
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Diphosphine-protected ultrasmall gold nanoclusters: opened icosahedral Au13 and heart-shaped Au8 clusters†
Shan-Shan
Zhang‡
a,
Lei
Feng‡
a,
Ravithree D.
Senanayake‡
b,
Christine M.
Aikens
 b,
Xing-Po
Wang
a,
Quan-Qin
Zhao
a,
Chen-Ho
Tung
a and
Di
Sun
*a
b,
Xing-Po
Wang
a,
Quan-Qin
Zhao
a,
Chen-Ho
Tung
a and
Di
Sun
*a
aKey Lab of Colloid and Interface Chemistry, Ministry of Education, School of Chemistry and Chemical Engineering, Shandong University, Jinan, 250100, P. R. China. E-mail: dsun@sdu.edu.cn
bDepartment of Chemistry, Kansas State University, Manhattan, Kansas 66506, USA
First published on 4th December 2017
Abstract
Due to distinctive quantum confinement effects, ultrasmall gold nanoparticles usually exhibit interesting electronic structure and molecular-like properties. However, the lack of atomically-precise structural information makes the understanding of them almost impossible, such as understanding the relationships between their compositions and unique properties. Herein, by reducing a diphosphine AuI precursor (Au2(dppm)2Cl2; dppm = Ph2PCH2PPh2) with or without a S2− releasing reagent, we enriched our knowledge of the members in the families of Au13 and Au8 by the structural determinations of two new dppm-protected gold nanoclusters, [Au13(dppm)6]5+ (SD/Au1) and [Au8(dppm)4S2]2+ (SD/Au2), respectively. Within SD/Au1, the Au13 kernel significantly deviates from the ideal Ih icosahedron by the elongation of three surface Au–Au bonds to ∼3.5 Å, giving it C3 symmetry, whereas SD/Au2 has a novel heart-shaped C2 symmetric Au8S2 core (central Au4 tetrahedron + two Au2S units) protected by four μ2-dppm ligands in the outer shell. Of note, SD/Au1 represents a rare Au13 nanocluster with an opened icosahedral geometry, and SD/Au2 shows a new edge-shared “core + 4exo” structure type that has never been observed before. The electronic structures and optical absorption spectra of these systems are correlated with time-dependent density functional theory (TDDFT) calculations. Based on the spherical jellium model, the stability of the Au13 and Au8 nanoclusters can be ascribed to 8- and 2-electron superatoms with 1S21P6 and 1S2 configurations, respectively. Interestingly, the cluster SD/Au2 exhibits bright yellow luminescence with an emission maximum at 591 nm that slightly hypsochromically shifts to 581 nm upon cooling to 93 K. Our findings not only enrich the family of diphosphine-protected ultrasmall gold nanoclusters, but also demonstrate the rich variations of gold kernels during the transformation from a simple AuI precursor to Au nanoclusters.
Introduction
Gold nanoclusters with defined nuclearity and configurations have attracted considerable interest due to their nuclearity-selective optical, electronic, and catalytic properties which diverge significantly from those of their bulk metal counterparts.1 The total structure characterization of gold nanoclusters by X-ray crystallography is a prerequisite to the better understanding of their stability, metal–ligand interfacial bonding, as well as the aforementioned properties.2 Until now, phosphine,3 thiolate,4 and alkynyl-protected5 gold nanoclusters have been well identified based on total structure elucidations; however, it is still a challenge to acquire their X-ray quality single crystals and there are also many difficult questions still to be answered in this field, such as whether the face centered-cubic (fcc) structure in bulk gold would be preserved in these ultrasmall nanoclusters and how the organic ligands influence the configuration of the gold kernel, even for systems with the same nuclearity.6 Compared to the rapid progress of gold–thiolate nanoclusters starting from the seminal X-ray structure identification of the Au102 cluster,7 phosphine-protected gold nanoclusters are severely underdeveloped,8 although the first X-ray single crystal structure of a phosphine-protected cluster (i.e. Au11) dates back to 1969.9 The well-recognized phosphine-protected gold nanoclusters include undecagold Au11 and icosahedral Au13 protected by phosphine and halide.10Larger nanoclusters such as [Au20(PPhpy2)10Cl4] and [Au20(PP3)4] were recently characterized based on new phosphine ligands [PPhpy2 = bis(2-pyridyl)-phenylphosphine; and PP3 = tris[2-(diphenylphosphino)ethyl]phosphine].11 Among many of the reported gold nanoclusters, Au13 can be seen as a common ‘seed’ in the growth of larger ones from monoicosahedral Au13, biicosahedral Au25, to triicosahedral Au37, i.e. a ‘cluster of clusters’ motif.12 Of note, the configuration of the Au13 kernel usually deviates from that of the ideal Ih icosahedron depending on the protecting ligands; at the same time, such a variation in the metallic kernel may dramatically influence the physical and chemical properties.13 With these considerations in mind, herein, we revisited the dppm-protected gold nanoclusters and isolated two new members, [Au13(dppm)6]5+ (SD/Au1) and [Au8(dppm)4S2]2+ (SD/Au2), in this family (Scheme 1). Although 1 initially looks like its cousin with nitrate counteranions reported in 1981 (ref. 14) at first glance, in 1 the metallic kernel severely deviates from an ideal Ih icosahedron by the elongation of three Au–Au contacts to approximately 3.5 Å, giving it C3 symmetry. After introducing a S2− releasing reagent, another novel octanuclear gold nanocluster was isolated that possessed a heart-shaped C2 symmetric Au8S2 core (central Au4 tetrahedron + two Au2S units).
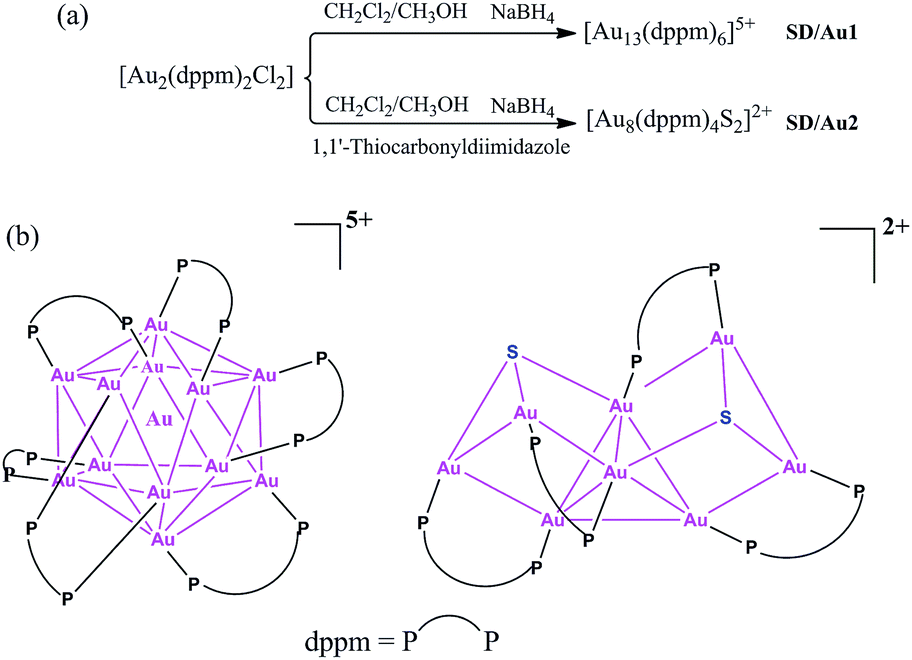 | ||
| Scheme 1 (a) Synthetic routes for [Au13(dppm)6]5+ and [Au8(dppm)4S2]2+ and (b) a chemical structure illustration of the trideca- and octagold nanoclusters. | ||
Results and discussion
The X-ray structures of [Au13(dppm)6]Cl5 (SD/Au1·5Cl) and [Au8(dppm)4S2]Cl2 (SD/Au2·2Cl)
Details of the synthesis are shown in the ESI.† Briefly, to 12 mL of methanol and dichloromethane suspension containing 0.02 mmol Au2(dppm)2Cl2,15 a freshly prepared solution of NaBH4 (0.60 mmol in 1 mL of cold water) was added dropwise under vigorous stirring. The color immediately changed from colorless to dark green, and then to black. The reaction continued for 12 h at 0 °C in air under the exclusion of light. The crude black solid obtained by rotary evaporation was dissolved in 6 mL of CH2Cl2, and the resulting solution was subject to diffusion of benzene to afford black crystals (Scheme S1†). The identity was determined to be [Au13(dppm)6]Cl5 (SD/Au1·5Cl). Similarly, in the presence of a S2− releasing reagent during a similar synthesis as for SD/Au1·5Cl, we could obtain [Au8(dppm)4S2]Cl2 (SD/Au2·2Cl) as yellow block crystals. The difference in the kernel structures between the two systems may arise from the etching effect of S2− on the Au core surface due to its strong coordination ability toward Au atoms. Therefore, the introduction of S2− in the synthesis of Au nanoclusters can significantly influence the final core structure.The single crystal X-ray diffraction (SCXD) analysis of SD/Au1·5Cl at 173 K indicates that it crystallizes in the trigonal space group P31c and that the asymmetric unit contains only one sixth of the Au13 nanocluster. The overall structure of SD/Au1 is a distorted icosahedral Au13 kernel with six μ2-dppm ligands bound to its six edges of the outer shell (Fig. 1a). The Au1 (denoted as Auc hereafter) site possesses one C3 axis and 3 C2 axes, giving it an occupancy of 1/6, whereas all of the Cl− ions are located on the C3 axis and Cl2 additionally sits on the inversion center. Thus, in total, five uncoordinated Cl− ions are found in the lattice corresponding to each Au13 cluster, making the charge state of the cationic part of SD/Au1 +5. As shown in Fig. 1b, the positive mode of the high-resolution electrospray ionization mass spectrometry (HR-ESI-MS) shows a main peak centered at m/z 1622.0950 corresponding to [Au13(dppm)6]3+ (calcd. 1622.0949). The different charge state between the X-ray diffraction and mass spectroscopy results may be attributed to the trapping of two additional electrons by SD/Au1 under the mass spectroscopy conditions.
 | ||
| Fig. 1 (a) The X-ray crystal structure of cationic [Au13(dppm)6]5+. Thermal contours are drawn at the 50% probability level. Color labels: purple, Au; orange, P; and gray, C. H atoms and Cl− ions are omitted. (b) The positive mode of the HRESI-MS of SD/Au1·5Cl dissolved in CH2Cl2. Inset: comparison of the measured (black trace) and simulated (red trace) isotopic patterns of [Au13(dppm)6]3+. (c and e) Top view of the kernel structure of Au13 along the C3 axis with the crystallographically unique Au atoms labelled. (d and f) Side view of the kernel structure of Au13. The three cyan edges denote the longer Au–Au separations (3.4995(5) Å). | ||
The views of the Au13 kernel of SD/Au1 drawn orthogonal with respect to the C3 axis are shown in Fig. 1c–f. The Au13 kernel consists of Au3 trigons perpendicular to the C3 axis at two poles and a chair-like Au6 ring (or Au7 if considering the central Au as well) sandwiched by them at the equatorial position. Each dppm ligand bridges two Au atoms, one at the pole and another at the equator (Au–P: 2.282(2) and 2.346(2) Å). The distortion of the Au13 kernel in SD/Au1 is mainly reflected by three extraordinarily long Au–Au contacts of nearly 3.5 Å and a larger acute bond angle around the central Au atom (73.519(12)°), which makes the symmetry of the metallic kernel differ substantially from that of an ideal Ih icosahedron. The central Au atom (Auc) binds to twelve surface Au atoms (Aus), giving twelve radial bonds of 2.7024(4) and 2.9236(4) Å, whereas 27 of the 30 Aus–Aus bonds fall in the range of 2.7155(7)–3.0272(5) Å, leaving the remaining three Aus–Aus separations to be 3.4995(5) Å, which are much longer than those observed in many other phosphine-protected Au nanoclusters and thus do not belong in the classic bonding range for aurophilic interactions.16 Although phosphines are deemed as one of the best types of ligand to stabilize gold nanoclusters, and the one mostly used is triphenylphosphine,17 diphosphine-protected (hereafter Ph2P(CH2)mPPh2 = Lm for short) gold nanoclusters with atomically-precise structural identification are still few, such as [Au11(L2)6]3+,18 [Au13(L2)5Cl2]3+,19a [Au6(L3)4]2+,20 [Au8(L3)4]2+,21 [Au11(L3)5]3+,22 and the largest is [Au22(L8)6].23 Dppm, as the shortest diphosphine ligand, has rarely been used to build gold nanoclusters; only two were reported previously, Au5 and Au13.14
The most interesting feature of SD/Au1·5Cl compared to the previous [Au13(dppm)6](NO3)4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 14 structure is the configuration of the Au13 kernel, which is an opened icosahedral cage in SD/Au1 instead of a closed one. For a perfect icosahedron, it should have twofold, threefold, and fivefold rotation axes, but the latter is missing in SD/Au1 due to the distortion. It is also worth noting that in [Au13(dppm)6](NO3)4, both the Aus–Aus and Auc–Aus bond lengths are below 3.0 Å (2.75–2.98 Å). By comparing opened icosahedral SD/Au1 and closed icosahedral [Au13(dppm)6](NO3)4 and [Au13(dppe)5(C
14 structure is the configuration of the Au13 kernel, which is an opened icosahedral cage in SD/Au1 instead of a closed one. For a perfect icosahedron, it should have twofold, threefold, and fivefold rotation axes, but the latter is missing in SD/Au1 due to the distortion. It is also worth noting that in [Au13(dppm)6](NO3)4, both the Aus–Aus and Auc–Aus bond lengths are below 3.0 Å (2.75–2.98 Å). By comparing opened icosahedral SD/Au1 and closed icosahedral [Au13(dppm)6](NO3)4 and [Au13(dppe)5(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CPh)2]3+,19b we found that SD/Au1 and [Au13(dppm)6](NO3)4 have the same ligand shell but different electron counts in the Au13 core, whereas SD/Au1 and [Au13(dppe)5(CCPh)2]3+ have different ligand shells but isoelectronic Au13 cores, which facilitated us to reasonably assign the distortion of the Au13 core to the synergistic effects of the electron count of the Au13 core and ligand size as well as the ligand arrangement on the surface. The current Au13 cluster carries five positive charges with eight valence electrons delocalized in “superatomic orbitals” (1S21P6), which matches a major shell closing in the electron shell model.24 Thus, SD/Au1·5Cl is the first gold nanocluster with an opened icosahedral Au13 kernel.
CPh)2]3+,19b we found that SD/Au1 and [Au13(dppm)6](NO3)4 have the same ligand shell but different electron counts in the Au13 core, whereas SD/Au1 and [Au13(dppe)5(CCPh)2]3+ have different ligand shells but isoelectronic Au13 cores, which facilitated us to reasonably assign the distortion of the Au13 core to the synergistic effects of the electron count of the Au13 core and ligand size as well as the ligand arrangement on the surface. The current Au13 cluster carries five positive charges with eight valence electrons delocalized in “superatomic orbitals” (1S21P6), which matches a major shell closing in the electron shell model.24 Thus, SD/Au1·5Cl is the first gold nanocluster with an opened icosahedral Au13 kernel.
Considering the high oxidation state of SD/Au1, we also tried to reduce it by adding NaBH4 in CH2Cl2 in the presence of nBu4NPF6, and as a result we isolated another Au135+ cluster, SD/Au3·4PF6·Cl, which has almost the same structure as that of SD/Au1·5Cl, except for the difference in the counteranions. The synthesis and structural graphic of SD/Au3·4PF6·Cl are shown in the ESI.† These results indicate that an 8e Au135+ nanocluster cannot be reduced to a 10e Au133+ nanocluster and in turn support the oxidation state of SD/Au1. Due to the large thermal ellipsoids of two of the four PF6− in the X-ray structure of SD/Au3·4PF6·Cl, we re-determined the number of PF6− using 31P NMR of HCl-digested SD/Au3·4PF6·Cl (Fig. S2†), which clearly showed the ratio of dppm (singlet, δ = 28.40 ppm) and PF6− (heptet, δ = −133.36, −137.01, −140.80, −144.19, −147.41, −151.17, and −154.62 ppm) to be 1:0.31 (calcd. 1:0.33), indicating a total of four PF6− anions in SD/Au3 and verifying the +5 oxidation state of SD/Au3.
In the presence of a S2− releasing reagent during a similar synthesis as for SD/Au1·5Cl, we isolated crystals of SD/Au2·2Cl in benzene or CH2Cl2 solvent. The single crystal structure analysis of SD/Au2·2Cl at 173 K reveals that it crystallized in an orthorhombic Ccca space group with a half molecule in the asymmetric unit. This nanocluster contains eight Au atoms, four dppm ligands, and two S2− ions (Fig. 2a). The composition and charge state were further confirmed by the good comparison of the HR-ESI-MS which gave one intense peak at m/z 1588.5774 that is assigned to the parent [Au8(dppm)4S2]2+ with a simulated isotopic distribution pattern and no other fragments were observed in the m/z range of 1000–2000 (Fig. 2b). A C2 axis passes through the Au8 cluster through the mid points of Au1–Au1i and Au2–Au2i (symmetric code: i = −x + 1.5, −y + 1, z). SD/Au2 is not a Au-centered polyhedral skeleton but has a “core + 4exo” structure exhibiting a heart shape. The octanuclear kernel in SD/Au2 has a tetrahedral core and two pairs of exo gold atoms attached at the opposite edges through edge-sharing, thus being described as a Au4 + 2(Au2) type structure, as shown in Fig. 2c and d. The four exo Au atoms form rectangles by sharing an edge of the Au4 tetrahedron. The pure metallic kernel is further capped by two μ3-S2− ions with Au–S distances of 2.297(4)–2.600(4) Å, giving the overall core structure of Au8S2. The Au–Au bonds within the tetrahedron are in the range of 2.6223(13)–2.8155(13) Å, which are shorter than that in metallic gold (2.88 Å). The core-to-exo distances (Au1–Au4 = 2.9907(9) Å and Au2–Au3 = 2.9269(9) Å) are markedly longer than those in the tetrahedron. Each dppm ligand as a bidentate bridge links one Au atom in the tetrahedron and one in an exo position to consolidate this “core + 4exo” structure (Au–P: 2.263(4)–2.320(4) Å). Several Au8 nanoclusters with different kernel geometries have been previously reported, such as capped centered chair-like [Au8(PPh3)7]2+25a and [Au8(PPh3)8]2+,25b [bitetrahedron + two]-type [Au8(dppp)4Cl2]2+,21 [Au8(dppp)4(CCPh)2]2+,26 and tritetrahedral [Au8(dppp)4]2+;21 however, a “core + 4exo” structure type has not been isolated hitherto. The closest case is [Au8(PMes3)6]2+ reported by Sharp et al. in 1994,27 and it also has a tetrahedral core like in SD/Au2 but with two pairs of Au atoms appended on the vertices, and not the edges, of the tetrahedron. The current Au8 cluster carries two positive charges, thus the total number of free valence electrons is calculated to be 2 (n = 8 − 4 − 2), the smallest magic electron count. The formation of such a special Au8 cluster different from the other Au8 cousins found before may be caused by the coordination of the S2− ligand, and its stability should be attributed to the jellium electronic shell closing of 1s2.
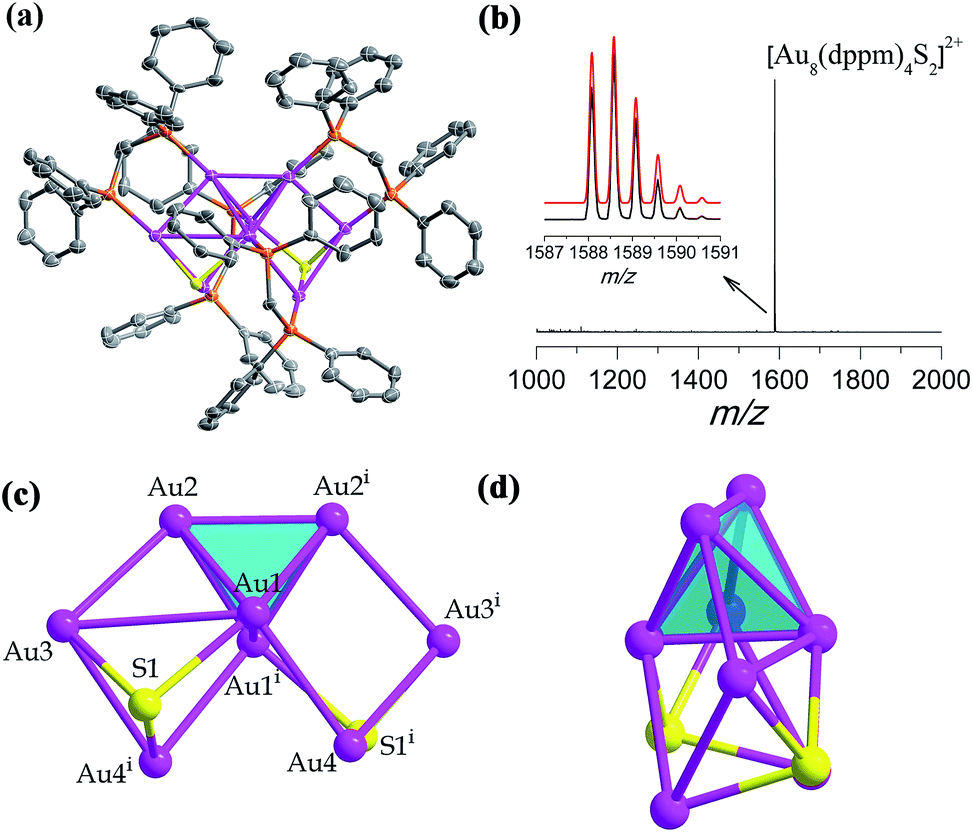 | ||
| Fig. 2 (a) The X-ray crystal structure of cationic [Au8(dppm)4S2]2+. Thermal contours are drawn at the 50% probability level. Color labels: purple, Au; orange, P; and gray, C. H atoms and Cl− ions are omitted. (b) The positive mode of the HRESI-MS of SD/Au2 dissolved in CH2Cl2. Inset: comparison of the measured (black trace) and simulated (red trace) isotopic patterns of [Au8(dppm)4S2]2+. (c and d) Orthogonal views of the kernel structure of Au8S2. | ||
The optical properties and time-dependent DFT (TDDFT) calculations of SD/Au1·5Cl and SD/Au2·2Cl
SD/Au1·5Cl can be dissolved in CH2Cl2 and ethanol, whereas SD/Au2·2Cl can be dissolved in methanol, acetonitrile and CH2Cl2. In these solvents, both SD/Au1 and SD/Au2 keep their cluster integrity without any disassembly, as shown by their parent ion peaks in the HR-ESI-MS (Fig. S3†). As revealed by time-dependent UV-vis spectra, no obvious changes were observed after their solutions were stored under ambient conditions for two weeks (Fig. S4†), indicating that both SD/Au1 and SD/Au2 are quite stable in solution. The optical absorption spectra of SD/Au1 and SD/Au2 were recorded in CH2Cl2 solution and are shown in Fig. 3. The absorption spectrum of SD/Au1 showed tail-and-hump spectral features that comprised a strong absorption band in the UV region and a relatively weak shoulder peak at 440 nm tailing to the red region of the spectrum, whereas only two peaks at 327 and 342 nm were observed in the absorption spectrum of SD/Au2 and no obvious peaks could be detected in the visible region. As discussed by Konishi,10b the absorption spectra of gold clusters are mainly influenced by the kernel nuclearity but far less so by the phosphine ligands. As shown in Table S7,† the absorption bands of Au13 clusters reported by Mingos and Konishi are very similar at ∼340 and ∼430 nm, whereas the absorption peak of SD/Au1 shifted to 440 nm, which suggested that the absorption spectra of gold clusters are not only influenced by the kernel nuclearity but also the configuration of the core even with the same nuclearity.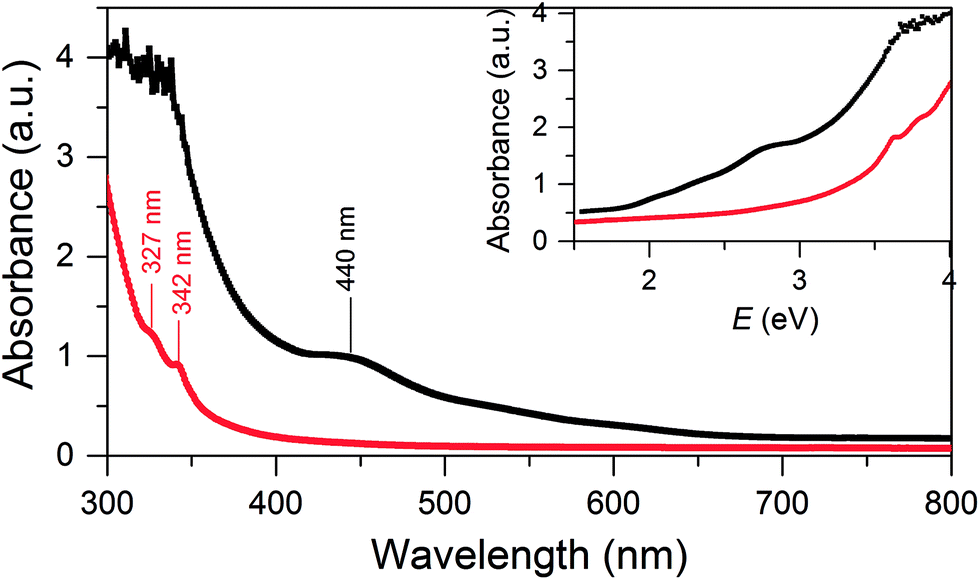 | ||
| Fig. 3 UV-vis absorption spectra of the Au nanoclusters SD/Au1 (black) and SD/Au2 (red). Inset: optical absorbance vs. photon energy (eV). | ||
Density functional theory (DFT) calculations were performed to analyze the electronic structures and optical absorptions of the clusters SD/Au1 and SD/Au2. The calculated time-dependent DFT (TDDFT) spectrum for SD/Au1 is shown in Fig. 4a. According to Fig. 4a and Table S3,† it is clear that there is a set of high and medium intensity peaks in the range of 401–484 nm (2.5–3 eV). The experimental peak observed at 440 nm is also clearly visible in the calculated absorption spectrum with an oscillator strength of 0.021–0.023 (Table S3†). The most probable transitions involved in the excitation around 440 nm are HOMO−2 → LUMO+2 and HOMO−2 → LUMO+3 with other transitions being less significant. The HOMO−2, LUMO+2, and LUMO+3 orbitals are gold core-based 6sp mixed orbitals with small contributions from the p orbitals from phosphorus (Fig. 4b). The HOMO−2 orbital has superatomic P character, as expected from the electron count of 8 in the core; similarly, the LUMO+3 has superatomic D-like character. A few low energy peaks were also observed in the calculated spectrum between 500–700 nm (1.7–2.4 eV) which could correspond to the very small peaks observed experimentally in a similar energy range. All of the relevant most probable transitions are shown in Table S3.† Apart from that, the theoretical calculation could identify a peak around ∼0.97 eV (1278 nm) which is the HOMO → LUMO transition (Fig. 4a and Table S3†). The HOMO and LUMO orbitals arise from gold 6s orbitals based in the core.
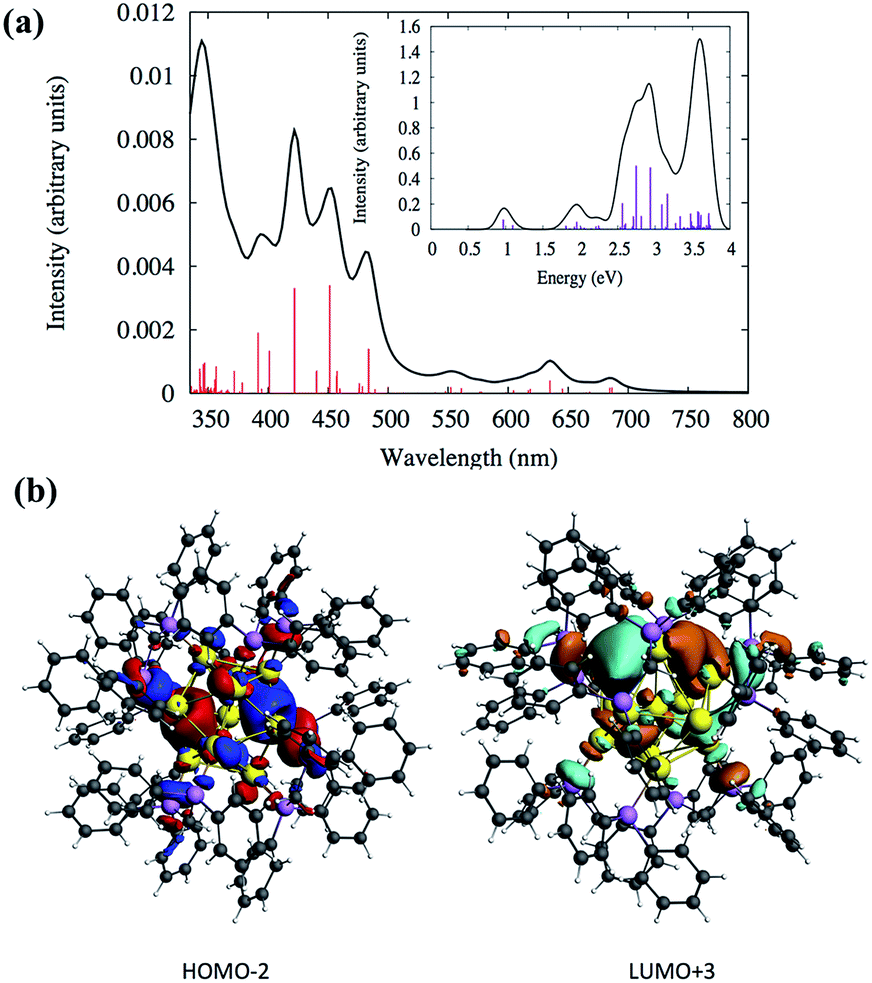 | ||
| Fig. 4 (a) Calculated TDDFT UV-vis absorption spectra of the Au nanocluster SD/Au1. Inset: optical absorbance vs. photon energy (eV). (b) The calculated HOMO−2 and LUMO+3 orbitals of SD/Au1. | ||
Similarly, the TDDFT absorption spectrum for the cluster SD/Au2 was also analyzed, as shown in Fig. 5 and Table S4.† The experimental absorption spectrum could only identify two peaks around 327 and 342 nm, whereas the theoretically calculated spectrum demonstrates similar high intensity peaks around the same energy range (Table S4†). According to the calculated spectrum (Fig. 5 and Table S4†), a high intensity peak is predicted to arise around 408 nm (∼3.0 eV) with a high oscillator strength of 0.033 which has HOMO → LUMO+25 (Table S4†) as the most probable transition contributing to that peak. Due to the underestimation expected for the level of theory used in this work, this peak could correspond to the 342 nm peak observed experimentally. The deviation in the peak positions is similar to the underestimation/overestimation observed for this level of theory in previous work.28 This HOMO → LUMO+25 transition originates out of an orbital based on atomic p orbitals from sulfur atoms mixed with d orbitals of the gold core into the π* orbitals of the phenyl rings (Fig. 5b). The HOMO is not a superatomic orbital because the superatomic S orbital (corresponding with 2 core electrons) usually lies lower in energy than the gold d and ligand atomic orbitals.
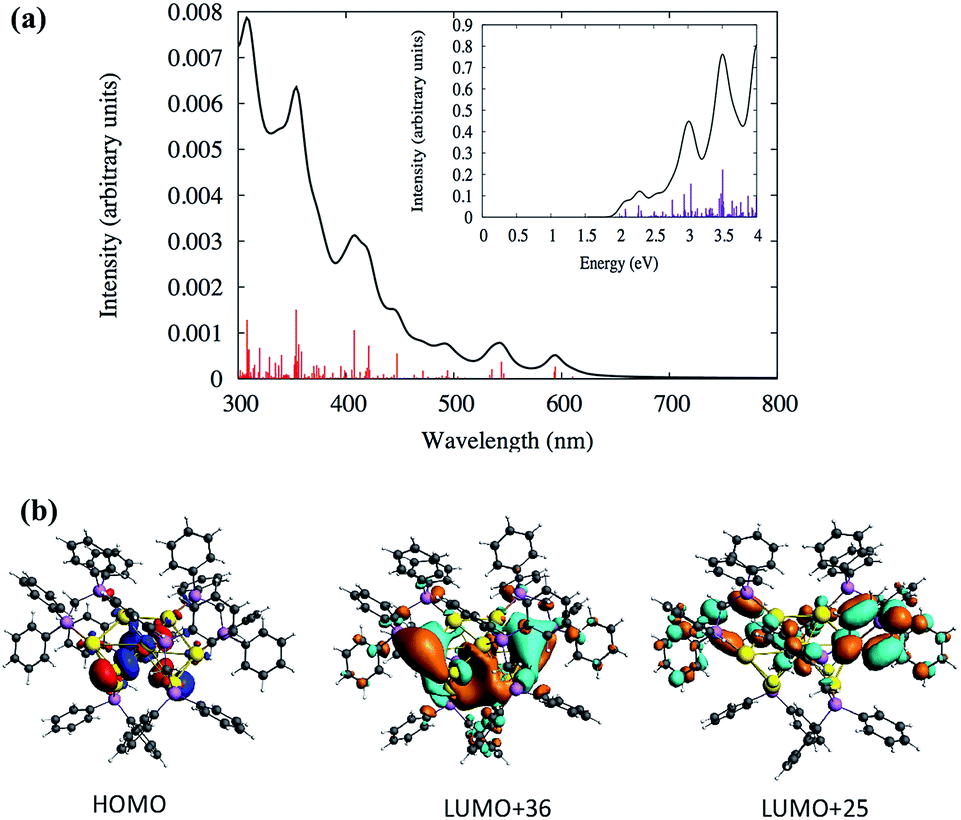 | ||
| Fig. 5 (a) Calculated TDDFT UV-vis absorption spectra of the Au nanocluster SD/Au2. Inset: optical absorbance vs. photon energy (eV). (b) The calculated HOMO, LUMO+36 and LUMO+25 orbitals of the cluster SD/Au2. | ||
From Fig. 5a, two other high intensity peaks are visible around 308 nm (4.0 eV) and 354 nm (3.5 eV) with higher oscillator strengths. The peak around 308 nm has HOMO−7 → LUMO+5 and HOMO → LUMO+36 as the most probable transitions. The occupied orbitals have p atomic orbital character from sulfur which is mixed with d orbitals of the gold core while the unoccupied orbitals have π* character from the phenyl rings which is mixed with the p and s orbitals of gold and p of phosphorus and sulfur. Fig. 5b shows the shapes of some orbitals involved in the transitions. The HOMO−1 → LUMO+34 and HOMO−2 → LUMO+24 transitions are mainly contributing to the peak at 345 nm. Here, the HOMO−2 and HOMO−1 have the p character from phosphorus and sulfur and have a small contribution from the d orbitals of the gold core atoms. The unoccupied orbitals have π* character from the phenyl rings with the p and s orbitals of gold mixed with p of phosphorus. The experimental spectrum did not identify any peaks in the visible range (400–700 nm) for the cluster SD/Au2. Even so, the calculated spectrum for SD/Au2 shows that several other weak peaks are located at 421, 447, 544 and 594 nm, although these may be too broadened to be noticeable in experiment. All of the relevant transitions are shown in Table S4.†
The photoluminescence properties of SD/Au2·2Cl
We investigated the photoluminescence properties of SD/Au1·5Cl and SD/Au2·2Cl in the solid state at room temperature, and only SD/Au2·2Cl was actively luminescent, displaying yellow emission at 591 nm under an excitation of 370 nm (Fig. 6a). The absolute luminescence quantum yield obtained by using an integrating sphere is about 4.57% with the emission decay lifetimes on the microsecond scale (τ1293 K = 1.43 and τ2293 K = 7.10 μs), indicating a phosphorescent nature (Fig. S5†). Upon gradual cooling to 93 K, the emission maxima blue-shift from 591 to 581 nm along with a 3-fold intensity enhancement. The yellow emission can be tentatively assigned to a S2− (or dppm) → Au charge transfer triplet state, probably with some mixing of a metal-centered (ds/dp) state.29 A linear correlation with the function of Iem = −808.031T + 250600 and a correlation coefficient of 0.98 was constructed between the emission intensity and temperature (Fig. 6b). To clarify the mechanism of the temperature-dependent emission behaviors of SD/Au2·2Cl, we collected variable-temperature SCXD data for the same crystal at 93, 183, 243, 273, and 293 K (Table S5†). The results firstly ruled out the possible phase transition dependent emission changes as indicated by the similar unit cell parameters as well as the invariable space group. Upon cooling, we found that the average Au–Au bond distances in SD/Au2 stay almost constant within the margin of error in the 93–293 K range (Fig. S6†). Such small variations of the Au–Au separation may not significantly influence the aurophilicity-related metal-centered (ds/dp) state, thus no red-shifted emission upon cooling was observed. Therefore, the cooling-induced emission blue-shift and intensity boosting should be caused by the enhanced rigidity of the Au8 cluster, which is in turn supported by the elongated lifetime (τ193 K = 10.08 and τ293 K = 19.60 μs) at low temperature.30 Compared to non-emissive Au13 with the same dppm protection shell, we can find that both the core structures and the incorporation of S2− can affect the radiative path and efficiency of the luminescence.31
 | ||
| Fig. 6 (a) The luminescence spectra of the cluster SD/Au2 recorded in the solid state from 293 to 93 K under an excitation of 370 nm. Inset in the top right corner: photographs of the solid sample of SD/Au2 excited by a hand-held UV lamp (365 nm) at room temperature and at 77 K. (b) The correlation between the temperature and emission intensity at 153–293 K. | ||
Electrochemistry of SD/Au1·5Cl and SD/Au2·2Cl
Electrochemical study can provide rich information related to the properties of highly monodisperse metal nanoparticles, especially with regards to HOMO–LUMO energy gaps determined from the difference value between the first oxidation and reduction peaks in differential pulse voltammograms or, equivalently, the E1/2 potentials for these redox couples in cyclic voltammograms.32 Differential pulse voltammetry (DPV) was performed to investigate the electrochemical properties of the nanocluster SD/Au1·5Cl and SD/Au2·2Cl at room temperature in CH2Cl2 solution (0.1 M nBu4NPF6 as electrolyte). As shown in Fig. 7a, the electrochemical energy gap of 1.66 V is in excellent agreement with that derived from the UV-vis spectroscopy data of 1.68 eV (440 nm) for SD/Au1·5Cl. We note that such an energy gap of Au13 is 0.10 eV smaller than that of a fully-closed icosahedral Au13(PPh3)4(SC12H25)2Cl2 cluster.33 The energy gap deduced from the DPV (Fig. 7b) of the Au8 cluster is 1.82 V, which is significantly smaller than 2.86 eV (342 nm) deduced from the experimental UV-vis spectrum of SD/Au2·2Cl. The theoretical calculation for cluster 2 predicted a HOMO–LUMO gap of 2.019 eV using the LB94/DZ level of theory. An optical gap of 2.032 eV is calculated for the first excited energy state (S1 state). Due to its low oscillator strength, it is not observed in the experimental spectrum, but lies close to the energy gap deduced from DPV. The difference between the electrochemical gaps and optical absorption of SD/Au2·2Cl is ascribed to the fact that the lowest energy excited state is not observed in the experimental UV-vis spectrum.34 The electrochemical energy gap of the Au13 cluster is smaller than that of the Au8 cluster, suggesting that their electronic structures are distinctively different and depend on the nuclearity and geometry of the metallic cores.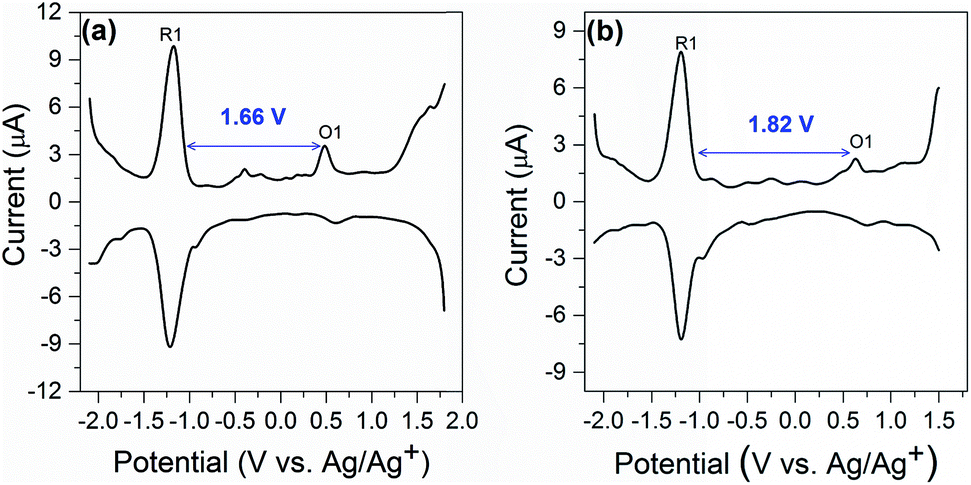 | ||
| Fig. 7 Differential pulse voltammetry spectrum of SD/Au1·5Cl (a) and SD/Au2·2Cl (b). | ||
Conclusions
In conclusion, we have successfully synthesized two new small gold nanoclusters: [Au13(dppm)6]5+ and [Au8(dppm)4S2]2+. The atomically-precise structures of these two nanoclusters were determined by single-crystal X-ray crystallography. The frameworks of SD/Au1 and SD/Au2 contain an opened C3-symmetric icosahedral Au13 kernel and a heart-shaped C2 symmetric Au8S2 core with a new “core + 4exo” structure type, respectively. The correlations between the electronic structures and optical absorption spectra are revealed by TDDFT calculations. The stability of the Au13 and Au8 nanoclusters can be ascribed to 8- and 2-electron superatoms with 1S21P6 and 1S2 configurations, respectively. More interestingly, the cluster SD/Au2 exhibits bright yellow luminescence and a temperature-induced hypsochromic shift. This study thus not only enriches the structures of ultrasmall gold nanoclusters but also provides fundamental insights into their electronic structures and luminescence properties.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the NSFC (Grant No. 21571115), the Natural Science Foundation of Shandong Province (No. ZR2014BM027 and ZR2017BM061), the Young Scholars Program of Shandong University (2015WLJH24), and the Fundamental Research Funds of Shandong University (104.205.2.5 and 2015JC045). R. D. S. and C. M. A. are grateful to the US Department of Energy (DE-SC0012273) for financial support.References
- (a) G. Schmid, Chem. Soc. Rev., 2008, 37, 1909–1930 RSC; (b) R. C. Jin, Nanoscale, 2010, 2, 343–362 RSC; (c) J. F. Parker, C. A. Fields-Zinna and R. W. Murray, Acc. Chem. Res., 2010, 43, 1289–1296 CrossRef CAS PubMed; (d) Y. Lu and W. Chen, Chem. Soc. Rev., 2012, 41, 3594–3623 RSC; (e) H. F. Qian, M. Z. Zhu, Z. K. Wu and R. C. Jin, Acc. Chem. Res., 2012, 45, 1470–1479 CrossRef CAS PubMed; (f) P. Maity, S. Xie, M. Yamauchi and T. Tsukuda, Nanoscale, 2012, 4, 4027–4037 RSC; (g) G. Li and R. C. Jin, Acc. Chem. Res., 2013, 46, 1749–1758 CrossRef CAS PubMed; (h) S. Yamazoe, K. Koyasu and T. Tsukuda, Acc. Chem. Res., 2014, 47, 816–824 CrossRef CAS PubMed; (i) S. Knoppe and T. Bürgi, Acc. Chem. Res., 2014, 47, 1318–1326 CrossRef CAS PubMed.
- R. C. Jin, C. J. Zeng, M. Zhou and Y. X. Chen, Chem. Rev., 2016, 116, 10346–10413 CrossRef CAS PubMed.
- (a) K. Konishi, Structure and Bonding, in Gold Clusters, Colloids and Nanoparticles I, ed. D. M. P. Mingos, Springer Inter-national Publishing, Berlin, 2014, vol. 161, pp. 49–86 Search PubMed; (b) B. S. Gutrath, I. M. Oppel, O. Presly, I. Beljakov, V. Meded, W. Wenzel and U. Simon, Angew. Chem., Int. Ed., 2013, 52, 3529–3532 CrossRef CAS PubMed; (c) X.-K. Wan, S.-F. Yuan, Z.-W. Lin and Q.-M. Wang, Angew. Chem., Int. Ed., 2014, 53, 2923–2926 CrossRef CAS PubMed; (d) B. K. Teo, X. B. Shi and H. Zhang, J. Am. Chem. Soc., 1992, 114, 2743–2745 CrossRef CAS; (e) L. Y. Yao and V. W.-W. Yam, J. Am. Chem. Soc., 2016, 138, 15736–15742 CrossRef CAS PubMed.
- (a) C. J. Zeng, Y. X. Chen, K. Kirschbaum, K. J. Lambright and R. C. Jin, Science, 2016, 354, 1580–1584 CrossRef CAS PubMed; (b) R. C. Jin, Nanoscale, 2015, 7, 1549–1565 RSC; (c) R. C. Jin, Nanoscale, 2010, 2, 343–362 RSC; (d) M. Z. Zhu, C. M. Aikens, F. J. Hollander, G. C. Schatz and R. C. Jin, J. Am. Chem. Soc., 2008, 130, 5883–5885 CrossRef CAS PubMed; (e) D. Crasto, G. Barcaro, M. Stener, L. Sementa, A. Fortunelli and A. Dass, J. Am. Chem. Soc., 2014, 136, 14933–14940 CrossRef CAS PubMed; (f) M. W. Heaven, A. Dass, P. S. White, K. M. Holt and R. W. Murray, J. Am. Chem. Soc., 2008, 130, 3754–3755 CrossRef CAS PubMed; (g) C. J. Chen, Y. X. Chen, K. Kirschbaum, K. Ap-pavoo, M. Y. Sfeir and R. C. Jin, Sci. Adv., 2015, 1, e1500045 Search PubMed; (h) W. Kurashige, Y. Niihori, S. Sharma and Y. Negishi, Coord. Chem. Rev., 2016, 320–321, 238–250 CrossRef CAS; (i) H. F. Qian, M. Z. Zhu, Z. K. Wu and R. C. Jin, Acc. Chem. Res., 2012, 45, 1470–1479 CrossRef CAS PubMed; (j) S. Chen, S. X. Wang, J. Zhong, Y. B. Song, J. Zhang, H. T. Sheng, Y. Pei and M. Z. Zhu, Angew. Chem., Int. Ed., 2015, 54, 3145–3149 CrossRef CAS PubMed; (k) D. Crasto, S. Malola, G. Brosofsky, A. Dass and H. Häkkinen, J. Am. Chem. Soc., 2014, 136, 5000–5005 CrossRef CAS PubMed; (l) Y. X. Chen, C. J. Zeng, C. Liu, K. Kirschbaum, C. Gayathri, R. R. Gil, N. L. Rosi and R. C. Jin, J. Am. Chem. Soc., 2015, 137, 10076–10079 CrossRef CAS PubMed; (m) H. F. Qian, W. T. Eckenhoff, Y. Zhu, T. Pintauer and R. C. Jin, J. Am. Chem. Soc., 2010, 132, 8280–8281 CrossRef CAS PubMed.
- (a) X.-K. Wan, Q. Tang, S.-F. Yuan, D.-e. Jiang and Q.-M. Wang, J. Am. Chem. Soc., 2015, 137, 652–655 CrossRef CAS PubMed; (b) X. K. Wan, S. F. Yuan, Q. Tang, D.-e. Jiang and Q. M. Wang, Angew. Chem., Int. Ed., 2015, 54, 5977–5980 CrossRef CAS PubMed; (c) X. K. Wan, W. W. Xu, S. F. Yuan, Y. Gao, X. C. Zeng and Q. M. Wang, Angew. Chem., Int. Ed., 2015, 54, 9683–9686 CrossRef CAS PubMed.
- C. J. Zeng and R. C. Jin, Structure and Bonding, in Gold Clusters, Colloids and Nanoparticles I, ed. D. M. P. Mingos, Spring-er International Publishing, Berlin, 2014, vol. 161, pp. 87–116 Search PubMed.
- P. D. Jadzinsky, G. Calero, C. J. Ackerson, D. A. Bushnell and R. D. Kornberg, Science, 2007, 318, 430–433 CrossRef CAS PubMed.
- (a) K. P. Hall and D. M. P. Mingos, Prog. Inorg. Chem., 1985, 237–325 Search PubMed; (b) J. M. M. Smits, J. J. Bour, F. A. Vollenbroek and P. T. J. Beurskens, J. Crystallogr. Spectrosc. Res., 1983, 13, 355–363 CrossRef CAS; (c) C. E. Briant, B. R. C. Theobald, J. W. White, L. K. Bell, D. M. P. Mingos and A. J. Welch, J. Chem. Soc., Chem. Commun., 1981, 5, 201–202 RSC.
- M. McPartlin, R. Mason and L. Malatesta, J. Chem. Soc., Chem. Commun., 1969, 7, 334 RSC.
- (a) Y. Shichibu, Y. Kamei and K. Konishi, Chem. Commun., 2012, 48, 7559–7561 RSC; (b) Y. Shichibu, K. Suzuki and K. Konishi, Nanoscale, 2012, 4, 4125–4129 RSC.
- (a) X.-K. Wan, Z.-W. Lin and Q.-M. Wang, J. Am. Chem. Soc., 2012, 134, 14750–14752 CrossRef CAS PubMed; (b) J. Chen, Q.-F. Zhang, P. G. Williard and L.-S. Wang, Inorg. Chem., 2014, 53, 3932–3934 CrossRef CAS PubMed.
- (a) C. E. Briant, B. R. C. Theobald, J. W. White, L. K. Bell and D. M. P. Mingos, J. Chem. Soc., Chem. Commun., 1981, 201–202 RSC; (b) H. F. Qian, W. T. Eckenhoff, M. E. Bier, T. Pintauer and R. C. Jin, Inorg. Chem., 2011, 50, 10735–10739 CrossRef CAS PubMed; (c) R. X. Jin, C. Liu, S. Zhao, A. Das, H. Z. Xing, C. Gayathri, Y. Xing, N. L. Rosi, R. R. Gil and R. C. Jin, ACS Nano, 2015, 9, 8530–8536 CrossRef CAS PubMed; (d) B. K. Teo, M. C. Hong, H. Zhang and D. B. Huang, Angew. Chem., Int. Ed. Engl., 1987, 26, 897–899 CrossRef.
- S. B. Tian, Y.-Z. Li, M.-B. Li, J. Y. Yuan, J. L. Yang, Z. K. Wu and R. C. Jin, Nat. Commun., 2015, 6, 8667 CrossRef CAS PubMed.
- J. W. A. Van der Velden, F. A. Vollenbroek, J. J. Bour, P. I. Beuskens, J. M. M. Smits and W. P. Bosman, Recl.: J. R. Neth. Chem. Soc., 1981, 100, 148–152 CAS.
- I. J. B. Lin, J. M. Hwang, D.-F. Feng, M. C. Cheng and Y. Wang, Inorg. Chem., 1994, 33, 3469–3472 Search PubMed.
- (a) H. Schmidbaur and A. Schier, Chem. Soc. Rev., 2012, 41, 370–412 RSC; (b) F. A. Vollenbroek, W. P. Bosman, J. J. Bour, J. H. Noordik and P. T. J. Beurskens, J. Chem. Soc., Chem. Commun., 1979, 387–388 RSC; (c) F. Wen, U. Englert, B. Gutrath and U. Simon, Eur. J. Inorg. Chem., 2008, 106–111 CrossRef CAS; (d) J. M. M. Smits, P. T. Beurskens, J. J. Bour and F. A. Vollenbroek, J. Crystallogr. Spectrosc. Res., 1983, 13, 365–372 CrossRef CAS; (e) M. Manassero, L. Naldini and M. San-soni, J. Chem. Soc., Chem. Commun., 1979, 385–386 RSC.
- (a) C. E. Briant, K. P. Hall, D. M. P. Mingos and A. C. Wheeler, J. Chem. Soc., Dalton Trans., 1986, 3, 687–692 RSC; (b) B. S. Gutrath, U. Englert, Y. Wang and U. Simon, Eur. J. Inorg. Chem., 2013, 2002–2006 CrossRef CAS.
- Y. Shichibu, Y. Kamei and K. Konishi, Chem. Commun., 2012, 48, 7559–7561 RSC.
- (a) Y. Shichibu and K. Konishi, Small, 2010, 6, 1216–1220 CrossRef CAS PubMed; (b) M. Sugiuchi, Y. Shichibu, T. Nakanishi, Y. Hasegawa and K. Konishi, Chem. Commun., 2015, 51, 13519 RSC.
- J. W. A. Van der Velden, J. J. Bour, J. J. Steggerda, P. T. Beurskens, M. Roseboom and J. H. Noordik, Inorg. Chem., 1982, 21, 4321–4324 CrossRef CAS.
- N. Kobayashi, Y. Kamei, Y. Shichibu and K. Konishi, J. Am. Chem. Soc., 2013, 135, 16078–16081 CrossRef CAS PubMed.
- J. M. M. Smits, J. J. Bour, F. A. Vollenbroek and P. T. Beurskens, J. Crystallogr. Spectrosc. Res., 1983, 13, 355–363 CrossRef CAS.
- J. Chen, Q. F. Zhang, T. A. Bonaccorso, P. G. Williard and L. S. Wang, J. Am. Chem. Soc., 2014, 136, 92–95 CrossRef CAS PubMed.
- M. Walter, J. Akola, O. Lopez-Acevedo, P. D. Jadzinsky, G. Calero, C. J. Ackerson, R. L. Whetten, H. Gronbeck and H. Hakkinen, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 9157–9162 CrossRef CAS PubMed.
- (a) J. W. A. Van der Velden, J. J. Bour, W. P. Bosman and J. H. Noordik, J. Chem. Soc., Chem. Commun., 1981, 23, 1218–1219 RSC; (b) M. Amasser, L. Naldini and M. Sansoni, J. Chem. Soc., Chem. Commun., 1979, 9, 385–386 Search PubMed.
- N. Kobayashi, Y. Kamei, Y. Shichibu and K. Konishi, J. Am. Chem. Soc., 2013, 135, 16078–16081 CrossRef CAS PubMed.
- Y. Yang and P. R. Sharp, J. Am. Chem. Soc., 1994, 116, 6983–6984 CrossRef CAS.
- (a) C. M. Aikens, J. Phys. Chem. A, 2009, 113, 10811–10817 CrossRef CAS PubMed; (b) C. M. Aikens, S. Li and G. C. Schatz, J. Phys. Chem. C, 2008, 112, 11272–11279 CrossRef CAS; (c) G.-T. Bae and C. M. Aikens, J. Phys. Chem. C, 2012, 116, 10356–10367 CrossRef CAS; (d) K. L. D. M. Weerawardene and C. M. Aikens, J. Phys. Chem. C, 2016, 120, 8354–8363 CrossRef CAS.
- (a) T. K. M. Lee, N. Y. Zhu and V. W. W. Yam, J. Am. Chem. Soc., 2010, 132, 17646–17648 CrossRef CAS PubMed; (b) S. Y. Yu, Q. F. Sun, T. K. M. Lee, E. C. C. Cheng, Y. Z. Li and V. W. W. Yam, Angew. Chem., Int. Ed., 2008, 47, 4551–4554 CrossRef CAS PubMed.
- V. W.-W. Yam, V. K.-M. Au and S. Y.-L. Leung, Chem. Rev., 2015, 115, 7589–7728 CrossRef CAS PubMed.
- Y. Kamei, Y. Shichibu and K. Konishi, Angew. Chem., Int. Ed., 2011, 50, 7442–7445 CrossRef CAS PubMed.
- (a) S. Chen, R. S. Ingram, M. J. Hostetler, J. J. Pietron, R. W. Mur-ray, T. G. Schaaff, J. T. Khoury, M. M. Alvarez and R. L. Whetten, Science, 1998, 280, 2098–2101 CrossRef CAS PubMed; (b) R. L. Donkers, D. Lee and R. W. Murray, Langmuir, 2004, 20, 1945–1952 CrossRef CAS; (c) V. L. Jimenez, D. G. Georganopoulou, R. J. White, A. S. Harper, A. J. Mills, D. Lee and R. W. Murray, Langmuir, 2004, 20, 6864–6870 CrossRef CAS PubMed; (d) S. K. Haram, B. M. Quinn and A. J. Bard, J. Am. Chem. Soc., 2001, 123, 8860–8861 CrossRef CAS PubMed; (e) D. Lee, R. L. Donkers, J. M. DeSimone and R. W. Murray, J. Am. Chem. Soc., 2003, 125, 1182–1183 CrossRef CAS PubMed.
- L. D. Menard, S.-P. Gao, H. Xu, R. D. Twesten, A. S. Harper, Y. Song, G. Wang, A. D. Douglas, J. C. Yang, A. I. Frenkel, R. G. Nuzzo and R. W. Murray, J. Phys. Chem. B, 2006, 110, 12874–12883 CrossRef CAS PubMed.
- (a) S. Chen, R. S. Ingram, M. J. Hostetler, J. J. Pietron, R. W. Mur-ray, T. G. Schaaff, J. T. Khoury, M. M. Alvarez and R. L. Whetten, Science, 1998, 280, 2098–2101 CrossRef CAS PubMed; (b) D. Lee, R. L. Donkers, G. Wang, A. S. Harper and R. W. Murray, J. Am. Chem. Soc., 2004, 126, 6193–6199 CrossRef CAS PubMed; (c) A. Franceschetti and A. Zunger, Appl. Phys. Lett., 2000, 76, 1731–1733 CrossRef CAS; (d) A. Fran-ceschetti and A. Zunger, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 62, 2614–2623 CrossRef CAS; (e) U. Banin, Y. Cao, D. Katz and O. Millo, Nature, 1999, 400, 542–544 CrossRef CAS; (f) S. K. Haram, B. M. Quinn and A. J. Bard, J. Am. Chem. Soc., 2001, 123, 8860–8861 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed synthesis procedure, computational details, IR, 31P NMR, ESI-MS, details of the data collection and structure refinements, and detailed analysis of the computed spectra. Additional figures (Fig. S1–S5 and Scheme S1) and tables (Tables S1–S4). CCDC 1562504, 1562505 and 1577669 for SD/Au1, SD/Au2, and SD/Au3. CCDC 1577877–1577881 for SD/Au2 at 93, 183, 243, 273, and 293 K. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc03566g |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2018 |