
Facile one-pot synthesis of MOF supported gold pseudo-single-atom catalysts for hydrogenation reactions†
Zumin
Wang
 a,
Lin
Gu
b,
Li
Song
c,
Hao
Wang
d and
Ranbo
Yu
*ad
a,
Lin
Gu
b,
Li
Song
c,
Hao
Wang
d and
Ranbo
Yu
*ad
aDepartment of Physical Chemistry, School of Metallurgical and Ecological Engineering, University of Science and Technology Beijing, Beijing 100083, P. R. China. E-mail: ranboyu@ustb.edu.cn; Fax: +86-10-62332525; Tel: +86-10-62332525
bInstitute of Physics, Chinese Academy of Sciences, Beijing 100190, China
cNational Synchrotron Radiation Laboratory, CAS Center for Excellence in Nanoscience, University of Science and Technology of China, Hefei, Anhui 230029, P. R. China
dCentre for Future Materials, University of Southern Queensland, Queensland, Australia
First published on 12th March 2018
Abstract
Catalysts based on extremely small (single atoms or pseudo single atoms) noble metals can lead to much more efficient use through enhanced reactivity and selectivity. However, fabrication of practical and stable (pseudo) single-atom catalysts on various supports remains a significant challenge. Herein, we report a facile co-precipitation and conversion strategy to fabricate a series of gold pseudo single-atom supported metal organic framework catalysts (PSAC Au/MOF) on a large scale. By well controlled synthesis of precursors, up to 1.0 wt% surface clean Au atoms can be atomically dispersed and stabilized on the support. The PSAC Au/MOF exhibited superior catalytic activity in hydrogenation of p-nitrophenol, exceeding the turnover frequency of most reported catalysts by more than one order of magnitude. No obvious decline in activity was observed after 5 cycles.
Introduction
Dispersing extremely small metal particles on supported catalysts is the ultimate pursuit in heterogeneous catalysis, since most catalytic reactions take place on the surface of metal species or at their interfaces with the supports. To achieve the utmost utilization of metals, the concept of a single atom catalyst (SAC) was put forward, which was defined as a catalyst that consists of exclusively atomic isolated metal atoms uniformly dispersed on a support.1 Downsizing metal species to the limit facilitates the activation of these metal species, thus generating more dangling bonds and empty d orbitals of metal species on the surface. Accordingly, a precisely controlled structure is highly desirable for assisting in optimizing and designing a new catalyst with high activity2 and/or selectivity.3 For practical applications, SACs offer maximum atom efficiency and minimum cost, and provide an ideal strategy to create highly efficient catalysts. So SACs have been proved promising to be a major form of catalysts for future applications.Recent developments of SACs have attracted extensive attention both experimentally3–6 and theoretically.7–9 However, two major challenges still remain in the field of SACs: (i) to prepare them with a loading content high enough for practical applications; (ii) to keep the individual metal sites from sintering/reunion under catalytic conditions. A possible effective solution to resolve these problems is to prepare pseudo single atom catalysts (PSACs) instead of SACs. PSACs consist of atomically dispersed single atoms and slightly clustered ones (like dimers, and trimers). As a result, PSACs can contain a higher metal loading density, which is enough for practical applications, while still maintaining the majority of metal species in single atom form to avoid the formation of NPs. It is a compromise between size and loading amount, which comes to a harmonious balance in PSACs. So compared with SACs, PSACs may show similar catalytic performance but more potential for promising practical industrial applications.
On the other hand, for both SACs and PSACs, the supports, which offer many anchoring sites to stably bind the metal through strong interactions for atomically dispersed catalysts, are deliberately chosen. However there are only a few known examples of supported single-atom catalysts and pseudo single atom catalysts so far, and most of them have focused on supporting single-atoms on oxides,1,5,6,9,10 zeolites11 or carbon based materials.2,3,12,13 This very limited selection of supported materials presents a significant obstacle to the application of SACs and PSACs for specific reactions. There is thereby an urgent need but it is still a significant challenge to prepare SACs and PSACs containing a larger amount of single atom active sites with greater scope of typical supports.
Currently, metal−organic frameworks (MOFs) have been emerging as a class of promising multifunctional materials owing to their nanosized channels and cavities, ultrahigh surface area, chemical tunability and structure flexibility.14 Compared with other porous materials, MOFs show tremendous advantages as platforms of supported catalysts.15–18 To name a few, loading metal species into the pores of MOFs is expected to control the migration and growth of active sites in the confined cavities and produce stable catalytic active centers, which could further increase their catalytic activities and stabilities.19,20 By incorporating functional moieties with high affinities for certain molecules, modifying the pore chemistry can be easily achieved.21,22 Thus, the selectivity of reactant adsorption and product or intermediate desorption can be tuned.23,24 Moreover, the three-dimensional topological pore structures and interactions of metal sites make MOFs possess unique advantages in terms of chemoselectivity, especially for shape selective catalysis.25–27 The introduction of MOFs into the field of SACs may open the door to effective selectivity regulators for a wide variety of important yet challenging transformations, which paves the way for future research.
Moreover, although various synthetic protocols have been applied to prepare SACs and PSACs, such as mass-selected soft landing,28 high temperature vapor transport,10 atomic layer deposition13 and combustion/pyrolysis synthesis,2 they are far from satisfactory due to two problems: (1) too low loadings limit overall performance; and (2) too complex steps and professional equipment are hard to achieve. Thus, it still remains a big challenge to develop an easy method to construct SACs/PSACs with a loading content high enough for practical applications. Herein, we report a room-temperature co-precipitation and conversion strategy to fabricate a high yield, atomically dispersed Au catalyst (pseudo single atom Au/MOF) on a series of conventional MOFs with Au loading up to about 1.0 wt%. This unique method demonstrates its effectiveness for producing ultra-small atoms on MOF supports and provides a powerful route to create clean and neat active surfaces for both metal and support without further treatment. The PSACs exhibit extremely high catalytic activities and stabilities in hydrogenation of p-nitrophenol. A turnover frequency (TOF) greater than that of surface Au atoms on reported analogous catalysts by a factor of >55 was demonstrated on the PSACs in the hydrogenation of p-nitrophenol at room temperature. And no decay in the catalytic activity was observed during catalysis.
Experimental
Materials and methods
All chemicals were of analytical grade and used as received without further purification. Trimesic acid (TA, 1,3,5-benzene-tricarboxylic acid) was purchased from J&K Scientific Ltd. Sodium sulfate (Na2SO4) and sodium hydroxide (NaOH) were from Beijing Chemical Works. Ferrous chloride (FeCl2·4H2O), chloroauric acid (HAuCl4·4H2O) and absolute ethanol were from Sinopharm Chemical Reagent Co. Ltd. Aluminium oxide (γ-Al2O3) was from Alfa Aesar. TiO2 (P25, 20% rutile and 80% anatase) was purchased from Degussa.Synthetic procedures
Characterization
Transmission electron microscope (TEM) images were obtained on a JEOL JEM-2100 (UHR) operated at 200 kV. Spherical aberration-corrected STEM was carried out using a JEOL 2100F (JEOL) transmission electron microscope with a CEOS (Heidelberg) probe aberration corrector, operated at 200 keV. Samples were prepared by placing a drop of a dilute toluene dispersion of nanocrystals onto a 200 mesh carbon-coated copper grid and evaporated immediately at ambient temperature. Scanning electron microscopy (SEM) images were obtained using a ZEISS SUPRA 55 microscope operating at 5.0 kV. The samples were prepared by placing a few drops of the suspensions on small pieces (about 5 mm × 5 mm) of copper wafer. The X-ray diffraction patterns (XRD) of the samples were obtained on a Panalytical X’Pert PRO MPD X-ray diffractometer operated at 1600 W power (40 kV, 40 mA) and equipped with a Cu Kα radiation source (λ = 1.54 Å). Thermogravimetry-differential thermal analysis (TG-DTA) curves were recorded on a TG/DTA6300 thermo-analyzer at a linear heating rate of 5 °C min−1 under an air atmosphere. The nitrogen adsorption–desorption isotherm curves were measured on a Quantachrome Instrument NOVA4000 at 77 K. The loading content of gold was determined by ICP-AES (IRIS, Intrepid II XSP, Thermo Electron, USA). The X-ray photoelectron spectra (XPS) were recorded on a Kratos spectrometer (AXIS Ultra DLD, Shimadzu/Kratos, Ltd) with monochromatic Al Kα radiation (hν = 1486.6 eV). The binding energies for all spectra were calibrated with respect to the C 1s reference signal at 284.8 eV. The XAFS spectra at the Au L III-edge of the samples were measured at the 1W1B beamline at the Beijing Synchrotron Radiation Facility (BSRF) and beamline BL11U of the National Synchrotron Radiation Laboratory (NSRL). The output beam was selected by an Si(111) monochromator. The energy was calibrated by the Au foil. The data were collected at room temperature under fluorescence mode by using a solid-state detector. Au foil as a standard compound was measured simultaneously by using the third ionization chamber so that energy calibration could be performed scan by scan. The Athena software package was employed to process the data.Catalytic test
The catalytic test was directly taken in a quartz cuvette in the UV-vis spectrophotometer at room temperature. The aqueous solutions of p-nitrophenol (1 mmol L−1) and NaBH4 solution (0.1 mol L−1) were freshly prepared. In a typical process (as has been reported32), 150 μL of nitrophenol and 2 mL of NaBH4 were mixed. Then 20 μL of the aqueous solution containing the catalysts (1.4 mg mL−1) was quickly injected into the cuvette to trigger the reaction. The intensity of the absorption peak at 400 nm for p-nitrophenol was monitored and recorded by UV-vis spectroscopy along with the corresponding time. All the samples were thoroughly washed and purified by centrifugation before the next catalytic cycle.Results and discussion
Synthesis and characterization
The mechanism for the formation of the pseudo single atoms supported on MOF structures has been demonstrated as depicted in Scheme 1. The material formation experienced three stages, that is, the nucleation–growth of layered metal hydroxides and the intercalation of the anions. In the nucleation–growth stage, M(OH)x was obtained through the reaction of M2+ and NaOH. And the anions between the layers reacted as a blocking barrier to avoid further oxidation and dehydration. Then the following introduced AuCl4− anions would be firmly attracted on the M(OH)x surface via electrostatic interactions and replaced some of the anions (i.e. sulfate ion) and still maintained its former structures.33 As a consequence, when the auto redox reaction occurred, ultra small Au atoms were formed and every one of them has been firmly enclosed between layers, forming the original Au–hydroxide hybrid structures (i.e. doped processors).34 The mutual inhibition effect between hydroxides and Au makes their hybrids rather stable and able to keep their size, shape, and structure. Afterward the organic linker was added to trigger the conversion of the hydroxide into a MOF through a weakening of the structure through progressive breaking of M–O(H)–M bonds by coordination of the carboxylic organic linker.35 Finally, single or pseudo single Au atoms were supported firmly on the formed MOF support, and were either captured by the benzene rings and the carboxyl groups from the organic motif or interacted with the metal clusters and open metal sites in the MOFs.36 And some of them can even be encapsulated or confined within the pores. More detailed characterization as well as model simulations were indispensable in order to classify their locations, and are still ongoing. During all procedures, the absence of surface modification on both Au and MOFs means that they are naked on their surfaces and are thus ultra-active for catalytic reactions.17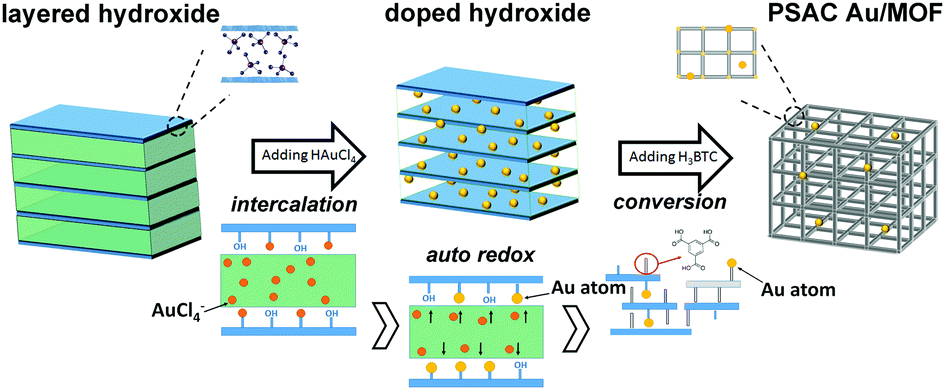 | ||
| Scheme 1 Schematic illustration of the preparation procedures and plausible mechanism of the formation of PSAC Au/MOF. | ||
It should be noted that the whole process was continuous and tandem, and carried through in one reactor. We describe this process in three divided steps only to clearly define the purpose of each operation. Moreover, as shown in Fig. S1 (ESI†), this approach is very productive (gram level production per synthetic batch) and has a good yield rate (>62%). Notably, our clean and fast method can easily realize the above procedure on a large scale, just with larger volumes of the solutions being used. The other conditions were the same as in the above procedures. And it is universal for many other Au/MOF catalysts (namely, CoBTC, NiBTC, CuBTC, as describe in the ESI†). It is a promising synthesis route due to its fast kinetics, mild conditions, high space–time–yields and low waste generation, which makes it a remarkably accessible, affordable and environmentally-friendly process. A patent application has been submitted for the synthesis method for catalyst preparation (the application number is CN2018102016905).
The scanning electron microscopy (SEM) images in Fig. S2a (ESI†) show that the as-obtained Au/MOF hybrids exhibited nanosized (∼250 nm) crystal aggregates with no residual crystals of precursors or impurities observed. The TEM image (Fig. S2, ESI†) also showed the typical MOF morphology; no Au NPs beneath the loose structure can be observed by their deeper contrast from MOFs. So to verify the existence of Au atoms, high-resolution high-angle annular dark-field (HAADF-STEM) imaging (Fig. 1a) and EDS element mapping (Fig. 1b) have been carried out, which revealed that single or pseudo single Au atoms were evenly dispersed in the sample. The PXRD patterns of the materials (Fig. 1c) matched well with the simulated result, indicating the lack of the presence of any other phrases, and the framework structure was retained upon the loading of Au species. On the other hand, peaks due to Au NPs at 38.27°, 44.60° and 64.68° were also not detected, indicating the tiny size of the Au species. The permanent porosity of all these samples is preserved as confirmed by measurement of the N2 gas-adsorption isotherm (Fig. 1d), which exhibits a Type I behavior similar to the reported results. The corresponding Langmuir surface areas of the MOF and Au/MOF were calculated to be 723 and 500 m2 g−1, respectively; values that are also similar to those found in previous reports.35 ICP-AES revealed that the amounts of Au for all samples were found to be around 1.0 wt%. To further clarify the existing state of Au species in the sample, we performed X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) spectrometry (Fig. 2a and b), considering the poor resolution of X-ray photoelectron spectroscopy (XPS) signals for the detection of a small amount of rather tiny Au species (Fig. S3, ESI†).37 For comparison, an Au nanoparticle supported catalyst was also synthesized (described in the ESI†) and tested. There was only one notable peak (with a certain degree of separation as a result of the relativistic effect caused potential well distortion) in the region 2 to 3 Å from the Au–Au contribution, and no peak in the region before and after the Au–Au bond from the Au–Fe, Au–C, and Au–Cl contribution, confirming the absence of any other atoms around the dispersed Au atoms in the samples. The occurrence of Au–Au long coordination at a distance of about 2.57 Å may suggest the presence of loosely clustered Au single atoms as observed in the HAADF-STEM images. Obviously, the shapes of the Au peaks of the PSAC and NP supported one were quite different, suggesting the difference in their coordination environment and electronic state distribution. Therefore, the structure and behaviour of the as-prepared PSACs, in essence, resembles those of catalysts with metal atoms in totally isolated single form, and therefore they can be considered as PSACs. Moreover, Fig. 2b demonstrated that the white-line intensity of the XANES spectra of the pseudo single atom and nanoparticle supported catalysts were almost exactly the same. Both of them matched well with Au foil (only very slightly higher), suggesting that the Au species were in a zero valent state and consisted of uncharged Au atoms. The similarity between the Au foil and our sample strongly suggested that the Au atoms were surface naked and in pure metallic form.
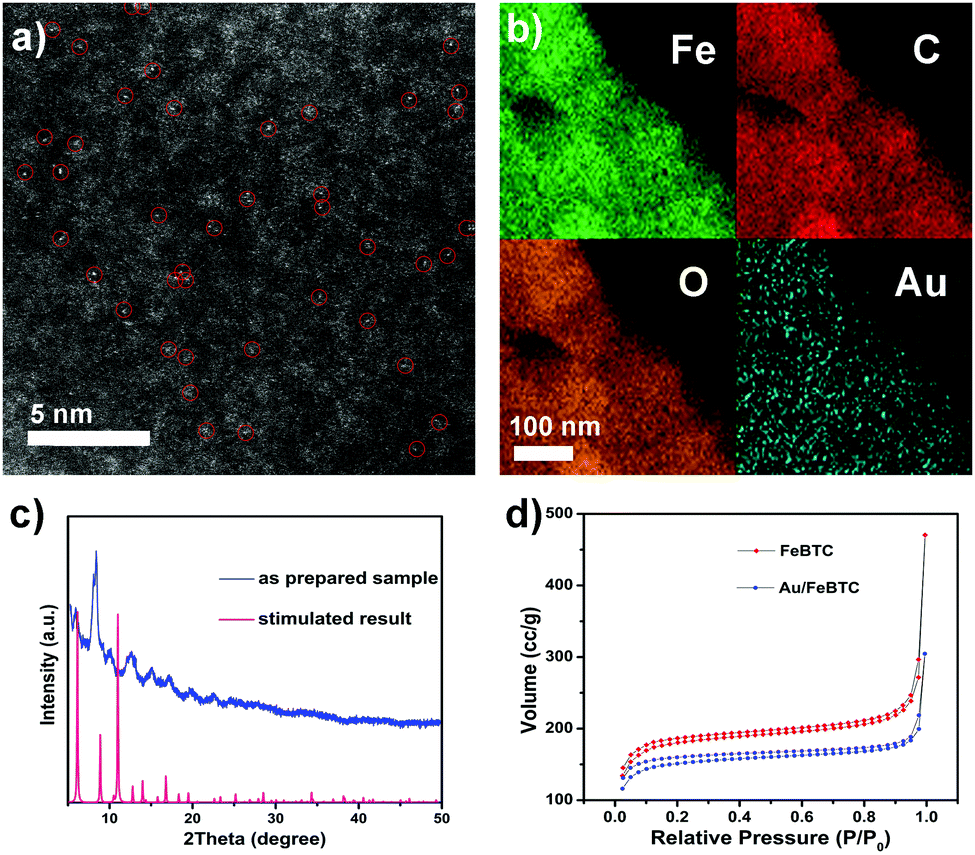 | ||
| Fig. 1 (a) High-resolution high-angle annular dark-field (HAADF) STEM image; (b) EDS elemental mapping; (c) XRD; and (d) N2 gas-adsorption isotherm of the as-prepared PSAC Au/FeBTC. | ||
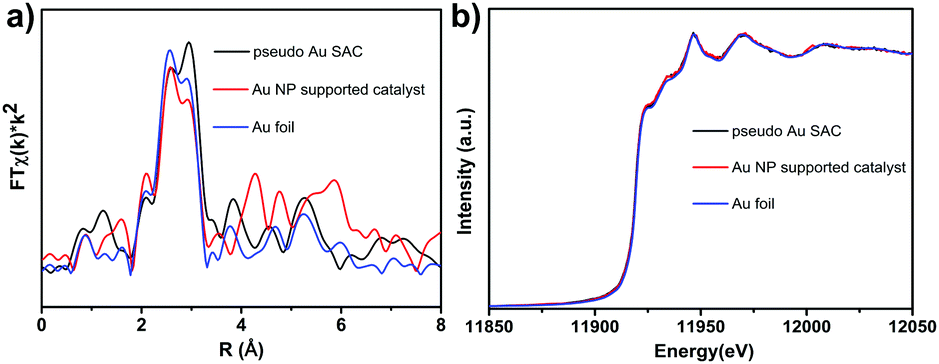 | ||
| Fig. 2 (a) FT-EXAFS spectra of Au/MOF and bulk Au foil at the Au K-edge, showing the surrounding atoms adjacent to Au atoms; (b) normalized XANES spectra at the Au LIII-edge of the Au/MOF samples. | ||
TG-DTA analysis (Fig. S4, ESI†) indicated that the product was thermally stable until nearly 320 °C. And the product obtained from this synthesis process was extremely clean with no mass loss before the decomposition temperature, owning to the absence of high boiling point organic solvents during all the synthesis procedures. This meant that there were no cumbersome and repeated solvent exchanges or calcination procedures necessary to remove solvents and unreacted materials and activate internal surfaces and tunnels.
The conversion of layered hydroxides into MOF based materials remains a promising synthesis route due to its fast kinetics, mild conditions, high space–time–yields and low waste generation, which makes it a remarkably accessible, affordable and environmentally-friendly process. The most exciting thing is that the obtained hybrid materials possessed a clean porous scaffold and bare active sites (ultra small pseudo single Au atoms) via an in situ redox reaction, which made them ideal choices for catalytic reactions.
Catalytic test
p-Nitrophenol is a serious toxic organic compound in industrial wastes, which can cause serious pollution in water, and it is difficult to completely decompose p-nitrophenol by traditional wastewater treatments such as biodegradation and physical methods. However, its hydrogenation product, p-aminophenol, is a commercially important chemical and medical intermediate for manufacturing agrochemicals, pharmaceuticals and dyes. So the catalytic hydrogenation of p-nitrophenol is an environmentally benign technology for the production of p-aminophenol, which can change a harmful chemical into a beneficial one. Besides practical applications, the catalytic hydrogenation of p-nitrophenol is often selected as a probe reaction to investigate the catalytic properties of studied samples. So we used the reduction of p-nitrophenol by NaBH4 as a model system to quantitatively evaluate the catalytic activity of PSACs Au/MOF. And the color changes along with the reduction process of p-nitrophenol also provide a simple way of monitoring the reaction kinetics based on UV-vis absorption spectroscopic measurements. It is well-known that a 4-NP solution exhibits a strong absorption peak at 317 nm under neutral or acidic conditions. Upon the addition of NaBH4, the alkalinity of the solution increases, and 4-NP ions become the dominant species, producing a spectral shift to 400 nm. After the catalyst was added, the absorption peak at 400 nm gradually dropped in intensity as the reduction reaction proceeded (Fig. 3a). At the same time, with the production of p-aminophenol, a new absorption peak started to increase as a shoulder at 315 nm. Because the peak at 400 nm was much stronger than that at 315 nm, we decided to measure the concentrations of p-nitrophenolate ions and thus to monitor the progress or kinetics of the reaction by recording the absorbance at 400 nm. For all experiments, since excess NaBH4 was used, the BH4− concentration remained essentially constant throughout the reaction. An isosbestic point between the two absorption bands was observed, indicating that only two principal species, p-nitrophenol and p-aminophenol, influence the reaction kinetics. Therefore, pseudo-first-order kinetics could be applied for the evaluation of rate constants. This pseudo-first-order kinetics can be written as ln(C0/Ct) = kt, where k is the kinetic rate constant, and C0 and Ct are the initial and apparent concentrations of 4-NP, respectively. The ratio of Ct and C0, where Ct and C0 are the p-nitrophenol concentrations at time t and 0, respectively, was measured from the relative intensity of the respective absorbance, At/A0. The linear relations of ln(Ct/C0) versus time were observed for all catalyst particles, indicating that the reactions followed first-order kinetics. The rate constants were estimated from diffusion-coupled first order reaction kinetics using the slopes of the straight lines in Fig. 3b.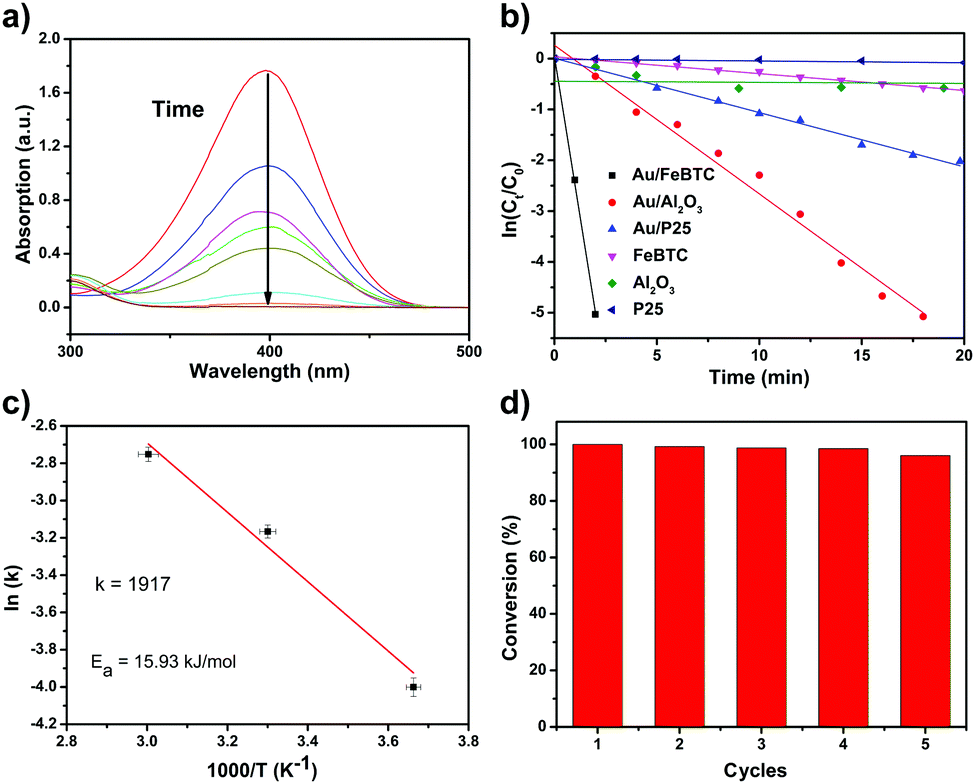 | ||
| Fig. 3 (a) The extinction spectra at different reaction times, indicating the disappearance of the peak for p-nitrophenol due to the reduction of −NO2 groups into −NH2 groups; (b) plots of ln(Ct/C0) vs. time using different samples under the same test conditions. (c) The Arrhenius plots for reactions catalyzed by different catalysts. The activation energy (Ea) can be calculated from the slope of the linear fitting in each case. (d) The stability test of the PSAC Au/MOF sample. | ||
In order to study the activities of all catalysts, the absorption effect of catalysts and non-catalytic reactions should be eliminated. Therefore, a set of control experiments were conducted. First of all, an experiment with all the same parameters except that no catalyst but 20 μL of water was added instead was carried out. As shown in Fig. S4 (ESI†), no obvious changes in the respective absorbance could be detected for as long as 2 h. So we could safely conclude that the non-catalytic hydrogenation reaction was rather slow and it could be neglected compared with the fast catalytic reaction. Secondly, a series of experiments was devised to eliminate the pure adsorption of the catalysts. So p-nitrophenol solution and catalyst solution were mixed without a hydrogen source (NaBH4), and NaOH was added to maintain the same alkalinity conditions. From the data observed (Fig. S5, ESI†), despite the ultra-large surface area of all the catalysts, the velocities of adsorption were at least two orders of magnitude lower than those of the catalytic reactions. So we can safely conclude that the observed fast decrease of the absorption peaks resulted mainly from the catalytic reaction triggered by our catalysts.
To obtain the activation energy (Ea) of reduction of p-nitrophenol catalyzed by PSACs Au/FeBTC, the catalytic experiments were carried out at temperatures ranging from 0 to 60 °C (Fig. S8, ESI†), and Ea was determined to be 15.9 kJ mol−1, which is among the best results of the reported values.38,39 In order to evaluate the basic properties of the catalysts, not only the kinetic rate constant but also the turnover frequency (TOF) was studied. The rate constant of PSACs Au/FeBTC hybrid was 2.49 min−1 which was among the highest values ever reported under the exact same conditions (at 25 °C). And the approximate turnover frequency (TOF),40 defined as moles of p-aminophenol molecules generated per moles of noble metal atoms per minute (as described by eqn (1)) during the first 2 min (conversion reached >90%), has been used to compare the catalytic activities of the samples.
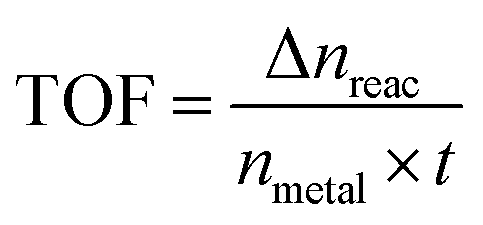 | (1) |
Additional comparison about the catalytic activity has been made between the present PSACs Au/MOF and other reported catalyst systems for the reduction of 4-NP (Table S1, ESI†). Compared with other traditional noble metal supported catalysts, the TOF of the metal/MOF hybrid catalyst was 581 min−1, which varied from hundreds to thousands of times the reported results. The detailed comparison is listed in Table S1 (ESI†). We believed that the strong synergistic interactions between the Au atoms and the MOF support were significantly important in achieving superior activity.
In order to demonstrate superiority of the fast conversion and in situ redox approach, we compared our samples with those synthesized by using traditional pre-synthesis and post-synthesis methods (see detailed information of those samples in Fig. S7 and S8, ESI†). On one hand, in terms of pre-synthesized Au NP-loaded MOF catalysts, it is always necessary to use surface protecting agents (or so-called stabilizers) to reduce the Au particle size and to prevent NPs aggregation. The presence of stabilizer (such as PolyVinyl Pyrrolidone or PVP and Sulfur Alcohol) inhibits the active sites of the metal NPs, resulting in low activity of the catalysts.41 On the other hand, in terms of post-synthesized Au NPs in and on MOF catalysts, the formation of Au NPs within the MOF matrix causes structure destruction, and the uncontrollability of Au nucleation and growth results in a larger average size and broader size distribution of Au NPs, which jeopardizes the catalytic performance. As for our in situ redox Au atoms, because of the clean surfaces and the strong binding force between the active sites and supports, no surface reconstruction was needed, and thus no induction time was observed (Table 1).38
| Noble metals loaded catalysts | T (K) | k (min−1) | TOF (min−1) | t ind (min) |
|---|---|---|---|---|
| a Traditional pre-synthesis Au stabilized with PVP. b Traditional pre-synthesis Au NPs stabilized with sulfur alcohol.46 c Post-synthesized Au NPs in and on MOF catalysts. | ||||
| Catalyst as-prepared | 298 | 2.49 | 581 | 0 |
| Catalyst method 1a | 298 | 0.0014 | 5.6 | 6.1 |
| Catalyst method 2b | 298 | 0.0051 | 10.5 | 2.3 |
| Catalyst method 3c | 298 | 0.638 | 163 | 1.5 |
Finally, stability tests were carried out in terms of durability in five cyclic usages. As shown in Fig. 3d, there was a slight decrease of activity after five cycles, but the conversion rate within two minutes was not changed. Additionally, after 10 cycles of catalytic reaction, the catalyst kept its original morphology and activity (Fig. S9, ESI†), indicating its excellent stability and long catalytic lifetime. Obviously, the as-synthesized Au/MOFs catalysts have shown excellent catalytic performance, for the following reasons:
(1) First, this novel synthetic process without using high-temperature solvent leads to clean MOFs with high nanoporosity and open pore network, which permits full accessibility and fast molecule diffusion of 4-NP as well as the products on and off the Au surface.
(2) Second, MOFs offer preferable specific adsorption for the enrichment of π-rich 4-NP from solution via π–π stacking interaction,42,43 leading to a high local concentration of reactants.
(3) Third, the pseudo single Au atoms on the MOF hybrid catalysts were synthesized in situ without any stabilizer or surfactant; therefore, the unsaturated surface atoms of Au species may coordinate in a better manner and adsorb 4-NP, thus speeding up the reaction.42,44,45 This well-studied reaction can be modelled by classical Langmuir–Hinshelwood kinetics, in which the surface coverage of metals by adsorbed reactants influenced the total activity greatly.38 So a clean and neat metal surface is the key to improvement of catalytic performance.
(4) Finally, the porous surface structure of the MOF may offer steric restriction to confine and effectively suppress the aggregation of Au atoms and their leaching and dissolution into a reaction mixture.
This unusual atomic constitution of supported metals is suggestive of a new approach to prepare extremely efficient pseudo single-atom catalysts.
Conclusions
In all, we have successfully prepared PSAC Au/MOF materials on a large scale via a facile wet chemistry route. Without addition of any reducing agents or surfactants, the whole reaction is a clean redox process that occurs between hydroxides and AuCl4− in an alkaline aqueous solution, then followed by a fast conversion from layered hydroxides into porous MOF materials through anion exchange in the aqueous ethanolic linker solution at room temperature. Such a fast, green, and large-scale synthetic strategy is of great significance. Using this promising clean synthesis strategy, the obtained hybrids possessed clean cavities and bare metal surfaces, which is favorable for catalytic applications. STEM and EXAFS data show the samples to be primarily supported pseudo single Au atoms. These PSACs showed extraordinary activities as well as maximum atom efficiency, with a TOF of about 600 for hydrogenation of p-nitrophenol as a probe reaction, exceeding the TOF of most reported catalysts by more than one order of magnitude. No obvious decline in activity was observed for 5 cycles. This work demonstrates that supported atomically dispersed metal sites can be preserved during the transformation of supports under mild conditions. This study may open a new avenue for the design and preparation of advanced SACs and PSACs with a larger loading amount, enhanced selectivity and activity on various supports.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The National Natural Science Foundation of China (No. 51472025 and 21671016) supported this work financially. We are thankful to Dr Shuangming Chen from University of Science and Technology of China (USTC) for the help in synchrotron radiation data collection and analysis, and Dr Qinghua Zhang from Institute of Physics, Chinese Academy of Science for the help in electron microscope observation and analysis.Notes and references
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS PubMed.
- H. Fei, J. Dong, M. J. Arellano-Jimenez, G. Ye, N. Dong Kim, E. L. Samuel, Z. Peng, Z. Zhu, F. Qin, J. Bao, M. J. Yacaman, P. M. Ajayan, D. Chen and J. M. Tour, Nat. Commun., 2015, 6, 8668 CrossRef CAS PubMed.
- H. Yan, H. Cheng, H. Yi, Y. Lin, T. Yao, C. Wang, J. Li, S. Wei and J. Lu, J. Am. Chem. Soc., 2015, 137, 10484–10487 CrossRef CAS PubMed.
- R. Lang, T. Li, D. Matsumura, S. Miao, Y. Ren, Y. T. Cui, Y. Tan, B. Qiao, L. Li, A. Wang, X. Wang and T. Zhang, Angew. Chem., Int. Ed., 2016, 55, 16054–16058 CrossRef CAS PubMed.
- J. Lin, A. Wang, B. Qiao, X. Liu, X. Yang, X. Wang, J. Liang, J. Li, J. Liu and T. Zhang, J. Am. Chem. Soc., 2013, 135, 15314–15317 CrossRef CAS PubMed.
- T. K. Ghosh and N. N. Nair, ChemCatChem, 2013, 5, 1811–1821 CrossRef CAS.
- K. Mao, L. Li, W. Zhang, Y. Pei, X. C. Zeng, X. Wu and J. Yang, Sci. Rep., 2014, 4, 5441 CrossRef CAS PubMed.
- M. Moses-DeBusk, M. Yoon, L. F. Allard, D. R. Mullins, Z. Wu, X. Yang, G. Veith, G. M. Stocks and C. K. Narula, J. Am. Chem. Soc., 2013, 135, 12634–12645 CrossRef CAS PubMed.
- F. Li, Y. Li, X. C. Zeng and Z. Chen, ACS Catal., 2014, 5, 544–552 CrossRef.
- J. Jones, H. Xiong, A. T. DeLaRiva, E. J. Peterson, H. Pham, S. R. Challa, G. Qi, S. Oh, M. H. Wiebenga, X. I. Pereira Hernandez, Y. Wang and A. K. Datye, Science, 2016, 353, 150–154 CrossRef CAS PubMed.
- M. Yang, S. Li, Y. Wang, J. A. Herron, Y. Xu, L. F. Allard, S. Lee, J. Huang, M. Mavrikakis and M. Flytzani-Stephanopoulos, Science, 2014, 346, 1498–1501 CrossRef CAS PubMed.
- G. Vile, D. Albani, M. Nachtegaal, Z. Chen, D. Dontsova, M. Antonietti, N. Lopez and J. Perez-Ramirez, Angew. Chem., Int. Ed., 2015, 54, 11265–11269 CrossRef CAS PubMed.
- S. Sun, G. Zhang, N. Gauquelin, N. Chen, J. Zhou, S. Yang, W. Chen, X. Meng, D. Geng, M. N. Banis, R. Li, S. Ye, S. Knights, G. A. Botton, T.-K. Sham and X. Sun, Sci. Rep., 2013, 3 Search PubMed.
- H. Furukawa, K. E. Cordova, M. O’Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- A. Dhakshinamoorthy and H. Garcia, Chem. Soc. Rev., 2012, 41, 5262–5284 RSC.
- L. Oar-Arteta, T. Wezendonk, X. Sun, F. Kapteijn and J. Gascon, Mater. Chem. Front., 2017, 1, 1709–1745 RSC.
- Q.-x. Luo, B.-w. An, M. Ji and J. Zhang, Mater. Chem. Front., 2018, 2, 219–234 RSC.
- J. Zhang, B. An, Y. Hong, Y. Meng, X. Hu, C. Wang, J. Lin, W. Lin and Y. Wang, Mater. Chem. Front., 2017, 1, 2405–2409 RSC.
- H.-L. Jiang, B. Liu, T. Akita, M. Haruta, H. Sakurai and Q. Xu, J. Am. Chem. Soc., 2009, 131, 11302–11303 CrossRef CAS PubMed.
- Y. Lei, Y. Wan, G. Li, X.-Y. Zhou, Y. Gu, J. Feng and R. Wang, Mater. Chem. Front., 2017, 1, 1541–1549 RSC.
- E. Virmani, O. Beyer, U. Lüning, U. Ruschewitz and S. Wuttke, Mater. Chem. Front., 2017, 1, 1965–1974 RSC.
- J. An and N. L. Rosi, J. Am. Chem. Soc., 2010, 132, 5578–5579 CrossRef CAS PubMed.
- L. Valenzano, B. Civalleri, S. Chavan, G. T. Palomino, C. O. Areán and S. Bordiga, J. Phys. Chem. C, 2010, 114, 11185–11191 CAS.
- D. Saha, Z. Bao, F. Jia and S. Deng, Environ. Sci. Technol., 2010, 44, 1820–1826 CrossRef CAS PubMed.
- M. Zhao, K. Deng, L. He, Y. Liu, G. Li, H. Zhao and Z. Tang, J. Am. Chem. Soc., 2014, 136, 1738–1741 CrossRef CAS PubMed.
- M. Zhao, K. Yuan, Y. Wang, G. Li, J. Guo, L. Gu, W. Hu, H. Zhao and Z. Tang, Nature, 2016, 539, 76–80 CrossRef CAS PubMed.
- S. C. King, R.-B. Lin, H. Wang, H. D. Arman and B. Chen, Mater. Chem. Front., 2017, 1, 1514–1519 RSC.
- S. Abbet, A. Sanchez, U. Heiz, W. D. Schneider, A. M. Ferrari, G. Pacchioni and N. Rösch, J. Am. Chem. Soc., 2000, 122, 3453–3457 CrossRef CAS.
- J. M. R. Génin, A. A. Olowe, P. Refait and L. Simon, Corros. Sci., 1996, 38, 1751–1762 CrossRef.
- J. Bernal, D. Dasgupta and A. Mackay, Clay Miner. Bull., 1959, 4, 15–30 CAS.
- Z. Wang, J. Qi, K. Zhao, L. Zong, Z. Tang, L. Wang and R. Yu, Mater. Chem. Front., 2017, 1, 1629–1634 RSC.
- Z. Wang, S. Jiang, Y. Li, P. Xu, K. Zhao, L. Zong, H. Wang and R. Yu, Sci. China Mater., 2017, 60, 646–653 CrossRef CAS.
- P. Huang, J. Liu, F. Wei, Y. Zhu, X. Wang, C. Cao and W. Song, Mater. Chem. Front., 2017, 1, 1550–1555 RSC.
- Y. Ma, B. Li and S. Yang, Mater. Chem. Front., 2018, 2, 456–467 RSC.
- G. Majano, O. Ingold, M. Yulikov, G. Jeschke and J. Pérez-Ramírez, CrystEngComm, 2013, 15, 9885 RSC.
- L. B. Vilhelmsen, K. S. Walton and D. S. Sholl, J. Am. Chem. Soc., 2012, 134, 12807–12816 CrossRef CAS PubMed.
- A. A. Herzing, C. J. Kiely, A. F. Carley, P. Landon and G. J. Hutchings, Science, 2008, 321, 1331–1335 CrossRef CAS PubMed.
- P. Herves, M. Perez-Lorenzo, L. M. Liz-Marzan, J. Dzubiella, Y. Lu and M. Ballauff, Chem. Soc. Rev., 2012, 41, 5577–5587 RSC.
- J. Zeng, Q. Zhang, J. Chen and Y. Xia, Nano Lett., 2010, 10, 30–35 CrossRef CAS PubMed.
- L. Gomathi Devi and R. Shyamala, Mater. Chem. Front., 2018 10.1039/c7qm00536a.
- S. Ikeda, S. Ishino, T. Harada, N. Okamoto, T. Sakata, H. Mori, S. Kuwabata, T. Torimoto and M. Matsumura, Angew. Chem., Int. Ed., 2006, 45, 7063–7066 CrossRef CAS PubMed.
- J. Cao, S. Mei, H. Jia, A. Ott, M. Ballauff and Y. Lu, Langmuir, 2015, 31, 9483–9491 CrossRef CAS PubMed.
- T. Zeng, X. Zhang, S. Wang, H. Niu and Y. Cai, Environ. Sci. Technol., 2015, 49, 2350–2357 CrossRef CAS PubMed.
- X. Liu, S. Cheng, J. Long, W. Zhang, X. Liu and D. Wei, Mater. Chem. Front., 2017, 1, 2005–2012 RSC.
- H. Yamamoto, H. Yano, H. Kouchi, Y. Obora, R. Arakawa and H. Kawasaki, Nanoscale, 2012, 4, 4148–4154 RSC.
- J. Wang, N. Goswami, T. Shu, L. Su and X. Zhang, Mater. Chem. Front., 2018 10.1039/c7qm00609h.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8qm00081f |
| This journal is © the Partner Organisations 2018 |