
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFemtosecond stimulated Raman evidence for charge-transfer character in pentacene singlet fission†
Stephanie M.
Hart
,
W. Ruchira
Silva
and
Renee R.
Frontiera
 *
*
Department of Chemistry, University of Minnesota, Minneapolis, MN 55455, USA. E-mail: rrf@umn.edu
First published on 20th December 2017
Abstract
Singlet fission is a spin-allowed process in which an excited singlet state evolves into two triplet states. We use femtosecond stimulated Raman spectroscopy, an ultrafast vibrational technique, to follow the molecular structural evolution during singlet fission in order to determine the mechanism of this process. In crystalline pentacene, we observe the formation of an intermediate characterized by pairs of excited state peaks that are red- and blue-shifted relative to the ground state features. We hypothesize that these features arise from the formation of cationic and anionic species due to partial transfer of electron density from one pentacene molecule to a neighboring molecule. These observations provide experimental evidence for the role of states with significant charge-transfer character which facilitate the singlet fission process in pentacene. Our work both provides new insight into the singlet fission mechanism in pentacene and demonstrates the utility of structurally-sensitive time-resolved spectroscopic techniques in monitoring ultrafast processes.
Introduction
Singlet fission is a photophysical process in which a singlet excitation on one molecule couples to a nearby ground state molecule and is converted into two triplet excitations. Currently, extensive research is focused on exploiting this process to generate two charge-carrier pairs from the absorption of a single photon in organic photovoltaics.1,2 The Shockley–Queisser limit constrains the efficiency of single-junction photovoltaic cells to just 32%, but when materials which undergo efficient singlet fission are incorporated into these devices the theoretical limit increases to 45%.3 This drastic increase in efficiency has been demonstrated with the use of the singlet fission sensitizer pentacene and a fullerene acceptor, resulting in a solar cell with external quantum efficiency of 109%.4Unfortunately, the molecular mechanism behind singlet fission is not yet well understood, hindering further optimization of high efficiency photovoltaics. The nature of the electronic states involved in this process, particularly the role of those with charge-transfer character, has been subject to extensive debate.2 Studies suggest that vibrational motions sculpting the potential energy landscape in these systems are critical in facilitating efficient singlet fission.5,6 However, little experimental work has focused on examining these structural changes on the relevant femtosecond timescale over which the singlet fission process occurs. Ultrafast vibrational spectroscopy can be used to examine the nuclear motions driving singlet fission, thus providing insight into the complex nature of the multidimensional reaction coordinate that remains challenging to access by computational methods. A more comprehensive understanding of these nuclear coordinates will in turn play a vital role in the molecular design of singlet fission materials for highly efficient photovoltaic systems.
Following the influx of singlet fission studies in the last decade, researchers have generally agreed upon two possible mechanisms for the singlet fission process. The one-step mechanism involves conversion of the excited singlet directly to a triplet without any observable intermediate. This process can occur through a direct or mediated interaction, the former of which involves coupling strictly between the initial excited state and final state, while the latter is driven by coupling of both the excited singlet and final triplet states to a high-lying virtual charge-transfer state. While the relative importance of these directed and mediated interactions has been subject to extensive literature debate, neither involves an observable intermediate with a measurable lifetime. The two-step mechanism involves conversion of the singlet to the triplet state through a structurally distinct and measureable intermediate species with substantial charge-transfer character. Here the two singlets are converted to species with strong cationic and anionic character, which are then converted to two triplets via back-electron transfer.2
Theoretical calculations by various groups have provided support for both direct and mediated one-step mechanisms as well as direct population of an intermediate species via a two-step mechanism. However, the one-step mechanism maintains a stronger computational backing for the pentacene system. Calculations examining electronic states associated with pentacene singlet fission determined that charge-transfer states are substantially higher lying in energy than singlet or triplet states, suggesting that population of charge-transfer states does not play a significant role in mediating the singlet fission process.7,8 Furthermore, this work showed that the direct mechanism could account for the nonadiabatic coupling strengths required to achieve sub 100 fs singlet fission.7 Computational work based on Redfield theory suggests that the mediated mechanism could proceed by coupling to a charge-transfer intermediate in a superexchange mechanism,9,10 or in some cases the charge-transfer state could be directly populated as described by a two-step mechanism.11,12 Additionally, Frenkel-Holstein models have shown that coupling to a charge-transfer state is necessary to reproduce features of the pentacene electronic spectra.13 Recent theoretical work examining coupling constraints in multiple acene systems confirmed that the coupling between the singlet and charge-transfer state is stronger than that between the singlet and triplet.14 This study also established that varying contributions of both the direct and mediated mechanisms play a significant role in determining the macroscopic kinetics of singlet fission. This collective body of work highlights the fact that despite extensive theoretical effort, there still is not a strong consensus as to the mechanism describing the molecular underpinnings of the singlet fission process.
Several experiments have supplemented the plethora of theoretical studies in examining the singlet fission mechanism, and have also provided support for various mechanisms. Transient absorption studies initially determined the timescale of singlet fission in evaporated polycrystalline thin films of pentacene (∼80 fs).15 These electronic absorption studies did not find evidence for intermediate states, however difficulty in the overlapping electronic band structure provides for a complex analysis to distinguish excited state features in one-dimension. In particular, given that the absorption spectrum of the pentacene anion and cation overlaps with the absorption spectrum of the pentacene triplet state in polycrystalline samples, any clear identification of a clearly defined intermediate with a finite lifetime is hindered by these isoenergetic electronic transitions.5,15–17 Two recent ultrafast studies using time-domain impulsive Raman spectroscopy18 and 2D electronic spectroscopy5 presented evidence for the direct mechanism of singlet fission by determining that vibrational coherences created upon photoexcitation survive to the triplet state. These authors attribute these coherences to a lack of a distinct intermediate state, and while they could be explained by coupling between the singlet and a charge-transfer intermediate characteristic of a superexchange mechanism, they provide indirect support for the one-step mechanism as the authors infer that survival of vibrational coherences with well-defined phases through multiple states would be unlikely. In contrast, two-photon photoemission experiments have observed a short-lived intermediate with singlet, charge-transfer, and multiexcitonic character, thus presenting evidence for the mediated one-step mechanism.19 This work formed the basis of the quantum coherent model for the mediated mechanism in the strong coupling limit, which describes optical excitation as creating a superposition of Frenkel exciton, multiexciton, and triplet states.20 The conclusions drawn by this work concerning quantum coherences have been scrutinized by some theoretical work,14 but other calculations support photoexcitation to a state with some charge-transfer character.21 Ultrafast transient absorption studies on a variety of nanoparticles of acene derivatives22 point to an important role of charge-transfer configurations in driving efficient singlet fission. To date, the few ultrafast experiments examining singlet fission have offered contradictory conclusions concerning the distinction between the direct and mediated mechanisms. This necessitates additional experiments to elucidate the singlet fission mechanism, particularly those capable of probing structural dynamics on the ultrafast timescale.
The role of charge-transfer states, both in direct formation of a structurally distinct intermediate as well as in mediation through a virtual state, has been investigated in a number other non-pentacene singlet fission systems. The role of solvent polarity has been shown to drive the rate of singlet fission in diphenylisobenzofuran23 as well as in intramolecular singlet fission in a number of solution phase pentacene dimers.24,25 These studies suggest either coupling to a charge-transfer intermediate via a super-exchange mechanism or direct formation of an observable intermediate. Recent work by Margulies and co-workers has also presented evidence of charge-transfer states via solvent stabilization and observation of the charge-transfer state acting as trap state in terrylenediimide dimers.26 In addition, direct spectroscopic evidence of a charge-transfer intermediate has been observed in solution phase TIPS pentacene derivatives27 and terrylenediimide thin films.28 The direct observation of a charge-transfer intermediate in a number of systems undergoing fission on the picosecond timescale highlights the role of a two-step charge-transfer mechanism for efficient singlet fission, although most of these studies have been performed in solution or with coupled dimers, and the applicability to device architecture remains to be determined.
Here we use femtosecond stimulated Raman spectroscopy (FSRS) to examine the ultrafast dynamics during singlet fission in crystalline pentacene. FSRS is an ultrafast vibrational spectroscopic technique that probes the transient structural response of a photoreactive system. FSRS provides structurally-sensitivity measurements with <10 cm−1 spectral resolution and sub-picosecond temporal resolution.29 When coupled with an actinic pump to promote photoexcitation, FSRS has been used to examine ultrafast excited state dynamics in systems such as solution phase biological samples, charge-transfer complexes, and solvatochromatic dyes.30–32 FSRS has also been used to study several solid state systems including polymeric semiconductors, polystyrene beads, and dye-sensitized inorganic colloids.33–35 As compared to time-domain ultrafast Raman techniques such as that used in ref. 18, frequency-domain Raman methods such as FSRS have to date been more successful in observing structural evolution on the sub-50 fs timescale.30,31 Here we perform time-resolved FSR spectroscopy in a microscope to enable FSRS measurements on pentacene crystals. FSRS offers both the spectral and temporal resolution necessary to probe the structural dynamics driving singlet fission directly in the frequency domain, and thus provides the empirical methodology to examine the multidimensional reaction coordinate underlying the singlet fission process.
Experimental
Femtosecond stimulated Raman spectroscopy
We generated the three pulses required for FSRS with the 4.4 W output of a Coherent Libra-F-1K femtosecond amplifier.32 We used a home-built non-collinear parametric amplifier to generate the actinic pump. The 2 μW white light seed continuum was generated in a 2 mm sapphire crystal. We generated the 56 mW 400 nm pump pulse via doubling of the 800 nm fundamental beam in a BBO crystal. These two pulses were combined non-collinearly in a 1 mm BBO crystal and the resultant visible pulse was compressed by a SF10 prism pair in order reduce linear chirp. The actinic pump used in these experiments had a Gaussian spectral profile with a central wavelength of 572 nm and a FWHM of 17 nm. We used a home-built grating filter to generate the 2.1 ps narrowband Raman pump from the 800 nm fundamental.36 We generated the broadband Raman probe by focusing 2.5 mW of the 800 nm fundamental through a 2 mm sapphire crystal and compressed it with a fused silica prism compressor. The continuum was filtered through an RG830 long pass filter prior to compression and an RG1000 long pass filter before the sample position. We used an inverted Olympus IX 73 microscope to focus the three beams non-collinearly to the sample. The microscope objective was an Olympus 20× with flat field optical correction and a numerical aperture of 0.40. We used a piezoelectric delay stage to temporally overlap the Raman probe and actinic pump, and the cross correlation time of these two pulses was 410 ± 10 fs as measured by optical Kerr effect with a 2 mm cuvette of cyclohexane at the sample position. After the sample, we focused the probe and stimulated Raman signal to an ISA HR 320 spectrograph (600 g mm−1 grating with 800 nm blaze), and a Princeton Instruments PIXIS 100F CCD detected spectra at the 1 kHz laser repetition rate. We acquired the femtosecond stimulated Raman spectra with a home-built Labview program and obtained the Raman gain by dividing the Raman pump-on spectrum by the Raman pump-off spectrum. Spectra were acquired for 20–50 seconds per time point with the actinic pump flux varying from 0.5 ± 0.1 to 5 ± 1 W cm−2 and Raman pump flux varying from 3 ± 0.3 to 8.0 ± 0.8 W cm−2.Continuous-wave Raman spectra
We obtained the spontaneous Raman spectra seen in Fig. 1c using a home-built Raman spectrometer. We illuminated the sample with 6.0 mW of light from a HeNe laser in a back-scattering geometry with an Olympus 10× objective for focusing. A Princeton Instruments Acton SP2500 spectrometer and a Princeton Instruments PIXIS:100BX were used for detection.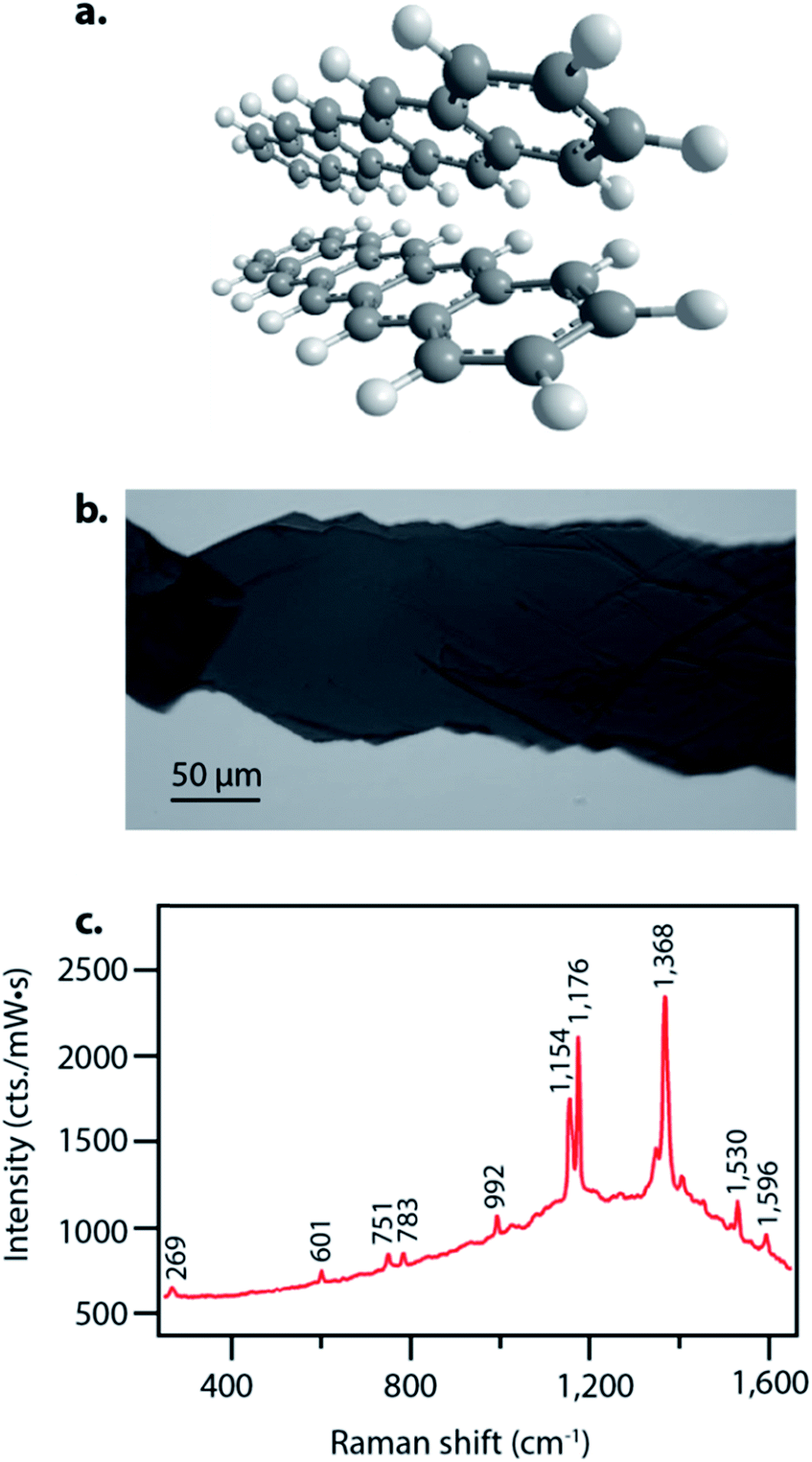 | ||
| Fig. 1 Characterization of pentacene samples used for FSRS experiments. (a) Structure of pentacene in a herringbone structure characteristic of this crystalline solid. (b) Image of crystal used in the FSRS experiments, grown by physical vapor transport. (c) Spontaneous ground state Raman spectrum of crystalline pentacene at 633 nm excitation. | ||
Sample preparation
We used sublimed grade 99.9% pentacene from Sigma-Aldrich and grew crystals by physical vapor transport according to previously published procedures.37 We annealed the furnace for 12 hours at 300 °C before the crystal growth. We then placed 20 mg of pentacene in a glass boat inside the furnace and heated it to 100 °C at a gauge pressure of 0.16 MPa with an argon flow rate of 40 mL min−1 for 45 minutes. We subsequently relieved the pressure and heated the furnace to 220 °C for 1 hour with an argon flow rate of 70 mL min−1. Then we heated the sample zone of the furnace to 310 °C and used a thermocouple to regulate the temperature to 272 °C 30 cm from the sample zone to create a temperature gradient. We then increased the argon flow rate to 93 mL min−1 and crystals began to form after 25 minutes in these conditions. After 1 hour and 45 minutes we stopped heating the furnace and allowed it to cool to room temperature before removing the pentacene crystals. The pentacene crystals were needle-like in shape and 3–7 mm long with optical densities at the photoexcitation wavelength of 572 nm varying from 0.8 to 1.1.DFT and TD-DFT calculations
We performed theoretical calculations to obtain molecular geometries and vibrational frequencies of neutral, anion, and cation pentacene molecules using restricted and unrestricted density functional theory. We used time-dependent density functional as implemented in Gaussian 09 for geometry optimization and frequency calculations on the excited state pentacene molecule.38 Time-dependent density functional theory (TD-DFT) is a reliable and cost effective method to obtain the excited state structure and vibrational frequencies for small organic molecules such as pentacene.43 As it gives accurate and widely accepted vibrational frequency values, we used the triple-split-valence basis set 6-311++G(d,p) with the B3LYP functional for all molecules.39 The neutral-ground state Raman spectrum of pentacene is in an agreement with experimental values after scaling by 0.967.44 Calculated vibrational frequencies for ground and excited state pentacene are summarized in the ESI.†Results
Pentacene singlet fission has been well-characterized electronically and occurs on a sub-picosecond timescale with nearly 200% quantum efficiency,15 thus making it an ideal system to examine structural dynamics of singlet fission with femtosecond stimulated Raman spectroscopy. Pentacene crystallizes in a herringbone structure with 50–55° angles between the planes, as seen in Fig. 1a, allowing for ideal chromophore interactions in facilitating efficient singlet fission.40 We grew the pentacene crystals used in these experiments by physical vapor transport, resulting in crystalline samples such as that shown in Fig. 1b, similar to those used to study triplet transport for photovoltaic applications by Poletayev et al.37 The ground state spontaneous Raman spectrum with 633 nm excitation is shown in Fig. 1c. The intense peaks from 1154 cm−1 to 1596 cm−1 arise from carbon–carbon stretching modes, while the features from 269 cm−1 to 992 cm−1 correspond to ring breathing and torsional modes.We used FSRS to probe the excited state structural dynamics in a pentacene crystal following photoexcitation. Our FSR spectrometer was coupled into an inverted microscope in order to provide facile probing of small pentacene crystals. As a result of the objective used for focusing, the time resolution of the experiment as measured by the optical Kerr effect was 410 ± 10 fs. FSRS generates signal from both the ground and excited state potential energy surfaces, and thus the raw FSR spectra acquired upon photoexcitation contain signals from both excited state and ground state features. In order to obtain strictly excited state features from the FSRS measurements, the ground state FSR spectrum was subtracted from each excited state spectrum, thus yielding difference spectra with significant contribution from ground state depletion. The frequency overlap between the excited state peaks and ground state depletion features necessitated adding ground state spectra back into the difference spectra in order to examine the excited state peaks. We added ground state spectra back into the excited state spectra such that no negative features remained, resulting in the FSR spectra seen in Fig. 2. A complete discussion of the ground state addition process including the original data after one-to-one subtraction, the ground state addition kinetics, and alternative methods of ground state addition is presented in the ESI.†
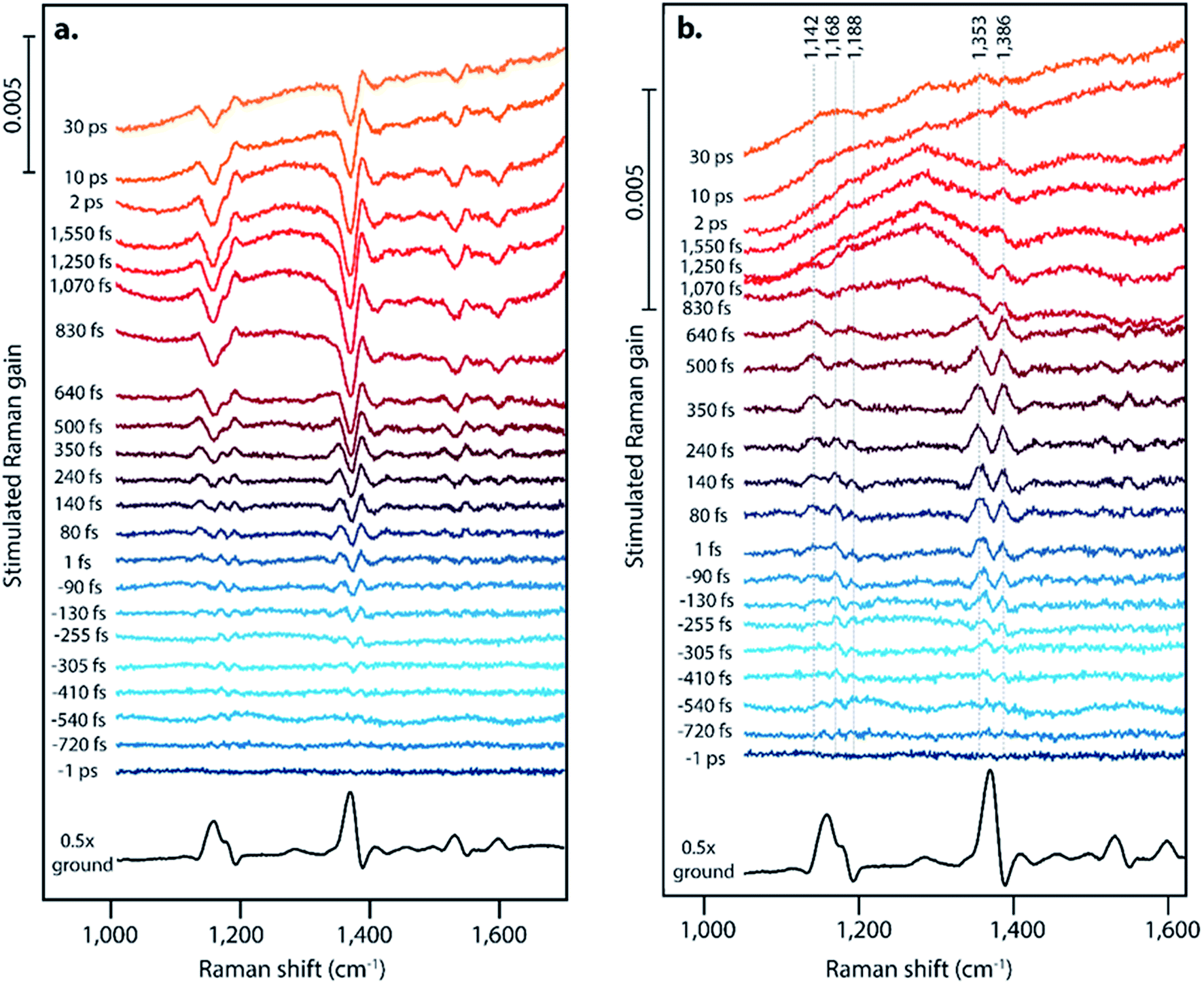 | ||
| Fig. 2 Differential femtosecond stimulated Raman spectra following photoexcitation (a) without ground state addition and (b) after ground state addition. New excited state peaks are seen at frequencies shifted from the ground state peaks at 1163, 1175, 1369, 1531, and 1599 cm−1. The sloped baseline at later time points is due to excited state transient absorption upon formation of triplets. | ||
Fig. 2 displays offset FSR spectra at the indicated time points following photoexcitation, along with the ground state spectrum. Fig. 2a presents the data following a one-to-one subtraction of the ground state spectrum, and Fig. 2b displays the transient spectra following subsequent scaled addition of the non-photoexcited ground state, as is customarily done in FSRS data analysis. The ESI† details the ground state addition process, including consideration of an alternate data analysis procedure and its mechanistic implications. At later time delays, clear evidence for triplet formation is visible through the large transient absorption background. Upon generation of the triplet state at later time delays, pentacene undergoes a change in resonance conditions such that a strong absorption feature arises from 750–950 nm.15 This gives rise to the strong transient absorption background seen in the FSR spectra. Based on the long lifetime and transient absorption features which are in agreement with those measured previously, we can assign spectra observed after ∼800 fs in our FSRS measurements to the triplet state in pentacene. While singlet fission on the order of hundreds of femtoseconds is slower than the fastest values previously observed in polycrystalline pentacene thin films by transient absorption methods (sub-100 fs),15 we attribute this to our use of a single pentacene crystal or to variations in pentacene crystal growth methods, which are known to significantly alter the rate of fission.41 The lack of Raman features corresponding to the triplet is likely due to the strong resonance of the triplet state and the 800 nm Raman pump, thus the high power picosecond Raman pump pulse likely promotes a large fraction of the T1 population to the higher lying triplet state prior to probe interaction.45–49 The lack of new Raman features upon triplet formation could also be attributed to minimal structural rearrangement from the ground state structure.
Surprisingly, we observe significantly different FSR spectra at early time delays, providing evidence for a structural intermediate during the singlet fission process, in which we consider an intermediate to be a state that is structurally distinct from the Franck–Condon region and from the triplet state potential energy surface. For the first ∼1 ps, the transient Raman spectra shown in Fig. 2 show spectral splitting of each ground state peak into two distinct peaks on the excited state. This phenomenon is clearly evident for the ground state peaks at 1163, 1175, and 1369 cm−1. For each ground state peak, two excited state peaks are visible, one upshifted and one downshifted by 5–25 cm−1 from the ground state. These peaks shift in frequency as a function of time, and the magnitude of the two peaks in each pair is not identical. Excitation at varying excitation wavelengths of 532 nm and 600 nm, in addition to the data at 570 nm excitation shown in Fig. 2, does not change the structure or dynamics of this intermediate feature, as discussed in Section 10 of the ESI.† Interestingly, evidence for an intermediate in this time regime has not been found with ultrafast transient absorption spectroscopy, indicating that these states may either have very low optical absorption cross sections, or have absorption transitions in the shortwave infrared region. One major difference between the polycrystalline thin films used for most transient absorption experiments and the single crystal used here is the anisotropy of the transition dipoles. It is possible that the orientation of the excited state transition dipole of the charge-transfer intermediate is much more sensitive to crystal orientation in the as compared to the triplet state transition dipole, which could explain the lack of transient absorption features during the charge-transfer intermediate lifetime in this single crystal work.
Fig. 3A shows the transient evolution of the femtosecond stimulated Raman spectra in the 1330–1400 cm−1 region. Here we see the excited state peaks split spectrally from the ground state with both the magnitude of splitting and peak width increasing with time. The 1369 cm−1 ground state peak splits into two distinct peaks with initial frequencies of 1365 cm−1 and 1382 cm−1. These peaks shift to 1346 cm−1 and 1387 cm−1 respectively after 1 ps. Similar splitting is seen for other ground state modes, including those at 1163, 1175, 1531, and 1599 cm−1. As these features precede the formation of triplet state they are clearly signatures of an intermediate in the singlet fission process. Fig. 3B displays the kinetics of the features seen by FSRS. Here we plot the Raman peak amplitude of the 1365 cm−1 peak (blue), the triplet transient absorption signal at 840 nm (black), and the instrument response function as measured by the optical Kerr effect (grey). The kinetics show a clear progression from the CT state to the triplet state. These frequency shifts occurring on the femtosecond timescale give insight into high temporal resolution features of the highly anharmonic potential energy surface that have not been previously accessible by the ultrafast methods used to examine singlet fission in pentacene.
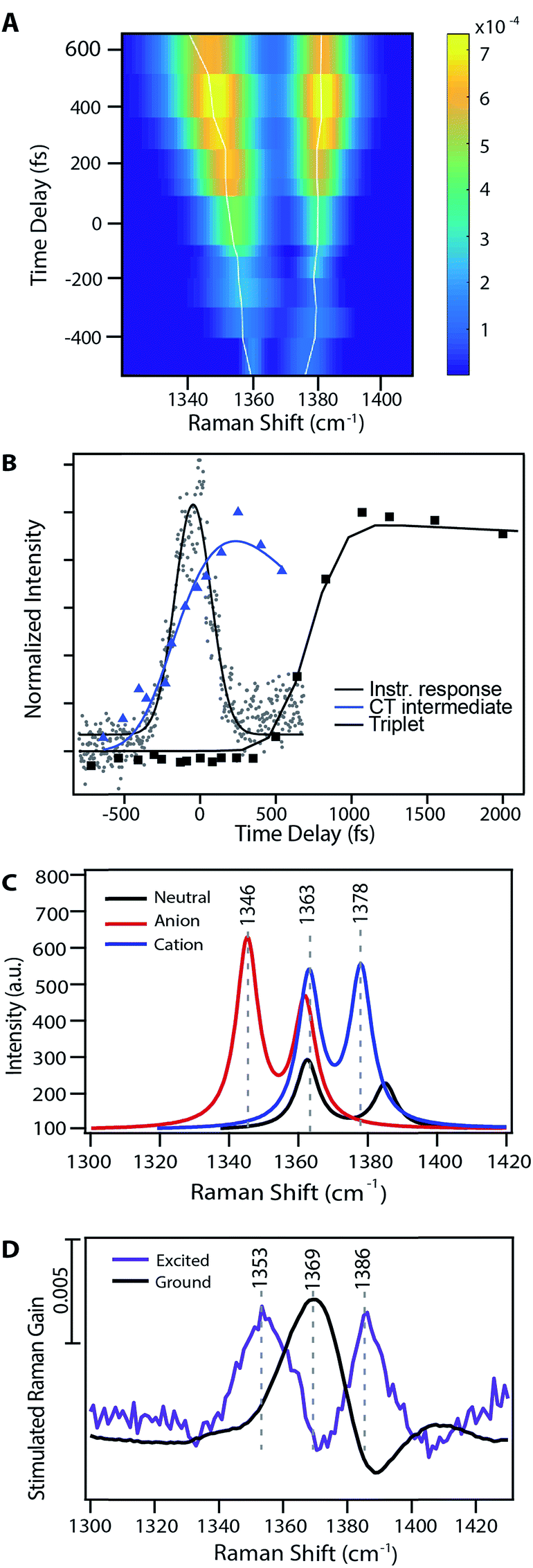 | ||
| Fig. 3 Evidence for a charge-transfer intermediate in pentacene singlet fission. (A) Excited state peak fits from Fig. 2 at nominal time points relative to photoexcitation. Traces show the maximum amplitude peak frequency dynamics. (B) Charge-transfer state Raman peak and triplet population excited state absorption kinetics with associated kinetic fits. (C) TD-DFT Raman frequency calculations for the pentacene neutral singlet, anion doublet, and cation doublet. (D) Experimental data for both the ground and excited state spectra in a similar frequency region as that shown in part (B). | ||
Discussion
Spectral splitting on the femtosecond timescale upon photoexcitation could arise from either a rapid molecular structural change or the redistribution of electron density of neighboring molecules. The spectral splitting seen in Fig. 2 occurs at all ground state frequencies, therefore the new structure formed on the excited state potential energy surface would need to give rise to two distinct modes for each ground state mode. It would be unusual for a single new structure to precisely double the number of modes seen in the ground state configuration, therefore we suggest that the Raman features seen in Fig. 2 do not arise from strictly nuclear conformational changes.Analysis of the peak frequency dynamics and the peak integrated areas offers evidence for the presence of two distinct species formed upon photoexcitation. As seen in Fig. 2 and 3, the magnitude of transient frequency shifts varies between red-shifted and blue-shifted peaks. This suggests that the species giving rise to the red-shifted and blue-shifted peaks originate from different localized regions of the potential energy surface, likely corresponding to two distinct but coupled species. Furthermore, the difference in amplitude of the red-shifted and blue-shifted peaks offers additional support for the presence of two distinct species. For example, in Fig. 3A, the red-shifted peak is slightly larger in area than the corresponding blue-shifted peak. This suggests that our Raman pump couples more strongly to and thus more effectively resonantly enhances the species giving rise to the red-shifted peak than the species giving rise to the blue-shifted peak, or that the polarizability of vibrations in the two species is slightly different in magnitude, assuming equal sized populations of each species are formed. Additionally, the frequency dynamics for the two peaks shown in Fig. 3A are different, with the lower frequency peak experiencing a larger change in frequency as compared to the higher frequency peak, suggesting that the two features are evolving on separate potential energy surfaces.
Thus, we assign the spectral splitting features in Fig. 2 to a partial charge transfer amongst pairs of pentacene molecules. Here the absorption features of the pentacene anion and cation are both in the near-IR regime and are resonantly enhanced with our 800 nm stimulated Raman pump.16,17 Additionally, these electronic features overlap with those of the triplet, thus hindering their direct observation in previous transient absorption and 2D electronic spectroscopy studies on polycrystalline thin films.5,15
In this case, the donation of electron density from one molecule to a neighbouring molecule in the pentacene pair forms species with cationic and anionic character. The changes in electron density subsequently drive the Raman mode energies associated with the cationic and anionic species to higher and lower frequencies, respectively. As such, the formation of a charge-transfer pair could easily give rise to the spectral splitting features seen Fig. 2. Based on the frequency dynamics and peak size ratio along the direction of peak splitting, we assign the spectral features seen immediately after photoexcitation to the presence of a structurally distinct intermediate with significant charge-transfer character. As displayed in Fig. 3B, this feature occurs temporally before the appearance of the triplet excited state absorption, thus we hypothesize that pentacene singlet fission proceeds through this charge-transfer intermediate state.
Interestingly, the excited state FSRS spectral response of a TIPS-pentacene polycrystalline thin film (shown in Fig. S11†) is markedly different than that of pentacene and shows only ground state bleaching features, suggesting that singlet fission proceeds by an alternate pathway in these systems. We emphasize that the pentacene results presented here arise from a single crystal sample, and the photophysics between single crystals and polycrystalline samples may be fundamentally different. Compared to polycrystalline thin films, single crystals have minimal defect sites, no interfaces or ‘hot spots’ where singlet fission may be accelerated, and are easier to compare to existing theoretical models. Furthermore, the lack of similarity between the pentacene and TIPS-pentacene FSRS data minimizes the possibility of these features arising from any experimental artefacts. Similar excited state features in pentacene were also observed in experiments using an etalon to shape the picosecond Raman pump pulse (ESI†), further suggesting that the excited state spectral splitting response is not an experimental artefact.
We utilized DFT and TD-DFT computational methods to confirm our assignment of the excited state spectral splitting to a charge-transfer intermediate and directly assign the transient Raman peaks to species with cationic and anionic character. Although DFT calculations clearly do not completely capture the complex structural dynamics occurring during singlet fission in these crystalline systems, they provide reliable and cost-effective methods for calculating vibrational frequencies on selected potential energy surfaces of individual pentacene molecules.42 DFT is known to be inaccurate for charge-transfer processes due to issues with accurate consideration of exchange correlation, but here we use it to calculate the vibrational spectra for individual radical anions and cations in vacuo, as has been done previously for similar acenes.43Table 1 summarizes relevant experimental and calculated frequencies for selected vibrational modes. All reported DFT frequencies have been scaled by 0.967.44 In particular, the excited state features seen in FSRS around 1360 cm−1 at 240 fs are typically within 10 cm−1 of the calculated frequencies for the anion and cation states of pentacene, indicating that our assignment of the intermediate state to the formation of anionic and cationic species is supported by computational frequency calculations. Other mode assignments are presented in the ESI,† and while the agreement between experiment and theory is not quantitative, the calculations are consistent with the charge-transfer intermediate mechanism.
| Mode description | FSRS frequencies (cm−1) | DFT frequencies (cm−1) | ||||
|---|---|---|---|---|---|---|
| Ground | Excited | S0 | S1 | Anion | Cation | |
| In-plane H wag on terminal rings | 993 | 984 | 1011 | 997 | 1004 | |
| In-plane H wag | 1163 | 1142, 1168 | 1160 | 1157 | 1137 | 1158 |
| In-plane H wag | 1175 | 1168, 1188 | 1167 | 1177 | 1168 | 1178 |
C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretch C stretch |
1369 | Obscured, 1386 | 1363 | 1378 | 1362 | 1378 |
| CC stretch and in-plane H wag |
Obscured | 1353, obscured | 1385 | 1337 | 1345 | 1363 |
| CC stretch and in-plane H wag |
1531 | 1516, 1548 | 1522 | 1523 | 1508 | 1524 |
| CC stretch on central rings and in-plane H wag |
1599 | 1586, 1614 | 1578 | 1540 | — | — |
Fig. 3 examines the frequency response in the 1300–1400 cm−1 region of the Raman spectrum, displaying theoretical frequency values in part (C) and the experimental ground state spectrum and transient spectrum at 240 fs following photoexcitation in part (D). There are two ground state modes in this region, both corresponding to CC stretches. Scaled DFT frequencies for these modes are 1363 and 1385 cm−1. Experimentally we observe one mode at 1369 cm−1, and the higher frequency ground state mode is obscured by a Raman loss side wing seen frequently in FSRS.44 Theoretical predictions for these same vibrational modes in the anion and cation states include peaks at 1345 and 1378 cm−1, which are in agreement with the experimentally observed values of 1353 and 1386 cm−1. Modes predicted to be at 1362 and 1363 cm−1 are obscured by overlapping ground state features, as shown in Fig. 3. Due to the ground state addition process detailed in the ESI,† we do not use regions which overlap with ground state features in our analysis, as the uncertainty of the signal magnitude in these spectral regions is large. Thus, we can assign the 1353 cm−1 peak transient FSR spectra to a CC stretching mode in pentacene with anionic character, and the 1386 cm−1 peak to a different CC stretching mode in pentacene with cationic character. Similar assignments can be made for other modes, as shown in Table 1. Interestingly, the near quantitative agreement between DFT calculations and femtosecond stimulated Raman spectra indicate that the degree of charge transfer between pentacene pairs is quite large, and may approach complete charge transfer.
In recent years, singlet fission work in both theory and multidimensional spectroscopy has emerged suggesting that fission could proceed by excitation to mixed bright singlet and charge-transfer state or proceed through a multiexcitonic state.5,13 In this work, we only observe spectral signatures of the charge-transfer state evolve between the cross-correlation time and formation of the triplet, assigning it to a structural distinct intermediate, but do not observe features of an initially formed singlet species following direct excitation to the Franck–Condon region. As such, our data could also support the hypothesis that the photoexcited state corresponds to an admixture with charge-transfer and singlet character, and thus the state charge-transfer state spectral splitting features could be a signature of not an intermediate, but instead the initially photoexcited state with some amount of charge-transfer character. Here we define an intermediate state as a measureable state which is structurally distinct from the reactant and product, and thus this work supports the charge-transfer intermediate mechanism of singlet fission in crystalline pentacene.
Support of the charge-transfer spectral features by theoretical calculations allows us to further examine this new insight into the singlet fission mechanism. We have observed evidence for a charge-transfer intermediate immediately after photoexcitation as identified by red- and blue-shifted vibrational features in the transient FSR spectra. This intermediate appears before any signatures of the triplet state, and is fairly short-lived in nature, but maintains a measurable lifetime. We therefore assert that this charge-transfer intermediate occurs on the reaction pathway to triplet formation. Our data provide evidence for a two-step mechanism of singlet fission as we have provided experimental evidence for intermediate states with charge-transfer character that exist on the timescale of singlet to triplet conversion. Furthermore, these data suggest that the highly anharmonic potential energy surfaces associated with the anionic and cationic species could play a critical role in facilitating the fast and efficient singlet fission seen in pentacene.
Conclusions
In this study we have observed the formation of a state with significant charge-transfer character in pentacene singlet fission with femtosecond stimulated Raman spectroscopy. This species with charge-transfer character exists in the excited state Raman spectra prior to formation of triplet species with characteristic splitting in peak frequency from the ground state features. Additionally, the transient peak frequency shifts that occur on the femtosecond timescale provide insight into the evolution of the highly anharmonic potential energy surfaces associated with the pentacene singlet fission process. Thus, FSRS is a valuable tool to examine singlet fission structural dynamics in various morphological systems. A more comprehensive understanding of the temporal and spatial structural features driving singlet fission in pentacene and related systems is important in constructing a mechanistic picture of singlet fission. This in turn will play a vital role in the molecular design of new sensitizers for organic photovoltaic systems.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Department of Energy, DE-SC0018203. The authors thank Professor James Johns (U. Minnesota) for use of the PVT furnace and helpful discussions, and the Minnesota Supercomputing Institute (MSI) at the University of Minnesota for providing resources that contributed to the research results reported in this paper. We thank Matthew Vollmer and Connie Lu for assistance with synthesis.References
- M. B. Smith and J. Michl, Chem. Rev., 2010, 110, 6891–6936 CrossRef CAS PubMed.
- M. B. Smith and J. Michl, Annu. Rev. Phys. Chem., 2013, 64, 361–386 CrossRef CAS PubMed.
- M. C. Hanna and A. J. Nozik, J. Appl. Phys., 2006, 100, 074510 CrossRef.
- D. N. Congreve, J. Lee, N. J. Thompson, E. Hontz, S. R. Yost, P. D. Reusswig, M. E. Bahlke, S. Reineke, T. Van Voorhis and M. A. Baldo, Science, 2013, 340, 334–337 CrossRef CAS PubMed.
- A. A. Bakulin, S. E. Morgan, T. B. Kehoe, M. W. Wilson, A. W. Chin, D. Zigmantas, D. Egorova and A. Rao, Nat. Chem., 2016, 8, 16–23 CrossRef CAS PubMed.
- J. Zheng, Y. Xie, S. S. Jiang and Z. G. Lan, J. Phys. Chem. C, 2016, 120, 1375–1389 CAS.
- P. M. Zimmerman, F. Bell, D. Casanova and M. Head-Gordon, J. Am. Chem. Soc., 2010, 133, 19944–19952 CrossRef PubMed.
- N. Renaud, P. A. Sherratt and M. A. Ratner, J. Phys. Chem. Lett., 2013, 4, 1065–1069 CrossRef CAS PubMed.
- T. C. Berkelbach, M. S. Hybertsen and D. R. Reichman, J. Chem. Phys., 2013, 138, 114103 CrossRef PubMed.
- R. W. A. Havenith, H. D. de Gier and R. Broer, Mol. Phys., 2012, 110, 2445–2454 CrossRef CAS.
- E. C. Greyson, J. Vura-Weis, J. Michl and M. A. Ratner, J. Phys. Chem. B, 2010, 114, 14168–14177 CrossRef CAS PubMed.
- T. Zeng, R. Hoffmann and N. Ananth, J. Am. Chem. Soc., 2014, 136, 5755–5764 CrossRef CAS PubMed.
- D. Beljonne, H. Yamagata, J. L. Bredas, F. C. Spano and Y. Olivier, Phys. Rev. Lett., 2013, 110, 226402 CrossRef CAS PubMed.
- S. R. Yost, J. Lee, M. W. Wilson, T. Wu, D. P. McMahon, R. R. Parkhurst, N. J. Thompson, D. N. Congreve, A. Rao, K. Johnson, M. Y. Sefeir, M. G. Bawendi, T. M. Swagger, R. H. Friend, M. A. Baldo and T. Van Voorhis, Nat. Chem., 2014, 6, 492–497 CrossRef CAS PubMed.
- M. W. B. Wilson, A. Rao, J. Clark, R. S. S. Kumar, D. Brida, G. Cerullo and R. H. Friend, J. Am. Chem. Soc., 2011, 133, 11830–11833 CrossRef CAS PubMed.
- T. M. Halasinski, D. M. Hudgins, F. Salama, L. J. Allamandola and T. Bally, J. Phys. Chem. A, 2000, 104, 7484–7491 CrossRef CAS.
- J. Szczepanski, C. Wehlburg and M. Vala, Chem. Phys. Lett., 1995, 232, 221–228 CrossRef CAS.
- A. J. Musser, M. Liebel, C. Schnedermann, T. Wende, T. B. Kehoe, A. Rao and P. Kukura, Nat. Phys., 2015, 11, 352–357 CrossRef CAS.
- W.-L. Chan, M. Ligges, A. Jailaubekov, L. Kaake, L. Miaja-Avila and X.-Y. Zhu, Science, 2011, 334, 1541–1545 CrossRef CAS PubMed.
- N. Monahan and X.-Y. Zhu, Annu. Rev. Phys. Chem., 2015, 66, 601–618 CrossRef CAS PubMed.
- Y. Fujihashi and A. Ishizaki, J. Phys. Chem. Lett., 2016, 7, 363–369 CrossRef CAS PubMed.
- R. D. Pensack, A. J. Tilley, S. R. Parkin, T. S. Lee, M. M. Payne, D. Gao, A. A. Jahnke, D. G. Oblinsky, P. F. Li, J. E. Anthony, D. S. Seferos and G. Scholes, J. Am. Chem. Soc., 2015, 137, 6790–6803 CrossRef CAS PubMed.
- J. C. Johnson, A. J. Nozik and J. Michl, Acc. Chem. Res., 2013, 6, 1290–1299 CrossRef PubMed.
- J. Zirzlmeier, D. Lehnherr, P. B. Coto, E. T. Chernick, R. Casillas, B. S. Basel, M. Thoss, R. R. Tykwinski and D. M. Guldi, Proc. Natl. Acad. Sci. U. S. A., 2015, 17, 5325–5330 CrossRef PubMed.
- S. Lukman, A. J. Musser, K. Chen, S. Athanasopoulos, C. K. Yong, Z. Zeng, Q. Ye, C. Chi, J. M. Hodgkiss, J. Wu, R. H. Friend and N. C. Greenham, Adv. Funct. Mater., 2015, 25, 5452–5461 CrossRef CAS.
- E. A. Margulies, C. E. Miller, Y. Wu, L. Ma, G. C. Schatz, R. M. Young and M. R. Wasielewski, Nat. Chem., 2016, 8, 1120–1125 CrossRef CAS PubMed.
- S. Lukman, K. Chen, J. M. Hodgkiss, D. H. P. Turban, N. D. M. Hine, S. Dong, J. Wu, N. C. Greenham and A. J. Musser, Nat. Commun., 2016, 7, 1–13 Search PubMed.
- E. A. Margulies, J. L. Logsdon, C. E. Miller, L. Ma, E. Simonoff, R. M. Young, G. C. Schatz and M. R. Wasielewski, J. Am. Chem. Soc., 2017, 139, 663–671 CrossRef CAS PubMed.
- R. R. Frontiera and R. A. Mathies, Laser Photonics Rev., 2011, 5, 102–113 CrossRef CAS.
- P. Kukura, D. W. McCamant, S. Yoon, D. B. Wandschneider and R. A. Mathies, Science, 2005, 310, 1006–1009 CrossRef CAS PubMed.
- C. Fang, R. R. Frontiera, R. Tran and R. A. Mathies, Nature, 2009, 462, 200–274 CrossRef CAS PubMed.
- W. R. Silva and R. R. Frontiera, Phys. Chem. Chem. Phys., 2016, 18, 20290–20297 RSC.
- F. Provencher, N. Bérubé, A. W. Parker, G. M. Greetham, M. Towrie, C. Hellmann, M. Côté, N. Stingelin, C. Silva and S. C. Hayes, Nat. Commun., 2014, 5, 4288 CAS.
- E. Ploetz, S. Laimgruber, S. Berner, W. Zinth and P. Gilch, Appl. Phys. B: Lasers Opt., 2007, 87, 389–393 CrossRef CAS.
- R. R. Frontiera, J. Dasgupta and R. R. Mathies, J. Am. Chem. Soc., 2009, 131, 15630–15632 CrossRef CAS PubMed.
- D. W. McCamant, P. Kukura, S. Yoon and R. A. Mathies, Rev. Sci. Instrum., 2004, 75, 4971–4980 CrossRef CAS PubMed.
- A. D. Poletayev, J. Clark, M. W. B. Wilson, A. Rao, Y. Makino, S. Hotta and R. Friend, Adv. Mater., 2014, 26, 919–924 CrossRef CAS PubMed.
- M. J. Frisch, et al., Gaussian 09 Rev. E.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- R. F. Li, J. J. Zheng and D. G. Truhlar, Phys. Chem. Chem. Phys., 2010, 12, 12697–12701 RSC.
- D. Nabok, P. Puschnig, C. Ambrosch-Draxl, O. Werzer, R. Resel and D. M. Smilgies, Phys. Rev. B, 2007, 76, 235322 CrossRef.
- D. H. Arias, J. L. Ryerson, J. D. Cook, N. H. Damrauer and J. C. Johnson, Chem. Sci., 2016, 7, 1185–1191 RSC.
- M. Dierksen and S. Grimme, J. Chem. Phys., 2004, 120, 3544–3554 CrossRef CAS PubMed.
- S. R. Langhoff, J. Phys. Chem., 1996, 100, 2819–2841 CrossRef CAS.
- Precomputed Vibrational Scaling Factors in Computation Chemistry Comparison and Benchmark Database Vol. 2016, ed. R. D. Johnson III, National Institute of Standards and Technology, 2015 Search PubMed.
- S. Yoon, D. W. McCamant, P. Kukura, R. A. Mathies, D. Zhang and S.-Y. Lee, J. Chem. Phys., 2005, 122, 9 CrossRef PubMed.
- R. R. Frontiera and R. A. Mathies, J. Chem. Phys., 2007, 127, 124501 CrossRef PubMed.
- S. Umapathy, B. Mallick and A. Lakshmanna, J. Chem. Phys., 2010, 133, 024505 CrossRef CAS PubMed.
- Z. Sun, J. Lu, D. H. Zhang and S.-Y. Lee, J. Chem. Phys., 2008, 128, 144114 CrossRef PubMed.
- D. W. McCamant, P. Kukura and R. A. Mathies, J. Phys. Chem. B, 2005, 109, 10449–10457 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Actinic pump spectrum, discussion on ground state addition process, peak fitting procedure, transient absorption data, power dependence measurements, etalon pulse shaping, TIPS-pentacene FSRS data, and optimized geometry and frequency calculation results. See DOI: 10.1039/c7sc03496b |
| This journal is © The Royal Society of Chemistry 2018 |