
A direct synthetic method for (nitronyl nitroxide)-substituted π-electronic compounds via a palladium-catalyzed cross-coupling reaction with a zinc complex†
Shuichi
Suzuki
 *,
Fumiya
Nakamura
and
Takeshi
Naota
*
*,
Fumiya
Nakamura
and
Takeshi
Naota
*
Department of Chemistry, Graduate School of Engineering Science, Osaka University, Toyonaka, Osaka 560-8531, Japan. E-mail: suzuki-s@chem.es.osaka-u.ac.jp; naota@chem.es.osaka-u.ac.jp
First published on 18th January 2018
Abstract
We have developed an efficient synthetic method for (nitronyl nitroxide)-substituted (NN-substituted) π-electronic compounds via palladium-catalyzed cross-coupling reactions with a zinc complex of the parent nitronyl nitroxide radical anion. Various aryl iodides can be directly converted to the desired coupling compounds with high efficiency. The utility of the present method has been demonstrated by direct synthesis of a newly prepared radical species, NN-substituted phenothiazine 15P, which could not be obtained from the corresponding aldehyde compound by typical condensation-oxidation methods. The molecular structure and characteristic electronic nature of 15P were examined by single-crystal X-ray diffraction analysis and cyclic voltammetry, indicating that 15P is a promising candidate as a precursor for a triplet ground state species by one-electron oxidation.
Introduction
Open-shell π-electronic compounds are promising components for next-generation electronic devices because of their wide diversity in molecular design, structural flexibility, and processability.1–3 In the past few decades, nitronyl nitroxides (NN-Rs) and their related radicals have been especially utilized for organic spin sources and their unique spin-related properties have been investigated in view of purely organic ferromagnets,1 hybrid ferrimagnets,4 and negative magnetoresistance.5 NN-Rs are generally prepared via the condensation reactions of N,N′-dihydroxy-2,3-dimethylbutane-2,3-diamine with various aldehydes and subsequent oxidation with a stoichiometric amount of oxidant (Scheme 1a).6 However, the condensation reactions with highly electron-donating5,6b,7 and electron-accepting6b,8 aromatic aldehydes were sometimes unsuccessful, probably due to the low reactivity of the substrates and low stability of the products. Some modified methods using the corresponding dimethyl acetals7 and butanediamine4b,9 have been reported for the syntheses of NN-Rs with specified functionalities. One of the significant aspects to be improved in these conventional methods is the stoichiometric use of strong oxidants, such as lead dioxide6 and m-chloroperbenzoic acid,4b,9 since the use of these oxidants frequently leads to the undesired decomposition of product NN-Rs, especially with electron-rich π-electronic systems. | ||
| Scheme 1 Synthetic methods of NN derivatives: (a) typical condensation–oxidation method and (b) palladium-catalyzed cross-coupling method with NN-Zn. | ||
The direct introduction of the NN group to organic platforms using a parent NN (NN-H) as a synthetic unit is an alternative but promising method for the unlimited synthesis of a variety of NN derivatives. A number of nucleophilic coupling reactions of NN-H have been performed with various electrophiles such as MeOCH2Cl,10 MeOTf,11 CO2,11 pyridine-oxide and related-N-oxide,12 2-methyl-2-nitrosopropane,13 nitrone,14 and pentafluorocyanobenzene,15 although the methods are limited to using well-developed π-electronic systems due to preparations of reactants with suitable substituents. Recently, Okada and coworkers reported that a gold(I) complex of the NN radical anion can be used as the cross-coupling reagent for introducing NN moieties to π-electronic systems using the corresponding aryl halides.16 Herein, we have developed a one-pot synthesis of NN-substituted π-electronic compounds from NN-Hvia a zinc complex of the NN radical anion (NN-Zn), which works as a good nucleophile for palladium-catalyzed Negishi-reactions (Scheme 1b).17 The method has afforded various NN derivatives with high efficiency including a new radical species that could not be effectively obtained by typical condensation–oxidation methods.
Results and discussion
Cross-coupling reaction of aryl iodides with NN-H
We initially examined the reaction of 4-iodoacetophenone (1a) (1a) with NN-Zn in the presence of a tetrakis(triphenylphosphine)palladium(0) [Pd(PPh3)4] catalyst (Table 1, entry 1). After the generation of lithiated NN (NN-Li) upon the treatment of NN-H with lithium hexamethyldisilazane (LHMDS) at −78 °C in tetrahydrofuran (THF),11–15 the resulting bluish purple mixture was allowed to react with ZnCl2 at −78 °C to afford the reddish purple solution of NN-Zn. To the solution of NN-Zn, a THF solution of two equivalents of 1a and a catalytic amount of Pd(PPh3)4 was added and refluxed for 2 h.18 The solvent was removed under reduced pressure, then the residue was purified by silica gel column chromatography to afford the product 1P with a good yield (78% yield based on NN-H).
Since a marked color change was observed during the consumption and generation of various NN species, the reaction could be periodically monitored by means of UV-vis analysis (Fig. 1a and Fig S15, ESI†). In the initial state, the spectrum of NN-H showed an absorption band at approximately 540 nm. After the addition of LHMDS, the absorption was shifted to a longer wavelength region (λmax = 635 nm). Then, the absorption band was blue-shifted to 541 nm (and 426 nm) after the subsequent addition of ZnCl2. Finally, the solution color turned to purple (λmax = 605 nm), indicating the generation of the coupling product 1P. Also, the reaction could be monitored by ESR spectra. The ten-line ESR spectrum that can be attributed to NN-H (|aN| (hyperfine coupling of 14N nuclei) = 0.743 mT and |aH| (hyperfine coupling of 1H nucleus) = 0.358 mT, in dichloromethane) was converted to a five-line spectrum attributed to NN-Li (|aN| = 0.752 mT, in THF) (Fig. 1b). The hyperfine coupling constant was similar to the reported value of the NN radical anion generated from NN-H and potassium tert-butoxide in dimethylsulfoxide.10,19 The ESR spectrum of 1P was identical to the reported spectrum.16a
 | ||
| Fig. 1 (a) UV-Vis spectra change during the coupling reaction of NN-H and 1a in THF: initial state of the reaction (red solid line), the mixture after addition of LHMDS (blue dashed line), the mixture after addition of ZnCl2 (purple dashed line), and the isolated product 1P (blue solid line). (b) ESR spectra of NN-H (red line, in dichloromethane), NN-Li generated by addition of LHMDS (blue-dashed line, in THF), and 1P (blue line, in dichloromethane). | ||
The catalytic activity of various palladium complexes was examined for the reaction of 1a with an NN-Zn reagent under the same conditions. Pd(PPh3)4 was the best catalyst among the catalysts examined. The reactions using other palladium catalyst systems such as bis(triphenylphosphine)palladium(II) dichloride [Pd(PPh3)2Cl2], 1,1′-bis((diphenylphosphino)ferrocene)palladium(II) dichloride [Pd(dppf)Cl2], (palladium(II) acetate)–(2-dicyclohexylphosphino-2′,4′,6′-triisopropylbiphenyl) [Pd(OAc)2–XPhos], and tris(dibenzylideneacetone)dipalladium(0)–chloroform adduct [Pd2(dba)3·CHCl3] were not effective to give the desired product (Table 1, entry 4–7). The use of toluene as a solvent alternated to THF under similar conditions to entry 1 gave trace amounts of the desired product.
Table 2 summarizes the representative results of the one-pot Pd(PPh3)4-catalyzed coupling reactions of various aryl halides with NN-Zn. Various aryl iodides could be converted into the corresponding NN-coupled products efficiently by stirring in boiling THF for 2 h (entries 2, 4–10 and 12–15), while a similar treatment of aryl bromides gave unsatisfactory results (entries 1 and 3). NN compounds bearing phenyl, naphthyl, and thiophenyl moieties (3P, 9P, and 11P) can be obtained from the corresponding aryl iodides (3a, 9a, 11a) (entries 4, 10, and 12). Phenyl iodides with various electron-donating and electron-withdrawing groups (4a–8a) could be converted into the desired NN derivatives (4P–8P) (entries 5–9), while the reaction with pentafluorophenyl iodide (10a) hardly occurred under similar conditions, probably due to its strong electron-withdrawing property (entry 11). Additionally, diiodo compounds 12a–14a were selectively converted to the corresponding mono-substituted NN compounds 12P–14P, although excess equivalents of NN-H (Table 2, entries 13–15) were used.20 A molecular structure of 14P was unequivocally established by single-crystal X-ray diffraction (XRD) analysis of the crystal obtained from dichloromethane/hexane (Fig. 2a).21 Notably, monoiodo compounds 12P, 13P, and 14P were employed as convenient synthetic intermediates for various NN π-electronic molecules by means of Suzuki cross-coupling reactions.22
|
|
|||
|---|---|---|---|
| Entry | Ar-X | Product | Yield/% |
| a Conditions: 1 equiv. NN-H, 0.45 equiv. aryl iodide, 10 mol% Pd(PPh3)4. b Detected by TLC. c Isolated yield based on NN-H. d Isolated yield based on aryl diiodide. | |||
| 1 | 1b | 1P | Traceb |
| 2 | 2a | 2P | 77c |
| 3 | 2b | 2P | Traceb |
| 4 | 3a | 3P | 85c |
| 5 | 4a | 4P | 76c |
| 6 | 5a | 5P | 81c |
| 7 | 6a | 6P | 69c |
| 8 | 7a | 7P | 72c |
| 9 | 8a | 8P | 65c |
| 10 | 9a | 9P | 48c |
| 11 | 10a | 10P | Traceb |
| 12 | 11a | 11P | 61c |
| 13a | 12a | 12P | 65d |
| 14a | 13a | 13P | 53d |
| 15a | 14a | 14P | 71d |

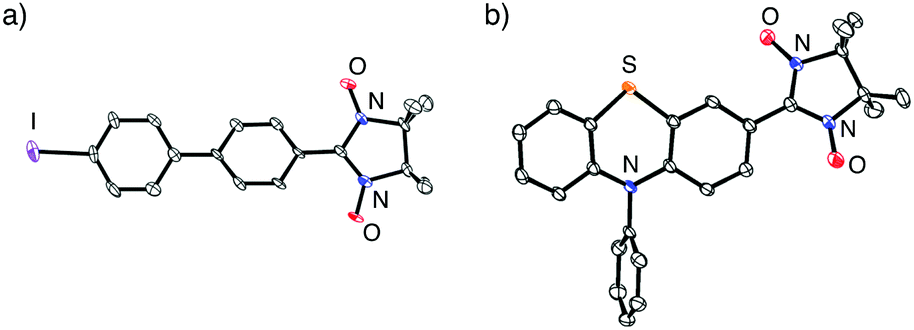 | ||
| Fig. 2 ORTEP views of (a) 14P and (b) 15P (50% probabilities). Hydrogen atoms were omitted for clarity. | ||
Synthesis of NN-substituted phenothiazine
The present method can be applied to the synthesis of the newly designed NN-substituted phenothiazine 15P, the one electron oxidation species of which could be an ideal molecular system for a radical-substituted radical cation in the triplet ground state through the spin polarization mechanism from the viewpoint of spin density distribution, as shown in Fig. 3a.4,5,23 The theoretical calculation suggests that 15P+ was expected to be in a triplet ground state with large intramolecular interactions (Jintra-calc./kB = +1032 K) (Table S1, ESI†).23,24 It was found that the yields of NN-substituted phenothiazine derivatives by condensation methods with hydroxylamine (Scheme 1a) were quite low, partially because sulfur atoms on the phenothiazine derivatives were easily oxidized by strong oxidants, such as mCPBA, to give S-oxide and S-dioxide derivatives.25 | ||
| Fig. 3 (a) Spin density map of diradical cation 15P+ in the triplet state estimated by DFT (UB3LYP/6-31G*) calculation. Red and green orbitals denote positive and negative spin densities, respectively. (b) Cyclic voltammogram of 15P in dichloromethane. | ||
The desired compound 15P could be prepared from the corresponding iodophenothiazine 15a and NN-H in 60% yield under similar reaction conditions (Scheme 2). The radical 15P is quite stable even under aerated conditions in both solution and the solid states. The molecular structure of 15P was determined by single-crystal XRD analysis of crystals obtained by recrystallization from dichloromethane/hexane (Fig. 2b).21
 | ||
| Scheme 2 Synthetic procedure of NN-substituted phenothiazine 15P. | ||
In order to clarify the possibility of the generation of the triplet species 15P+, we measured a cyclic voltammogram of 15P in dichloromethane (Fig. 3b). Compound 15P had two reversible redox waves at +0.32 V vs. Fc/Fc+ and +0.52 V vs. Fc/Fc+. The former and latter waves are attributed to the oxidation processes of the phenothiazine moiety and the NN moiety, respectively, from the comparison of the results of cyclic voltammograms for N-phenyl-10H-phenothiazine (15R) (EOX = +0.26 V vs. Fc/Fc+) and NN-H (EOX = +0.39 V vs. Fc/Fc+). This result suggests that the one-electron oxidation species of 15P has the electronic structure of an NN-radical-substituted phenothiazine radical cation, which is expected to be in the triplet ground state. The oxidation potential of the phenothiazine moiety for 15P was slightly higher than that of 15R due to the electron-withdrawing property of the NN moiety. The large shift in the oxidation potential of the NN moiety was due to the on-site Coulombic repulsions in the dicationic state 15P2+.
Conclusions
A new method of introducing the NN moiety to π-electronic systems via a palladium-catalyzed cross-coupling reaction with NN-Zn was developed. Various aryl iodides were converted to NN-substituted π-electronic compounds by the present one-pot reactions. Mono-substituted NN compounds 12P–14P, building blocks for further modifications of NN-substituted π-electronic systems, were obtained from the corresponding aryl diiodide, selectively. The utility of the present method was demonstrated by the direct synthesis of NN-substituted phenothiazine 15P, which could not be prepared via typical condensation–oxidation methods. The radical species 15P is a promising candidate as a precursor for a triplet ground state species by one-electron oxidation. The preparations and investigations of diradical cation 15P+ including other phenothiazine derivatives substituted with NN moieties are underway. We believe that the reported method will pave the way for the efficient direct access of molecular conversions from closed shell systems to open shell ones.Experimental
General information
UV-vis spectra were recorded on a V650 spectrometer. IR spectra were recorded on a JASCO FT/IR-400 spectrometer. IR absorption intensities are designed as follows: s (strong), m (medium), w (weak). FAB-MS and APCI-MS spectra were recorded on a JEOL JMS-700 MStation spectrometer and a Bruker micrOTOF II spectrometer. ESR spectra were recorded on a Bruker EMX spectrometer. All the samples for the ESR measurements were degassed by a freeze–pump–thaw method, and then the sample tubes were sealed before the measurements. Cyclic voltammogram measurements were carried out on an ALS Electrochemical Analyzer Model CH600, and recorded with a platinum electrode as a working electrode, a platinum wire as a counter electrode, and a Ag/AgCl electrode as a reference electrode. The measurements were carried out with a sweep rate of 100 mV s−1 in dichloromethane containing 0.1 M tetra-n-butylammonium hexafluorophosphate as electrolyte. The redox potentials were finally corrected by the ferrocene/ferrocenium couple (Fc/Fc+). The X-ray data were collected by a Rigaku XtaLAB P200 diffractometer with graphite monochromated Mo-Kα radiation.Silica gel 60 (230–400 mesh) was used for column chromatography. All commercially available compounds were reagent grade and used without further purification. 4,4,5,5-Tetramethylimidazoline-3-oxide-1-oxyl (NN-H)10 and 3-iodo-10-phenyl-10H-phenothiazine (15a)26 were prepared according to the literature. Known compounds 1P–13P were identified by ESR and TLC.
General synthetic procedures
4,4,5,5-Tetramethylimidazoline-3-oxide-1-oxyl (NN-H) was dissolved in THF under argon and a THF solution of lithium bis(trimethylsilyl)amide (LHMDS) (1 eq.) was added to the solution at −78 °C. After stirring for 20 min, a THF solution of zinc chloride (1 eq.) was added at −78 °C and stirred for 10 min. Then, a THF solution of aryl iodide (2 eq.) and palladium catalyst (10 mol%) was added at −78 °C. The reaction mixture was gradually warmed up to 80 °C and stirred for 2 h. After being cooled to room temperature, the mixture was concentrated under reduced pressure. The product was purified by silica gel column chromatography, to afford the corresponding cross-coupling product.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 as the eluent. TLC (silica gel) Rf 0.40; ESR g = 2.007 (ν0 = 9.815013 GHz), |aN| = 0.752 mT (in CH2Cl2 at room temperature); MS (FAB+): m/z 275 [M+], 276 [(M + H)+], 277 [(M + 2H)+].
:1 as the eluent. TLC (silica gel) Rf 0.79; ESR g = 2.007 (ν0 = 9.813534 GHz), |aN| = 0.761 mT (in CH2Cl2 at room temperature); MS (APCI+): m/z 309 [M+], 310 [(M + H)+], 311 [(M + 2H)+].
:1 as the eluent. TLC (silica gel) Rf 0.57; ESR g = 2.007 (ν0 = 9.608108 GHz), |aN| = 0.769 mT in (CH2Cl2 at room temperature); MS (FAB+): m/z 233 [M+], 234 [(M + H)+], 235 [(M + 2H)+].
:1 as the eluent. TLC (silica gel) Rf 0.79; ESR g = 2.007 (ν0 = 9.811410 GHz), |aN| = 0.773 mT (in CH2Cl2 at room temperature); MS (FAB+): m/z 247 [M+], 248 [(M + H)+], 249 [(M + 2H)+].
:1 as the eluent. TLC (silica gel) Rf 0.69; ESR g = 2.007 (ν0 = 9.598689 GHz), |aN| = 0.752 mT (in CH2Cl2 at room temperature); MS (FAB+): m/z 278 [M+], 279 [(M + H)+].
:1 as the eluent. TLC (silica gel) Rf 0.82; ESR g = 2.007 (ν0 = 9.812717 GHz), |aN| = 0.773 mT (in CH2Cl2 at room temperature); MS (FAB+): m/z 258 [M+], 259 [(M + H)+], 260 [(M + 2H)+].
:1 as the eluent. TLC (silica gel) Rf 0.76; ESR g = 2.007 (ν0 = 9.813716 GHz), |aN| = 0.748 mT (in CH2Cl2 at room temperature); MS (FAB+): m/z 261 [M+], 262 [(M + H)+], 263 [(M + 2H)+].
:1 as the eluent. TLC (silica gel) Rf 0.49; ESR g = 2.007 (ν0 = 9.616935 GHz), |aN| = 0.769 mT (in CH2Cl2 at room temperature); MS (FAB+): m/z 263 [M+], 264 [(M + H)+], 265 [(M + 2H)+].
:1 as the eluent. TLC (silica gel) Rf 0.53; ESR g = 2.007 (ν0 = 9.595996 GHz), |aN| = 0.734 mT (in CH2Cl2 at room temperature); MS (FAB+): m/z 283 [M+], 284 [(M + H)+], 285 [(M + 2H)+].
:1 as the eluent. TLC (silica gel) Rf 0.67; ESR g = 2.007 (ν0 = 9.599547 GHz), |aN| = 0.787 mT (in CH2Cl2 at room temperature); MS (FAB+): m/z 239 [M+], 240 [(M + H)+], 241 [(M + 2H)+].
:1 as the eluent. TLC (silica gel) Rf 0.81; ESR g = 2.007 (ν0 = 9.810618 GHz), |aN| = 0.752 mT (in CH2Cl2 at room temperature); MS (APCI+): m/z 359 [M+], 360 [(M + H)+].
:1 as the eluent. TLC (silica gel) Rf 0.81; ESR g = 2.007 (ν0 = 9.813807 GHz), |aN| = 0.752 mT (in CH2Cl2 at room temperature); MS (APCI+): m/z 359 [M+], 360 [(M + H)+], 361 [(M + 2H)+].
:1 as the eluent. TLC (silica gel) Rf 0.82; mp: 124–125 °C; ESR g = 2.007 (ν0 = 9.812223 GHz), |aN| = 0.757 mT (in CH2Cl2 at room temperature); MS (APCI+): m/z 435 [M+], 436 [(M + H)+], 437 [(M + 2H)+]; high resolution MS (APCI+): found m/z 436.0624; calculated for C19H24N2O2I m/z 436.0642; IR (KBr, cm−1): 2925 (m), 2853 (w), 1605 (m), 1472 (m), 1361 (m), 1310 (m), 1166 (m), 1001 (s), 807 (s).
:1 as the eluent. TLC (silica gel) Rf 0.40; ESR g = 2.007 (ν0 = 9.815013 GHz), |aN| = 0.752 mT (in CH2Cl2 at room temperature); MS (FAB+): m/z 275 [M+], 276 [(M + H)+], 277 [(M + 2H)+].
:1 as the eluent. TLC (silica gel) Rf 0.79; ESR g = 2.007 (ν0 = 9.813534 GHz), |aN| = 0.761 mT (in CH2Cl2 at room temperature); MS (APCI+): m/z 309 [M+], 310 [(M + H)+], 311 [(M + 2H)+].
:1 as the eluent. TLC (silica gel) Rf 0.57; ESR g = 2.007 (ν0 = 9.608108 GHz), |aN| = 0.769 mT in (CH2Cl2 at room temperature); MS (FAB+): m/z 233 [M+], 234 [(M + H)+], 235 [(M + 2H)+].
:1 as the eluent. TLC (silica gel) Rf 0.79; ESR g = 2.007 (ν0 = 9.811410 GHz), |aN| = 0.773 mT (in CH2Cl2 at room temperature); MS (FAB+): m/z 247 [M+], 248 [(M + H)+], 249 [(M + 2H)+].
:1 as the eluent. TLC (silica gel) Rf 0.69; ESR g = 2.007 (ν0 = 9.598689 GHz), |aN| = 0.752 mT (in CH2Cl2 at room temperature); MS (FAB+): m/z 278 [M+], 279 [(M + H)+].
:1 as the eluent. TLC (silica gel) Rf 0.82; ESR g = 2.007 (ν0 = 9.812717 GHz), |aN| = 0.773 mT (in CH2Cl2 at room temperature); MS (FAB+): m/z 258 [M+], 259 [(M + H)+], 260 [(M + 2H)+].
:1 as the eluent. TLC (silica gel) Rf 0.76; ESR g = 2.007 (ν0 = 9.813716 GHz), |aN| = 0.748 mT (in CH2Cl2 at room temperature); MS (FAB+): m/z 261 [M+], 262 [(M + H)+], 263 [(M + 2H)+].
:1 as the eluent. TLC (silica gel) Rf 0.49; ESR g = 2.007 (ν0 = 9.616935 GHz), |aN| = 0.769 mT (in CH2Cl2 at room temperature); MS (FAB+): m/z 263 [M+], 264 [(M + H)+], 265 [(M + 2H)+].
:1 as the eluent. TLC (silica gel) Rf 0.53; ESR g = 2.007 (ν0 = 9.595996 GHz), |aN| = 0.734 mT (in CH2Cl2 at room temperature); MS (FAB+): m/z 283 [M+], 284 [(M + H)+], 285 [(M + 2H)+].
:1 as the eluent. TLC (silica gel) Rf 0.67; ESR g = 2.007 (ν0 = 9.599547 GHz), |aN| = 0.787 mT (in CH2Cl2 at room temperature); MS (FAB+): m/z 239 [M+], 240 [(M + H)+], 241 [(M + 2H)+].
:1 as the eluent. TLC (silica gel) Rf 0.81; ESR g = 2.007 (ν0 = 9.810618 GHz), |aN| = 0.752 mT (in CH2Cl2 at room temperature); MS (APCI+): m/z 359 [M+], 360 [(M + H)+].
:1 as the eluent. TLC (silica gel) Rf 0.81; ESR g = 2.007 (ν0 = 9.813807 GHz), |aN| = 0.752 mT (in CH2Cl2 at room temperature); MS (APCI+): m/z 359 [M+], 360 [(M + H)+], 361 [(M + 2H)+].
:1 as the eluent. TLC (silica gel) Rf 0.82; mp: 124–125 °C; ESR g = 2.007 (ν0 = 9.812223 GHz), |aN| = 0.757 mT (in CH2Cl2 at room temperature); MS (APCI+): m/z 435 [M+], 436 [(M + H)+], 437 [(M + 2H)+]; high resolution MS (APCI+): found m/z 436.0624; calculated for C19H24N2O2I m/z 436.0642; IR (KBr, cm−1): 2925 (m), 2853 (w), 1605 (m), 1472 (m), 1361 (m), 1310 (m), 1166 (m), 1001 (s), 807 (s).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Grant-in-Aid for Scientific Research on Innovative Areas “π-System Figuration: Control of Electron and Structural Dynamism for Innovative Functions (2601)” (JSPS KAKENHI Grant Number JP26102005 for S. S.), and Grant-in-Aid for Scientific Research (JSPS KAKENHI Grant Number JP17K05783 (S. S.), JP15H03796 (T. N.), and JP16H06516 (T. N.)). We thank Professor Dr Keiji Okada and Professor Dr Masatoshi Kozaki (Osaka City University) for helpful discussion of phenothiazine compounds.Notes and references
- (a) O. Kahn, in Molecular Magnetism, Wiley-VCH, New York, 1993, pp. 1 Search PubMed; (b) Molecular Magnetism, ed. K. Itoh and M. Kinoshita, Kodansha & Gordon and Breach, Tokyo, 2000 Search PubMed; (c) Magnetism: Molecules to Materials II–IV, ed. J. S. Miller and M. Drillon, Wiley-VCH, Weinheim, 2001–2005 Search PubMed; (d) π-Electron Magnetism From Molecules to Magnetic Materials, ed. J. Veciana, Springer, Berlin, 2001 Search PubMed; (e) D. Gatteschi, R. Sessoli and J. Villain, in Molecular Nanomagnets, Oxford University Press, New York, 2006, pp 1 Search PubMed.
- Stable Radicals: Fundamentals and Applied Aspects of Odd-Electron Compounds, ed. R. G. Hicks, Wiley, Chichester, 2010 Search PubMed.
- Y. Morita, S. Suzuki, K. Sato and T. Takui, Nat. Chem., 2011, 3, 197 CrossRef CAS PubMed.
- (a) Y. Masuda, M. Kuratsu, S. Suzuki, M. Kozaki, D. Shiomi, K. Sato, T. Takui, Y. Hosokoshi, X.-Z. Lan, Y. Miyazaki, A. Inaba and K. Okada, J. Am. Chem. Soc., 2009, 131, 4670 CrossRef CAS PubMed; (b) M. Kuratsu, S. Suzuki, M. Kozaki, D. Shiomi, K. Sato, T. Takui, T. Kanzawa, Y. Hosokoshi, X.-Z. Lan, Y. Miyazaki, A. Inaba and K. Okada, Chem. – Asian J., 2012, 7, 1604 CrossRef CAS PubMed.
- (a) M. M. Matsushita, H. Kawakami, Y. Kawada and T. Sugawara, Chem. Lett., 2007, 110 CrossRef CAS; (b) M. M. Matsushita, H. Kawakami, T. Sugawara and M. Ogata, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 195208 CrossRef; (c) T. Sugawara, H. Komatsu and K. Suzuki, Chem. Soc. Rev., 2011, 40, 3105 RSC.
- (a) J. H. Osiecki and E. F. Ullman, J. Am. Chem. Soc., 1968, 90, 1078 CrossRef CAS; (b) E. F. Ullman, J. H. Osiecki, D. G. B. Boocock and R. Darcy, J. Am. Chem. Soc., 1972, 94, 7049 CrossRef CAS.
- S. Hiraoka, T. Okamoto, M. Kozaki, D. Shiomi, K. Sato, T. Takui and K. Okada, J. Am. Chem. Soc., 2004, 126, 58 CrossRef CAS PubMed.
- (a) Y. Hosokoshi, M. Tamura, H. Sawa, R. Kato and M. Kinoshita, J. Mater. Chem., 1995, 5, 41 RSC; (b) O. V. Koreneva, G. V. Romanenko, Y. G. Shvedenkov, V. N. Ikorskii and V. I. Ovcharenko, Polyhedron, 2003, 22, 2487 CrossRef CAS.
- C. Hirel, K. E. Vostrikova, J. Pécaut, V. I. Ovcharenko and P. Rey, Chem. – Eur. J., 2001, 7, 2007 CrossRef CAS.
- D. G. B. Boocock, R. Darcy and E. F. Ullman, J. Am. Chem. Soc., 1968, 90, 5945 CrossRef CAS.
- R. Weiss, N. Kraut and F. Hampel, J. Organomet. Chem., 2001, 617–618, 473 CrossRef CAS.
- (a) O. N. Chupakhin, I. A. Utepova, M. V. Varaksin, E. V. Tretyakov, G. V. Romanenko, D. V. Stass and V. I. Ovcharenko, J. Org. Chem., 2009, 74, 2870 CrossRef CAS PubMed; (b) V. I. Ovcharenko, O. N. Chupakhin, I. S. Kovalev, E. V. Tretyakov, G. V. Romanenko and D. V. Stass, Russ. Chem. Bull., 2008, 57, 2227 CrossRef CAS.
- (a) S. Suzuki, T. Furui, M. Kuratsu, M. Kozaki, D. Shiomi, K. Sato, T. Takui and K. Okada, J. Am. Chem. Soc., 2010, 132, 15908 CrossRef CAS PubMed; (b) E. V. Tretyakov, S. E. Tolstikov, G. V. Romanenko, A. S. Bogomyakov, D. V. Stass, A. G. Maryasov, N. P. Gristan and V. I. Ovcharenko, Russ. Chem. Bull., 2011, 60, 2608 CrossRef CAS.
- (a) M. V. Varaksin, E. V. Tretyakov, I. A. Utepova, G. V. Romanenko, A. S. Bogomyakov, D. V. Stass, R. Z. Sagdeev, V. I. Ovcharenko and O. N. Chupakhin, Russ. Chem. Bull., 2012, 61, 1469 CrossRef CAS; (b) S. Tolstikov, E. Tretyakov, S. Fokin, E. Suturina, G. Romanenko, A. Bogomyakov, D. Stass, A. Maryasov, M. Fedin, N. Gritsan and V. Ovcharenko, Chem. – Eur. J., 2014, 20, 2793 CrossRef CAS PubMed.
- E. V. Tretyakov, P. A. Fedyushim, E. V. Panteleeva, D. V. Stass, I. Y. Bagryanskaya, I. V. Beregovaya and A. S. Bogomyakov, J. Org. Chem., 2017, 82, 4179 CrossRef CAS PubMed.
- (a) R. Tanimoto, S. Suzuki, M. Kozaki and K. Okada, Chem. Lett., 2014, 43, 678 CrossRef CAS; (b) S. Suzuki, S. Kira, M. Kozaki, M. Yamamura, T. Hasegawa, T. Nabeshima and K. Okada, Dalton Trans., 2017, 46, 2563 Search PubMed; (c) E. Tretyakov, K. Okada, S. Suzuki, M. Baumgarten, G. Romanenko, A. Bogomyakov and V. Ovcharenko, J. Phys. Org. Chem., 2016, 29, 725 CrossRef CAS; (d) E. Tretyakov, A. Keerthi, M. Baumgarten, S. Veber, M. Fedin, D. Gorbunov, I. Shundrina and N. Gritsan, ChemistryOpen, 2017, 6, 642 CrossRef CAS PubMed; (e) M. Haraguchi, E. Tretyakov, N. Gritsan, G. Romanenko, D. Gorbunov, A. Bogomyakov, K. Maryunina, S. Suzuki, M. Kozaki, D. Shiomi, K. Sato, T. Takui, S. Nishihara, K. Inoue and K. Okada, Chem. – Asian J., 2017, 12, 2929 CrossRef CAS PubMed.
- (a) D. Hass, J. M. Hammann, R. Greiner and P. Knochel, ACS Catal., 2016, 6, 1540 CrossRef; (b) E. Negishi, Angew. Chem., Int. Ed., 2011, 50, 6738 CrossRef CAS PubMed.
- The product was gradually decomposed in the case of longer reaction time under presence of Pd(PPh3)4 probably due to deoxygenated reaction.
- The ESR spectrum after addition of zinc chloride was complicated due to instability of NN-Zn.
- Mono-substituted NN compounds, 12P–14P, could not be converted to bisNN compounds by using the similar methods. Weak coordination properties of Mono-substituted NN compounds implied to inhibit the catalytic reactions.
- CCDC 1588474 (14P) and 1588475 (15P)†.
- (a) C. Stroh, M. Mayor and C. von Hänisch, Eur. J. Org. Chem., 2005, 3697 CrossRef CAS; (b) C. Stroh, M. Mayor and C. von Hänisch, Tetrahedron Lett., 2004, 45, 9623 CrossRef CAS.
- Calculated by Gaussian 09. See, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- K. Yamaguchi, T. Kawakami, Y. Takano, Y. Kitagawa, Y. Yamashita and H. Fujita, Int. J. Quantum Chem., 2002, 90, 370 CrossRef CAS.
- M. Toşa, C. Paizs, C. Majdik, L. Novák, P. Kolonits, F.-D. Irimiea and L. Poppeb, Tetrahedron: Asymmetry, 2002, 13, 211 CrossRef.
- R. Sugimura, S. Suzuki, M. Kozaki, K. Keyaki, K. Nozaki, H. Matsushita, N. Ikeda and K. Okada, Res. Chem. Intermed., 2013, 39, 185 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: ESR spectra of isolated products, detailed UV-vis spectra during reaction, and calculated results. CCDC 1588474 (14P) and 1588475 (15P). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7qm00565b |
| This journal is © the Partner Organisations 2018 |