
Fe/Fe3C@graphitic carbon shell embedded in carbon nanotubes derived from Prussian blue as cathodes for Li–O2 batteries†
Yanqing
Lai
 ,
Yifeng
Jiao
,
Junxiao
Song
,
Kai
Zhang
,
Jie
Li
and
Zhian
Zhang
*
,
Yifeng
Jiao
,
Junxiao
Song
,
Kai
Zhang
,
Jie
Li
and
Zhian
Zhang
*
School of Metallurgy and Environment, Central South University, Changsha Hunan 410083, China. E-mail: zza75@163.com
First published on 11th December 2017
Abstract
The effective design and facile preparation of noble metal-free catalysts is crucial for enhancing the performance of rechargeable Li–O2 batteries. Herein, we report a one-step controlled pyrolysis method to synthesize a Fe/Fe3C@graphitic carbon shell embedded in carbon nanotubes composite (F@NG-NCNTs) with Prussian blue as the single precursor. In these nanostructures, the Fe/Fe3C centres induce the formation of self-assembled N-doped carbon frameworks, which in turn offer superior connectivity as well as a protective coating. This novel architecture not only promotes the transfer of electrons but also uniformly encapsulates the catalytically active Fe/Fe3C nanoparticles, leading to a remarkably enhanced performance in Li–O2 batteries. As a result, cells catalyzed by the F@NG-NCNTs deliver a higher initial discharge capacity (6966 mA h g−1 at a current density of 0.1 mA cm−2) and demonstrate outstanding rate capability and cycle stability. This study verifies that F@NG-NCNTs can be a promising cathode material and provides a new avenue for the effective nanostructural design of electrocatalysts.
Introduction
Increasing energy demands and the exhaustion of unsustainable resources have led to extensive developments in renewable energy, as high-energy and eco-friendly energy-storage systems are required.1–3 Among several candidates, Li–O2 batteries have attracted great interest owing to their dramatically higher theoretical energy density of 3500 W h kg−1, based on the electrochemical reaction pathway of 2Li+ + O2 + 2e− ↔ Li2O2.4–9 However, a series of issues, including large polarization, poor rate capability and short lifespan,4,9–11 urgently need to be overcome before developing Li–O2 batteries for practical purposes. These aforementioned drawbacks mainly originate from the sluggish kinetics of the oxygen evolution reaction (OER) and oxygen reduction reaction (ORR).12,13 Thus, it is essential to develop desirable bifunctional catalysts that increase both the OER and ORR kinetics.From a reasonable design viewpoint, suitable catalysts should have well-distributed active sites and guarantee efficient electron transport to ensure the promotion of electrochemical reaction kinetics. Therefore, carbonaceous materials have generally been applied as cathode materials or valuable alternative substrates for Li–O2 batteries.10,14–18 Among various kinds of carbon materials, carbon nanotubes (CNTs) are considered a promising material for Li–O2 cathodes due to their high electro-conductivity, superior chemical stability, large accessibility of active sites and affordability.7,14,19,20 Additionally, nitrogen-doping has been confirmed as a feasible method to further enhance the electro-catalytic activity of carbon-based catalysts towards the ORR, owing to the nitrogen atoms modifying both the electron configurations and structural properties of the carbon materials.8,9 However, the poor OER activity of doped-carbon materials leads to high overpotentials during the charging process, which deteriorates the performance of the Li–O2 batteries. To overcome this disadvantage, different kinds of composites, such as noble metals9,18,21,22 and transition metal oxides,19,20,23–25 have been incorporated into carbonaceous materials to reduce the charge overpotentials. Nevertheless, issues like high cost and poor electrical conductivity also restrict their practical applications in Li–O2 batteries.
Recently, iron-based catalysts, especially iron/iron carbide (Fe/Fe3C) carbon composites, have been widely researched in various fields of electrocatalysis, demonstrating that the Fe/Fe3C–C composite could be a cost effective, efficient and robust catalyst for the ORR and OER.26–29 Yet few studies have reported using Fe/Fe3C as an oxygen electrode catalyst for Li–O2 batteries. Li et al. prepared a composite of Fe/Fe3C and carbon nanofibers by employing an electrospinning method for Li–O2 batteries.30 Our group synthesized Fe/Fe3C nanoparticles supported on porous N-doped graphene as a catalyst via carbonized MIL-100 (Fe).31 The improved electrochemical performance of these Li–O2 batteries indicates the superiority of Fe/Fe3C–C composites for Li–O2 batteries. However, agglomeration and inactivation of the catalyst during the charging/discharging process is an enormous hindrance for Li–O2 batteries.32,33 The catalysts are easily passivated during long-term use when directly exposed to oxygen in the electrolyte, due to the continuous accumulation of discharge and charge products during cycling. Moreover, the insulated Li2O2 can block the connection between Fe/Fe3C and the carbon framework, which causes a decrease in the electro-conductivity and activity of the catalyst. To the best of our knowledge, there have been no reports on a well-designed nanostructured Fe/Fe3C–C composite as a cathode catalyst for Li–O2 batteries.
Herein, we present a low-cost and facile one-step route for the fabrication of Fe/Fe3C–C composites. Fe/Fe3C nanoparticles coated by a graphitic shell are entirely encapsulated in carbon nanotubes (denoted as F@NG-NCNTs) through pyrolyzing the raw material Prussian Blue (PB) at a controlled temperature. Note that PB has a special chemical composition of Fe, N and C and is easily accessible. Embedding Fe/Fe3C@graphitic carbon shell (F@NG) into carbon nanotubes could be an effective strategy to stabilize the structure, provide more active sites and greatly facilitate fast electron transport. As a result, the cathodes of Li–O2 batteries with F@NG-NCNTs demonstrate high discharge capacities, stable rate properties and better cycling stabilities. The high electrochemical performance of the F@NG-NCNTs nano-composites can be attributed to the unique structural features as well as the synergistic effect generated from the Fe/Fe3C nanoparticles, graphitic carbon shell and carbon nanotubes.
Experimental
Preparation
In a typical process, Na4Fe(CN)6 was prepared by dehydration of commercial Na4Fe(CN)6·10H2O (SigmaAldrich, ≥99%) at 160 °C for 24 h. During the facile pyrolysis process, dehydrated Na4Fe(CN)6 was placed into a tube furnace and heated at 950 °C under Ar flow with a heating rate of 3 °C min−1. After 2 h, the sample was cooled naturally under inert atmosphere, and the F@NG-NCNTs were harvested. To explore the effect of temperature, the dehydration of Na4Fe(CN)6 was performed at selected temperatures of 850, 900 and 1000 °C, respectively. CNTs with a higher number of defects (DCNTs) were prepared through a KOH activation method.34 A physical mixture of CNTs and anhydrous KOH in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 weight ratio was heated at 800 °C for 3 h under argon flow. The process was carried out in a nickel boat placed in a 36 mm diameter quartz tube. The heating rate was 50 °C min−1 to 200 °C and then 10 °C min−1 to the final temperature. The resultant materials were washed repeatedly with distilled water to remove foreign ions and then dried at 80 °C for 12 h.
:4 weight ratio was heated at 800 °C for 3 h under argon flow. The process was carried out in a nickel boat placed in a 36 mm diameter quartz tube. The heating rate was 50 °C min−1 to 200 °C and then 10 °C min−1 to the final temperature. The resultant materials were washed repeatedly with distilled water to remove foreign ions and then dried at 80 °C for 12 h.
Materials characterization
The morphologies of the catalysts were characterized by transmission electron microscopy (TEM, Tecnai G2 20ST) and scanning electron microscopy (SEM, Nova NanoSEM 230). X-ray diffraction patterns (XRD) were recorded on a Rigaku-TTRIII (Cu Kα, radiation l = 0.154056 nm). Energy dispersive X-ray spectroscopy (EDX) was performed to detect the elemental distribution on the surface of the samples. Raman spectra were obtained from a Jobin-Yvon LabRAM HR-800 Raman spectrometer. The bond characteristics were measured by X-ray photoelectron spectroscopy (XPS, ESCA LAB 250Xi). Thermogravimetric analysis (TGA) was performed with NETZCH STA449F3 from 30 to 1000 °C under N2 flow with a heating rate of 10 °C min−1.Electrochemical measurements
Fabrication of the O2 electrodes: 60% F@NG-NCNTs, 30% Ketjen black (KB) and 10% polyvinylidene fluoride (PVDF) were mixed and added to N-methyl-2-pyrrolidone (NMP) to prepare a uniform slurry. Then the mixture was coated onto a nickel foam collector and was dried at 60 °C for 24 h to volatilize the residual solvent. The mass loading density of the catalysts was about 1 mg cm−2. The Li metal anode and prepared cathode were separated by a glass fiber separator (GF/D, Whatman). The organic electrolyte contained a solution of 1 M LiTFSI in tetraethylene glycol dimethyl ether (TEGDME). All the batteries were assembled in an Ar-filled glove box (MIKROUNA-Universal 2440/750, H2O < 1 ppm). For comparison, CNTs electrodes (6:3:1 mass ratio of CNTs:KB:PVDF), DCNTs electrodes (6:3:1 mass ratio of DCNTs:KB:PVDF) and KB electrodes (9:1 mass ratio of KB:PVDF) were also prepared by similar procedures, respectively. Galvanostatic charge–discharge tests were performed in an oxygen atmosphere using a LAND CT2001A testing system. Cyclic voltammograms (CVs) were collected by an electrochemical measurement system (PARSTAT 2273).
Results and discussion
The fabrication process for the F@NG-NCNTs is illustrated in Scheme 1. Firstly, the Na4Fe(CN)6 precursor was prepared by a simple dehydration method. Then the resulting powder was further annealed at 950 °C in a purified argon flow. As shown in Fig. S1 and S2 (ESI†), the phase structure and morphology of Na4Fe(CN)6 were observed by XRD and SEM, respectively. The XRD pattern shows that all the diffraction peaks match JCPDS No. 01-1026 and is in accordance with previously reported data.35,36 The SEM image reveals that the precursor has a cubic-like structure. As seen from the TGA curves (Fig. S4, ESI†), about 20 wt% of the initial precursor is retained. During this pyrolysis process in an inert atmosphere, PB gradually decomposes to carbon, iron, iron carbides (Fe7C3, Fe2C and Fe3C), nitrogen gas and carbon nitride gases (CN, C2N2+, C3N2+ and C3N3+). The metallic iron or iron carbide nanoparticles can catalyze the deposition of N-doped graphitic carbon layers from the carbon nitride gases derived from PB. They can also graphitize the amorphous carbon through a solid state transformation.37–39 To illuminate the temperature effect, a range of 850–1000 °C was employed to treat the precursor. The final product was denoted as sample-X, where X is the annealing temperature.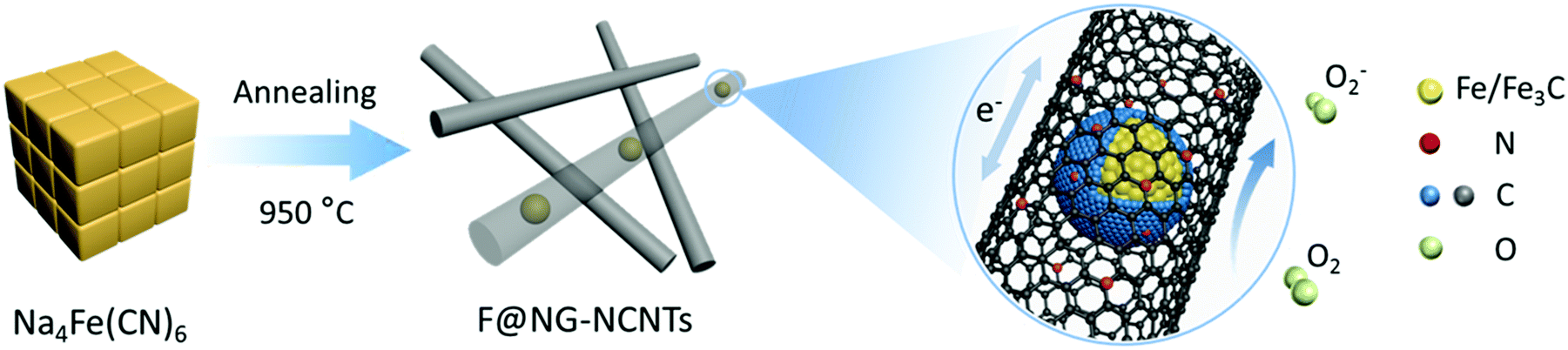 | ||
| Scheme 1 Synthetic scheme for the fabrication process of F@NG-NCNTs. | ||
The crystal phases of the samples at various pyrolysis temperatures were characterized by XRD (Fig. 1a and Fig. S3, ESI†). As shown in Fig. 1a, aside from the diffraction peak at about 26.5° that is ascribed to the (0 0 2) facet of graphitic carbon, two sharp humps can be observed at 44.7° and 65.1°, corresponding to the (1 1 0) and (2 0 0) planes of a-Fe (JCPDS, No. 87-0722), respectively. The rest reflections of 37°–60° are in good agreement with those of the Fe3C phase (JCPDS, No. 89-2867), which demonstrates the successful synthesis of Fe/Fe3C through the pyrolysis route. From the XRD observations, it was found that the phase of the Fe/Fe3C–C composite shows no change with rising temperature, while the carbon peaks become sharper, indicating that their crystallinity has increased. The XRD pattern of sample-850 is shown in Fig. S3 (ESI†). Apart from the peaks of carbon, Fe and Fe3C, other peaks are detected at 30.3° and 43.4°, which can be assigned to the NaCN phase (JCPDS, No. 04-0667). This result suggests that sodium ions are still retained at a relatively low temperature, which is in good agreement with the thermogravimetric analysis (TGA) (Fig. S4, ESI†), where the curve reveals that visible weight loss occurs at 830–920 °C, corresponding to the evaporation of sodium ions (Fig. S5, ESI†). Fig. 1b is the Raman spectrum, which exhibits a well-documented D band and G band located at 1328 and 1575 cm−1, respectively. As is known, the D band originates from disorders and defects in graphitic carbon, while the G band corresponds to the E2g vibration of the sp2-bonded carbon atoms in a two-dimensional hexagonal lattice.31,40 The proportional integrated intensity of the G and D bands (IG/ID) is widely employed to evaluate the degree of graphitization in carbon materials. The values of IG/ID tend to increase (1.03 to 1.18) with increasing temperature, suggesting the generation of a more ordered carbon structure at a higher annealing temperature. This result is consistent with the observation from XRD. In addition, as shown in Fig. S7 (ESI†), the DCNTs show a higher ID/IG value of 1.23 than the pristine CNTs (0.87), suggesting a greater degree of defects and disorder in the DCNTs, which can be attributed to the activation process. The higher degree of graphitization implies a higher electrical conductivity, which promotes charge transfer. On the other hand, the defects formed in the carbon matrix provide more active sites and also a pathway for oxygen mass transfer.31,41 Thus, according to the Raman result, sample-950 could have a well-built carbon nanostructure to keep a balance between the two sides. Finally, the asymmetric dwarf 2D peak at around 2690 cm−1 further confirms the presence of a multilayer graphitic structure in the prepared samples.31,35,42
 | ||
| Fig. 1 (a) XRD patterns and (b) Raman spectra of samples at different pyrolysis temperatures. | ||
Fig. 2 shows the morphology and nanostructure of the F@NG-NCNTs. After the heating process, the cubic-like precursors are mostly converted to intertwined carbon nanotubes with a diameter of around 100–400 nm (Fig. 2a and Fig. S2b, ESI†). For further inspection of the inner structure of the F@NG-NCNTs, transmission electron microscopy (TEM) images are presented in Fig. 2b–d. Notably, bamboo-type tubular structures, a feature of nitrogen-doped carbon nanotubes, are clearly observed (Fig. 2b).43–45 As can be seen from the typical carbon nanotube TEM image (Fig. 2c), the well-distributed and ellipsoid-like Fe/Fe3C particles are successfully encapsulated inside the channels of the CNTs (with a size of about 150 nm in external diameter and 90 nm in inner diameter, depending on the size of the Fe/Fe3C nanoparticles). From Fig. 2d, it can be seen that a Fe/Fe3C nanoparticle appears at one closed end of the carbon nanotube, suggesting that Fe/Fe3C is well protected by the tube wall and functions as a catalyst to foster the growth of carbon nanotubes. More detailed characteristics of the nanoparticles are shown in Fig. 2e. It is worth mentioning that the Fe/Fe3C nanoparticles (magenta area) are intimately surrounded by the graphitic carbon shell (yellow area), which are entirely encapsulated in the carbon nanotubes (light green area), forming a well-defined hierarchical structure. The high-resolution TEM (HRTEM) image of the yellow ring-like area shows clear lattice fringes with an interlayer spacing of 0.340 nm (bottom left inset of Fig. 2e), corresponding to the typical (0 0 2) plane of the graphitic carbon layer.28,46,47 Close observation (Fig. 2f, selected areas in Fig. 2d) further reveals the features of the carbon shell. Multi-layered graphitic layers with a thickness of about 5 nm can be observed, which is supported by the Raman result. In this coating shell, the non-continuous structure is interrupted by amorphous carbon (red arrows). The rich defects provide enough active sites for catalytic reactions while the remaining parts of the layer still act as a valid shield.28 As shown in Fig. 2g, the HRTEM image confirms that the lattice distances of the two directions with a crystal plane angle of 68.4° are both 0.210 nm, which match well with the (2 1 1) and (−2 1 1) planes of Fe3C. From this image, 0.207 nm can also be seen, which is attributed to the (1 1 0) plane of Fe. This result indicates the existence of Fe/Fe3C species, which is consistent with the XRD analysis. The derived elemental mapping images in Fig. 2h further demonstrate the nanostructure of F@NG-NCNTs and show that N atoms are homogeneously distributed on the whole surface of the carbon nanotubes. Additionally, the TEM observation (Fig. S8, ESI†) provides clear evidence that the activation process made more defects in the CNTs.
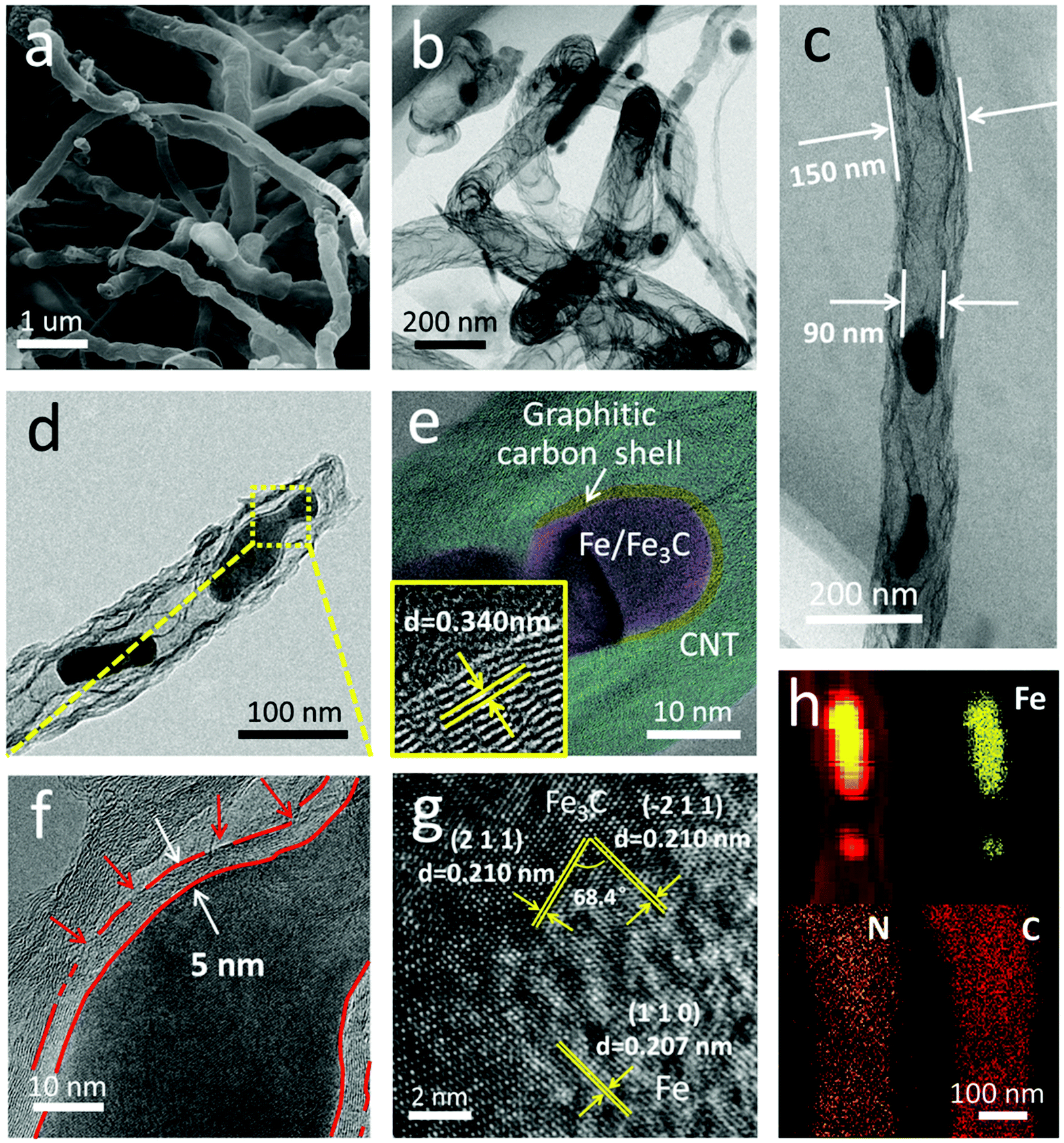 | ||
| Fig. 2 (a) SEM image, (b–f) TEM images, (g) high-resolution TEM image, and (h) corresponding elemental mapping images of the F@NG-NCNTs. The bottom left inset in (e) is an enlarged HRTEM. | ||
Samples of different pyrolysis temperatures were also investigated by SEM. As displayed in Fig. S6 (ESI†), the SEM image at 850 °C shows that the seriously aggregated particles are covered by thick carbon layers and no carbon nanotubes can be observed (Fig. S6a, ESI†) since the temperature does not meet the requirements to foster carbon nanotubes. When the temperature is raised to 900 °C, the particles disperse and the carbon layers become thin, tending to a core–shell structure (Fig. S6b, ESI†). However, the disordered structure still has few carbon nanotubes. Elevating the temperature to 950 °C, carbon nanotubes are formed. When the temperature is further increased to 1000 °C, CNTs with a larger diameter and smoother surface can be observed. (Fig. S6c and d, ESI†), which suggests fewer defects and is consistent with the XRD and Raman observations. According to the results above, it could be concluded that 950 °C is favorable for the fabrication of the F@NG-NCNTs composite.
The surface chemical compositions and atom states of the F@NG-NCNTs were further elucidated by XPS. The survey spectrum reveals the existence of elemental Fe, C, N and O, shown in Fig. 3a, which coincides with the result obtained by elemental mapping. Fig. 3b shows the high-resolution C 1s spectrum of the composite, in which three individual peaks can be further assigned. The main peak at 284.5 eV refers to the graphite-like carbon, suggesting that most of the C atoms are arranged in a conjugated hexagonal lattice. The two peaks centered at 285.7 and 287.7 eV correspond to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N (N–sp2 C) and C–N (N–sp3 C) bonds,48–50 respectively. The high-resolution spectrum of N 1s (Fig. 3c) can be deconvoluted into four peaks located at 398.4, 400.4, 401.2 and 403.3 eV, which are ascribed to pyridinic N (33.3%), pyrrolic N (31.4%), graphitic N (34.2%) and oxidized N (1.1%), respectively.51,52 As previously reported,31,49 doping nitrogen atoms into the carbon framework could create more defects, which may enhance the active sites and boost oxygen reduction. In addition, graphitic N and pyridinic N make the greatest contribution among all of these N species. The above results demonstrate that the composite may have excellent ORR catalytic activity. Fig. 3d displays the high-resolution spectrum of Fe 2p, which is fitted to five peaks. The photoelectron peak of metallic iron (Fe0) at 707.5 eV can be identified.27,53 Meanwhile, the peaks at 711.1 eV and 722.0 eV, corresponding to Fe(II) 2p, and the peaks at 714.7 eV and 726.1 eV, corresponding to Fe(III) 2p, could confirm the presence of Fe2+ and Fe3+ in the composite.28,53,54 Furthermore, the weak Fe peaks and low Fe content indicate that most of the Fe species are well protected by carbon layers, which is in agreement with the TEM results.
N (N–sp2 C) and C–N (N–sp3 C) bonds,48–50 respectively. The high-resolution spectrum of N 1s (Fig. 3c) can be deconvoluted into four peaks located at 398.4, 400.4, 401.2 and 403.3 eV, which are ascribed to pyridinic N (33.3%), pyrrolic N (31.4%), graphitic N (34.2%) and oxidized N (1.1%), respectively.51,52 As previously reported,31,49 doping nitrogen atoms into the carbon framework could create more defects, which may enhance the active sites and boost oxygen reduction. In addition, graphitic N and pyridinic N make the greatest contribution among all of these N species. The above results demonstrate that the composite may have excellent ORR catalytic activity. Fig. 3d displays the high-resolution spectrum of Fe 2p, which is fitted to five peaks. The photoelectron peak of metallic iron (Fe0) at 707.5 eV can be identified.27,53 Meanwhile, the peaks at 711.1 eV and 722.0 eV, corresponding to Fe(II) 2p, and the peaks at 714.7 eV and 726.1 eV, corresponding to Fe(III) 2p, could confirm the presence of Fe2+ and Fe3+ in the composite.28,53,54 Furthermore, the weak Fe peaks and low Fe content indicate that most of the Fe species are well protected by carbon layers, which is in agreement with the TEM results.
 | ||
| Fig. 3 XPS spectra of the F@NG-NCNTs: (a) survey scan, (b) C 1s spectrum, (c) N 1s spectrum, and (d) Fe 2p spectrum. | ||
The performances of Li–O2 batteries with F@NG-NCNTs were investigated by using 2032-type coin cells with 1 M LiTFSI in TEGDME as an electrolyte, a lithium metal anode and a home-made oxygen electrode. Cyclic voltammetry (CV) within a voltage range of 2.0–4.5 V at a scan rate of 0.2 mV s−1 was applied to estimate the electrocatalytic activity of the F@NG-NCNTs. As shown in Fig. 4a, the F@NG-NCNTs displays a higher ORR and lower OER onset potential than the pure KB electrode. Furthermore, a larger ORR/OER peak current is also observed from the F@NG-NCNTs electrode, revealing high catalytic activity for the corresponding process. The galvanostatic discharge–charge was measured with a voltage window from 2.2 to 4.5 V and all the results were evaluated in accordance with the total weight of the coating materials. For comparison, pure KB and CNTs were employed as catalysts for the air electrode. Fig. 4b shows that the F@NG-NCNTs composite achieves the highest discharge specific capacity of 6966 mA h g−1 at a constant current density of 0.1 mA cm−2, which is superior to that of the CNTs (4977 mA h g−1) and KB (2293 mA h g−1) electrodes. The charge capacity also delivers 6947 mA h g−1, demonstrating a coulombic efficiency of 99.7%, which is much higher than that of CNTs (90.1%) and KB (87.3%). More importantly, the F@NG-NCNTs cathode reveals a markedly lower overpotential than CNTs and KB, not only upon the charge but also the discharge process, indicating excellent conductivity and catalytic activity. As a contrast, the electrode composed solely of carbon nanotubes shows poor OER catalytic activity, which further indicates that the catalyst is stimulated by the presence of Fe/Fe3C nanoparticles. Several studies have described the effects of Fe3C, and although the encapsulated Fe3C nanoparticles are not directly exposed to oxygen and the electrolyte, they still play a key role in the nanostructure. This role may be defined by the synergetic interaction between the carbide and protective carbon layers. In other words, the encased Fe3C cores activate the surrounding graphitic shells, making the outer surface of the carbon layer active towards catalysis. Moreover, when metal/carbide nanoparticles are confined inside CNTs, unique host–guest electronic interactions decrease the local work function on the surface of the CNT walls.55–57 These conclusions are consistent with the above results. The discharge–charge curves of the F@NG-NCNTs electrode at various current densities of 0.1, 0.2 and 0.4 mA cm−2 are displayed in Fig. 4c. As the current density increases, the capacities of the cell gradually decrease and the charge overpotential rises but the discharge potential is nearly steady, which can be ascribed to the high ORR activity for the F@NG-NCNTs composite, even at a high current density. It is notable that the charge and discharge capacities at different current densities are almost equivalent, suggesting that the coulombic efficiency nears 100% (Fig. 4d). Accordingly, the above results demonstrate that the F@NG-NCNTs cathode has a superior rate performance, which is due to the improved ORR/OER kinetics that benefit rechargeable Li–O2 cells.
 | ||
| Fig. 4 (a) CV curves of F@NG-NCNTs and KB in a dry O2 atmosphere at a scanning rate of 0.2 mV s−1. (b) Initial discharge–charge curves at 0.1 mA cm−2 current density with F@NG-NCNTs, CNTs and KB electrodes. (c) Discharge–charge curves and (d) corresponding coulombic efficiency of Li–O2 batteries with F@NG-NCNTs at various current densities. | ||
In order to evaluate the cyclability of the oxygen electrode for Li–O2 batteries, the test was performed at a constant current density of 0.1 mA cm−2 with a fixed capacity of 500 mA h g−1, and the cut-off voltage ranged from 2.2–4.5 V. As illustrated in Fig. 5, the F@NG-NCNTs catalyst can extend the cycle life to 37 times with stable reversible capacities, while the CNTs electrode only experiences 23 cycles and the KB electrode rapidly deteriorates after 17 cycles. In the initial stage, the F@NG-NCNTs cathode displays an overvoltage gap of about 1.20 V, which is lower than that of the KB electrode (1.51 V) and CNTs electrode (1.44 V). Additionally, from the charge profile (Fig. 5a), it can clearly be seen that there is no loss of charge capacity and the charge voltage plateau remains as low as 4.12 V during the initial 20 cycles, which exhibits better performance than the comparison electrodes. This result demonstrates that the complete decomposition of the discharge product is related to the excellent OER properties. After 30 cycles, the polarization becomes serious, which may be attributed to a series of reasons, including the accumulation of irreversible discharge products, the formation of side products, degradation of the Li anode and carbon corrosion in a highly oxidizing environment.58–64Fig. 5d shows that the KB electrode only runs up to 10 cycles, followed by a sharp drop in discharge voltage upon termination, and the CNTs electrode decreases after 20 cycles. Using the F@NG-NCNTs catalyst, the stability is improved to 30 cycles and the terminal discharge potential falls slightly from 2.71 to 2.5 V (vs. Li+/Li), which further indicates the stable activity for oxygen reduction. As shown in Fig. S9 (ESI†), the discharge/charge capacities of the pure DCNTs cathode are 5461/5006 mA h g−1 with 91.6% round-trip efficiency. The cycling performances are presented in Fig. S10 (ESI†). The DCNTs electrode achieved more cycle numbers than CNTs. Therefore, the DCNTs electrode showed better electrochemical performance than CNTs, indicating the positive effect of defects on catalysis.
 | ||
| Fig. 5 Cycling performances of the Li–O2 cells at 0.1 mA cm−1 with a fixed specific capacity of 500 mA h g−1: (a) F@NG-NCNTs, (b) CNTs, and (c) KB. (d) Discharge capacity and terminal discharge voltage versus cycle number. | ||
To further identify the reversibility of discharge products on the F@NG-NCNTs cathode, ex situ SEM characterization was carried out to inspect the morphology of the electrode. The charge/discharge profile with a capacity limitation of 2000 mA h g−1 at a current density of 0.2 mA cm−2 is shown in Fig. 6a. Four batteries were disassembled at the pristine (c), half discharge (d), after discharge (e) and after charge (f) states. XRD analysis was applied to identify the composition of the reaction products (Fig. 6b). For the pristine electrode, the F@NG-NCNTs composites have a clean surface and are loosely embraced by KB particles (Fig. 6c). Ni peaks from the nickel foam and some weak Fe3C peaks from the F@NG-NCNTs could be detected in the XRD spectrum. After the discharge process at 1000 mA h g−1, it can be distinctly seen in Fig. 6d that the close-packed insoluble discharged products have precipitated on the surface of the electrode. Then, when the delivered capacity is increased to 2000 mA h g−1, the surface is almost fully covered by massive worm-like agglomerates which consist of discharged products (yellow ellipses). According to the XRD results, the characteristic Li2O2 peaks can be clearly observed, implying that the dominant crystalline product during the discharge process is Li2O2, which is consistent with previous reports.65–67 The accumulation of insulating Li2O2 hinders the pathway for O2, Li+ ions and electrons, leading to the termination of the discharge process.68,69 Interestingly, after subsequent recharge, the thick discharged products disappear and the electrode can nearly regain its original structure. Moreover, XRD analysis provides further evidence to support the sufficient decomposition of Li2O2. Notably, the SEM results indicate that the structure of the F@NG-NCNTs is finely preserved and the XRD spectra show no shift in the peak characteristics of Fe3C, demonstrating the extraordinary stability of the catalyst against oxygen attack. On the basis of these results, the outstanding electrochemical performance can be attributed to the synergistic effect of the inherent catalytic ability of the Fe/Fe3C species and the well-designed nanostructure of the carbon framework, which accelerates the transport of electrons and protects the Fe/Fe3C cores.
 | ||
| Fig. 6 (a) Initial discharge and charge profiles of the Li–O2 battery with a F@NG-NCNTs electrode at a constant current density of 0.2 mA cm−2. (b) Ex situ XRD patterns and (c–f) ex situ SEM images of different states of the electrode. | ||
Conclusions
In summary, F@NG embedded in NCNTs has been successfully fabricated by means of one-step pyrolysis at an optimal temperature of 950 °C. The N-doped composite consists of graphitic carbon-wrapped Fe/Fe3C nanocores evenly encapsulated in carbon nanotubes. In comparison with Ketjen black (KB) and carbon nanotubes (CNTs), the as-prepared composite reveals a low overpotential and a high initial discharge capacity of 6966 mA h g−1 at a current density of 0.1 mA cm−2. An enhanced rate capability, cycle stability and a stable coulombic efficiency close to 100% could also be achieved even at a high current density. Meanwhile, the efficient formation and decomposition of Li2O2 was demonstrated by ex situ XRD and SEM. The superior electrochemical performance can be attributed to the fact that: (i) the Fe/Fe3C nanoparticles stimulate the catalytic activity of the carbon frameworks; (ii) the Fe/Fe3C cores are well encapsulated by the graphitic carbon shell, preventing agglomeration and inactivation; and (iii) the high degree of graphitization and abundant defects provide superior conductivity and enough active sites to enhance the ORR and OER kinetics simultaneously. Furthermore, this research sheds new light on the design and fabrication of efficient and noble metal-free catalysts for Li–O2 batteries.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the National Natural Science Foundation of China (Grant No. 51674297), the Natural Science Foundation of Hunan Province (Grant No. 2016JJ2137), and the Project of Innovation-driven Plan in Central South University (2015CX001 and 2015CXS018) for financial support. This work was supported by the Fundamental Research Funds for the Central Universities of Central South University (Grant No. 2017zzts677) and the Postgraduate research and innovation project of Central South University (1053320170451).Notes and references
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J.-M. Tarascon, Nat. Mater., 2012, 11, 19–29 CrossRef CAS PubMed.
- G. Girishkumar, B. McCloskey, A. C. Luntz, S. Swanson and W. Wilcke, J. Phys. Lett., 2010, 1, 2193–2203 CAS.
- M. M. Ottakam Thotiyl, S. A. Freunberger, Z. Peng, Y. Chen, Z. Liu and P. G. Bruce, Nat. Mater., 2013, 12, 1050–1056 CrossRef CAS PubMed.
- X. Lu, Y. Yin, L. Zhang, L. Xi, S. Oswald, J. Deng and O. G. Schmidt, Nano Energy, 2016, 30, 69–76 CrossRef CAS.
- W. Yang, Z. Qian, C. Du, C. Hua, P. Zuo, X. Cheng, Y. Ma and G. Yin, Carbon, 2017, 118, 139–147 CrossRef CAS.
- K. Zhang, X. Han, Z. Hu, X. Zhang, Z. Tao and J. Chen, Chem. Soc. Rev., 2015, 44, 699–728 RSC.
- X. Hu, J. Wang, Z. Li, J. Wang, D. H. Gregory and J. Chen, Nano Lett., 2017, 17, 2073–2078 CrossRef CAS PubMed.
- X. Ren, B. Liao, Y. Li, P. Zhang, L. Deng and Y. Gao, Electrochim. Acta, 2017, 228, 36–44 CrossRef CAS.
- M. Nazarian-Samani, H.-D. Lim, S. Haghighat-Shishavan, H.-K. Kim, Y. Ko, M.-S. Kim, S.-W. Lee, S. F. Kashani-Bozorg, M. Abbasi, H.-U. Guim, D.-I. Kim, K.-C. Roh, K. Kang and K.-B. Kim, J. Mater. Chem. A, 2017, 5, 619–631 CAS.
- J. Shui, Y. Lin, J. W. Connell, J. Xu, X. Fan and L. Dai, ACS Energy Lett., 2016, 1, 260–265 CrossRef CAS.
- L. Li, C. Chen, J. Su, P. Kuang, C. Zhang, Y. Yao, T. Huang and A. Yu, J. Mater. Chem. A, 2016, 4, 10986–10991 CAS.
- H. Wu, W. Sun, Y. Wang, F. Wang, J. Liu, X. Yue, Z. Wang, J. Qiao, D. W. Rooney and K. Sun, ACS Appl. Mater. Interfaces, 2017, 9, 12355–12365 CAS.
- X. Lu, G.-P. Hao, X. Sun, S. Kaskel and O. G. Schmidt, J. Mater. Chem. A, 2017, 5, 6284–6291 CAS.
- H. Kim, H. Lee, M. Kim, Y. Bae, W. Baek, K. Park, S. Park, T. Kim, H. Kwon, W. Choi, K. Kang, S. Kwon and D. Im, Carbon, 2017, 117, 454–461 CrossRef CAS.
- H.-D. Lim, Y. S. Yun, Y. Ko, Y. Bae, M. Y. Song, H. J. Yoon, K. Kang and H.-J. Jin, Carbon, 2017, 118, 114–119 CrossRef CAS.
- H.-D. Lim, Y. S. Yun, S. Y. Cho, K.-Y. Park, M. Y. Song, H.-J. Jin and K. Kang, Carbon, 2017, 114, 311–316 CrossRef CAS.
- F. Wu, Y. Xing, L. Li, J. Qian, W. Qu, J. Wen, D. Miller, Y. Ye, R. Chen, K. Amine and J. Lu, ACS Appl. Mater. Interfaces, 2016, 8, 23635–23645 CAS.
- F. Tu, J. Hu, J. Xie, G. Cao, S. Zhang, S. A. Yang, X. Zhao and H. Y. Yang, Adv. Funct. Mater., 2016, 26, 7725–7732 CrossRef CAS.
- W. Ni, S. Liu, Y. Fei, Y. He, X. Ma, L. Lu and Y. Deng, ACS Appl. Mater. Interfaces, 2017, 9, 14749–14757 CAS.
- X. Zhang, X. Zhang, X.-G. Wang, Z. Xie and Z. Zhou, J. Mater. Chem. A, 2016, 4, 9390–9393 CAS.
- F. Wu, Y. Xing, X. Zeng, Y. Yuan, X. Zhang, R. Shahbazian-Yassar, J. Wen, D. J. Miller, L. Li, R. Chen, J. Lu and K. Amine, Adv. Funct. Mater., 2016, 26, 7626–7633 CrossRef CAS.
- V. R. Chitturi, M. Ara, W. Fawaz, K. Y. S. Ng and L. M. R. Arava, ACS Catal., 2016, 6, 7088–7097 CrossRef CAS.
- Z. Huang, B. Chi, L. Jian, S. Youyi and Y. Liu, J. Alloys Compd., 2017, 695, 3435–3444 CrossRef CAS.
- H. Xue, X. Mu, J. Tang, X. Fan, H. Gong, T. Wang, J. He and Y. Yamauchi, J. Mater. Chem. A, 2016, 4, 9106–9112 CAS.
- H. Gong, H. Xue, T. Wang, H. Guo, X. Fan, L. Song, W. Xia and J. He, ACS Appl. Mater. Interfaces, 2016, 8, 18060–18068 CAS.
- T. Zeng, M. Yu, H. Zhang, Z. He, J. Chen and S. Song, Catal. Sci. Technol., 2017, 7, 396–404 CAS.
- J. Yang, J. Hu, M. Weng, R. Tan, L. Tian, J. Yang, J. Amine, J. Zheng, H. Chen and F. Pan, ACS Appl. Mater. Interfaces, 2017, 9, 4587–4596 CAS.
- J.-S. Li, S.-L. Li, Y.-J. Tang, M. Han, Z.-H. Dai, J.-C. Bao and Y.-Q. Lan, Chem. Commun., 2015, 51, 2710–2713 RSC.
- B. K. Barman and K. K. Nanda, Green Chem., 2016, 18, 427–432 RSC.
- J. Li, M. Zou, L. Chen, Z. Huang and L. Guan, J. Mater. Chem. A, 2014, 2, 10634–10638 CAS.
- Y. Lai, W. Chen, Z. Zhang, Y. Qu, Y. Gan and J. Li, Electrochim. Acta, 2016, 191, 733–742 CrossRef CAS.
- F. S. Gittleson, R. C. Sekol, G. Doubek, M. Linardi and A. D. Taylor, Phys. Chem. Chem. Phys., 2014, 16, 3230–3237 RSC.
- F. S. Gittleson, W.-H. Ryu, M. Schwab, X. Tong and A. D. Taylor, Chem. Commun., 2016, 52, 6605–6608 RSC.
- K. Kierzek, E. Frackowiak, G. Lota, G. Gryglewicz and J. Machnikowski, Electrochim. Acta, 2004, 49, 515–523 CrossRef CAS.
- D. Su, M. Cortie and G. Wang, Adv. Energy Mater., 2017, 7 DOI:10.1002/aenm.201602014.
- Y. You, X.-L. Wu, Y.-X. Yin and Y.-G. Guo, Energy Environ. Sci., 2014, 7, 1643–1647 CAS.
- C. Aparicio, L. Machala and Z. Marusak, J. Therm. Anal. Calorim., 2012, 110, 661–669 CrossRef CAS.
- Z. Wen, S. Ci, F. Zhang, X. Feng, S. Cui, S. Mao, S. Luo, Z. He and J. Chen, Adv. Mater., 2012, 24, 1399–1404 CrossRef CAS PubMed.
- G. C. Loh and D. Baillargeat, J. Appl. Phys., 2013, 114, 033534 CrossRef.
- R. Zhou and S. Z. Qiao, Chem. Commun., 2015, 51, 7516–7519 RSC.
- L. Cao, Z.-H. Li, Y. Gu, D.-H. Li, K.-M. Su, D.-J. Yang and B.-W. Cheng, J. Mater. Chem. A, 2017, 5, 11340–11347 CAS.
- L. Wang, C. Tian, H. Wang, Y. Ma, B. Wang and H. Fu, J. Phys. Chem. C, 2010, 114, 8727–8733 CAS.
- W. Yang, X. Yue, X. Liu, L. Chen, J. Jia and S. Guo, Nanoscale, 2016, 8, 959–964 RSC.
- Z. Wang, R. Jia, J. Zheng, J. Zhao, L. Li, J. Song and Z. Zhu, ACS Nano, 2011, 5, 1677–1684 CrossRef CAS PubMed.
- W. Yang, X. Liu, X. Yue, J. Jia and S. Guo, J. Am. Chem. Soc., 2015, 137, 1436–1439 CrossRef CAS PubMed.
- G. Tan, L. Chong, R. Amine, J. Lu, C. Liu, Y. Yuan, J. Wen, K. He, X. Bi, Y. Guo, H.-H. Wang, R. Shahbazian-Yassar, S. Al Hallaj, D. J. Miller, D. Liu and K. Amine, Nano Lett., 2017, 17, 2959–2966 CrossRef CAS PubMed.
- Y. Hou, T. Huang, Z. Wen, S. Mao, S. Cui and J. Chen, Adv. Energy Mater., 2014, 4, 1400337 CrossRef.
- A. Mitra, P. Howli, D. Sen, B. Das and K. K. Chattopadhyay, Nanoscale, 2016, 8, 19099–19109 RSC.
- Q. Li, P. Xu, W. Gao, S. Ma, G. Zhang, R. Cao, J. Cho, H.-L. Wang and G. Wu, Adv. Mater., 2014, 26, 1378–1386 CrossRef CAS PubMed.
- G. Nie, X. Lu, Y. Zhu, M. Chi, M. Gao, S. Chen and C. Wang, ChemElectroChem, 2017, 4, 1095–1100 CrossRef CAS.
- R. Nie, H. Jiang, X. Lu, D. Zhou and Q. Xia, Catal. Sci. Technol., 2016, 6, 1913–1920 CAS.
- Q. Wang, X. Zhang, R. Lv, X. Chen, B. Xue, P. Liang and X. Huang, J. Mater. Chem. A, 2016, 4, 12387–12391 CAS.
- X. Zhang, L. Chen, J. Yun, X. Wang and J. Kong, J. Mater. Chem. A, 2017, 5, 10986–10997 CAS.
- B. Wu, L. Ge, D. Yu, L. Hou, Q. Li, Z. Yang and T. Xu, J. Mater. Chem. A, 2016, 4, 14545–14549 CAS.
- Y. Hu, J. O. Jensen, W. Zhang, L. N. Cleemann, W. Xing, N. J. Bjerrum and Q. Li, Angew. Chem., Int. Ed., 2014, 53, 3675–3679 CrossRef CAS PubMed.
- D. Deng, L. Yu, X. Chen, G. Wang, L. Jin, X. Pan, J. Deng, G. Sun and X. Bao, Angew. Chem., Int. Ed., 2013, 52, 371–375 CrossRef CAS PubMed.
- F. Zhang, X. Pan, Y. Hu, L. Yu, X. Chen, P. Jiang, H. Zhang, S. Deng, J. Zhang, T. B. Bolin, S. Zhang, Y. Huang and X. Bao, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 14861–14866 CrossRef CAS PubMed.
- J. Lu, L. Li, J.-B. Park, Y.-K. Sun, F. Wu and K. Amine, Chem. Rev., 2014, 114, 5611–5640 CrossRef CAS PubMed.
- Z. Ma, X. Yuan, L. Li, Z.-F. Ma, D. P. Wilkinson, L. Zhang and J. Zhang, Energy Environ. Sci., 2015, 8, 2144–2198 CAS.
- Y. Liu, L. Wang, L. Cao, C. Shang, Z. Wang, H. Wang, L. He, J. Yang, H. Cheng, J. Li and Z. Lu, Mater. Chem. Front., 2017, 1, 2495–2510 RSC.
- P. Tan, W. Shyy, M. C. Wu, Y. Y. Huang and T. S. Zhao, J. Power Sources, 2016, 326, 303–312 CrossRef CAS.
- J.-J. Xu, Q.-C. Liu, Y. Yu, J. Wang, J.-M. Yan and X.-B. Zhang, Adv. Mater., 2017, 29 DOI:10.1002/adma.201606552.
- J. K. Papp, J. D. Forster, C. M. Burke, H. W. Kim, A. C. Luntz, R. M. Shelby, J. J. Urban and B. D. McCloskey, J. Phys. Lett., 2017, 8, 1169–1174 CAS.
- S. Song, W. Xu, R. Cao, L. Luo, M. H. Engelhard, M. E. Bowden, B. Liu, L. Estevez, C.-M. Wang and J.-G. Zhang, Nano Energy, 2017, 33, 195–204 CrossRef CAS.
- Q. Zhao, C. Wu, L. Cong, Y. Zhang, G. Sun, H. Xie, L. Sun and J. Liu, J. Mater. Chem. A, 2017, 5, 544–553 CAS.
- J. Wang, L. Liu, S. Chou, H. Liu and J. Wang, J. Mater. Chem. A, 2017, 5, 1462–1471 CAS.
- W. Zhang, Y. Shen, D. Sun, Z. Huang, J. Zhou, H. Yan and Y. Huang, Nano Energy, 2016, 30, 43–51 CrossRef CAS.
- D. Sharon, D. Hirsberg, M. Salama, M. Afri, A. A. Frimer, M. Noked, W. Kwak, Y.-K. Sun and D. Aurbach, ACS Appl. Mater. Interfaces, 2016, 8, 5300–5307 CAS.
- H. Nie, C. Xu, W. Zhou, B. Wu, X. Li, T. Liu and H. Zhang, ACS Appl. Mater. Interfaces, 2016, 8, 1937–1942 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional XRD, SEM and TGA data. See DOI: 10.1039/c7qm00503b |
| This journal is © the Partner Organisations 2018 |