DOI:
10.1039/C7QM00553A
(Research Article)
Mater. Chem. Front., 2018,
2, 597-602
Solid-state self-inclusion complexation behaviour of a pillar[5]arene-based host–guest conjugate†
Received
3rd December 2017
, Accepted 24th January 2018
First published on 24th January 2018
Abstract
A host–guest conjugate consisting of a pillar[5]arene and an ethylene moiety containing a triazole group at one end and a perfluorooctyl group at the other end was synthesized. The host–guest conjugate displayed unusual real-time scale solid-state self-inclusion complexation behaviour. The host–guest conjugate formed a de-threaded supramolecular structure only in solution, even at high concentrations. However, heating the solid-state host–guest conjugate led to the formation of a self-inclusion complex structure at a real-time scale; at 100 °C, it took ca. 20 h to form the self-inclusion complex at the equilibrium state at a conversion of 85%. The self-inclusion complex converted to the de-threaded form at a real-time scale when it was stored in solvents with small molecules, which worked as competitive guests.
Introduction
A self-inclusion complex, pseudo[1]rotaxane, which consists of a macrocyclic host–guest conjugate, is one of the simpler supramolecular structures among various supramolecular structures such as rotaxanes, catenanes, polyrotaxanes, and polycatenanes.1–12 The host–guest conjugates exhibit a fast or slow exchange between the self-included and de-threaded supramolecular structure when exposed to different external stimuli. As the concentration of the host–guest conjugate increases, self-assembly normally leads to a mixture of a self-inclusion complexes and linear and cyclic supramolecular oligomers and polymers. Crown ethers,13–15 cyclodextrins,16–18 cucurbit[n]urils,19,20 calix[n]arenes,21–23 and pillar[n]arenes24–34 have been utilized as hosts to make host–guest conjugates. The kinetics of the self-inclusion complexation process and stability of the self-inclusion complex can be controlled by tuning the relationship between the cavity size of the macrocyclic hosts and length, size, and functional groups on the guest. In most cases, the change in the supramolecular structure in solution can be monitored using 1H NMR and optical measurements such as UV, fluorescence and CD measurements. In this study, we discovered an unusual solid-state self-inclusion complexation behaviour when we synthesized a new pillar[5]arene-based host–guest conjugate.
Pillar[n]arenes,35–37 which were first reported by our group,38 form stable host–guest complexes with linear alkanes containing electron-withdrawing groups at both ends.39–43 Thus, we used an ethylene moiety with two different electron-withdrawing groups, a triazole and a perfluorooctyl group, at each end as the guest to make the host–guest conjugate 1 with 9 cyclohexyl groups (Cy, Fig. 1). The host–guest conjugate 1 only formed the de-threaded structure 1d in solution. Interestingly, heating 1d in the solid state led to formation of a self-inclusion complex structure (1s). The change in the supramolecular structure in the solid state occurred at a real-time scale: it took ca. 20 h to reach the equilibrium state by heating at 100 °C. By controlling the temperature, 85% conversion of 1d to 1s was achieved at the equilibrium state. Reports of changes to a supramolecular structure in the solid-state are very rare. Horie and co-workers reported pseudorotaxanes that act as a thermal- and photo-driven molecular switch in the single-crystal state.44,45 Monitoring of macrocyclic ring motion of [2]rotaxane using 2H solid state NMR in metal–organic frameworks was reported by Loeb and co-workers.46 Takata and co-workers reported thermal-induced solid-state ring-shuttling in side-chain-type polyrotaxanes.47 However, to the best of our knowledge, there are no examples of solid-state supramolecular structural change of host–guest conjugates from a de-threaded to a self-inclusion complex structure by heating. Furthermore, 1s was very stable; even at high concentrations, 1s did not change to other supramolecular structures such as linear and cyclic supramolecular oligomers and polymers. This is an unusual phenomenon because supramolecular structures change from self-inclusion complexes to liner and cyclic supramolecular oligomers and polymers with increasing concentration.22–26 The selective synthesis of self-inclusion complexes has been reported using cyclodextrins and pillar[5]arenes as wheels,17,18,30–34 but the complexation to form the self-inclusion complexes was mainly in solution. To the best of our knowledge, this is the first example of self-inclusion complexation by external stimuli in the solid state, while there are reports of self-inclusion complexes in solid state.30–34 Furthermore, 1s was converted back to 1d by storing 1s in solution. The structural change also occurred at a real-time scale; it took ca. 20 h to reach the equilibrium state at 25 °C. In this study, we investigated the mechanism of the unusual self-inclusion complexation in the solid state and de-threading behaviour in solution.
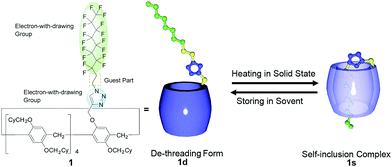 |
| | Fig. 1 Chemical structure of pillar[5]arene-based host–guest conjugate and supramolecular structural changes between de-threading form 1d and self-inclusion complex 1s. Cy: cyclohexyl group. | |
Experimental section
Materials and methods
Pillar[5]arene containing a monoalkyne group (2) and a monoazide containing ethylene and perfluorooctyl moieties (3) were synthesized according to previous reports.24,48 Solution 1H NMR spectra were recorded at 500 MHz with a JEOL-ECA500 spectrometer. Powder X-ray diffraction (PXRD) measurements were performed on a Rigaku Smart Lab high-resolution diffractometer. Differential scanning calorimetry (DSC) was performed using a PerkinElmer Jade DSC calorimeter equipped with an Intra-cooler II under a flow of dry nitrogen and with a heating rate of 10 °C min−1.
Synthetic procedures
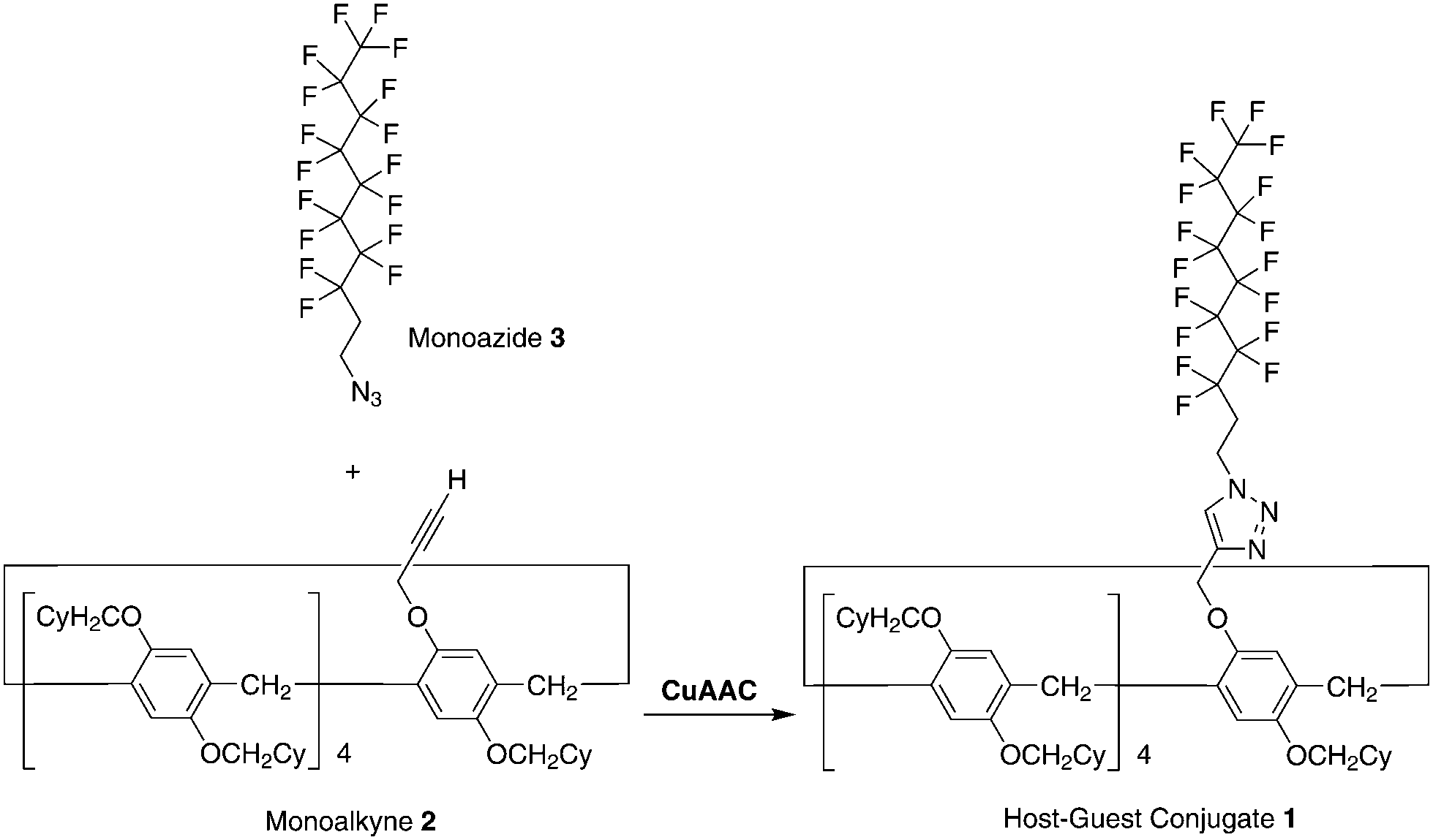
The synthesis of the pillar[5]arene-based host–guest conjugate 1 is shown in Scheme 1. Tris[(1-benzyl-1H-1,2,3-triazol-4-yl) methyl]amine (173 mg, 0.321 mmol) and Cu(CH3CN)4PF6 (121 mg, 0.321 mmol) were added to a solution of the monoalkyne 224 (485 mg, 0.321 mmol) and monoazide 348 (200 mg, 4.10 mmol) in dichloromethane (20 mL). The mixture was stirred at 25 °C for 48 h. The resulting solution was concentrated under vacuum. Column chromatography (silica gel; hexane/dichloromethane = 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) afforded a white solid in the de-threaded form (1d, 155 mg, 0.0774 mmol, yield: 24%). 1H NMR (CDCl3, 500 MHz, ppm): δ = 7.63 (s, 1H, triazole unit), 6.74–7.00 (m, 10H, phenyl), 5.25, 5.23, 5.08, 5.06 (dd, 2H, methylene), 4.65, 4.67, 4.67 (t, 2H, methylene), 3.36–3.88 (m, 28H, methylene), 2.81–2.91 (m, 2H, methylene), 1.05–2.05 (m, 99H, cyclohexyl group). 13C NMR (CDCl3, 125 MHz, ppm): δ = 150.6, 149.9, 148.7, 145.8, 128.8, 128.6, 128.5, 128.4, 128.3, 128.2, 128.1, 127.9, 127.8, 122.6, 115.4, 115.1, 115.0, 114.9, 114.7, 74.2, 74.1, 73.9, 62.8, 42.3, 38.6, 38.5, 38.4, 32.0, 31.9, 30.5, 30.4, 30.2, 30.0, 29.5, 29.2, 29.0, 26.7, 26.1, 26.0. HRESIMS: m/z calcd for C111H200F17N3Na1O10 [M + Na]+: 2025.0478, found 2025.0474.
:1) afforded a white solid in the de-threaded form (1d, 155 mg, 0.0774 mmol, yield: 24%). 1H NMR (CDCl3, 500 MHz, ppm): δ = 7.63 (s, 1H, triazole unit), 6.74–7.00 (m, 10H, phenyl), 5.25, 5.23, 5.08, 5.06 (dd, 2H, methylene), 4.65, 4.67, 4.67 (t, 2H, methylene), 3.36–3.88 (m, 28H, methylene), 2.81–2.91 (m, 2H, methylene), 1.05–2.05 (m, 99H, cyclohexyl group). 13C NMR (CDCl3, 125 MHz, ppm): δ = 150.6, 149.9, 148.7, 145.8, 128.8, 128.6, 128.5, 128.4, 128.3, 128.2, 128.1, 127.9, 127.8, 122.6, 115.4, 115.1, 115.0, 114.9, 114.7, 74.2, 74.1, 73.9, 62.8, 42.3, 38.6, 38.5, 38.4, 32.0, 31.9, 30.5, 30.4, 30.2, 30.0, 29.5, 29.2, 29.0, 26.7, 26.1, 26.0. HRESIMS: m/z calcd for C111H200F17N3Na1O10 [M + Na]+: 2025.0478, found 2025.0474.
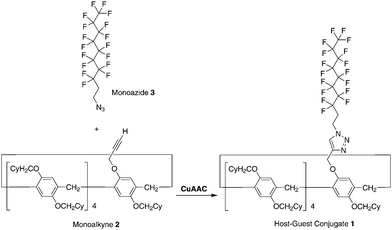 |
| | Scheme 1 Synthesis of the host–guest conjugate 1 by copper(I)-catalysed alkyne–azide cycloaddition (CuAAC) ‘‘click’’ reaction between the monoalkyne 2 and monoazide 3. | |
Monitoring the self-complexation and de-threading behaviour
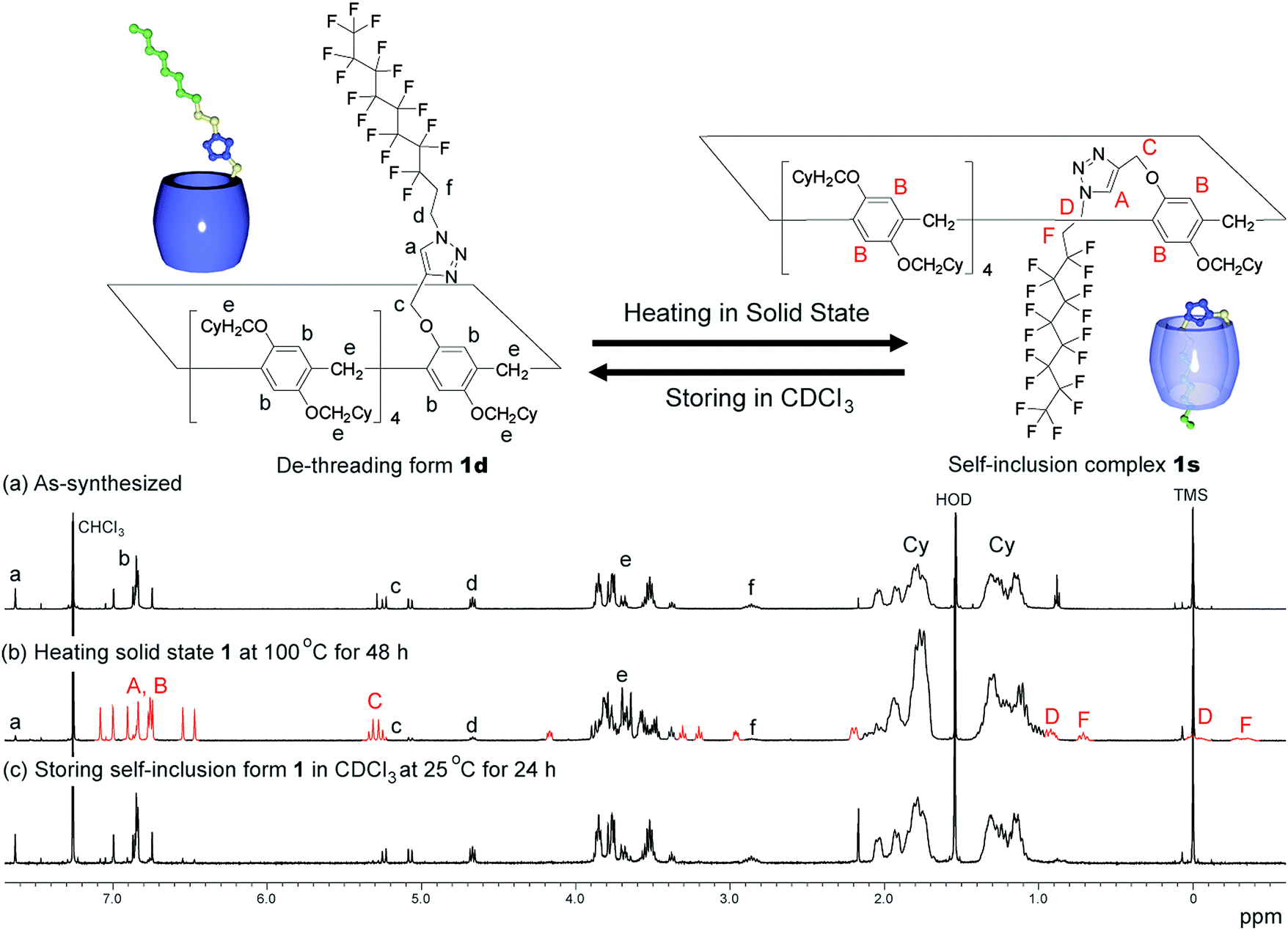
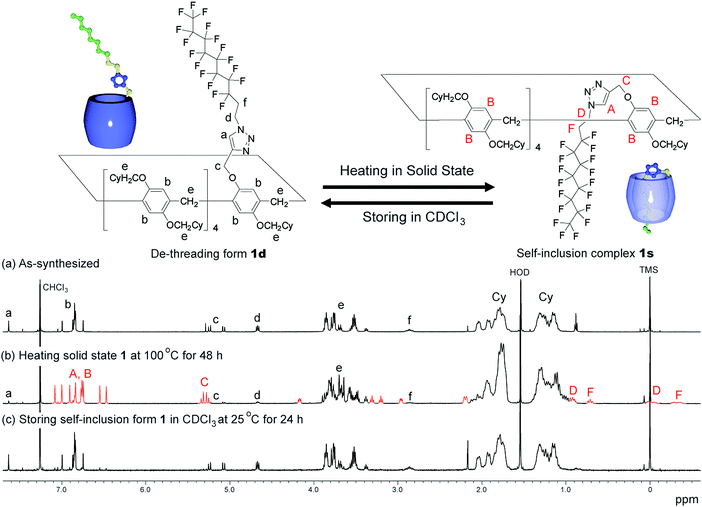
An as-synthesised sample of 1d in the solid state was heated at various temperatures. After heating the sample in the solid state, the sample was cooled to 25 °C, dissolved in CDCl3, then the 1H NMR spectrum of the sample was obtained after 3 min. De-threading occurred in CDCl3, but it took several hours (ca. 20 h at 25 °C) to reach equilibrium; therefore, 1H NMR spectra obtained quickly after dissolving the sample in CDCl3 can be used to determine the supramolecular structure formed in the solid state. The de-threading process was monitored by 1H NMR measurements by storing the mixture (1s/1d = 85/15), which was prepared by heating the solid state 1d at 100 °C for 48 h, at various temperatures in CDCl3. The ratio between 1s and 1d was determined by calculating the integration ratio between the 1H NMR signal C from 1s and the signal c from 1d (Fig. 2).
 |
| | Fig. 2
1H NMR spectra of the host–guest conjugate 1. As-synthesized (a), after heating of 1 in the solid state at 100 °C for 48 h, dissolving the solid sample in CDCl3, and obtaining the spectrum after 3 min (b), and 24 h (c). | |
Results and discussion
Unusual solid-state self-inclusion complexation behaviour of pillar[5]arene-based host–guest conjugate
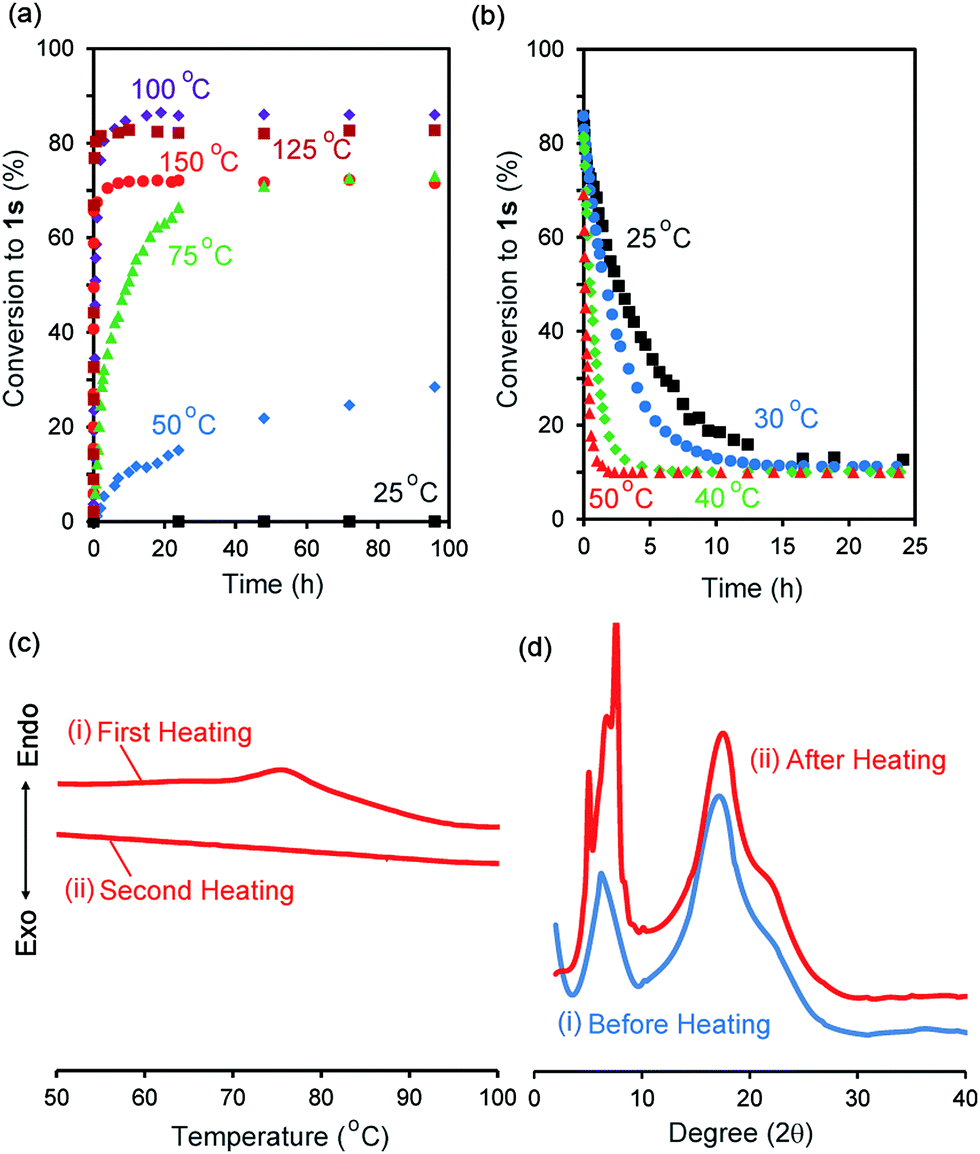
We synthesized the host–guest conjugate 1 by the copper(I)-catalysed alkyne–azide cycloaddition (CuAAC) click reaction between a pillar[5]arene containing a monoalkyne group (2) and a monoazide containing ethylene and perfluorooctyl moieties (3) (Scheme 1). To investigate the assembled structure of 1, we obtained the 1H NMR spectra of 1 at various concentrations (Fig. S3, ESI†). However, the proton peaks of 1 did not change even at a high concentration, which indicated that supramolecular polymerization and self-inclusion complexation did not take place. The as-synthesized 1 formed the de-threaded structure 1d. This was owing to the bulkiness of the perfluorooctyl moieties in 1. The cavity size of pillar[5]arenes is ca. 4.7 Å, which accommodates a linear hydrocarbon.49,50 The size of the perfluorooctyl moiety is slightly larger than the cavity size of a pillar[5]arene (see ESI,† in details). The other possible reason for the de-threaded structure of 1d is the electron repulsion force between the electron-rich cavity of the pillar[5]arene43 and the electronegativity of the perfluorooctyl moiety. The electron repulsion force would prevent threading of the perfluorooctyl moiety into the pillar[5]arene cavity to form a self-inclusion complex and cyclic and linear supramolecular oligomers and polymers. However, surprisingly, new proton signals (red) appeared along with the original proton signals of 1d after heating of 1d at 100 °C for 48 h in the solid state, which indicated that complexation occurred. No solvent molecules were included in the cavity of the pillar[5]arene host–guest conjugate 1, which confirmed by 1H NMR measurement of 1d (Fig. 1S). To investigate the supramolecular structure after the heating, we investigated the concentration-dependent 1H NMR spectra after heating 1d in the solid state. However, even at high concentrations, the new set of proton signals did not change (Fig. S6, ESI†), which indicated that the supramolecular structure of 1 after the heating was the self-inclusion complex 1s, which was very stable; 1s did not form cyclic and linear supramolecular oligomers and polymers even at high concentrations. We investigated the effect of the temperature on the conversion from 1d to 1s in the solid state (Fig. 3a). At 25 °C, 1s was not formed even after a significant period (150 h). 1s was slowly formed, but the reaction did not reach a thermodynamic equilibrium upon heating of 1d at 50 °C for 95 h. At 100 °C, it took 20 h to form 1s in a thermodynamic equilibrium; the conversion to 1s at the equilibrium state was 85%. The conversion to 1s decreased with increasing reaction temperature above 100 °C. The time required to reach a thermodynamic equilibrium became shorter with increasing reaction temperature. These results were used to determine the thermodynamic (ΔH‡ and ΔS‡) parameters based on Eyring plots (Fig. S4, ESI†). ΔH‡ and ΔS‡ were 70.8 kJ mol−1 and −124.2 J mol−1 K−1, respectively. The calculated ΔG‡ and half-life time t1/2 at 50 °C were 110.9 kJ mol−1 and 23.4 h, respectively. We could monitor the self-inclusion complexation behaviour of 1 in the solid state at a real time-scale. To understand the reason why 1 formed the self-inclusion complex 1s by heating, we calculated the electrostatic potential profile of the guest (ESI,† in details). The ethylene moieties in the guest are positive because triazole and perfluorooctyl groups at both ends of the guest part are negative. In the case of pillar[5]arenes, the inner surfaces of the cavities are negative, and the outer surfaces of the cavities and rims are almost neutral.43 Thus, pillar[5]arenes capture the positive moieties of the ethylene moieties in the guest part. This was consistent with the experimental data: the proton signals from the ethylene moieties in the guest part (signals d and f) showed a large up-field shift after the formation of the self-inclusion complex. However, the fluoro atoms have high negative charge (Fig. S12, ESI†); therefore, there would be electron repulsion between the perfluorooctyl group and the negatively charged pillar[5]arene cavity. In addition, the size of the perfluorooctyl group was slightly larger than the cavity size of pillar[5]arenes, because the cavity size of the pillar[5]arenes was just able to accommodate the linear hydrocarbons.49 Thus, heating was necessary to form the self-inclusion complex to induce threading of the perfluorooctyl groups into the pillar[5]arene cavity.
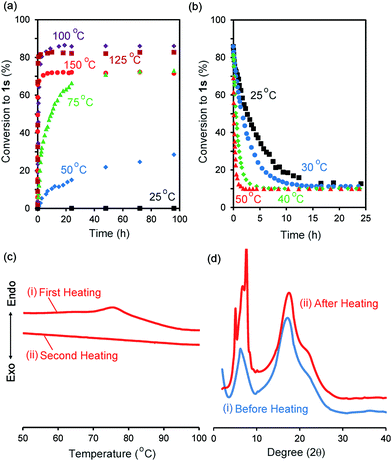 |
| | Fig. 3 Percentage conversions to 1sversus time (a) in solid state 1d at 25 °C (black squares), 50 °C (blue diamonds), 75 °C (green triangles), 100 °C (purple diamonds), 125 °C (brown squares), 150 °C (red circles) and (b) in CDCl3 at 25 °C (black squares), 30 °C (blue circles), 40 °C (green diamonds), 50 °C (red triangles). (c) DSC heating curves in (i) first and (ii) second heating processes. (d) PXRD patterns (i) before and (ii) after heating at 100 °C for 48 h. | |
Assembled structural change by self-inclusion complex in solid state by heating
To investigate the solid-state structural change of the supramolecular assembly from 1d to 1s, DSC measurements were carried out (Fig. 3c). In the first heating of 1d (Fig. 3c(i)), an endothermic peak was observed from 70 °C to 90 °C. However, an endothermic peak was not observed in the second heating (Fig. 3c(ii)). No endothermic peak was observed in the first and second heating of a mixture of 1s and 1d (1s/1d = 85/15, Fig. S5, ESI†). Thus, the endothermic peak observed in the first heating of 1d resulted from formation of 1s. Fig. 3d shows the powder X-ray diffraction patterns before and after heating of 1d in the solid state at 100 °C for 48 h. New diffraction peaks appeared in the small angle region around 7–10 degrees upon heating, which indicated that the structure of the assemblies change from 1d to 1s even in the solid state.
De-threading in solution
Assembly 1s was stable in the solid state, but de-threading slowly occurred when 1s was stored in CDCl3. Fig. 2b and c show the 1H NMR spectra after dissolving 1s (1s/1d = 85/15) in CDCl3 after 3 min and 24 h. The new set of the proton signals from 1s was observed after 3 min, but almost disappeared after 24 h, indicating that 1s changed to 1d by dissolving of 1s over several hours in CDCl3. 1s was slowly converted to 1d at 25 °C (Fig. 3b). The time required to reach thermodynamic equilibrium became shorter with increasing reaction temperature. The de-threading was accelerated by heating in solution. These results were used to determine the thermodynamic (ΔH‡ and ΔS‡) parameters based on Eyring plots (Fig. S8, ESI†). ΔH‡ and ΔS‡ were 72.3 kJ mol−1 and −82.5 J mol−1 K−1, respectively. The calculated ΔG‡ and half-life time t1/2 at 25 °C were 90.7 kJ mol−1 and 2.89 h, respectively. We also monitored the de-threading behaviour of the complex in CDCl3 in at a real-time scale.
The effect of different solvents on the supramolecular structure change was also investigated. When CD2Cl2 was used as a solvent (Fig. S9, ESI†), the set of signals attributed to 1s also disappeared after 24 h, as observed in CDCl3 (Fig. 2c), which indicated that de-threading occurred in CD2Cl2. In contrast, the 1H NMR signals of 1s did not change even after 24 h when 1s was dissolved in deuterated 1,1,2,2-tetrachloroethane and cyclohexane (Fig. S10 and S11, ESI†). The affinity of the solvent molecules for the pillar[5]arene cavity depends not only on the polarity of the solvent but also on the molecular size of the solvent molecules. The cavity size of pillar[5]arenes is 4.7 Å; thus, pillar[5]arenes can form complexes with solvents with a small molecule size but cannot form complexes with solvents with a large molecule size.51 The inclusion of the small CH2Cl2 molecules into the cavity of pillar[5]arenes has been completely characterized by single X-ray crystal analysis.52 The size of the CD2Cl2 and CDCl3 molecules is small enough to form complexes with pillar[5]arenes; thus, de-threading occurred in these solvents. In contrast, the 1,1,2,2-tetrachloroethane and cyclohexane molecules were too bulky to fit into the pillar[5]arene cavity. Therefore, no de-threading occurred in these solvents.
Conclusions
Unusual real-time scale solid-state self-inclusion complexation behaviour was observed when a pillar[5]arene-based host–guest conjugate was heated in the solid state. The inclusion of a superhydrophobic perfluorooctyl group in the guest in the solid state would be used in surface chemistry to control contact angles. The self-inclusion complex converted to the de-threaded form at a real-time scale when it was stored in solution in solvents with small molecules, which work as competitive guests. The real-time scale structural changes would be useful for controlling supramolecular assembly and systems at a real-time scale. This is now under investigation.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was partially supported by Grant-in-Aid for Scientific Research on Innovative Areas (2601): π-System Figuration (15H00990 and 17H05148), Kiban B (16H04130) from MEXT Japan and Kanazawa University Chozen Project.
Notes and references
- P. L. Anelli, N. Spencer and J. F. Stoddart, J. Am. Chem. Soc., 1991, 113, 5131–5133 CrossRef CAS PubMed.
- W. R. Dichtel, O. Š. Miljanić, W. Zhang, J. M. Spruell, K. Patel, I. Aprahamian, J. R. Heath and J. F. Stoddart, Acc. Chem. Res., 2008, 41, 1750–1761 CrossRef CAS PubMed.
- A. Harada, A. Hashidzume, H. Yamaguchi and Y. Takashima, Chem. Rev., 2009, 109, 5974–6023 CrossRef CAS PubMed.
- F. M. Raymo and J. F. Stoddart, Chem. Rev., 1999, 99, 1643–1664 CrossRef CAS PubMed.
- S. A. Nepogodiev and J. F. Stoddart, Chem. Rev., 1998, 98, 1959–1976 CrossRef CAS PubMed.
- Z. Niu and H. W. Gibson, Chem. Rev., 2009, 109, 6024–6046 CrossRef CAS PubMed.
- G. Wenz, B.-H. Han and A. Müller, Chem. Rev., 2006, 106, 782–817 CrossRef CAS PubMed.
- M. Xue, Y. Yang, X. Chi, X. Yan and F. Huang, Chem. Rev., 2015, 115, 7398–7501 CrossRef CAS PubMed.
- S. Dong, B. Zheng, F. Wang and F. Huang, Acc. Chem. Res., 2014, 47, 1982–1994 CrossRef CAS PubMed.
- M. Fujita, Acc. Chem. Res., 1999, 32, 53–61 CrossRef CAS.
- M. J. Langton and P. D. Beer, Acc. Chem. Res., 2014, 47, 1935–1949 CrossRef CAS PubMed.
- J.-P. Sauvage, Acc. Chem. Res., 1998, 31, 611–619 CrossRef CAS.
- H. V. Schroder, J. M. Wollschlager and C. A. Schalley, Chem. Commun., 2017, 53, 9218–9221 RSC.
- B. Zheng, F. Wang, S. Dong and F. Huang, Chem. Soc. Rev., 2012, 41, 1621–1636 RSC.
- Y. Kohsaka, Y. Koyama and T. Takata, Angew. Chem., Int. Ed., 2011, 50, 10417–10420 CrossRef CAS PubMed.
- Y. Inoue, P. Kuad, Y. Okumura, Y. Takashima, H. Yamaguchi and A. Harada, J. Am. Chem. Soc., 2007, 129, 6396–6397 CrossRef CAS PubMed.
- A. Harada, A. Miyawaki, P. Kuad, Y. Takashima and H. Yamaguchi, J. Am. Chem. Soc., 2008, 130, 17062–17069 CrossRef PubMed.
- Y. Inoue, M. Miyauchi, H. Nakajima, Y. Takashima, H. Yamaguchi and A. Harada, J. Am. Chem. Soc., 2006, 128, 8994–8995 CrossRef CAS PubMed.
- N. Zhao, G. O. Lloyd and O. A. Scherman, Chem. Commun., 2012, 48, 3070–3072 RSC.
- B. Vinciguerra, L. Cao, J. R. Cannon, P. Y. Zavalij, C. Fenselau and L. Isaacs, J. Am. Chem. Soc., 2012, 134, 13133–13140 CrossRef CAS PubMed.
- C. Gargiulli, G. Gattuso, A. Notti, S. Pappalardo and M. F. Parisi, Tetrahedron Lett., 2011, 52, 6460–6464 CrossRef CAS.
- C. Capici, Y. Cohen, A. D'Urso, G. Gattuso, A. Notti, A. Pappalardo, S. Pappalardo, M. F. Parisi, R. Purrello, S. Slovak and V. Villari, Angew. Chem., Int. Ed., 2011, 50, 11956–11961 CrossRef CAS PubMed.
- R. M. Yebeutchou, F. Tancini, N. Demitri, S. Geremia, R. Mendichi and E. Dalcanale, Angew. Chem., Int.
Ed., 2008, 47, 4504–4508 CrossRef CAS PubMed.
- T. Ogoshi, T. Furuta and T. Yamagishi, Chem. Commun., 2016, 52, 10775–10778 RSC.
- N. L. Strutt, H. C. Zhang, M. A. Giesener, J. Y. Lei and J. F. Stoddart, Chem. Commun., 2012, 48, 1647–1649 RSC.
- Z. Zhang, Y. Luo, J. Chen, S. Dong, Y. Yu, Z. Ma and F. Huang, Angew. Chem., Int. Ed., 2011, 50, 1397–1401 CrossRef CAS PubMed.
- Y. Wang, J. F. Xu, Y. Z. Chen, L. Y. Niu, L. Z. Wu, C. H. Tung and Q. Z. Yang, Chem. Commun., 2014, 50, 7001–7003 RSC.
- L. Liu, L. Wang, C. Liu, Z. Fu, H. Meier and D. Cao, J. Org. Chem., 2012, 77, 9413–9417 CrossRef CAS PubMed.
- Z. Zhang, G. Yu, C. Han, J. Liu, X. Ding, Y. Yu and F. Huang, Org. Lett., 2011, 13, 4818–4821 CrossRef CAS PubMed.
- X. Wu, M. Ni, W. Xia, X.-Y. Hu and L. Wang, Org. Chem. Front., 2015, 2, 1013–1017 RSC.
- Y. F. Guan, P. Y. Liu, C. Deng, M. F. Ni, S. H. Xiong, C. Lin, X. Y. Hu, J. Ma and L. Y. Wang, Org. Biomol. Chem., 2014, 12, 1079–1089 CAS.
- Y. Chen, D. R. Cao, L. Y. Wang, M. Q. He, L. X. Zhou, D. Schollmeyer and H. Meier, Chem. – Eur. J., 2013, 19, 7064–7070 CrossRef CAS PubMed.
- L. Chen, Z. Li, Z. Chen and J. L. Hou, Org. Biomol. Chem., 2013, 11, 248–251 CAS.
- Y. Han, G.-F. Huo, J. Sun, J. Xie, C.-G. Yan, Y. Zhao, X. Wu, C. Lin and L. Wang, Sci. Rep., 2016, 6, 28748 CrossRef PubMed.
- T. Ogoshi, T. Yamagishi and Y. Nakamoto, Chem. Rev., 2016, 116, 7937–8002 CrossRef CAS PubMed.
- M. Xue, Y. Yang, X. D. Chi, Z. B. Zhang and F. H. Huang, Acc. Chem. Res., 2012, 45, 1294–1308 CrossRef CAS PubMed.
- N. L. Strutt, H. C. Zhang, S. T. Schneebeli and J. F. Stoddart, Acc. Chem. Res., 2014, 47, 2631–2642 CrossRef CAS PubMed.
- T. Ogoshi, S. Kanai, S. Fujinami, T. Yamagishi and Y. Nakamoto, J. Am. Chem. Soc., 2008, 130, 5022–5023 CrossRef CAS PubMed.
- C. Li, Chem. Commun., 2014, 50, 12420–12433 RSC.
- X. Shu, S. Chen, J. Li, Z. Chen, L. Weng, X. Jia and C. Li, Chem. Commun., 2012, 48, 2967–2969 RSC.
- K. Han, Y. Zhang, J. Li, Y. Yu, X. Jia and C. Li, Eur. J. Org. Chem., 2013, 2057–2060 CrossRef CAS.
- X. Y. Shu, J. Z. Fan, J. Li, X. Y. Wang, W. Chen, X. S. Jia and C. J. Li, Org. Biomol. Chem., 2012, 10, 3393–3397 CAS.
- T. Ogoshi and T. Yamagishi, Chem. Commun., 2014, 50, 4776–4787 RSC.
- K.-J. Chen, Y.-C. Tsai, Y. Suzaki, K. Osakada, A. Miura and M. Horie, Nat. Commun., 2016, 7, 13321 CrossRef CAS PubMed.
- M. Horie, Y. Suzaki, D. Hashizume, T. Abe, T. Wu, T. Sassa, T. Hosokai and K. Osakada, J. Am. Chem. Soc., 2012, 134, 17932–17944 CrossRef CAS PubMed.
- K. Zhu, V. N. Vukotic, C. A. O’Keefe, R. W. Schurko and S. J. Loeb, J. Am. Chem. Soc., 2014, 136, 7403–7409 CrossRef CAS PubMed.
- N. Zhu, K. Nakazono and T. Takata, Chem. Commun., 2016, 52, 3647–3649 RSC.
- S. Périno, C. Contino-Pépin, S. Jasseron, M. Rapp, J.-C. Maurizis and B. Pucci, Bioorg. Med. Chem. Lett., 2006, 16, 1111–1114 CrossRef PubMed.
- T. Ogoshi, R. Sueto, K. Yoshikoshi, Y. Sakata, S. Akine and T. Yamagishi, Angew. Chem., Int. Ed., 2015, 54, 9849–9852 CrossRef CAS PubMed.
- Z. B. Zhang, B. Y. Xia, C. Y. Han, Y. H. Yu and F. H. Huang, Org. Lett., 2010, 12, 3285–3287 CrossRef CAS PubMed.
- T. Ogoshi, T. Akutsu, D. Yamafuji, T. Aoki and T. Yamagishi, Angew. Chem., Int. Ed., 2013, 52, 8111–8115 CrossRef CAS PubMed.
- T. Boinski and A. Szumna, Tetrahedron, 2012, 68, 9419–9422 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra of 1, concentration-dependent 1H NMR spectra of 1 in the de-threaded form and self-inclusion complex, de-threading behaviour of 1s in various deuterated solvents and calculations. See DOI: 10.1039/c7qm00553a |
|
| This journal is © the Partner Organisations 2018 |

 *ab,
T.
Furuta
a,
Y.
Hamada
a,
T.
Kakuta
a and
T.
Yamagishi
a
*ab,
T.
Furuta
a,
Y.
Hamada
a,
T.
Kakuta
a and
T.
Yamagishi
a