 peri-lesional, 3 NAWM and 3 control). By correlating with pixel positions on the sCARS micrographs, clusters belonging to peri-lesional areas, NAWM and controls are highlighted, showing distinct populations for each group. Next we plotted a histogram of each individual cluster/image showing the distribution of their pathological scores to further extract any biochemical variations, with focus on the peri-lesional sCARS sequence of images beyond the lesion rim. Fig. 4e shows the cumulative distribution function of sample C, where the spectral clusters belonging to peri-lesional images fall within the gated spectral parameters of ‘control’ and ‘lesion/pathological’. Interestingly the NAWM clusters were different from control samples, indicating abnormality of lipid biochemistry. The final step involved plotting the pathological scores of each spectral cluster taken at 95% of the normalized frequency distribution. With this, each spectral cluster is now represented by a scalar index representing severity of disease. By arranging the x-axis by distance from the lesion border the resulting plot shows a clear pathological gradient as one moved away from the lesion (Fig. 4f). Running this data analysis on the SPMS tissue showed clear differences in distributions of kernels in the density plots (Fig. 5 – first row). Distribution histograms analyzed by cluster/image showed further differences between samples (Fig. 5 – second row), which were further highlighted when plotting the pathological scores at the 95th percentile (Fig. 5 – third row). Individually, samples A to C have a similar pattern of decreasing pathology moving away from the lesion, however subtle differences in pathological scores are seen at various locations along their peri-lesional sequence. With its mostly still-myelinated lesion featuring an abundance of axonal spheroids and a diffuse rim, sample D showed high pathological scores or disease state that remained unchanged before and after passing through the diffuse border of the lesion. The one constant observed in all samples was an abnormal spectral signature for NAWM compared to non-neurological controls. | ||
| Fig. 4 Data analysis procedure: (a) typical sCARS image showing myelinated axons in green. (b) For analysis, only myelinated axons are considered. A blue spectral selection mask is applied based on spectral curve similarity. (c) Supervised classification of all relevant spectra using defined bracketing spectral parameters: control score = 0 and lesion/pathological score = 1. (d) Kernel density scatter plot of intensity vs. spectral index including peri-lesional series, NAWM and controls. Each sCARS image = 1 cluster. This example contains 17 clusters. (e) Cumulative distribution functions plotting the normalized frequency of pathological scores for each spectral image. Each line represents a certain distance away from the lesion border (peri-lesional) unless otherwise indicated. Dashed line indicates score 95% threshold of normalized distribution. (f) Bar graph plotting ‘Pathological Score’ of each image cluster taken at 95% of normalized frequency arranged by distance from lesion (peri-lesional) compared to averaged NAWM and control samples. Red dashed line represents lesion border. | ||
 | ||
| Fig. 5 Results of spectral scatter analysis (SSA) on SPMS tissue with patient samples arranged by column. First row: Kernel density scatter plot of all relevant spectra as classified by bracketing parameters. Rectangles show groupings of peri-lesional, NAWM and control clusters. Second row: normalised frequency histogram of each sCARS image by pathological score. The sCARS image closest to lesion, NAWM and control areas are highlighted. Third row: plots of the 95th percentile of kernels by pathological score arranged by distance from lesion. | ||
Discussion
Although the definitive cause of MS is unknown, the prevailing theory implicates an aberrant peripheral immune system prompting myelin-reactive T lymphocytes to enter the CNS, attack myelin, and result in multifocal inflammatory demyelination.60,61 This immune-centric approach has driven development of a number of therapies, which have proven highly effective at reducing inflammatory lesions.62,63 However, most MS patients assume a progressive course after several decades, and immune-suppressive agents offer little benefit in this stage, where most irreversible disability accrues.4,5,8 This has raised questions about the completeness of the immune-centric model, leading to an alternative hypothesis whereby a primary degeneration of CNS white matter secondarily entrains recurrent inflammatory demyelinating attacks; later, once immune deregulation subsides with age, the progressive degeneration continues unmasked.5,20,64 Novel more sensitive methods of detecting biochemical white matter pathology, before overt demyelination, and in the absence of inflammatory infiltration, could help unravel fundamentally important questions regarding MS pathogenesis. Using our sCARS system we directly targeted the inherent molecular vibrational signatures of the various lipids that are major components of myelin by probing the carbon-hydrogen (CH) spectral region.This CH region (2800–3010 cm−1) is assigned to the stretching modes of CH vibrations and H–C–H deformation overtones67 and is commonly used to probe the ordering of the long hydrocarbon chains of lipids (Table 1). The interplay between the various vibrational modes in this region is extremely sensitive to lipid molecular packing and system perturbation causing disorder both within and between molecules.23,68–70 With the high sensitivity of the technique to various aspects of lipid abnormalities and packing disorder, we intentionally focused on areas beyond conventional (by standard neuropathology) MS white matter lesions, particularly NAWM, representing regions far removed from demyelinated plaques.
| Raman shift (cm−1) | Vibrational assignment |
|---|---|
| 2845 | CH2 symmetric stretch |
| 2886 | CH2 asymmetric stretch/Fermi resonance CH2 stretch |
| 2935 | CH3 chain end symmetric stretch |
| 2960 | Out-of-plane chain end asymmetric CH3 stretch/protein |
| 2970 | Cholesterol/cholesterol ester |
| 3010 | Unsaturated = CH stretch |
Detailed studies of the NAWM suggest that MS white matter is affected more broadly than previously thought.21 As noted above, studies focusing on lipid abnormalities in NAWM report notable changes6,25,26,28,29,71 suggesting that alterations in myelin lipid molecular ordering may represent important and proximal steps in the complex pathogenesis of this disorder. Currently, there are no reliable histochemical means to specifically interrogate biochemical changes in myelin lipids. Additionally, the fundamental hydrophobic nature of lipids limits the methodologies that can be used to directly probe lipid-rich structures such as myelin without use of solvents and labels that may perturb the sample. Recent comparative studies of traditional lipophilic dyes have brought their specificity into question.32,33 To circumvent these issues, Raman spectroscopy and in particular, coherent anti-Stokes Raman scattering (CARS), has been proposed as an attractive alternative to probe the molecular constituents of myelin by interrogating the vibrational and rotational modes of molecules and their chemical bonds within the sample. The measurement is based on the interaction between the excitation light and the intrinsic molecular bonds in a given system, requiring no exogenous labels or sample treatment (other than simple fixation). Moreover, CARS is extremely sensitive to vibrational changes both within and between molecules.
Extraction and analysis of the molecular vibrational data afforded by our technique involved the parsing of every relevant spectral pixel of the complete CH wavenumber region through a classification system that provided a scalar index or pathological ‘score’. Through systematic analysis we show that peri-lesional white matter is biochemically abnormal in terms of lipid disorder, with such abnormalities sometimes extending for large distances (e.g. sample D, Fig. S1D†). The patterns of geographic abnormality were lesion and/or patient specific, suggesting that our technique could discern subtle biochemical pathology, revealing heterogeneity among subjects that cannot be revealed by traditional histological techniques. Importantly, all measures of NAWM far removed from lesions exhibited consistent lipid biochemical abnormalities compared to healthy controls, underscoring the notion that MS plaques detected by conventional pathology represent only the most obvious manifestation of a much more widespread CNS pathology in this disorder.
Conclusions
Unlike other techniques (e.g. MRI, bulk tissue biochemical analysis), sCARS provides chemically-specific information from biological tissue at a high spatial and spectral resolution, without requiring extrinsic probes or labels. The use of our spectral scatter analysis classification system provided an efficient way to collate, display and quantify the very large spectral datasets generated by sCARS. The method also provided a simplified quantifier through the assignment of a scalar index or ‘score’ to any region, generating a pathological grading index. Such an index may prove very useful in providing a more objective measure of pathology over the arbitrary nature of conventional staining techniques and visual inspection, while increasing the sensitivity and specificity of MS histopathology. Our preliminary data demonstrate how sCARS may be a powerful adjunct to conventional pathological examination of biological tissues, yielding information about subtle biochemical pathology in the absence of overt morphological abnormalities, which may further our fundamental understanding of pathogenesis. As it concerns human MS, our sCARS data suggest that early widespread biochemical abnormalities of myelin lipids may be an important early degenerative change, that could further promote autoimmune inflammation by providing a chronic source of biochemically modified antigenic lipids. Beyond multiple sclerosis, the sensitivity of sCARS to lipid changes has the potential to be applied to investigate a broad range of neurological disorders where lipid abnormalities play a pathophysiological role. Examples include CNS disorders such as various leukodystrophies, neuromyelitis optica, acute disseminated encephalomyelitis, PNS disorders such as Charcot–Marie–Tooth disease, Guillain–Barré syndrome and even towards tracking remyelination and repair treatments for traumatic spinal cord injuries.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported in part by: Leblanc Spine Chair, Canadian Institutes of Health Research, Alberta Innovates Health Solutions, Multiple Sclerosis Society of Canada, Canada Research Chairs.Notes and references
- L. M. D. Steinman, Cell, 1996, 85, 299–302 CrossRef CAS PubMed.
- Multiple Sclerosis International Federation, Atlas of MS, 2013, http://www.msif.org/wp-content/uploads/2014/09/Atlas-of-MS.pdf Search PubMed.
- B. D. Trapp and P. K. Stys, Lancet Neurol., 2009, 8, 280–291 CrossRef CAS PubMed.
- P. K. Stys, G. W. Zamponi, J. van Minnen and J. J. G. Geurts, Nat. Rev. Neurosci., 2012, 13, 507–514 CrossRef CAS PubMed.
- B. D. Trapp and K.-A. Nave, Annu. Rev. Neurosci., 2008, 31, 247–269 CrossRef CAS PubMed.
- W. T. Norton and W. Cammer, in Myelin, ed. P. Morell, Springer US, Boston, MA, 1984, pp. 369–403 Search PubMed.
- A. Scalfari, A. Neuhaus, A. Degenhardt, G. P. Rice, P. A. Muraro, M. Daumer and G. C. Ebers, Brain, 2010, 133, 1914–1929 CrossRef PubMed.
- A. Scalfari, A. Neuhaus, M. Daumer, P. A. Muraro and G. C. Ebers, J. Neurol., Neurosurg. Psychiatry, 2013, 85, 67–75 CrossRef PubMed.
- F. D. Lublin, S. C. Reingold, J. A. Cohen, G. R. Cutter, P. S. Sorensen, A. J. Thompson, J. S. Wolinsky, L. J. Balcer, B. Banwell, F. Barkhof, B. Bebo, P. A. Calabresi, M. Clanet, G. Comi, R. J. Fox, M. S. Freedman, A. D. Goodman, M. Inglese, L. Kappos, B. C. Kieseier, J. A. Lincoln, C. Lubetzki, A. E. Miller, X. Montalban, P. W. O'Connor, J. Petkau, C. Pozzilli, R. A. Rudick, M. P. Sormani, O. Stuve, E. Waubant and C. H. Polman, Neurology, 2014, 83, 278–286 CrossRef PubMed.
- J. H. Noseworthy, C. Lucchinetti, M. Rodriguez and B. G. Weinshenker, N. Engl. J. Med., 2000, 343, 938–952 CrossRef CAS PubMed.
- J. J. G. Geurts, P. K. Stys, A. Minagar, S. Amor and R. Zivadinov, J. Neurol. Sci., 2009, 282, 12–20 CrossRef PubMed.
- H. Lassmann, J. van Horssen and D. Mahad, Nature Publishing Group, 2012, 8, 647–656 CAS.
- C. Confavreux and S. Vukusic, Brain, 2006, 129, 606–616 CrossRef PubMed.
- J. M. Charcot, Histologie de la sclerose en plaques, 1868, vol. 41 Search PubMed.
- V. L. Stevenson, G. J. Parker, G. J. Barker, K. Birnie, P. S. Tofts, D. H. Miller and A. J. Thompson, J. Neurol. Sci., 2000, 178, 81–87 CrossRef CAS PubMed.
- M. A. van Buchem, J. C. McGowan, D. L. Kolson, M. Polansky and R. I. Grossman, Magn. Reson. Med., 1996, 36, 632–636 CrossRef CAS PubMed.
- M. A. Horsfield, M. Lai, S. L. Webb, G. J. Barker, P. S. Tofts, R. Turner, P. Rudge and D. H. Miller, Magn. Reson. Med., 1996, 36, 393–400 CrossRef CAS PubMed.
- N. M. Moll, A. M. Rietsch, S. Thomas, A. J. Ransohoff, J.-C. Lee, R. Fox, A. Chang, R. M. Ransohoff and E. Fisher, Ann. Neurol., 2011, 70, 764–773 CrossRef PubMed.
- M. Filippi, M. A. Rocca, G. Martino, M. A. Horsfield and G. Comi, Ann. Neurol., 1998, 43, 809–814 CrossRef CAS PubMed.
- S. K. Ludwin and J. Neuropathol, Exp. Neurol., 2006, 65, 305–318 Search PubMed.
- I. V. Allen, S. McQuaid, M. Mirakhur and G. Nevin, Neurol. Sci., 2001, 22, 141–144 CrossRef CAS PubMed.
- J. A. Benjamins, A. K. Hajra and B. W. Agranoff, in Basic Neurochemistry, ed. G. J. Siegel, R. W. Albers, S. T. Brady and D. L. Price, Basic Neurochemistry, edn. 7, 2006, pp. 33–50 Search PubMed.
- M. R. Bunow and I. W. Levin, Biochim. Biophys. Acta, Lipids Lipid Metab., 1977, 487, 388–394 CrossRef CAS.
- M. Pézolet and D. Georgescauld, Biophys. J., 1985, 47, 367–372 CrossRef.
- B. Gerstl, L. F. Eng, M. Tavaststjerna, J. K. Smith and S. L. Kruse, J. Neurochem., 1970, 17, 677–689 CrossRef CAS PubMed.
- J. N. Cumings and H. Goodwin, Lancet, 1968, 292, 664–665 CrossRef.
- C. Alling, M. T. Vanier and L. Svennerholm, Brain Res., 1971, 35, 325–336 CrossRef CAS PubMed.
- J. N. Cumings, Brain, 1955, 78, 554–563 CrossRef CAS PubMed.
- J. L. Kanter, S. Narayana, P. P. Ho, I. Catz, K. G. Warren, R. A. Sobel, L. Steinman and W. H. Robinson, Nat. Med., 2005, 12, 138–143 CrossRef PubMed.
- A. Diestel, O. Aktas, D. Hackel, I. Hake, S. Meier, C. S. Raine, R. Nitsch, F. Zipp and O. Ullrich, J. Exp. Med., 2003, 198, 1729–1740 CrossRef CAS PubMed.
- R. M. Lycette, W. F. Danforth, J. L. Koppel and J. H. Olwin, Stain Technol., 1970, 45, 155–160 CrossRef CAS PubMed.
- K. Yen, T. T. Le, A. Bansal, S. D. Narasimhan, J.-X. Cheng and H. A. Tissenbaum, PLoS One, 2010, 5, e12810 Search PubMed.
- T. Hellerer, C. Axäng, C. Brackmann, P. Hillertz, M. Pilon and A. Enejder, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 14658–14663 CrossRef CAS PubMed.
- C. H. Camp Jr and M. T. Cicerone, Nat. Photonics, 2015, 9, 295–305 CrossRef.
- J.-X. Cheng and X. S. Xie, J. Phys. Chem. B, 2004, 108, 827–840 CrossRef CAS.
- F. P. Henry, D. Côté, M. A. Randolph, E. A. Z. Rust, R. W. Redmond, I. E. Kochevar, C. P. Lin and J. M. Winograd, Plast. Reconstr. Surg., 2009, 123, 123S–130S CrossRef CAS PubMed.
- E. Belanger, F. P. Henry, R. Vallee, M. A. Randolph, I. E. Kochevar, J. M. Winograd, C. P. Lin and D. Cote, Biomed. Opt. Express, 2011, 2, 2698–2708 CrossRef CAS PubMed.
- T. B. Huff and J. X. Cheng, J. Microsc., 2007, 225, 175–182 CrossRef CAS PubMed.
- Y. Jung, J. H. Ng, C. P. Keating, P. Senthil-Kumar, J. Zhao, M. A. Randolph, J. M. Winograd and C. L. Evans, PLoS One, 2014, 9, e94054 Search PubMed.
- G. de Vito, I. Tonazzini, M. Cecchini and V. Piazza, Opt. Express, 2014, 22, 13733–13743 CrossRef CAS PubMed.
- Y. Fu, H. Wang, T. B. Huff, R. Shi and J.-X. Cheng, J. Neurosci. Res., 2007, 85, 2870–2881 CrossRef CAS PubMed.
- Y. Shi, D. Zhang, T. B. Huff, X. Wang, R. Shi, X.-M. Xu and J.-X. Cheng, J. Biomed. Opt., 2011, 16, 106012 Search PubMed.
- H. Wang, Y. Fu, P. Zickmund, R. Shi and J.-X. Cheng, Biophys. J., 2005, 89, 581–591 CrossRef CAS PubMed.
- Y. Shi, S. Kim, T. B. Huff, R. B. Borgens, K. Park, R. Shi and J.-X. Cheng, Nat. Nanotechnol., 2009, 5, 80–87 CrossRef PubMed.
- C.-S. Liao and J.-X. Cheng, Annu. Rev. Anal. Chem., 2016, 9, 69–93 CrossRef CAS PubMed.
- E. Belanger, S. Begin, S. Laffray, Y. De Koninck, R. Vallee and D. Cote, Opt. Express, 2009, 17, 18419–18432 CAS.
- Y. Shi, W. Sun, J. J. McBride, J.-X. Cheng and R. Shi, J. Neurochem., 2011, 117, 554–564 CrossRef CAS PubMed.
- J. Imitola, D. Co té, S. Rasmussen, X. S. Xie, Y. Liu, T. Chitnis, R. L. Sidman, C. P. Lin and S. J. Khoury, J. Biomed. Opt., 2011, 16, 021109 CrossRef PubMed.
- Y. Fu, T. J. Frederick, T. B. Huff, G. E. Goings, S. D. Miller and J.-X. Cheng, J. Biomed. Opt., 2011, 16, 106006 CrossRef PubMed.
- G. de Vito, A. Bifone and V. Piazza, Opt. Express, 2012, 20, 29369 CrossRef CAS PubMed.
- Y. Fu, T. B. Huff, H.-W. Wang, H. Wang and J.-X. Cheng, Opt. Express, 2008, 16, 19396–19409 CrossRef CAS PubMed.
- Y. Ozeki, W. Umemura, Y. Otsuka, S. Satoh, H. Hashimoto, K. Sumimura, N. Nishizawa, K. Fukui and K. Itoh, Nat. Photonics, 2012, 6, 1–7 CrossRef.
- C. H. Camp Jr, Y. J. Lee, J. M. Heddleston, C. M. Hartshorn, A. R. H. Walker, J. N. Rich, J. D. Lathia and M. T. Cicerone, Nat. Photonics, 2014, 8, 627–634 CrossRef CAS PubMed.
- A. F. Pegoraro, A. Ridsdale, D. J. Moffatt, Y. Jia, J. P. Pezacki and A. Stolow, Opt. Express, 2009, 17, 2984 CAS.
- Y. Fu, W. Sun, Y. Shi, R. Shi and J.-X. Cheng, PLoS One, 2009, 4, e6705 Search PubMed.
- A. Seewann, E.-J. Kooi, S. D. Roosendaal, F. Barkhof, P. van der Valk and J. J. G. Geurts, Acta Neurol. Scand., 2009, 119, 349–355 CrossRef CAS PubMed.
- V. Popescu, R. Klaver, P. Voorn, Y. Galis-de Graaf, D. L. Knol, J. W. R. Twisk, A. Versteeg, G. J. Schenk, P. van der Valk, F. Barkhof, H. E. de Vries, H. Vrenken and J. J. G. Geurts, Mult. Scler. J., 2015, 21, 1280–1290 CrossRef CAS PubMed.
- W. Langbein, I. Rocha-Mendoza and P. Borri, J. Raman Spectrosc., 2009, 40, 800–808 CrossRef CAS.
- V. Piazza, G. de Vito, E. Farrokhtakin, G. Ciofani and V. Mattoli, PLoS One, 2016, 11, e0156371 Search PubMed.
- E. M. Frohman, M. K. Racke and C. S. Raine, N. Engl. J. Med., 2006, 354, 942–955 CrossRef CAS PubMed.
- A. Compston and A. Coles, Lancet, 2008, 372, 1502–1517 CrossRef CAS.
- O. Aktas, B. Kieseier and H.-P. Hartung, Trends Neurosci., 2010, 33, 140–152 CrossRef CAS PubMed.
- M. S. Dirk Fitzner, Curr. Neuropharmacol., 2010, 8, 305 CrossRef PubMed.
- D. Wheeler, V. V. R. Bandaru, P. A. Calabresi, A. Nath and N. J. Haughey, Brain, 2008, 131, 3092–3102 CrossRef PubMed.
- C. Lee and C. D. Bain, Biochim. Biophys. Acta, Lipids Lipid Metab., 2005, 1711, 59–71 CrossRef CAS PubMed.
- C. Krafft, L. Neudert, T. Simat and R. Salzer, Spectrochim. Acta, Part A, 2005, 61, 1529–1535 CrossRef PubMed.
- S. P. Verma and D. F. H. Wallach, Biochim. Biophys. Acta, Lipids Lipid Metab., 1977, 486, 217–227 CrossRef CAS.
- S. P. Verma, D. F. H. Wallach and J. D. Sakura, Biochemistry, 1980, 19, 574–579 CrossRef CAS PubMed.
- N. Yellin and I. W. Levin, Biochim. Biophys. Acta, 1977, 489, 177–190 CrossRef CAS.
- N. V. Surovtsev and S. A. Dzuba, J. Chem. Phys., 2014, 140, 235103 CrossRef CAS PubMed.
- M. L. Cuzner and W. T. Norton, Brain Pathol., 1996, 6, 231–242 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc03992a |
| This journal is © The Royal Society of Chemistry 2018 |