
Ion extraction applications of bilayer-structured hybrid silicas†
R.
Besnard
,
R.
Winkler
,
G.
Arrachart
 *,
J.
Cambedouzou
and
S.
Pellet-Rostaing
*
*,
J.
Cambedouzou
and
S.
Pellet-Rostaing
*
ICSM, CEA, CNRS, ENSCM, Univ Montpellier, Bagnols sur Cèze Cedex, France. E-mail: stephane.pellet-rostaing@cea.fr
First published on 15th March 2018
Abstract
The self-assembly behaviour of amino-undecyl-triethoxysilane, which can be considered as an amphiphilic organosilane, has been used for the preparation of nanostructured materials. Due to the possibilities offered by this approach, lamellar hybrid materials bearing amine groups can be achieved. In this study, the reactive amino groups at the surface of the hybrid silica were modified for introducing ligands. For this purpose, various glycolamic acid groups have been introduced at the interface between two bilayers on the hybrid material obtained after condensation of silylated amines. The availability of the amino groups and the efficiency of the chemical immobilization of amic acid ligands onto the materials were followed by FTIR, NMR and TGA analysis, and the structural modifications induced by grafting were evaluated by SAXS experiments. The resulting materials exhibit a large number of amic acid groups. The adsorption capacities for rare earth elements were also investigated.
Introduction
There is increasing interest in the development of new solid phase extraction materials based on organosilica. Silica-based organic–inorganic hybrids are attractive materials due to their versatility, which originates from the combination of their inorganic silica network properties and their organic constituents.1Organosilica and mainly mesoporous organosilica sorbents have been actively investigated with success for the removal of several inorganic species as heavy metal ions,2 toxic oxyanions,3 lanthanide/actinides,4,5 radionuclides6 or organic pollutants.7,8
For these applications, there are three major strategies for the synthesis of organosilica sorbents.9 The first strategy involves the chemical grafting of specific ligands on the surface of mesoporous silica. An alternative synthesis procedure consists of a “one-pot” approach, in which the mesoporous silica is formed and functionalized by the simultaneous co-condensation of tetraethoxysilane (TEOS) with an organosiloxane precursor in the presence of structure-directing agents. Higher organic loading can be achieved using bridging organic groups of the type (R′O)3Si–R–Si(OR′)3 as single-source organosilica precursors through the synthesis of periodic mesoporous organosilicas (PMOs), in which the organic bridges are incorporated in the three-dimensional network structure of the silica matrix.10,11
Depending on the selected approaches, drawbacks12,13 can be problematic for solid phase extraction applications as follows: low functionalization and inhomogeneous distribution of the organic group12,14 lead to a significant part of functional groups being non-accessible, and the mesoscopic ordering formation can be disturbed.
In this context, aiming to overcome these drawbacks and improve the amount of available functional groups, a different approach has been explored to generate organosilicas using organosilane surfactant precursors.15–17 This ‘‘all-in-one’’ or “self-templating” approach allows both dense functionalization and accessibility of the functionalities of the silica by taking advantage of the self-assembling and condensable properties of organosilane surfactant precursors.17
Different structural types of supramolecular materials could be obtained depending on the specific arrangement of the organosilane and on the amount of silica involved during the synthesis without the addition of more external surfactant.15
For example, various multi-layered organosilicate have been synthesized by a self-assembly process leading to hybrid materials with high adsorption capacity for transition metal cations.18,19
Recently, we took advantage of this approach using a simple amine (amino-undecyl-triethoxysilane). Organic–inorganic hybrid materials with high amine content or aminooxy groups were obtained either with a lamellar or hexagonal periodicity from the self-assembly properties of this precursor under specific conditions.20–25
A primary amino group is one of the most attractive surface functionalities owing to its use as a ligand for various metal ions.3,26 We demonstrated the accessibility of the functional amine groups in materials prepared from a ‘‘all-in-one’’ approach through the performance of the materials engaged in the solid phase extraction of platinum or palladium salts, which are able to form strong complex bonds with amino functions.27
Solid phase extraction may require a high concentration and availability of functional groups. Taking into account such requirements, the preparation of silica materials bearing a high number of available amines represents a considerable challenge. Indeed, amino functions are useful for further post-synthesis functionalization, allowing various functionalities to be introduced by chemical modifications such as alkylation3,28 or the formation of amides5,29,30 and imines.30,31
In this context, we first studied the availability of amines in amine-bearing silica lamellar materials and their post-synthesis functionalization for the introduction of selective ligands aimed at rare earth extraction. Inspired by the works of Ogata et al.,32,33 we focused on opening ring reactions of anhydride succinic (S), glutaric (G) and diglycolic (D) leading to their respective amic acids (see Fig. 1).
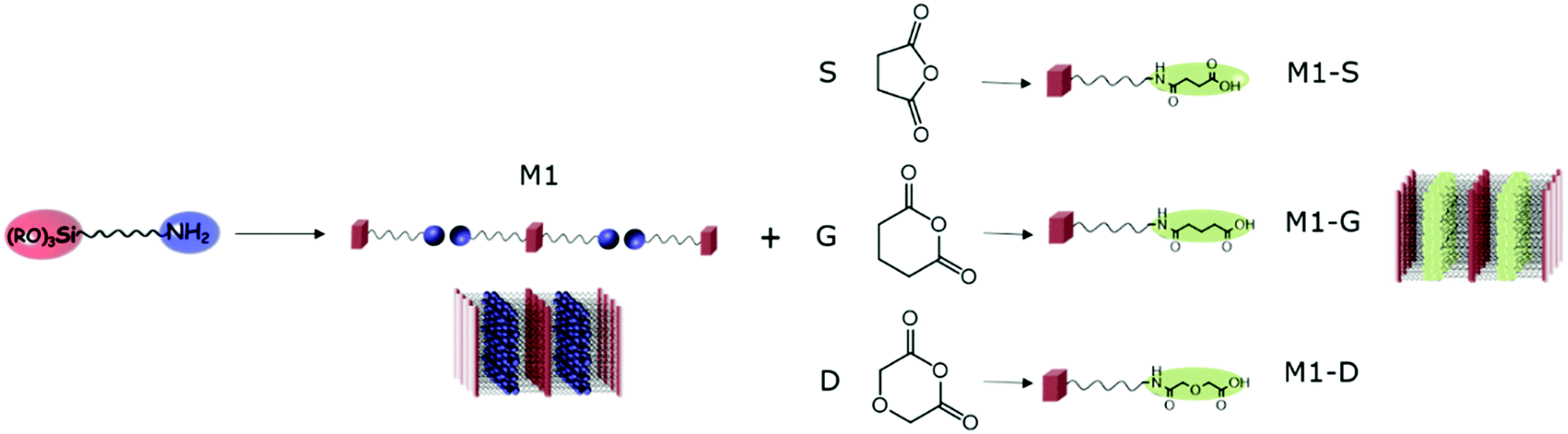 | ||
| Fig. 1 Schematic representation of opening ring reaction of anhydride by lamellar amino functionalized silica, leading to lamellar amic functionalized silica M1-S, M1-G and M1-D. | ||
The adsorption properties of amino functionalized materials was first compared to that of their analogous aminooxy functionalized materials. The discrepancies in extraction behaviours are discussed. The amine availability in regard to post-functional groups is calculated from the grafting ratio obtained by FTIR, NMR and TGA analyses. The structural modifications induced by grafting were evaluated by SAXS experiments. Then, the ability of the materials to extract metal ions was also studied by comparing the extraction behaviour in nitric media between d group elements, silver and rare earths elements (REEs).
Results and discussion
After the sol–gel process, thanks to the self-assembly behaviour of amino-undecyl-triethoxysilane, which can be considered as an amphiphilic organosilane, the preparation of lamellar hybrid material bearing easily available amine groups can be achieved. CO2 can be used as assembly agent leading to the formation of material M1 as bis-silylated-alkylammonium carbamate dimers arranged in a lamellar structure.23,27 The elimination of CO2 by heating allows obtaining materials with free amino groups in which the well-defined lamellar structure is maintained.The possibility of the post-functionalisation of the reactive amino groups at the surface of the hybrid silica was studied through opening ring reactions of anhydride succinic (S), glutaric (G) and diglycolic (D) resulting in the nanostructured M1-S, M1-G, and M1-D.
Post-grafting
FTIR and NMR were used to investigate the grafting to provide evidence of the opening ring on the amino functions. Additionally, TGA and an elemental analysis were used together to quantify the grafting.The FTIR spectra (Fig. 2) of the materials exhibited the characteristic bands at around 2920 and 2850 cm−1 corresponding to the νas(CH) and νs(CH) modes, respectively, of the alkyl chains. The intensity of these bands, comparatively to those corresponding to the silica matrix with Si–O–Si framework vibration (νas(Si–O)) between 985 and 1160 cm−1, is representative of the high density of organic parts in the hybrid materials. Fig. 2 shows a specific region included between 1450 and 1850 cm−1 of the FTIR spectra of the materials M1, M1-S, M1-G, and M1-D. The M1 spectrum does not exhibit any intense band in the region included between 1450 and 1850 cm−1, and grafted materials spectra present three broad bands located at 1550, 1630 and 1700 cm−1. The bands at 1550 and 1630 cm−1 are usually attributed to the N–H bending mode and the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching mode of secondary amides, respectively.34
O stretching mode of secondary amides, respectively.34
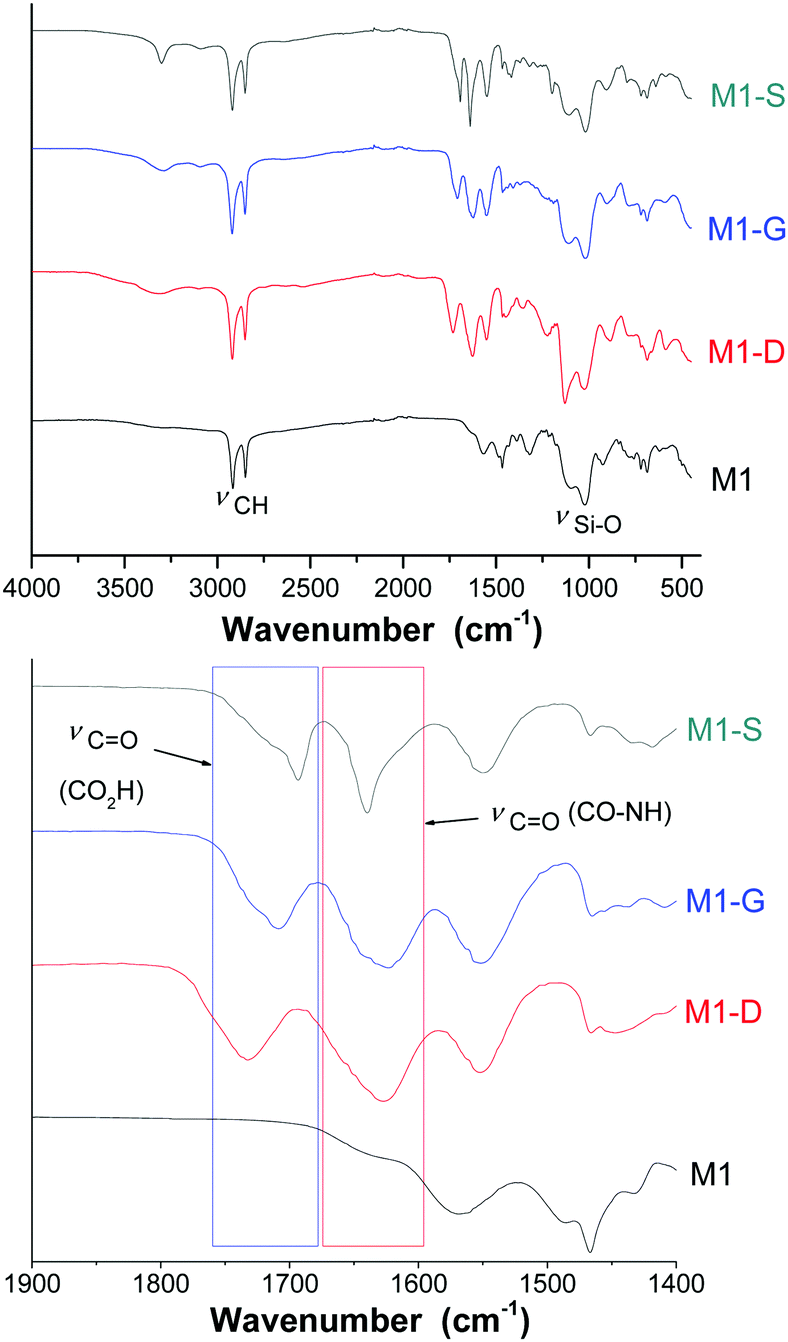 | ||
| Fig. 2 FTIR spectra of M1, M1-S, M1-G and M1-D. | ||
The band at 1700 cm−1 is known to be associated with the stretching mode of CO bound of a carboxylic acid. Notably, no band characterizing the carbonyl signal of the anhydride precursors was observed at 1800 cm−1, indicating that no residual trapped precursors are present in the material.
The solid-state 13C NMR leads to the same conclusion. Fig. 3 shows the 13C NMR spectrum of the M1-S material signals corresponding to the carbons of the different carbonyl groups previously mentioned. The carbon of the carboxylic acid is observed at 174 ppm, and the carbon of the amide is observed at 169 ppm.
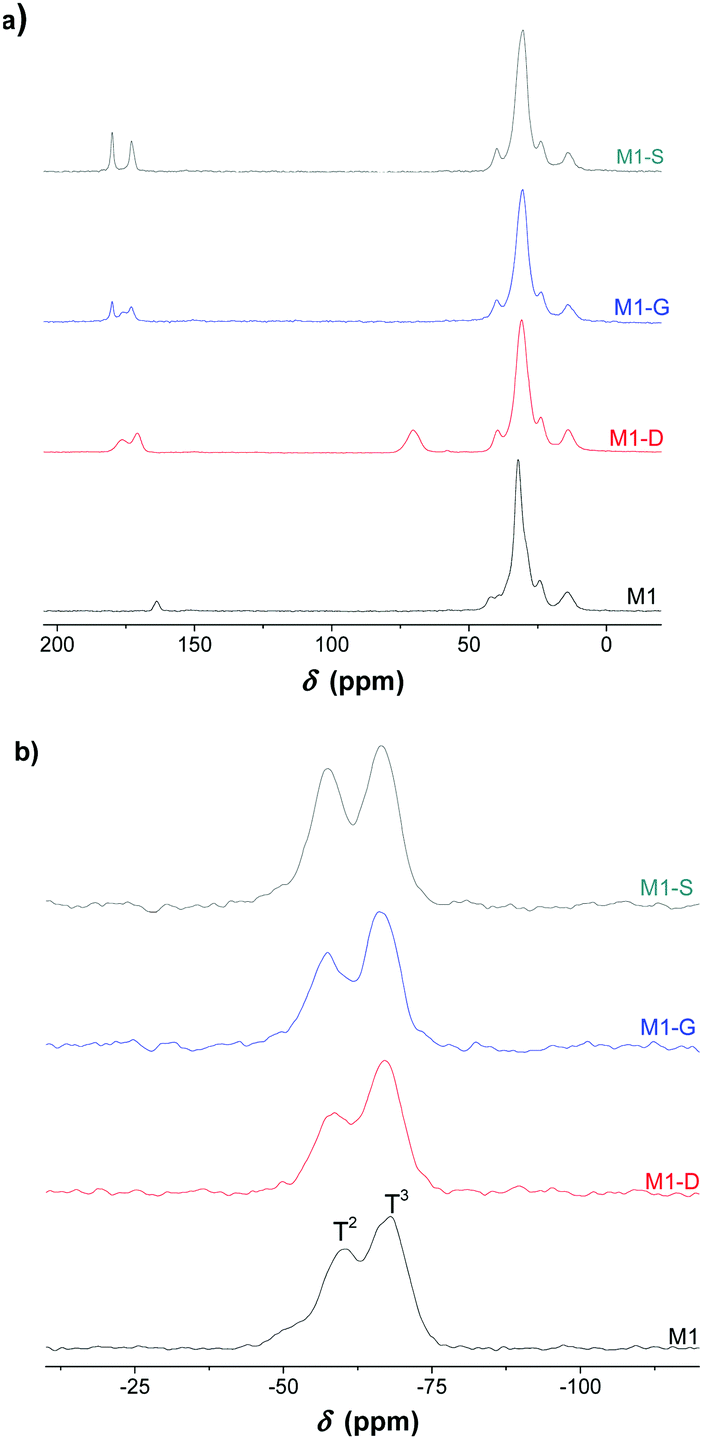 | ||
| Fig. 3 (a) 13C and (b) 29Si solid-state NMR spectra of the bulk silica hybrid material (M1, M1-D, M1-G, M1-S). | ||
The solid state NMR spectrum of M1 also presents a resonance signal located at 159 ppm, which is attributed to the carbamate carbonyl carbon, confirming the presence of ammonium carbamate salt as already observed in the literature.21
The solid-state 29Si CPMAS NMR spectra exhibits T2 (−55 ppm) and T3 (−65 ppm) silicate centres with the condensation level estimated at around 85%, which indicates that approximately five out of six silanol units in the material are condensed (see Table S1, ESI†).35,36 The materials consist of highly condensed siloxane networks with organic fragments covalently bonded to the silica network. This suggestion was supported by the solid-state 13C NMR spectra with the absence of signals attributable to residual ethoxy groups.
Spectroscopic measurements allow us to confirm the ring opening of the different precursors on the amine from the M1 material. Although information from these data seems to show that the grafting was complete, a quantitative analysis is required to provide solid evidence.
For this purpose, a thermogravimetric analysis was performed on materials from 30 °C to 1000 °C under air flow. The resulting weight losses for M1 and M1-S are shown in Fig. 4. The thermal profile of M1-G and M1-D are similar to that of M1-S (see Fig. S1, ESI†). The results for the M1 material show a CO2 and water release of approximately 10.5 wt% between 70 °C and 120 °C. The CO2 is initially used as the assembly agent leading to the formation of material M1 as bis-silylated-alkylammonium carbamate dimers arranged in a lamellar structure.23,27
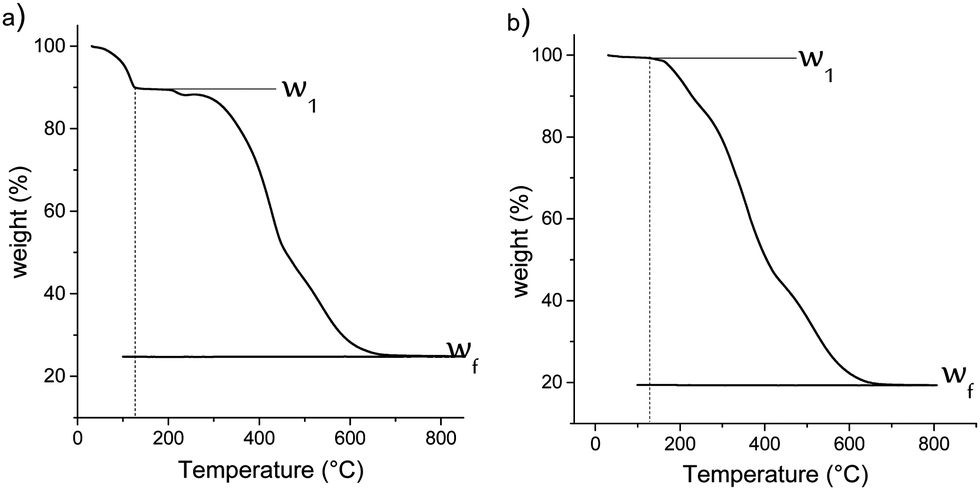 | ||
| Fig. 4 Thermogravimetric analysis of (a) M1 and (b) M1-S. | ||
This departure is no longer observed in the case of the M1-S material, reflecting the full substitution of the CO2 during the post-grafting step. This finding confirms that the amic acid groups have been introduced onto the M1 material.
In addition, the decrease of the residual weight wf at the end of the analysis from 24.7 wt% to 19.2 wt%, respectively, for the M1 and M1-S materials involves a higher organic content into the initial material. The residual masses after calcination wf can be considered as that of pure silica. Completely condensed silica presents a molar mass of 60 g mol−1. Considering that each silicon atom of the silica originates from condensed amino precursors or functionalized precursors of xOnSi(CH2)11NH2·yOnSi(CH2)11NH-R·zH2O, (where n is the number of oxygen molecules bounded on silicon, and R is the grafted moieties), the average molar mass Mt of the precursor can be deduced with eqn (1):
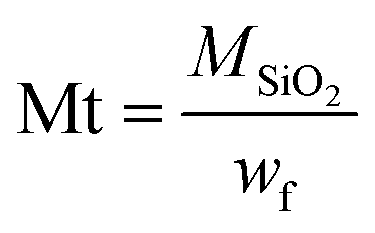 | (1) |
| Mt = xMM1 + yMM1−X + zMH2O | (2) |
Assuming that at 125 °C the silica matrix is also completely condensed and dried, the fraction w1 corresponding to the fraction at 125 °C corresponds to the material of formula xO1.5Si(CH2)11NH2·yO5Si(CH2)11NH-R of average molar mass Mt*.
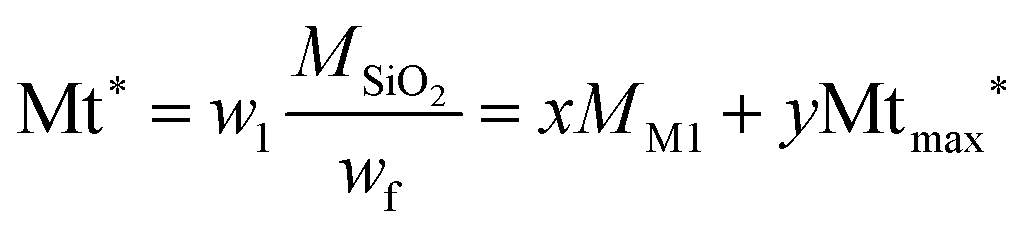 | (3) |
| Mt* = xMM1 + yMtmax* | (4) |
The conversion yield y is obtained by resolving eqn (5):
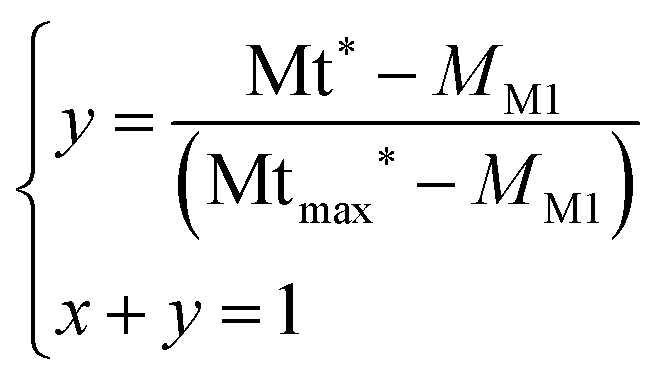 | (5) |
| w f (%) | w 1 (%) | Mt* (g mol−1) | Mtmax* (g mol−1) | y (%) | |
|---|---|---|---|---|---|
| M1 | 24.7 | 89.7 | 218 | 222 | — |
| M1-S | 19.2 | 99.8 | 312 | 322 | 90 |
| M1-G | 18.5 | 99.3 | 322 | 336 | 88 |
| M1-D | 17.6 | 98.3 | 335 | 338 | 98 |
Completely condensed silica is considered, and the calculated values and grafting yields are minimized. Therefore, as suggested by spectroscopic analysis, the thermal analysis demonstrates a quantitative grafting by ring opening reaction on the functionalized amino@silica. In addition, the elemental analysis on the materials leads to the same conclusion (see Table S2, ESI†).
Analyses allow for verification that a ring opening occurred in the quantitative yields. The introduction of such functional groups involves all the amine of the initial M1 material available from the reactional media. The availability of the amino groups located at the interfaces of bilayers in such material has been highlighted with the immobilization of platinoid salt complexes in the interlayer space.27 The chemical modification of the amino groups is therefore expected to involve structural modifications of the resulting grafted materials.
Structural effects
The amino functions are located at the interfaces of the bilayers built during the self-assembly of the aminoundecyltriethoxysilane precursor. The stacking of these bilayers corresponds to a lamellar structure highlighted in the SAXS profile of the material by structure peaks, for which the qn/q1 ratio is an integer n. Fig. 5 shows the SAXS profile of M1 and M1-S, and that of M1-G and M1-D are available in Fig. S2 (ESI†). We have identified the Bragg peaks originating from the lamellar structure. For the M1-S sample, five Bragg peaks denoted as qnS are clearly observed at scattering vectors of 1.6, 3.3, 4.8, 6.4 and 8.1 nm−1, corresponding to the five successive orders for a lamellar phase of parameter 3.9 nm.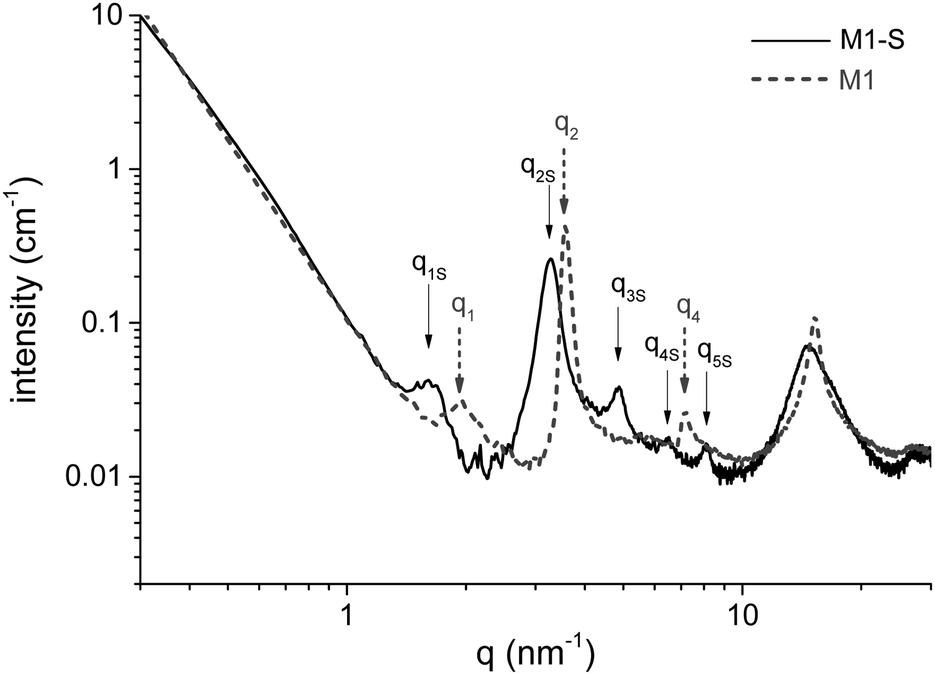 | ||
| Fig. 5 SAXS profiles of M1 and M1-S. | ||
In the case of M1, only 3 peaks are resolved, corresponding to the first, second and fourth diffraction order of a lamellar phase of lattice parameter of 3.4 nm.
To determine the consistency of the initial lamellar structure, SAXS measurements were performed on each material. All of the results reveal that the lamellar structure is preserved after grafting. However, the lattice parameter characterizing the distance between bilayers is larger than that of M1. The SAXS profile of M1-S is presented in Fig. 5 to illustrate the phenomenon; those of M1-G and M1-D are available in Fig. S2 (ESI†). Indeed, all the peaks are shifted towards weaker angles. For instance, we note that q2 peak shifts from 3.7 nm−1 for M1 to 3.3 nm−1.
From the q2 peak position, we calculated the swelling of the structure due to the chemical grafting. The interlayer distance Δd calculated from the q2 peak position, and the expected lengths of the grafted functions Df are summarized in Table 2.
| Δd | D f | |
|---|---|---|
| M1-S | 4 | 5 |
| M1-G | 4 | 6.3 |
| M1-D | 4 | 6.0 |
As previously demonstrated by thermal analysis, the CO2 constituting the carbamic acid of M1 has been eliminated during the grafting. Considering that the size of the carbamate groups is negligible and that they are all substituted by grafted functions, the expected swelling gap would be close to 2Df. However, the interlayer distances are close to Df for all of the grafted materials. These results suggest either a tilted lamellar structure or a bilayer structure where functional end chains are interpenetrated. The latest interpretation would likely be governed by hydrogen bounds between amides and acid groups.
We showed that the amino functions located at the bilayer interfaces were available for chemical modification. In addition, the functionalization has been completely performed for all the ring precursors without drastically damaging the initial lamellar structure. The ring opening reaction of cyclic anhydrides is thus an efficient way to functionalize amines from hybrid-structured silica. This reaction leads to multifunctional chemical patterns tunable by the nature of the grafted groups. Thus, we obtained amides coupled with terminal carboxylic acids bridged by two or three methylenes for M1-S and M1-G materials and a dimethylether for M1-D.
Extraction tests
In addition, the grafted ligands contain chemical moieties also known to have affinities with specific metal ions. Indeed, if carboxylic acids can complex a large range of metal ions,4,37 amides38,39 and amic acid40,41 seem peculiarly relevant as far as selective rare earth ion extraction is concerned.
Thus, we explore the extraction properties of materials in regard to the separation of Y3+, Nd3+, Eu3+ and Dy3+ representing the rare earth elements group. Usually, rare earth elements coming from ores of natural or urban origin are accompanied with iron and other d-block elements. Therefore, iron, copper and cobalt have been added to the simulated leach. Aiming at completing the study, silver has been considered as a monovalent cation.
The main aim of this study was to evaluate the affinities and the apparent selectivity of prepared materials in regard to these eight ions contained in different nitric acid solutions. The concentration in each ion was fixed at 0.1 mmol L−1. The mass of adsorbent material was 20 mg for M1 and M2 and 25 mg for the grafted material in order to obtain a ratio between ligands (L) and metal ions (M) L/M close to 10. Note that from pH = 3, iron ions precipitate and are eliminated from the solution before ICP-OES analysis. Therefore, the results for iron are not available for solutions containing 0.0001 M of nitric acid. First, we evaluate the extraction capacity of M1 in different acidic condition. To determine the pKa effects on the extraction phenomena, the results are compared to those of M2, which is the analogous lamellar material functionalized by an oxyamine.23Fig. 6a exhibits the extraction results of M1 for different nitric acid concentrations. We note that M1 extracts few elements in the two higher acidic concentration values. Only silver is significantly extracted, reaching 7% from the initial amount of this element for nitric acid concentration of 0.01 M, while at lower acid concentration (pH = 4), copper and silver are quantitatively extracted (93% and 97%, respectively) and heavy rare earths, albeit in lower proportion.
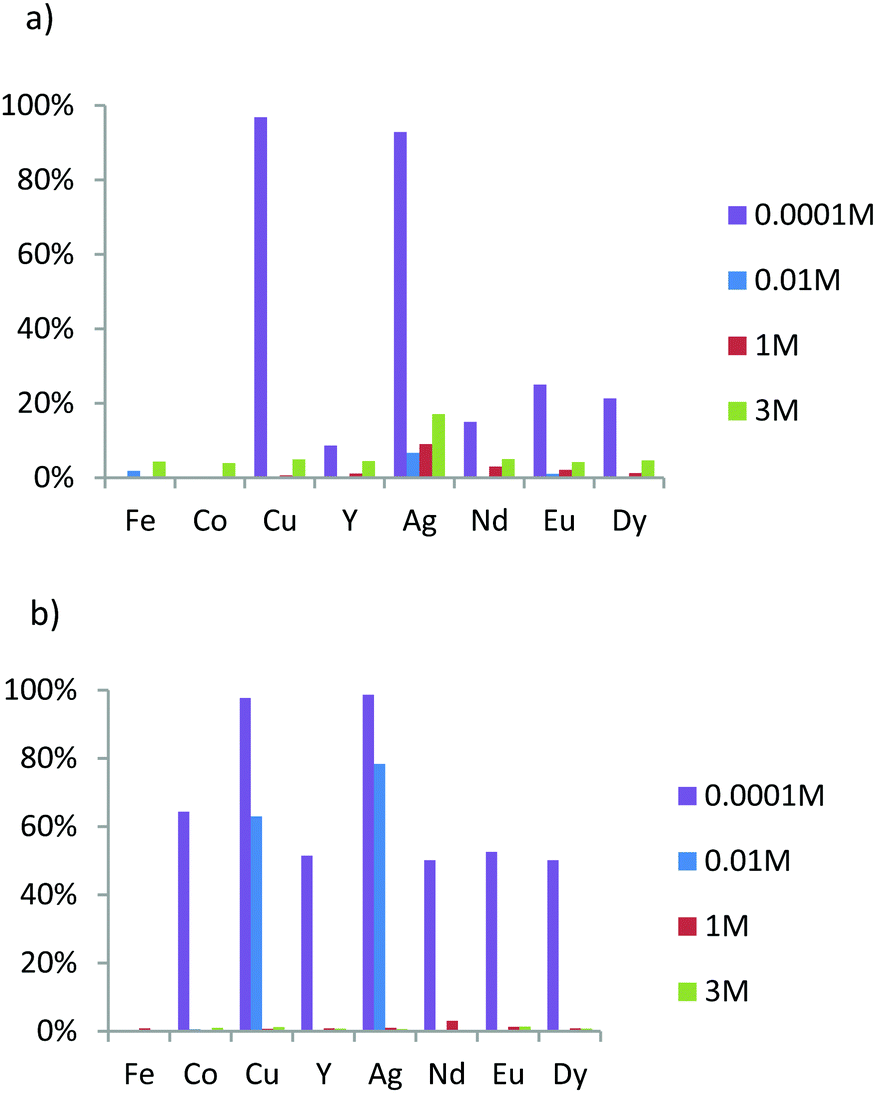 | ||
| Fig. 6 Acidity dependence of metal ion percent adsorption for adsorbents (a) M1 and (b) M2 in nitric acid solution with a concentration in each ion fixed at 0.1 mM. | ||
In comparison, in the higher acidic conditions, M2 appears to behave as M1 since no significant extraction was observed (Fig. 6a and b). However, while at 0.01 M HNO3, the extraction properties of M1 are low, those of M2 exhibit affinities specifically for both elements coming from block 11 of the periodic table, since only silver and copper are quantitatively extracted at 80% and 60%, respectively (Fig. 6b). For lower acid concentration (0.0001 M), the extraction of silver and copper was complete, but M2 also extracts 60% of cobalt ions and 50% of all of rare earth ions (Y3+, Nd3+, Eu3+ and Dy3+).
The use of silica-supported aliphatic amines reported by Soliman42 illustrated that the use of nitrogen donor atoms was an effective means of removing metals such as copper(II) ions.
The extraction properties of M1 and M2 seem to exhibit a similar behaviour to a certain extent, but the behaviours shifted with the pH of the aqueous media. The amino functions of M1 are initially under ammonium carbamate form, while oxyamino functions of M2 are supposed to be free. Moreover, the pKa of M1 amines is estimated to 10.6 against 5 for M2 oxyamines.
Two hypotheses can be expressed to explain these behaviour discrepancies between M1 and M2.
First, the ammonium carbamate, which hinders the bilayer interface constituting M1 material, is too stable in this thermal condition to allow extraction from solution. Second, in this acidic condition (pH = 2), amino and oxyamino functions are both mostly protonated under ammonium and oxyammonium form respectively for M1 and M2. However, the discrepancy between pKa values involves different equilibrium constants between the formation of ligand–metal species (non-protonated ligands) and non-chelating species (protonated ligands). Equilibrium promoting the formation of ligand–metal species would be observed for M2 at higher pH. Owing to the same shifted extraction behaviour, the second hypothesis is supported. Notably, at a pH value of 2, the calculated silver ion separation factor of M2 with respect to iron, and rare earth ions are included from 200 to 750, involving a higher selectivity.
We have previously demonstrated the accessibility of amine from carbamate interfaces between bilayers in regard to platinoid anionic salt27 and the possibility of chemical modifications. As previously shown by Alauzun et al.21 the availability of this function is also extended to cations in the ethanolic solution. Here, we demonstrate that amino functions from carbamate interfaces are also accessible in nitric aqueous media in a selective way. Owing to the shifted similar extraction behaviour, this selectivity towards copper and silver ions seems to be governed by the pKa of terminal functions and thus by the pH of the solution.
The extraction values for a nitric acid concentration of 0.01 M of these three materials are shown in Fig. 7. M1-S and M1-G have the same affinities in regard to metal ions. Indeed, both extracts have exclusively iron ions in the same proportion, namely, 75% of the initial quantity and, to a lesser extent, silver ions (6–8%). The increase by one carbon atom of the bridge between amide and carboxylic acid is not a dominant parameter to change the extraction properties. This result suggests that the extraction site of iron ion is not located between the abovementioned functions. While in M1-S and M1-G materials the bridges are composed of a methylene group, those of M1-D are ether groups. The M1-D material extracts iron ions in the same proportion, confirming the hypothesis that extraction sites of iron ions are not between the amide and carboxylic acid but rather on the carboxylic acid site, as suggested by the spectroscopic analysis of Ogata et al.33 M1-D has also significant affinities in regard to all the REEs. Indeed, the extraction of yttrium ions reaches 72%. Among the heavy rare earths, the extraction yields increase with the atomic number (Fig. 7).
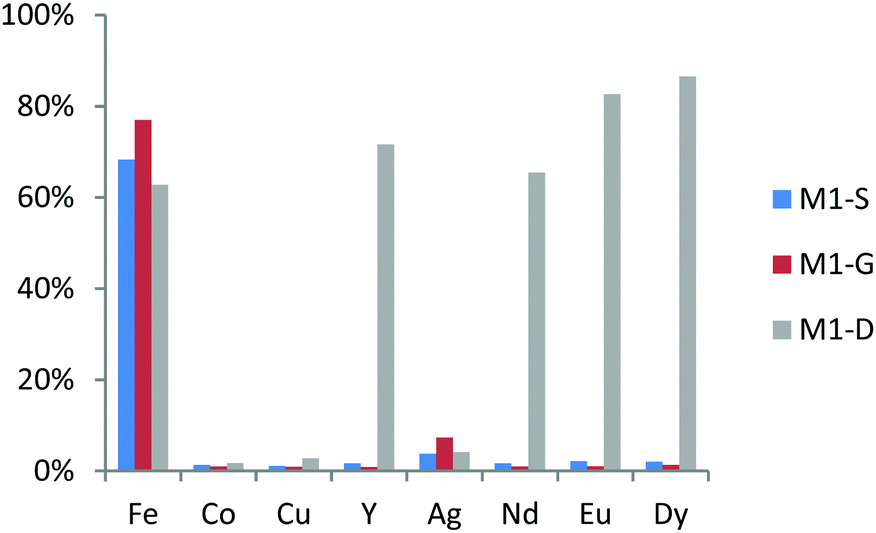 | ||
| Fig. 7 Metal ion percent adsorption for M1-S, M1-G and M1-D from a 0.01 M nitric acid solution with a concentration in each ion fixed at 0.1 mM. | ||
This result has been already observed by Ogata et al. in hydrochloric acid aqueous media.32,33
An interesting point in order to selectively extract the rare earth element is to first perform the extraction of the aqueous feed solution using M1-S or M1-G to remove the iron and then perform the extraction of the REEs thanks to the material M1-D.
Extraction experiments with a nitric acid concentration of 1 M have been performed and reveal negligible extraction of all the elements in the leach, suggesting that quantitative back-extraction is possible. Aiming at recovering the extracted rare earth ions from M1-D, we rinsed the material with a nitric acid aqueous solution at 3 M for 24 h at 25 °C. The reported results in Fig. 8 reveal recovery yields of the extracted elements greater than 86%, which could be considered quantitative.
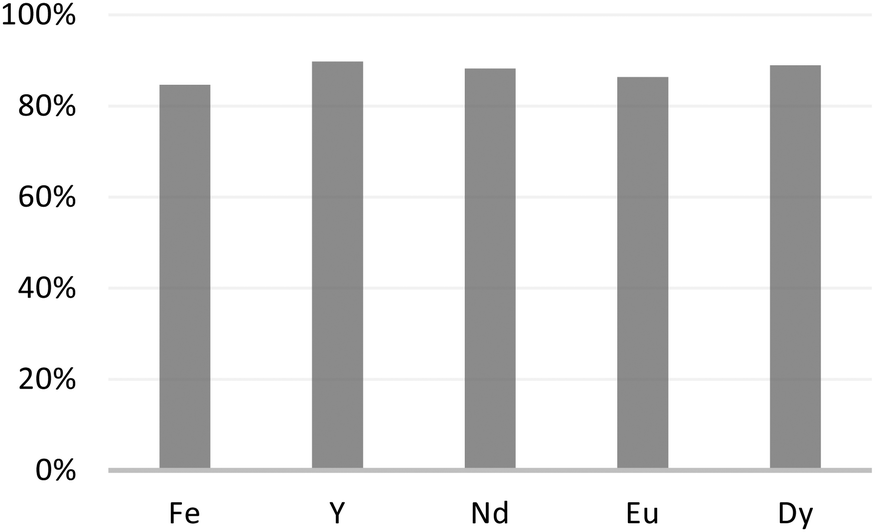 | ||
| Fig. 8 Metal ion percent recovery after rinsing the adsorbent M1-D with nitric acid aqueous solution at 3 M for 24 h at 25 °C. | ||
To determine the maximum adsorption capacity of M1-D material in regard to dysprosium ions, we performed an extraction test in nitric acid solution (0.1 mM) with an L/M ratio of 0.5. In light of a structural investigation of the grafted materials, the presumed interpenetrated bilayers structure could have limited the extraction or back-extraction. However, the maximum adsorption capacity of dysprosium ions for M1-D materials was measured at 0.4 mmol g−1 (65 mg g−1) involving an L/M ratio of 7.2. Such ratio involves complete availability and accessibility of organic functions to allow trapping ions.
Few results involving amic acid loaded onto inorganic supports have been published in regards to adsorption of cations. The introduction of diglycol amic group on silica gel has been proposed by Ogata et al. with adsorption capacity Q ≈ 20 mg g−1 for Dy. However, experiments have been realized at pH = 1 under hydrochloric acid.32 Similarly, adsorption capacity of about 8 mg g−1 with respect to Lu from HNO3 at pH 4 has been reported but using mesoporous sorbents functionalized by phthaloyl diamide.43 Zheng et al. reported a Q value between 50 and 80 mg g−1 with respect to Gd for an adsorbent based on modified mesoporous silica support with maleic anhydride under comparable conditions (HNO3 pH = 4).44
Therefore, as compared to the literature, the material M1-D obtained competitive Q values while the material is not based on the commonly modification of mesoporous silica support with chelating ligand.
Experimental
Technical details
NMR analyses were performed on a Bruker 400 ultrashield VS spectrometer. Displacements are reported in ppm using the solvent (CDCl3: 7.26 ppm for 1H; 77.16 ppm for 13C) as an internal reference.ESI-MS was performed on a Flexar SQ 300 MS instrument.
TGA analyses were performed on a Setaram Setsys Evolution 18 instrument under airflow (20 mL min−1) with a heating rate of (5 °C min−1). The coupled mass spectrometer was a Hiden analytical QGA instrument.
Solid-state FT-IR spectra were obtained using an ATR crystal on a Perkin-Elmer 100 spectrometer with a resolution of 4 cm−1.
Solid-state 13C CP MAS NMR spectra were performed on a Bruker 400 ultrashield VS spectrometer with a 12 kHz MAS spinning rate using Bruker MAS probes (4 mm rotors). The typical contact time was 2–3 ms, and the typical recycle delay was 5–30 s. The degrees of condensation were estimated from the 29Si CPMAS NMR spectra from the percentages of the different Tx units, which were obtained by deconvolution of the spectra using the Dmfit program.45
SAXS experiments were performed on a setup operating in transmission geometry. A Mo anode associated with a Fox2D multi-shell mirror (XENOCS) delivered a collimated beam of wavelength 0.71 Å. Two sets of scatterless slits46 delimitated the beam to a square section of side length of 0.8 mm. A MAR345 imaging plate detector allowed simultaneous recording of scattering vectors q ranging from 0.25 nm−1 to 25 nm−1. Samples were placed in glass capillaries with a diameter of 2 mm. Absolute intensities were obtained by measuring a calibration sample of high-density polyethylene (Goodfellow), for which the absolute scattering was already determined.
All SAXS profiles are plotted in log–log scale to highlight the dependency of the intensity versus a power law of the scattering vector q.
Metal concentrations were measured by inductively coupled plasma/atomic emission spectroscopy (ICP/AES) using a SPECTRO ARCOS spectrometer.
Precursor synthesis
All intermediate reactants and final products P1 and P2 were purified by distillation under a vacuum. 1H, 13C NMR, and mass spectrometry were used at each step to verify the structure and purity of the different products; all of the chemical characterizations were in agreement with the literature.47–49As shown in Scheme S1 (ESI†), 1 (34.058 g, 0.2 mol, 1 eq.) and NEt3 (25.5 g, 0.252 mol, 1.26 eq.) were added to 700 mL CHCl3. Over the course of 90 min, MsCl (27.494 g, 0.24 mol, 1.2 eq.) dissolved in 100 mL of CHCl3 was added dropwise into the stirred solution at room temperature. The temperature was controlled by a water bath. Addition of MsCl produced HCl, which was removed by frequent venting. After 20 h, the solution turned slightly yellow. Half of the reaction mixture was washed with 450 mL water. The aqueous phase was extracted with CHCl3 (2 × 100 mL). The same process was repeated for the second half. All the united aqueous phases were again extracted with CHCl3 (100 mL). The organic phases were reduced to 200 mL and washed with brine (100 mL). CHCl3 was evaporated, and the residue was distilled at 80 °C and 0.5 mbar to remove volatile impurities. The resulting dark oil was again distilled at 160 °C and 0.3 mbar to afford product 2 as translucent brown oil (45.12 g, 0.18 mol, 91%) in the distillate.
Under N2-atmosphere, Karstedt catalyst (2.1–2.4 wt% Pt, 0.462 g, 0.02 mmol, 3 × 10−4 eq.) was added to a stirred mixture of 2 (15 g, 60.05 mmol, 1 eq.) and triethoxysilane (22.99 g, 139.94 mmol, 2.33 eq.) at room temperature. The temperature was kept constant using a water bath. After 20 h, activated charcoal (2 g) was added, and the mixture was stirred at 40 °C for 1 h. The mixture was filtered, and the filtrate was distilled at 170 °C (0.3 mbar) to remove more volatile impurities. Product 3 was obtained as brown oil (21.87 g, 53.06 mmol, 88%).
Under N2-atmosphere, NaN3 (7.09, 109.14 mmol, 3 eq.) suspended in CH3CN (100 mL) was heated to 85 °C. 3 (15 g, 36.4 mmol, 1 eq.) in CH3CN (50 mL) was added dropwise to the heated mixture. After 48 h the mixture was cooled to 0 °C filtered, washing the residue with cold CH3CN (2 × 20 mL). After evaporation of CH3CN more volatile compounds were removed by distillation at 110 °C (0.6 mbar) for 2 h. The residue was distilled 200 °C (0.6 mbar) to afford the product 4 as clear, colourless oil (11.94 g, 31.75 mmol, 87%) in the distillate.
Reduction of the azido group with palladium (10% Pd/C, Sigma-Aldrich) in anhydrous ethanol under hydrogen atmosphere led to amino-undecyl-triethoxysilane P1. Under an Ar-atmosphere, anhydrous ethanol (500 mL) was added to Palladium supported on carbon (Pd/C, 10 w%) (0.2 g, 0.19 mmol, 0.01 eq.), and 4 balloons filled with H2 were bubbled through. A solution of 4 (7.9 g, 21 mmol, 1 eq.) dissolved in anhydrous ethanol (250 mL) was infused by bubbling through one balloon of H2. The solution containing 4 was then added dropwise under an H2 atmosphere over the course of 1 h. After 2 h of stirring at room temperature, the solution was filtered over Celite, and the solvent of the filtrate was evaporated. The residue was dissolved in pentane, and the resulting solution containing a white precipitate was again filtered over Celite. The pentane was evaporated, and the product P1 was obtained as a slightly yellow liquid (5.5 g, 16.4 mmol, 78%) after fractionated distillation at 200 °C (0.7 mbar).
Materials synthesis
Amino-undecyl-triethoxysilane (3 mmol) was introduced in a 50 mL flask under pure CO2 gas, resulting in a gel. After 30 min, 20 mL of pentane were poured in the flask and then hydrochloric acid solution was added (81 mL at pH 1.5). The material was aged for 3 days at 30 °C. The resulting material M1 was obtained after filtration followed by a washing step with ethanol and ether before drying under vacuum.
Post-grafting
The opening ring of the different cyclic anhydrides by the amino functionalities of M1 was performed as follows: the M1 material was added to a solution of THF containing 1.1 equivalent of cyclic anhydride (succinic, glutaric or diglycolic) in regard to the amino function contained in M1. The resulting materials are respectively denoted as M1-S, M1-G, and M1-D. The mixture was stirred for 12 hours at 70 °C. After stirring for 12 hours at 70 °C, the mixture was centrifuged at 4500 rpm for 15 min. Then, the resulting materials were rinsed with ethanol and ether. Finally, the powders were dried under a vacuum for 1 hour.Extraction test
All adsorption experiments were conducted using batch contact methodology in which the powdered adsorbent was contacted with solutions containing the target species in a volume/mass ratio of approximately 400–500 mL g−1. The adsorbent material (20–25 mg) was added to 10 mL of a solution containing metal ions, and the mixture was stirred at 298 K for 24 h. After centrifugation at 4500 rpm for 15 min, the concentration of metal ions in the filtrate was measured by inductively coupled plasma optical emission spectroscopy. The uptake of metal ions efficiency E% was calculated from the following equation, in which Ci and Cf are the initial and the final concentration of metal ions, respectively: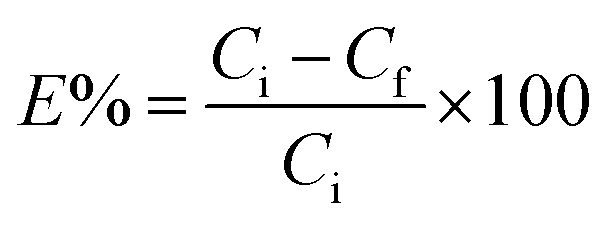 | (6) |
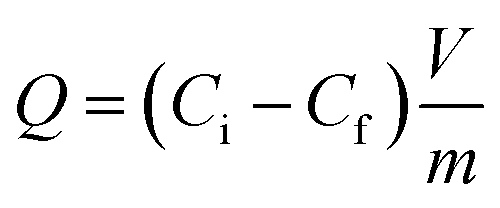 | (7) |
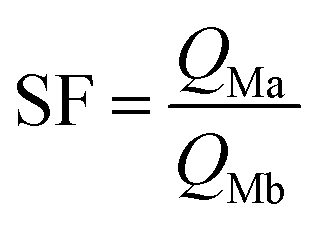 | (8) |
Conclusions
We demonstrated the complete accessibility and availability of amines from carbamate interfaces between bilayers in regard to metal ions and the ring opening reaction of anhydrides.Similar but shifted extraction behaviours of amino and oxyamino@silica materials suggest that their extraction properties are governed by the pKa of the amine moiety.
The ring opening reaction of anhydrides in the M1 material has been quantitatively performed, as indicated by complementary analyses such as TGA and elemental analyses, with three different anhydrides leading to succinamic, glutaramic, and diglycolamic acids.
The structure of the resulting materials is swelled by the grafted molecule hindrance but preserved.
As for the initial amino@silica material, the interactions at the amic bilayer interfaces do not prevent their accessibility regarding metal ions, allowing quantitative extraction and desextraction in nitric media.
Further work will be devoted to the selective extraction of REE with regards to Fe by first performing the extraction of the aqueous feed solution using M1-S or M1-G to remove the iron, and then performing the extraction of the REE thanks to the M1-D material. Indeed, the latter material, prepared by a simple and convenient approach, is a promising adsorbent in regards to its high extraction performance of REEs and their easy desorption.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge C. Rey and V. Dubois for their technical support.Notes and references
- C. Sanchez, K. J. Shea and S. Kitagawa, Chem. Soc. Rev., 2011, 40, 471–472 RSC.
- R. I. Nooney, M. Kalyanaraman, G. Kennedy and E. J. Maginn, Langmuir, 2001, 17, 528–533 CrossRef CAS.
- P. K. Jal, S. Patel and B. K. Mishra, Talanta, 2004, 62, 1005–1028 CrossRef CAS PubMed.
- G. Arrachart, I. Karatchevtseva, D. J. Cassidy, G. Triani, J. R. Bartlett and M. Wong Chi Man, J. Mater. Chem., 2008, 18, 3643–3649 RSC.
- A. Charlot, S. E. Mourabit, F. Goettmann, G. Arrachart, R. Turgis and A. Grandjean, RSC Adv., 2014, 4, 64138–64141 RSC.
- R. Turgis, G. Arrachart, C. Delchet, C. Rey, Y. Barré, S. Pellet-Rostaing, Y. Guari, J. Larionova and A. Grandjean, Chem. Mater., 2013, 25, 4447–4453 CrossRef CAS.
- A. Walcarius and L. Mercier, J. Mater. Chem., 2010, 20, 4478–4511 RSC.
- H. Yoshitake, J. Mater. Chem., 2010, 20, 4537–4550 RSC.
- F. Hoffmann, M. Cornelius, J. Morell and M. Fröba, Angew. Chem., Int. Ed., 2006, 45, 3216–3251 CrossRef CAS PubMed.
- W. J. Hunks and G. A. Ozin, J. Mater. Chem., 2005, 15, 3716–3724 RSC.
- P. Van Der Voort, D. Esquivel, E. De Canck, F. Goethals, I. Van Driessche and F. J. Romero-Salguero, Chem. Soc. Rev., 2013, 42, 3913–3955 RSC.
- M. H. Lim, C. F. Blanford and A. Stein, Chem. Mater., 1998, 10, 467–470 CrossRef CAS.
- M. H. Lim and A. Stein, Chem. Mater., 1999, 11, 3285–3295 CrossRef CAS.
- A. Walcarius and C. Delacôte, Chem. Mater., 2003, 15, 4181–4192 CrossRef CAS.
- E. Ruiz-Hitzky, S. Letaïef and V. Prévot, Adv. Mater., 2002, 14, 439–443 CrossRef CAS.
- R. Voss, A. Thomas, M. Antonietti and G. A. Ozin, J. Mater. Chem., 2005, 15, 4010–4014 RSC.
- W. Otani, K. Kinbara, Q. Zhang, K. Ariga and T. Aida, Chem. – Eur. J., 2007, 13, 1731–1736 CrossRef CAS PubMed.
- T. Chujo, Y. Gonda, Y. Oumi, T. Sano and H. Yoshitake, J. Mater. Chem., 2007, 17, 1372–1380 RSC.
- H. Yoshitake, H. Nakajima, Y. Oumi and T. Sano, J. Mater. Chem., 2010, 20, 2024–2032 RSC.
- J. Alauzun, A. Mehdi, C. Reyé and R. J. P. Corriu, J. Am. Chem. Soc., 2005, 127, 11204–11205 CrossRef CAS PubMed.
- J. Alauzun, E. Besson, A. Mehdi, C. Reyé and R. J. P. Corriu, Chem. Mater., 2008, 20, 503–513 CrossRef CAS.
- R. Besnard, J. Cambedouzou, G. Arrachart, O. Diat and S. Pellet-Rostaing, Langmuir, 2013, 29, 10368–10375 CrossRef CAS PubMed.
- R. Besnard, G. Arrachart, J. Cambedouzou and S. Pellet-Rostaing, RSC Adv., 2015, 5, 57521–57531 RSC.
- R. Besnard, G. Arrachart, J. Cambedouzou and S. Pellet-Rostaing, Langmuir, 2016, 32, 4624–4634 CrossRef CAS PubMed.
- R. Besnard, G. Arrachart, J. Cambedouzou and S. Pellet-Rostaing, J. Sol-Gel Sci. Technol., 2017, 81, 452–467 CrossRef CAS.
- T. Yokoi, H. Yoshitake, T. Yamada, Y. Kubota and T. Tatsumi, J. Mater. Chem., 2006, 16, 1125–1135 RSC.
- R. Besnard, J. Cambedouzou, G. Arrachart, X. F. Le Goff and S. Pellet-Rostaing, RSC Adv., 2015, 5, 77619–77628 RSC.
- J. H. Clark and D. J. Macquarrie, Chem. Commun., 1998, 853–860 RSC.
- Y. An, M. Chen, Q. Xue and W. Liu, J. Colloid Interface Sci., 2007, 311, 507–513 CrossRef CAS PubMed.
- G. T. Hermanson, Bioconjugate Techniques, Academic Press, New York, 2nd edn, 2008, pp. 169–212 Search PubMed.
- K. A. Utting and D. J. Macquarrie, New J. Chem., 2000, 24, 591–595 RSC.
- T. Ogata, H. Narita and M. Tanaka, Hydrometallurgy, 2015, 152, 178–182 CrossRef CAS.
- T. Ogata, H. Narita and M. Tanaka, Hydrometallurgy, 2016, 163, 156–160 CrossRef CAS.
- G. Careri, U. Buontempo, F. Galluzzi, A. C. Scott, E. Gratton and E. Shyamsunder, Phys. Rev. B: Condens. Matter Mater. Phys., 1984, 30, 4689–4702 CrossRef CAS.
- G. Cerveau, R. J. P. Corriu, C. Lepeytre and P. Hubert Mutin, J. Mater. Chem., 1998, 8, 2707–2713 RSC.
- G. Arrachart, C. Carcel, P. Trens, J. J. E. Moreau and M. Wong Chi Man, Chem. – Eur. J., 2009, 15, 6279–6288 CrossRef CAS PubMed.
- A. Mehdi, J. Mater. Chem., 2010, 20, 9281–9286 RSC.
- J.-C. Broudic, O. Conocar, J. J. E. Moreau, D. Meyer and M. Wong Chi Man, J. Mater. Chem., 1999, 9, 2283–2285 RSC.
- G. E. Fryxell, H. Wu, Y. Lin, W. J. Shaw, J. C. Birnbaum, J. C. Linehan, Z. Nie, K. Kemner and S. Kelly, J. Mater. Chem., 2004, 14, 3356–3363 RSC.
- T. Ogata, H. Narita and M. Tanaka, Chem. Lett., 2014, 43, 1414–1416 CrossRef CAS.
- A. S. Suneesh, K. V. Syamala, K. A. Venkatesan, M. P. Antony and P. R. Vasudeva Rao, J. Colloid Interface Sci., 2015, 438, 55–60 CrossRef CAS PubMed.
- E. M. Soliman, Anal. Lett., 1997, 30, 1739–1751 CrossRef CAS.
- Y. M. Hu, E. Drouin, D. Lariviere, F. Kleitz and F. G. Fontaine, ACS Appl. Mater. Interfaces, 2017, 9, 38584–38593 CAS.
- X. Zheng, C. Wang, J. Dai, W. Shi and Y. Yan, J. Mater. Chem. A, 2015, 3, 10327–10335 CAS.
- D. Massiot, F. Fayon, M. Capron, I. King, S. Le Calvé, B. Alonso, J.-O. Durand, B. Bujoli, Z. Gan and G. Hoatson, Magn. Reson. Chem., 2002, 40, 70–76 CrossRef CAS.
- Y. Li, R. Beck, T. Huang, M. C. Choi and M. Divinagracia, J. Appl. Crystallogr., 2008, 41, 1134–1139 CrossRef CAS.
- B. P. Pichon, M. Wong Chi Man, C. Bied and J. J. E. Moreau, J. Organomet. Chem., 2006, 691, 1126–1130 CrossRef CAS.
- S. Lambert, K. Y. Tran, G. Arrachart, F. Noville, C. Henrist, C. Bied, J. J. E. Moreau, M. Wong Chi Man and B. Heinrichs, Microporous Mesoporous Mater., 2008, 115, 609–617 CrossRef CAS.
- F. Martin, A. Diran, J.-O. Durand, M. Granier and R. Desmet, Tetrahedron Lett., 2005, 46, 7973–7975 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Material characterization: NMR, TGA, elemental analysis, SAXS. See DOI: 10.1039/c8qm00022k |
| This journal is © the Partner Organisations 2018 |