
Pyridine coupled mono and bisbenzimidazoles as supramolecular gelators: selective metal ion sensing and ionic conductivity†
Santanu
Panja
a,
Subhratanu
Bhattacharya
b and
Kumaresh
Ghosh
 *a
*a
aDepartment of Chemistry, University of Kalyani, Kalyani-741235, India. E-mail: ghosh_k2003@yahoo.co.in
bDepartment of Physics, University of Kalyani, Kalyani-741235, India
First published on 26th December 2017
Abstract
Pyridine coupled mono and bisbenzimidazoles 1–6 are synthesized and their gelation properties are examined in various polar as well as nonpolar solvents. Among the structures, compounds 1 and 2 form instant gels from DMSO/H2O and MeOH/H2O solvents. The gel states of both 1 and 2 are responsive to Ag+ and Cu2+ ions whereas the gel to sol transition of 2 in DMSO–H2O is additionally caused by Hg2+ ions. On the other hand, compounds 3 and 4 produce gels only in the presence of Ag+ ions under similar conditions and validate their visual readout. In relation to this, the nongelation behavior of p-isomers 5 and 6 (structural isomers) under identical conditions emphasizes the positional role of the pyridyl ring nitrogen with respect to the imidazole ring. Furthermore, metallogels of 3 and 4 exhibit thermally activated ionic conductivity due to movement of Ag+ ions within the gel network.
Introduction
Molecular architectures that are capable of forming dimensionally controlled aggregates in solution via non-covalent interactions (hydrogen bonds, π–π stacking, electrostatic and van der Waals interactions, metal coordination, etc.) draw attention in the area of supramolecular chemistry.1–6 Emphasis has been given to such a special class of compounds not only to correlate the fundamental processes of gelation with structural diversities3,5,7–9 but also to develop new materials with wide applications in biomedicine, drug delivery, reaction catalysis, tissue engineering, water purifiers, photophysics, etc.10–14 In addition, gelators that exhibit a gel to sol transition in the presence of different external stimuli such as light, redox, pH, ions, etc., have gained attention with a view to development of new materials.4,15–19 In this endeavor, metal and anion-induced phase transitions of gelators are worth mentioning. Many research groups in this field have shown interest especially to develop metal responsive supramolecular gels owing to their wide applications in catalysis, sensing, nanoparticle synthesis, optics and magnetic fields, etc.18–20 In this regard, a number of organogelators are known which contain pyridine-based ureas and amides in the structural backbones as they form directed and strong hydrogen bonds.21–26 In contrast, pyridine coupled benzimidazoles that are capable of forming self-aggregates are less reported in the literature27–29 and thus there is a surge of interest to examine these compounds in gelation with possible applications.As part of our ongoing research on the development of stimuli responsive supramolecular gelators,30–33 herein we report metal responsive behaviours of some pyridine coupled mono and bisbenzimidazoles 1–6 (Fig. 1).
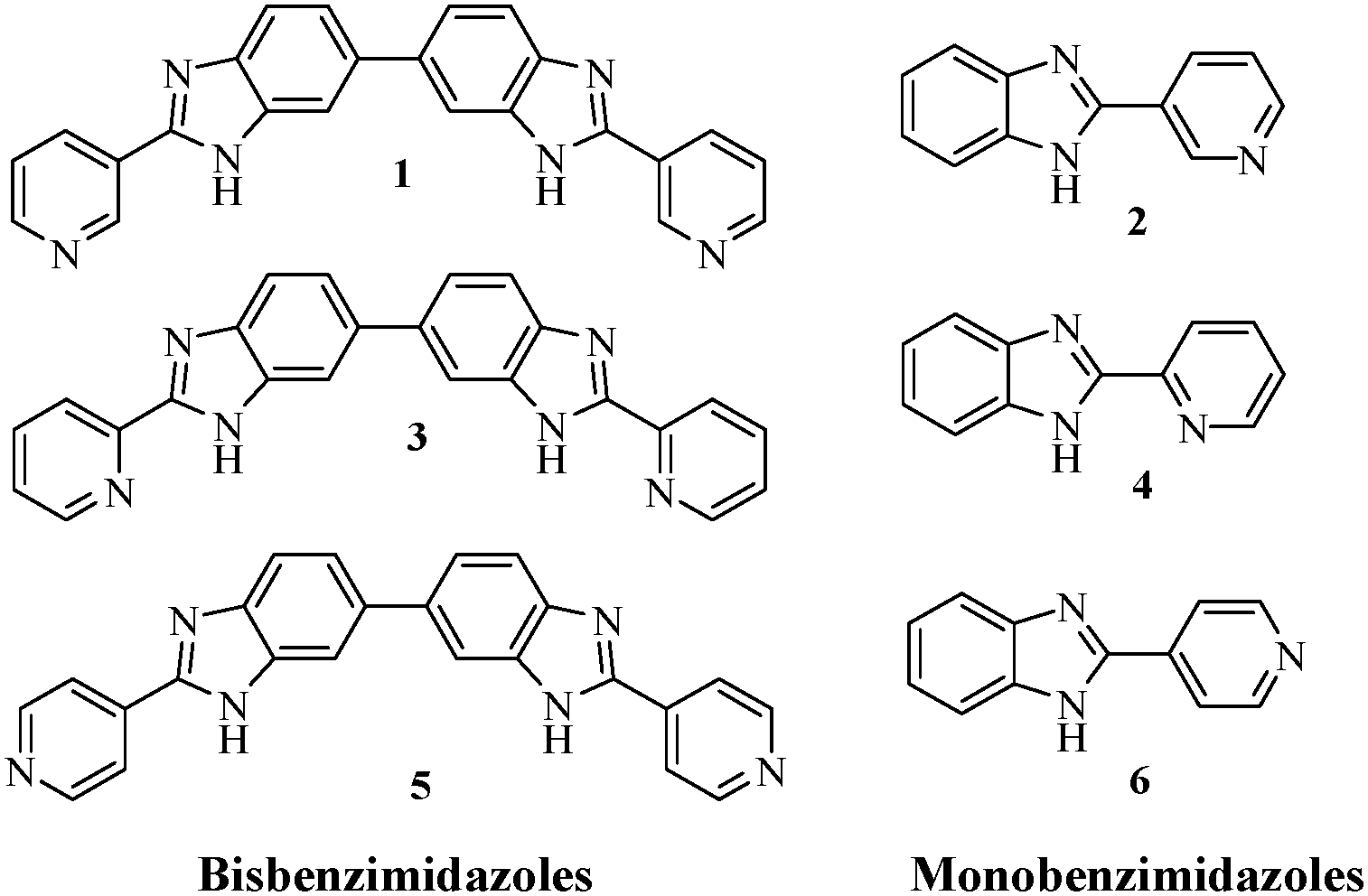 | ||
| Fig. 1 Structures of compounds 1–6. | ||
Importantly, the synthesis of compounds 1–6 and DNA binding studies of some of them are reported by Neidle et al. and other research groups.34–36 But no attempt on examining their gelation characteristics, which is the prime focus of this study, has been made yet.
Analysis of structures 1–6 reveals that the compounds are different due to the change in the position of the pyridine ring nitrogens. This change is deeply associated with their different gelation behaviours. As the gels are formed, they behave differently to metal ions. While the gel states of 1 and 2 are converted to sols in the presence of Ag+ and Cu2+ ions, the gel state of 2 additionally exhibits strong affinity for Hg2+ ions. The structural isomers 3 and 4, in comparison, exhibit gelation selectively in the presence of Ag+ ions under similar conditions. Along this direction, the nongelation behavior of 5 and 6 (p-derivatives) under identical conditions emphasized the positional role of the pyridyl ring nitrogen with respect to the imidazole ring.
Compounds 1–4 in solution also showed measurable interaction with the said metal ions as verified by fluorescence. Fluorescence sensing of metal ions draws attention due to their environmental and physiological significance.37 Of various metal ions, Ag+, Cu2+ and Hg2+ ions are of considerable importance. An Ag+ ion is associated with bioaccumulation and toxicity.38,39 Similarly, a Cu2+ ion is one of the essential elements in our life and is linked with a variety of fundamental physiological processes in organisms.40,41 It serves as a catalytic cofactor for a variety of metalloenzymes like superoxide dismutase, tyrosinase and cytochrome oxidase. Exposure to high levels of copper can also cause gastrointestinal disturbance and liver or kidney damage. This metal ion also causes environmental pollution.41 Mercury is another important analyte because of its toxicity to the ecosystem and damage to health,42 as demonstrated by Minamata disease.43 Mercury exposure can have a number of severe adverse effects on health, such as brain damage, kidney failure, and various cognitive and motion disorders.44 Therefore, selective recognition of these metal ions is of utmost interest. In spite of reasonable progress in the detection/sensing of these ions in solutions, the use of organogelators for the same is less explored and thus requires attention.
In addition to the metal sensing behavior in sol–gel medium, the metallogels obtained from 3 and 4 exhibited a good response in terms of ionic conductivity, because of the movement of the metal ions within the gel networks under an applied electrical field. Conducting gels are widely used in the simulation of electrochemical devices such as super capacitors, fuel cells, rechargeable batteries, solar cells, etc.20,45 In general, long range delocalization of charges is necessary for a gel medium to illustrate conducting properties. However, most of the examples found in the literature are based on polymer gels and the methods that are abruptly used in the fabrication of conducting gels basically involve either direct production of gels from conducting polymers or blending of conducting polymers into non-conducting gel networks.46 Not enough data are found regarding the fabrication of ion conducting supramolecular gels derived from simple metallo-organic architectures.47
Results and discussion
Synthesis
Compounds 1–6 were achieved according to Scheme 1.36 Oxidative condensation between 3,3′-diaminobenzidine and 2-, 3- and 4-pyridine carboxaldehyde in dry nitrobenzene at 140 °C resulted in one step synthesis of compounds 1, 3 and 5, respectively. On the other hand, condensation of o-phenylenediamine with the said aldehydes in dry DMSO at 90 °C produced compounds 2, 4 and 6 in appreciable yields. All the compounds were characterized by usual spectroscopic methods.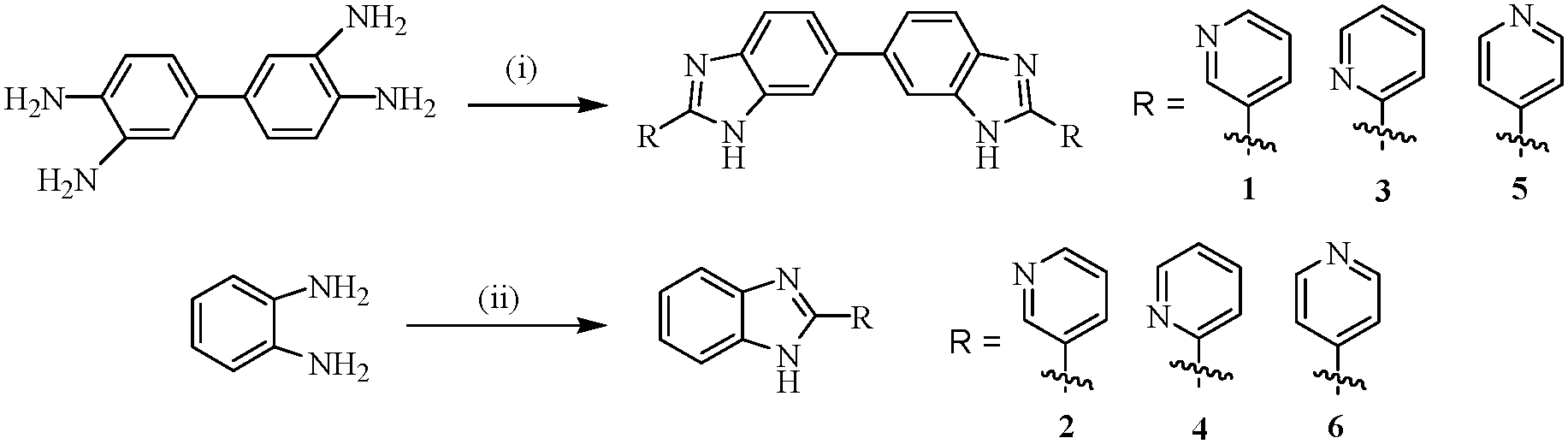 | ||
| Scheme 1 (i) 2-, 3-, or 4-pyridine carboxaldehyde, nitrobenzene, 140 °C, 20–24 h; (ii) 2-, 3-, or 4-pyridine carboxaldehyde, DMSO, 90 °C, 6–10 h. | ||
Gelation, thermal stability, morphology, rheology and metal ion sensing behaviors of the gels
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O (1:3, v/v), compound 1 formed a brown colored gel at a minimum concentration of 3.8 mg mL−1 whereas at least 5 mg compound was required for 2 to gelate 1 mL of the DMSO:H2O (1:6, v/v) solvent mixture to form a white colored gel. The gels were thermo-reversible and upon heating were transformed into clear solution which upon cooling again to room temperature reappeared as gels (Fig. S1, ESI†). The thermal stability of the gels derived from aqueous DMSO was studied. A sharp increase in the gel melting temperature was observed with an increase in the gelator concentration (Fig. S2, ESI†).
:H2O (1:3, v/v), compound 1 formed a brown colored gel at a minimum concentration of 3.8 mg mL−1 whereas at least 5 mg compound was required for 2 to gelate 1 mL of the DMSO:H2O (1:6, v/v) solvent mixture to form a white colored gel. The gels were thermo-reversible and upon heating were transformed into clear solution which upon cooling again to room temperature reappeared as gels (Fig. S1, ESI†). The thermal stability of the gels derived from aqueous DMSO was studied. A sharp increase in the gel melting temperature was observed with an increase in the gelator concentration (Fig. S2, ESI†).
The morphology of the gels was derived from SEM images that predicted a fibrous network (Fig. 2A). Structural difference of the two compounds tuned the morphology of the gels. Gel of 1 is composed of very thin nanofibers which are intertwined in a regular shape demonstrating well-organized molecular packing. In contrast, the gel state of 2 exhibited thin discrete particles.
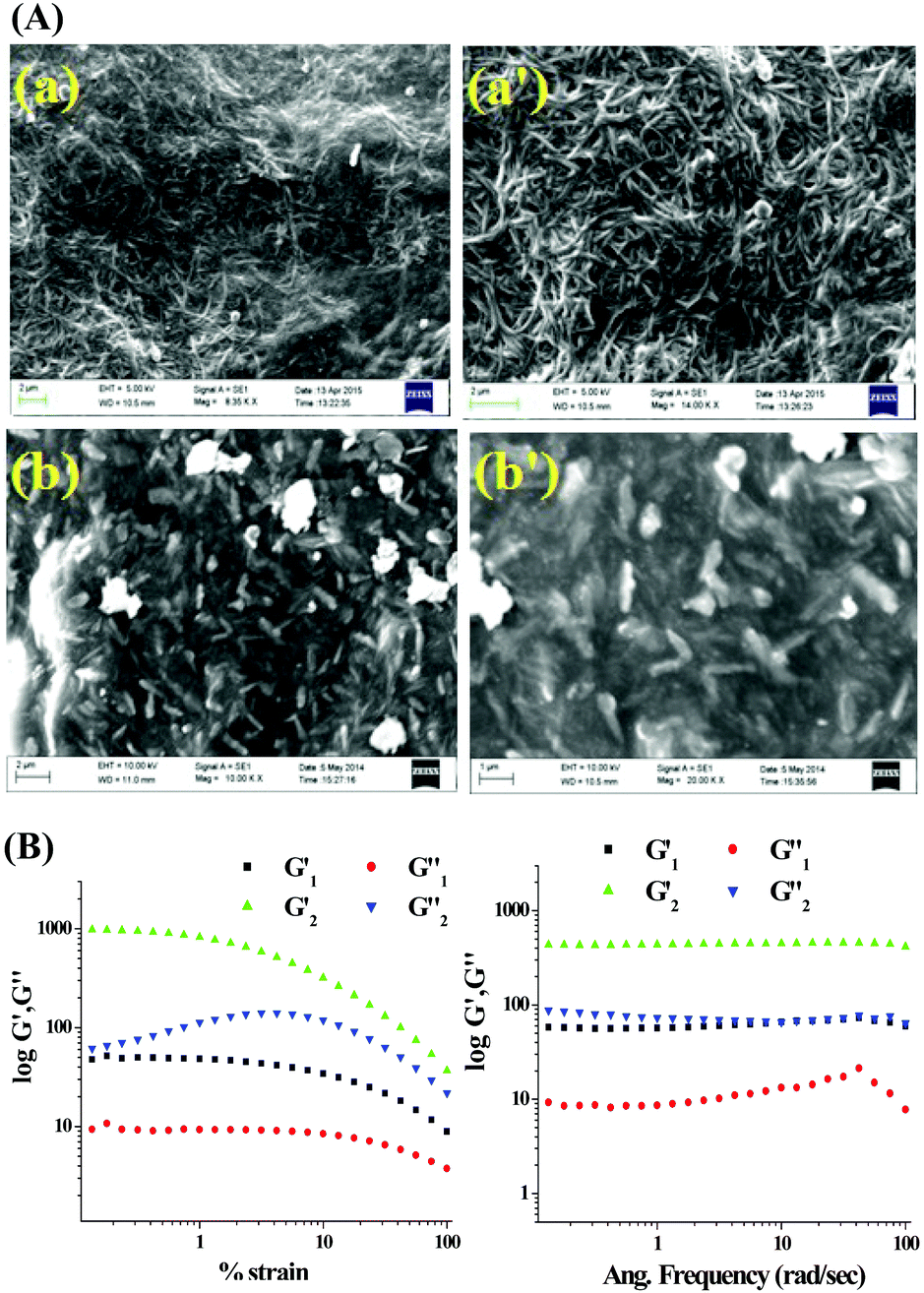 | ||
| Fig. 2 (A) SEM images of xerogel of 1 (a and a′) and 2 (b and b′) prepared from aqueous DMSO at different scale bars; (B) rheology experiments of the DMSO:H2O gels of 1 and 2. | ||
To examine the mechanical properties of the gels, rheology experiments (Fig. 2B) were carried out. For both the gels the elastic storage modulus (G′) was higher than the corresponding loss modulus (G′′). With a gradual increase in the applied stress at a fixed frequency (ω = 10 rad s−1), nonlinear characteristics were observed for both G′ and G′′ although no crossover point was found within the experimental stress region. However, in the case of 1, the values of G′ and G′′ were almost independent of stress compared to the case of 2, which demonstrated that gel 1 is stronger than gel 2. The plot of G′ and G′′ against the angular frequency ω at a constant strain of 1% indicated that both G′ and G′′ were frequency invariant over the entire frequency range and the values of G′ were almost 4 to 6 times higher than those of G′′, indicating their true gel behaviour.
Out of different possibilities, we believe that the imidazole ring nitrogen, pyridine nitrogen and the N–H groups of 1 and 2 are involved in intermolecular hydrogen bonding according to the modes described in Fig. 3 to form the aggregates. Water presumably acts as a bridging entity involving hydrogen bonding with the pyridine ring nitrogens and helps in establishing the cross-linked network in solution.31 In FTIR of 1 and 2, the stretching frequency for –NH in the amorphous states was higher than the gel states which explained the hydrogen bond formation during gelation (Fig. S3, ESI†). Additionally, the appearance of a new signal in the gel states of both in the region 2090 cm−1 to 2101 cm−1 is likely to be due to the involvement of pyridine nitrogen in the formation of strong hydrogen bonds in the network. In addition, the stretching of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N of benzimidazoles in 1 and 2 which appeared at 1577 cm−1 and 1575 cm−1, respectively underwent blue-shift.
N of benzimidazoles in 1 and 2 which appeared at 1577 cm−1 and 1575 cm−1, respectively underwent blue-shift.
 | ||
| Fig. 3 Suggested modes of interaction of (a) 1 and (b) 2 to form the networks during gelation. Comparison of absorption spectra of 1 (c) and 2 (d) in sol and gel states. | ||
To gain more insight into the aggregate formation, absorption spectra of 1 and 2 in sol and gel states were compared. A shoulder at 388 nm in the UV-vis spectrum of 1 in the gel state corroborated stacking induced aggregation (Fig. 3c). Additionally, a bathochromic shift of 14 nm of the absorbance at 330 nm is possibly due to planarization of the biphenyl backbone which in turn enhanced the effective π-conjugation length.53,54 The stacking-induced aggregation was much efficient in the case of gel of 2 that exhibited a new absorption at 342 nm (Fig. 3d). Such aggregation resulted in quenching of emission with red shifts of 33 nm and 28 nm in the case of 1 and 2, respectively (Fig. S4, ESI†).
The gel phases of 1 and 2 exhibited a sharp response to the metal ions, leading to the gel-to-sol transition. For this, we studied the gelation propensity of the compounds in the presence of a series of metal ions in aqueous DMSO. In the case of 1, the presence of 4 equiv. amounts of Ag+ and Cu2+ inhibited the gel formation (Fig. 4A) whereas other metal ions taken in the study left a marginal effect on gelation. We also investigated, on the other hand, the stimuli responsive nature of the gel by adding different metal ions to the gel state of 1. The sustainability of the gel state was checked upon addition of 2 equiv. amounts of Ag+, Cu2+, Hg2+, Cd2+, Zn2+, Co2+, Ni2+, Pb2+, Mg2+, and Fe2+ (as perchlorate salts) in DMSO:H2O (1:3, v/v). Only the gel phase of 1 in the presence of 2 equiv. amounts of Ag+ and Cu2+ ions was disrupted to the sol (resulting in precipitation) within 30 mins. Other metal ions, under similar conditions, did not show any effect on phase transition of 1 suggesting that gelator 1 is highly sensitive and selective towards Ag+ and Cu2+ ions over the other metal ions tested. The selectivity of 1 towards Ag+ and Cu2+ is presumed to be due to the positional role of pyridine nitrogen with respect to the imidazole nitrogens. Pyridine nitrogen away from the imidazole nitrogens is incapable of forming a metal chelate involving any collaborative effect. As a result, pyridine and imidazole will likely be involved in metal ion coordination individually. Although it is true for all the ions taken in the study, the pronounced effect of Ag+ and Cu2+ ions is due to their high affinity towards nitrogen atoms of either pyridine or imidazole rings to attain favourable geometries which, in turn, involve disruption of self-assemblies.55
 | ||
| Fig. 4 Photographs showing the changes of the gels of (A) 1 (8 mg mL−1, DMSO:H2O = 1:3 v/v) and (B) 2 (8 mg mL−1, DMSO:H2O = 1:6 v/v) when the gels were prepared with 4 equiv. amounts of different metal ions [c = 0.2 M in H2O as perchlorate salt] after 2 h. | ||
The metal-induced broken gels were found to be chemically reversible as tested by externally added chelating species (Fig. S5, ESI†). The Ag+-induced disrupted gel was recovered in the presence of excess KCl solution after 2 h. The scavenging of Ag+ ions through the formation of AgCl led to ligand 1 free to attain its original structural feature for which gelation took place. Likewise, the Cu2+-induced broken gel was recovered upon addition of ethylene diamine (in excess) that resulted in a blue color solution which ultimately became thick after 6 h. In this case, the color of the recovered gel was changed to blue rather than brown due to the presence of a Cu–ethylene diamine complex within the gel matrix.
Compound 2, which formed a gel in DMSO:H2O (1:6, v/v) in a similar study, was incapable of forming gels in the presence of Ag+, Cu2+ and Hg2+ ions (Fig. 4B). This highlighted the sensitivity of 2 towards Ag+, Cu2+ and Hg2+ ions. In the presence of 1 equiv. amount of Ag+ and Cu2+ ions individually, the gel state of 2 was disrupted at a faster rate (within 1 h) than the case with Hg2+ which almost took 2.5 h to transform the gel into a sol. The Ag+, Cu2+ and Hg2+-broken gels were not recovered in the presence of external chelators (e.g. Cl−, ethylene diamine and I− ions) and thereby revealed their chemical irreversibility. The nonappearance of the gel or disruption of the gel of gelator 2 in the presence of Ag+, Cu2+ and Hg2+ is presumably due to the same reason as given for gelator 1. Like 1, the pyridine and imidazole nitrogens in 2 interact strongly with Ag+ and Cu2+ ions to attain their favourable geometries. The mercury ion exhibits weak affinity in comparison to Ag+ and Cu2+ ions and therefore, takes longer time to break the self-assembly of 2.
Due to the presence of pyridyl and benzimidazole nitrogens as the basic centre36 we wished to study the effect of pH on the gel states of both 1 and 2 (Fig. S6, ESI†). The gels were stable within the pH range 6 to 9. At pH below 6 the gel state derived from 1 started to disintegrate, while at pH 5 there was partial gelation, and the gel was completely converted to a brown colored sol at pH 4. However, upon further change of pH to 6, the gel reappeared instantly. At above pH 9, the gel was ruptured to the brown colored solution presumably due to the deprotonation of benzimidazole –NH. Similar observations were noted in the case of gel 2. In this case, gel 2 was initially found to be stable at pH 5 for 30 h and then was converted to the sol slowly. These experimental observations underline the fact that the gels of 1 and 2 are stable within the pH window 6 to 9. Below pH 6 or above pH 9, the nonexistence of the gel states is due to either protonation or deprotonation phenomenon, respectively.
:H2O solvent (Table S1, ESI†), they formed thick gels instantly only in the presence of Ag+ ions. It is mentionable that in the presence of 2 equiv. amounts of Ag+ ions, compound 3 at a minimum concentration of 4.9 mg mL−1 formed a yellowish brown-colored gel in DMSO:H2O (1:1, v/v) whereas at least 4.5 mg compound was necessary for 4 to gelate 1 mL of DMSO:H2O (1:2, v/v) solvent to produce a white colored gel. The presence of less than 2 equiv. amounts of Ag+ ions resulted in partial gelation in both cases. We also investigated the effect of other metal ions during gelation of 3 and 4 separately (Fig. 5). Interestingly except for Ag+, other metal ions taken in the study were unable to convert sol states of 3 and 4 into their gel states. This indicated the selectivity of the gelators to Ag+ ions.
 | ||
| Fig. 5 Photographs showing the phase changes of the gels of (A) 3 (4.9 mg mL−1, DMSO:H2O = 1:1, v/v) and (B) 4 (4.5 mg mL−1, DMSO:H2O = 1:2, v/v) in the presence of 2 equiv. amounts of different metal ions [c = 0.2 M in H2O as perchlorate salt] after 2 h. | ||
The morphologies of the gels were characterised by SEM images. While close packed globular microstructures were noted for 3, intertwined tape like fibers were observed for the gel of 4 (Fig. 6).
 | ||
| Fig. 6 SEM images of xerogels derived from 3 (a and a′) and 4 (b and b′) with Ag+ from aqueous DMSO at different scale bars. | ||
The Ag+-induced gels of 3 and 4 exhibited a temperature dependent sol–gel reversible transition (Fig. S7, ESI†). At 56 °C, the gel state of 3 (at mgc) was converted to the sol state with brown color. However, the gel state of 4 at mgc was transformed into a clear solution at 64 °C. In both cases, Tgel varies directly with the gelator concentration (Fig. S8, ESI†). Such kind of gel to sol transition was also observed in the presence of Cl− ions at room temperature. Addition of 2 equiv. amounts of Cl− ions caused rapid destruction of the gels within 1 h giving a white precipitate of AgCl. Further addition of Ag+ ions assembles ligands 3 and 4 to attain the required structural features, responsible for gelation (Fig. S9, ESI†).
Fig. S10 (ESI†) represents the probable binding modes of 3 and 4 out of several possibilities during gel formation. The strong coordination of Ag+ ions involving the pyridyl and benzimidazole ring nitrogens and the hydrogen bonding interplaying involving water are presumably the directives to attain a cross-linked structure in the solution to afford the gels. In FTIR spectra of 3 and 4, from amorphous to their gel state there was a shifting of the –NH stretching to higher wavenumber (Fig. S11, ESI†). Moreover, the stretching of CN of the imidazole rings appearing at 1589 cm−1 and 1588 cm−1 in 3 and 4, respectively underwent a blue-shift and moved to 1595 cm−1 and 1592 cm−1, respectively. The blue-shift essentially reflects the strengthening of the CN bond in the presence of Ag+ and it is ascribed to the inhibition of electronic delocalization around the benzimidazolic unit.56
Moreover, due to aggregation, the gel states of 3 and 4 exhibited strong quenching of emission with redshifts of 40 nm and 28 nm of the emission bands, respectively (Fig. S12 and S13, ESI†).
Positional role of pyridine ring nitrogen with respect to benzimidazole in gelation
A detailed theoretical study on the molecules (1, 3 and 5) was put forward by Bhattacharya and co-workers.36 In their study, they have reported the conformational behaviour of the molecules in gas and solvent phases in detail. In 3 (ortho isomer), the intramolecular hydrogen bonding between benzimidazole –NH and pyridine ring nitrogen led to coplanarity between rings and thereby ruled out the possibility of intermolecular aggregation through hydrogen bonding. On moving from 3 to 1 (meta-isomer) and 5 (para-isomer), the case of intramolecular hydrogen bonding between benzimidazole –NH and pyridine ring nitrogen is ruled out. Consequently, the energy barrier related to the orientation of the pyridine ring with respect to benzimidazole is reduced to a significant extent and thus isomers 1 and 5 are conformationally mobile (Fig. 7).36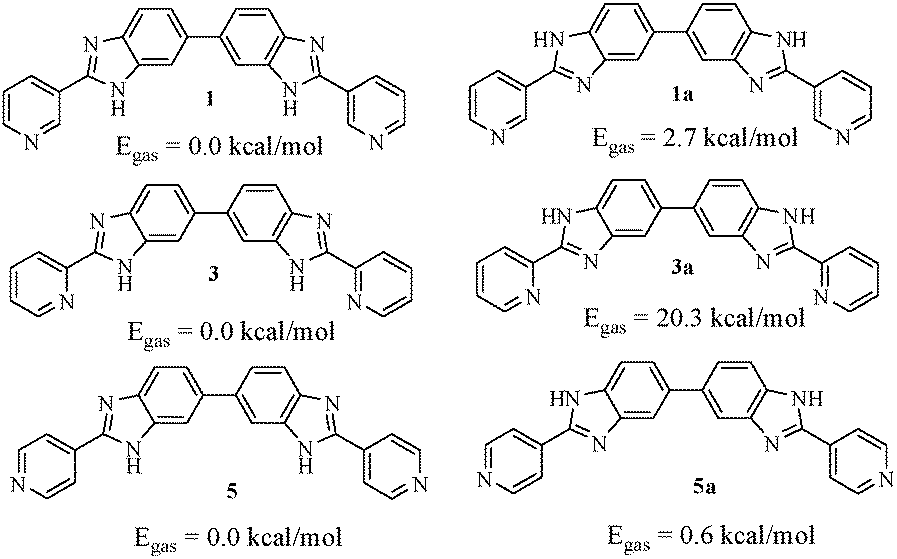 | ||
| Fig. 7 Two relative conformations of the isomeric compounds based on the protons attached to the benzimidazole N face and their relative gas phase energies as ascertained by Bhattacharya et al.,36 using the B3LYP/6-31G* level of theory. | ||
Therefore, in our opinion, isomers 1 and 5 may undergo aggregation through intermolecular hydrogen bonding in several ways and may form gels. But the experimental observation reveals that only isomer 1 exhibits aggregation-induced gelation from DMSO in the presence of water as cosolvent which most likely helps in cross-linking involving pyridine to establish a network for gelation. In 5, the involvement of water in setting up of a cross-linked network involving pyridine as that of 1 is likely to be less feasible and therefore, gelation does not occur.
The case of intramolecular hydrogen bonding in 3 and the role of water in linking the molecules were realized by considering the MMX calculation57 of the dimeric forms of the monomeric analogues 2, 4 and 6 (Fig. S14, ESI†). While in 2 the dimeric form is associated with water to form the network, structure 6 is assumed to be less efficient in producing suitable network for gelation. In comparison, the pyridine in dimeric form of 3 is unavailable for water coordination to form the network for entrapping solvent.
Metal ion binding in solution with compounds 1–4
Pyridyl benzimidazoles are established motifs in the formation of stable metal–ligand complexes.27–29,58–60 In order to verify the solution phase interactions of the pyridyl benzimidazoles of the present study, we performed UV-vis and fluorescence titrations of 1 and 2 (c = 2.5 × 10−5 M) in DMSO:H2O (1:9, v/v). In the fluorescence study, compound 1 showed strong emission at 442 nm upon excitation at 330 nm. Fig. S15 (ESI†) represents the change in fluorescence ratio of 1 at 442 nm in the presence of 10 equiv. amounts of metal ions. The result indicated the sensitivity of 1 towards Ag+ ions over the other metal ions considered in the study. During titration of 1, the intensity of the emission at 442 nm underwent significant quenching only in the presence of Ag+ ions (Fig. 8a) with a blue shift of 16 nm in emission spectra. Copper ion perturbed the emission moderately resulting in a blue shift of 5 nm (Fig. 8b), while other metal ions showed negligible effect on the emission of 1 (Fig. S16, ESI†). The binding constant values61 were found to be (2.78 ± 0.45) × 104 M−1 and (1.88 ± 0.28) × 104 M−1 for Ag+ and Cu2+, respectively with 1:1 stoichiometries62 (Fig. S17 and S18, ESI†). In this sensing process, the detection limits63 for Ag+ and Cu2+ were determined to be 3.69 × 10−6 M and 9.51 × 10−6 M, respectively (Fig. S19, ESI†). Ag+-Induced greater quenching of emission at 442 nm even in the presence of other metal ions corroborated the selectivity of 1 toward Ag+ ions (Fig. S20, ESI†) thereby indicating the noninterference of other metal ions.
 | ||
| Fig. 8 Change in emission of 1 (c = 2.5 × 10−5 M) upon addition of 10 equiv. amount of (a) Ag+ and (b) Cu2+ (c = 1.0 × 10−3 M) in DMSO:H2O (1:9, v/v) respectively, Change in emission of (c) 3 and (d) 4 (c = 2.5 × 10−5 M) upon addition of 1 equiv. and 5 equiv. amounts of Ag+ (c = 1.0 × 10−3 M) in DMSO:H2O (1:9, v/v), respectively. | ||
On the other hand, compound 2 in DMSO:H2O (1:9, v/v) upon excitation at 300 nm exhibited emission at 382 nm which was perturbed (quenching) almost equally upon interaction with metal ions such as Ag+, Cu2+ and Hg2+ (Fig. S21, ESI†). Upon gradual addition of these metal ions to the solution of 2, the emission intensity was gradually decreased without any other change in the emission spectra. Other metal ions taken in the study did not introduce any noticeable effect on the emission of 2 (Fig. S22, ESI†). In solution, compound 2 showed 1:2 (H:G) stoichiometric interactions with Ag+, Cu2+and Hg2+ ions (Fig. S23, ESI†). From fluorescence titration data, the binding constant values were calculated which described the stronger complexation of Ag+ [K1 = (1.05 ± 0.15) × 104 M−1 and K2 = (9.75 ± 4.01) × 102 M−1] than Cu2+ [K1 = (9.02 ± 2.44) × 103 M−1 and K2 = (6.57 ± 1.58) × 103 M−1] and Hg2+ [K1 = (6.01 ± 0.28) × 103 M−1 and K2 = (2.07 ± 0.04) × 103 M−1] ions (Fig. S24, ESI†). The detection limits of 2 for Ag2+, Cu2+ and Hg2+ were determined to be 3.34 × 10−6 M, 5.06 × 10−6 M and 2.02 × 10−6 M, respectively (Fig. S25, ESI†).
The solution phase interactions of 3 and 4 (c = 2.5 × 10−5 M) were also investigated with Ag+ (c = 1.0 × 10−3 M) ions under similar conditions. In the fluorescence study, the presence of only 1 equiv. amount of Ag+ resulted in almost complete quenching of emission of 3 with a 30 nm blue shift (Fig. 8c). In comparison, compound 4 exhibited complete fluorescence quenching in the presence of 5 equiv. amounts of Ag+ (Fig. 8d) with no spectral shift like 3. The binding constant values were calculated to be (1.30 ± 0.31) × 106 M−1 and (4.98 ± 0.98) × 104 M−1 for 1:1 complexation of Ag+ ions with 3 and 4, respectively (Fig. S26–S29, ESI†). From the fluorescence titration data, the detection limits for Ag+ with 3 and 4 were determined to be 1.93 × 10−7 M and 1.28 × 10−6 M, respectively (Fig. S30 and S31, ESI†).
In UV-vis titration of 1, in the presence of both Ag+ and Cu2+ ions, the intensity of the absorption at 330 nm was decreased with the simultaneous increase of a peak at 260 nm (Fig. 9a and b). In the case of Ag+, the ratiometric nature of the absorption spectra was observed only up to the addition of 2 equiv. amounts of Ag+. Further addition of silver ions caused a gradual increase in absorption at 330 nm. In the ground state, compound 1 interacted with both Ag+ and Cu2+ ions in a 1:1 stoichiometric manner with binding constant values of (2.13 ± 0.48) × 103 M−1 and (1.04 ± 0.15) × 104 M−1, respectively (Fig. S32–34S, ESI†). Compound 2 interacted more strongly with Ag+ in comparison to 1 in the ground state, showing a steady decrease in the absorption at 300 nm with a binging constant value of (3.60 ± 0.57) × 103 M−1 involving 1:1 complexation (Fig. S35–S37, ESI†). However, a red shift of 16 nm in the absorption spectra was noted during titration of 3 with Ag+ ions (Fig. 9c). Interestingly, in the presence of Ag+, a ratiometric change with a stable isosbestic point at 333 nm was observed in the absorption spectra of 4 (Fig. 9d). The Job method corroborated 1:1 complexation of Ag+ ions with both 3 and 4 with the binding constant values of (9.47 ± 1.07) × 104 M−1 and (2.46 ± 0.66) × 104 M−1, respectively (Fig. S26–S29, ESI†). Table S2 (ESI†) summarises the metal–ligand interaction profile in detail and the results indicate that the o-derivatives 3 and 4 chelate Ag+ ions more strongly both in ground and excited states probably due to the formation of a more stable five membered ring during complexation.
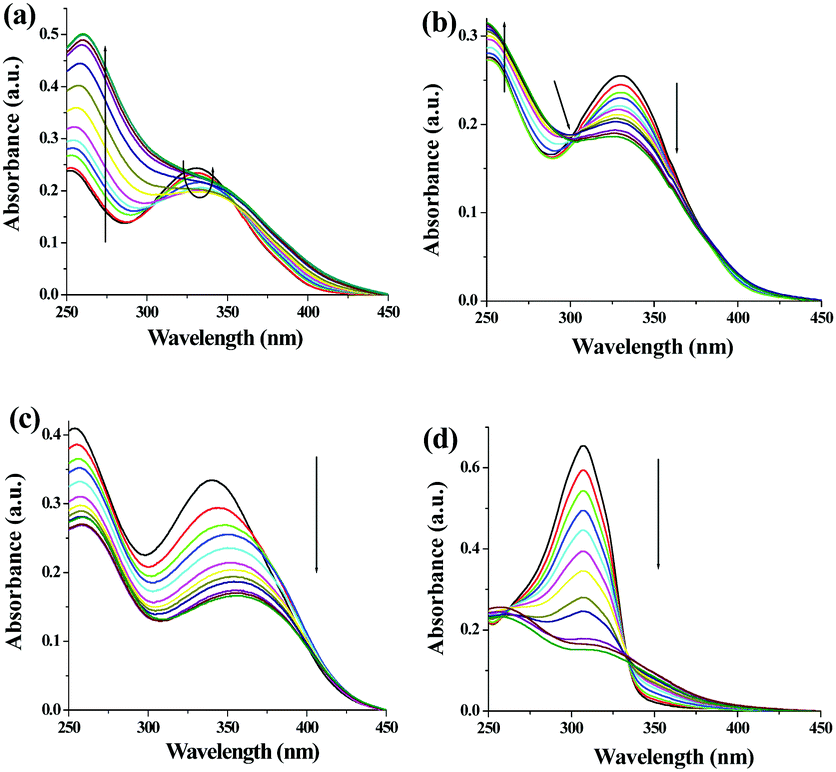 | ||
| Fig. 9 Change in absorbance of 1 (c = 2.5 × 10−5 M) upon addition of 6 equiv. amounts of (a) Ag+ and (b) Cu2+ (c = 1.0 × 10−3 M) in DMSO:H2O (1:9, v/v) respectively. Change in absorbance of (c) 3 and (d) 4 (c = 2.5 × 10−5 M) upon addition of 2 equiv. amounts of Ag+ (c = 1.0 × 10−3 M) in DMSO:H2O (1:9, v/v), respectively. | ||
In order to understand the metal ligand interactions, we performed 1H NMR experiments of 1–4 in the absence and presence of Ag+ in respective solvents. In 1H NMR of 1 and 2 with an equivalent amount of Ag+, the downfield chemical shifts of the imidazole –NHa (Δδ for 1 = 0.37 ppm and 2 = 0.54 ppm in d6-DMSO) indicated strong involvement of the imidazole ring nitrogen in complexation with Ag+ ions (Fig. S38 (ESI†) and Fig. 10A). Due to complexation, the benzimidazole ring proton He moved downfield by 0.67 ppm and 0.17 ppm for 1 (Fig. S38, ESI†) and 2 (Fig. 10A), respectively. Moreover, the downfield chemical shift of pyridyl ring protons in 1 and 2 indicated the simultaneous involvement of the pyridine rings in complexation (Fig. 10A). Such kind of dinuclear Ag+-coordination in M2L2 fashion is known in the literature.58,59 A similar characteristic feature in 1H NMR spectra was observed with 3 and 4. In 1H NMR spectra of 1:1 mixture of 3 and Ag+ in d6-DMSO, the aromatic protons became too broad to detect their exact positions except the imidazole–NHa that exhibited a downfield chemical shift of 0.79 ppm (Fig. S39, ESI†). However in the case of 4, in the presence of an equivalent amount of Ag+ in CDCl3, no significant shift of the pyridyl protons of type Hb and Hc was observed. Moreover, the disappearance of the signal for imidazole –NHa was attributed to the hydrogen bond mediated broadening (Fig. 10B).
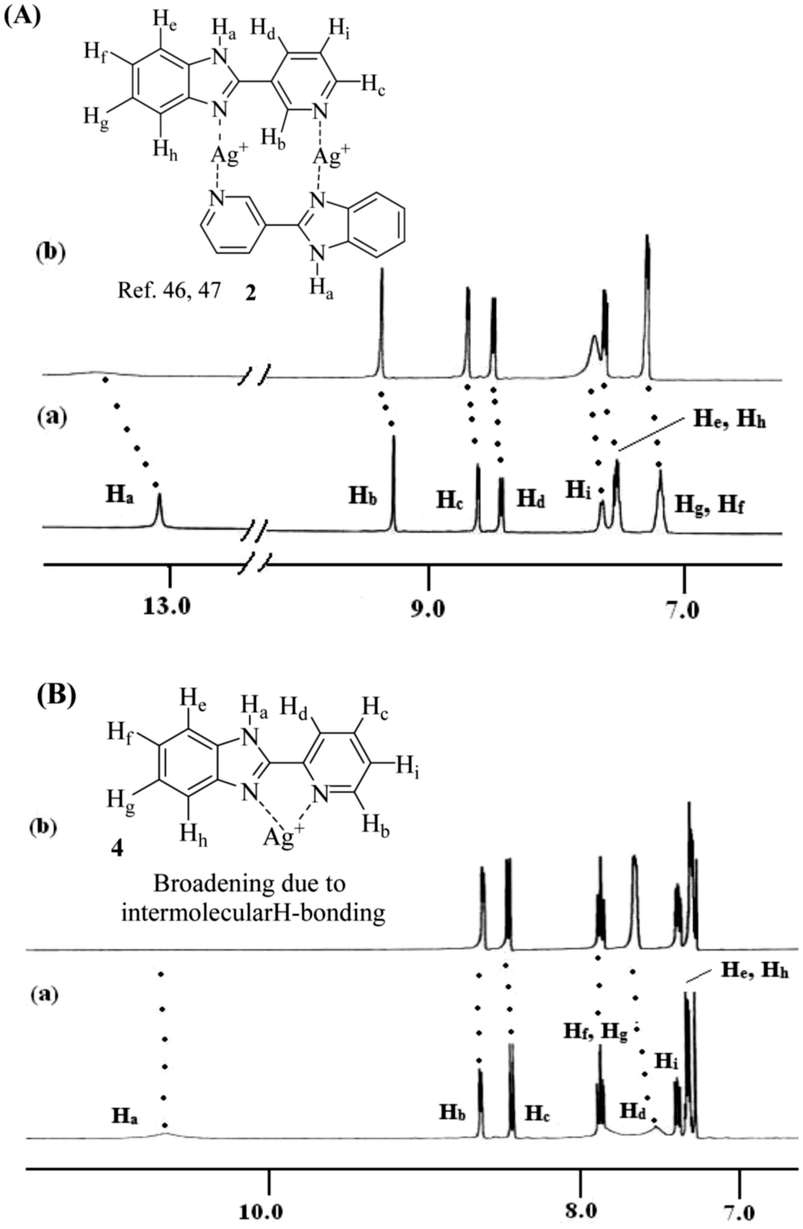 | ||
| Fig. 10 (A) Probable binding mode of 2 with Ag+ in solution along with 1H NMR (400 MHz, d6-DMSO) changes of (a) compound 2 (c = 2.56 × 10−2 M) and (b) 2 with 1 equiv. amount of AgClO4. (B) Probable binding mode of 4 with Ag+ in solution along with 1H NMR (400 MHz, CDCl3) changes of (a) compound 4 (c = 3.41 × 10−2 M) and (b) 4 with 1 equiv. amount of AgClO4. | ||
Ion conductivity
As application the metallogels were explored in an ion conductivity study. The Ag+-induced metallogels of 3 and 4 gave a good response in terms of electrical conductivity. The dielectric properties of a charge containing medium are usually measured by means of the impedance spectroscopy technique.64Fig. 11a and b, in this aspect, show the frequency and temperature dependence of the real part of complex total ionic conductivity of the gels of 3 and 4 (8 mg mL−1) respectively, which arises due to the migration of Ag+ ions within the gel matrices. It is observed from both the figures that the frequency dependence of ionic conduction within the gels can be characterized by two mutually correlated phenomena viz. low frequency electrode polarization and high frequency independent macroscopic charge conduction. For both the gels, it is worth mentioning that below an onset frequency (ωon) the conductivity σt(ω) slowly decreases with decreasing frequency. This corresponds to the electrode polarization due to the accumulation of charges at the electrolyte–electrode interface, commonly designated as the frequency dependent double layer capacitive effect (Cdl).65,66 Above the onset frequency of electrode polarizations σt remains frequency independent, which, in accordance with other reports,67,68 can be attributed to the macroscopic dc-conductivity of the materials obtained from the intersection of the extrapolated straight lines with the y-axis as depicted in the figures. It is observed from the figures that as the temperature decreases, the overall conductivity decreases and the onset of electrode polarization (ωon) shifts toward lower frequencies with reduction in the frequency range of electrode polarization. Fig. 11c depicts the comparison of the frequency dependent real part of ac-conductivity of the gels of 3 and 4 at room temperature (303 K). Fig. 11d displays the temperature dependence of the dc-conductivity obtained from Fig. 11a and b, respectively. It is interesting to note that σdc in both the gels shows thermally activated nature up to the respective Tgel. Also, as the Tgel approaches, the gels gradually lose their thermally activated conductive nature and conductivity tends to saturate. Above Tgel the conductivity decreases from the saturated value. The above nature of temperature dependence of conductivity for both the gels clearly indicates that the gel matrix favours Ag+ ion conduction to a greater extent than the sol due to the increase in defect states which actually enhances the hopping conductivity. In the vicinity of Tgel the gel structure is on the way of rupturing and simultaneously the defect states are diminished which hinders further increase of conductivity. Above Tgel the hopping conductivity is destroyed and the uncorrelated movement of cations and anions within the sol contributes to the conductivity which is less than the hopping conductivity. Both Fig. 11c and d clearly indicate that the gel matrix of 4 favours the Ag+ ion conduction more than the gel of 3 at each measured temperature. The hindrance to Ag+ ion conduction in the gel of 3 is likely to be due to the greater entanglement of Ag+ in the gel network of 3 for participation of more number of pyridyl benzimidazole units.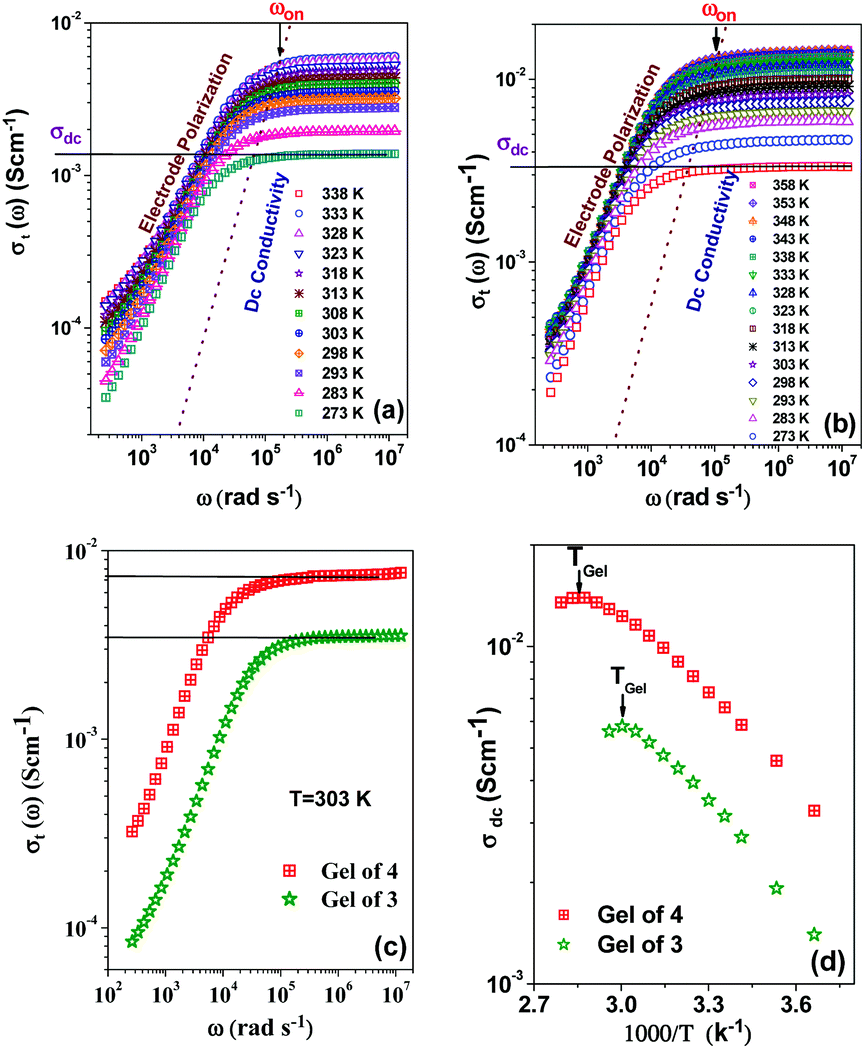 | ||
| Fig. 11 Frequency dependent real part of total ionic conductivity of (a) gel of 3, (b) gel of 4, (c) comparison of the frequency dependent real part of total ionic conductivity of the gels at room temperature, (d) temperature dependence of the dc conductivity of the gels. | ||
Conclusions
In conclusion, we have explored the possibilities of pyridine-coupled mono and bisbenzimidazoles 1–6 in gelation and their subsequent use in detection of metal ions. Of the benzimidazoles 1–6, compounds 1 and 2 formed instant gels from DMSO/H2O and MeOH/H2O solvents. The gels of 1 and 2 are pH responsive and thermoreversible. Moreover, the gel phases of 1 and 2 are rapidly transformed into the sol states in the presence of Ag+ and Cu2+ ions. Additionally, compound 2 exhibited a strong affinity for Hg2+ and recognized it via a gel to sol transition.On the other hand, the structural isomers 3 and 4 exhibited gelation tendency in the presence of Ag+ ions selectively from similar solvent systems and thus validated their visual sensing over a series of other metal ions. In relation to this, nongelation behavior of p-derivatives 5 and 6 (structural isomers) under identical conditions emphasized the positional role of the pyridyl ring nitrogen with respect to the imidazole ring. Thus the uniqueness of pyridyl benzimidazoles in gel chemistry is admirable. While they are easy-to-make, the presence of three N-atoms and imine –NH groups with extended π backbone is of great use in supramolecular self-aggregation either alone or in the presence of metal ions.
As an application, the metallogels derived from 3 and 4 were explored as conducting electrolytes as they responded well in terms of thermally activated ionic conductivity due to the movement of Ag+ ions within the gel matrix. This kind of conductivity study with pyridyl benzimidazoles is unexplored in the literature. Thus the present study in this report draws attention and is a new addendum to the existing merely explored pyridyl benzimidazole gelators for metal ion sensing (Table S3, ESI†).
Experimental
Materials
3-Amino pyridine was purchased from Spectrochem. 3,3′-Diaminobenzidine was obtained from Sigma-Aldrich. Metal salts used in the study were purchased from Sigma-Aldrich and were carefully handled. All solvents used in the synthesis were purified, dried and distilled as required. Solvents used in NMR experiments were obtained from Aldrich. Thin layer chromatography was performed on Merck precoated silica gel 60-F254 plates. 1H and 13C NMR spectra were recorded using a Bruker 400 MHz instrument using TMS as internal standard. High resolution mass data were acquired by the electron spray ionization (ESI) technique on a XEVO GS-2 QTOf Waters mass spectrometer. FTIR measurements of all the compounds and dried gels (xerogels) were carried out using a Perkin-Elmer L120-00A spectrometer (νmax in cm−1) using a KBr cell and KBr pellets, respectively. Scanning electron microscopy (SEM) images were obtained on a EVO LS-10 ZEISS instrument. Fluorescence and UV-Vis studies were performed using a Horiba Fluoromax 4C spectrofluorimeter and a Shimadzu UV-2450 spectrophotometer, respectively.Syntheses
Gelation test and SEM imaging
Respective amounts of compounds of 1–6 were dissolved in 1 mL of desired solvents, slightly warmed to form a homogeneous solution and then allowed to cool slowly to room temperature to form a gel. All the gels were tested by an inversion of vial method. Samples of gels for SEM imaging were dried under vacuum and then coated with a thin layer of gold metal.Determination of gel–sol transition temperature (Tg)
The gel-to-sol transition temperature (Tg) was measured by the dropping ball method. The Tgel is defined as the temperature at which the gel melted and started to flow. In this test, a small glass ball was carefully placed on the top of the gel to be tested, which was present in a test tube. The tube was slowly heated in a thermostated oil bath until the ball fell to the bottom of the test tube. The temperature at which the ball reaches the bottom of the test tube is taken as Tg of that system.General procedure for fluorescence and UV-vis titrations
Stock solutions of the compounds were prepared in DMSO:H2O (1:9, v/v) at a concentration of 2.50 × 10−5 M. Stock solutions of metal ions were also prepared in the same solvents at a concentration of 1.0 × 10−3 M. A solution of each compound (2 mL) was taken in the cuvette and to this solution different metal ions were individually added in different amounts. Upon addition of metal ions, the change in emission of the compound was recorded. The same stock solutions were used to perform the UV-vis titration experiment in the same way.
Binding constant determination61
Binding constant values for receptors with the ionic guests were determined by the nonlinear curve fitting procedure. The 1:1 [H:G] nonlinear fit was done using the equation I = I0 + (Ilim − I0)/2CH{CH + CG + 1/Ka − [(CH + CG + 1/Ka)2 − 4CHCG]1/2}, where I represents the intensity; I0 represents the intensity of pure host; CH and CG are the corresponding concentrations of the host and ionic guest; Ka is the binding constant. The binding constant (Ka) and correlation coefficients (R) were obtained from a non-linear least-squares analysis of I versus CH and CG.
Binding constant values for receptors with the ionic guests in 1:2 complexations were determined by the nonlinear curve fitting procedure using the equation: I = [I0 + (K1CGIm) + (K1K2AX2)]/(1 + K1X + K1K2X2), where I represents the intensity; I0 represents the intensity of pure host; Im represents the intensity of the host in the presence of an equiv. amount of guest; CG is the corresponding concentrations of the guest; K1 and K2 are the binding constant values. The binding constant values and correlation coefficients (R) were obtained from a non-linear least-squares analysis of I versus CG.
Method for the Job plot62
The stoichiometry was determined by the continuous variation method (Job plot). In this method, solutions of host and guests of equal concentrations were prepared in respective solvents. Then host and guest solutions were mixed in different proportions maintaining a total volume of 3 mL of the mixture. The related compositions for host:guest (v/v) were 3:0, 2.8:0.2; 2.5:0.5, 2.2:0.8, 2:1, 1.8:1.2, 1.5:1.5, 1:2, 0.8:2.2, 0.5:2.5, 0.2:2.8. All the prepared solutions were kept for 1 h with occasional shaking at room temperature. Then emission and absorbance of the solutions of different compositions were recorded. The concentration of the complex, i.e., [HG] was calculated using the equation [HG] = ΔI/I0 × [H] or [HG] = ΔA/A0 × [H] where ΔI/I0 and ΔA/A0 indicate the relative emission and absorbance intensities, respectively. [H] corresponds to the concentration of pure host. Mole fraction of the host (XH) was plotted against the concentration of the complex [HG]. In the plot, the mole fraction of the host at which the concentration of the host–guest complex [HG] is maximum gives the stoichiometry of the complex.
Calculation of the detection limit63
The detection limit was calculated using the fluorescence titration data. The emission of compounds was measured 5 times, and the standard deviation of the blank measurement was achieved. To obtain the slope, fluorescence intensities were plotted as concentrations of metal ions. The detection limits were calculated using the equation: detection limit = 3σ/k, where σ is the standard deviation of the blank measurement, and k is the slope.Ionic conductivity measurements
The ionic conductivity of the gels of 3 and 4 was measured by means of impedance spectroscopy (Hioki 3532-50 LCR Hi-Tester from 42 Hz–2 MHz (signal amplitude, 0.1 V)). For measurements, the sample was placed between two parallel steel electrodes with a Teflon ring spacer in a constant-volume cylindrical cell. The measurements of the gels were taken isothermally from 273 K to 358 K in temperature steps of 5 K. The temperature was controlled using a Eurotherm temperature controller and temperature constancy of ±0.2 K was achieved in the entire range of measurements. At each temperature, the gels were thermally equilibrated for 30 min before the measurement. To examine the arbitrary errors related to the changes in the environmental conditions and contact etc. the experiments were repeated several times under identical experimental conditions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
SP thanks CSIR, New Delhi, India, for a fellowship. KG thanks SERB, DST, New Delhi, for financial support (File No. EMR/2016/008005/OC). The authors thank Prof. P. Dastidar, IACS, Kolkata, for the rheological study.References
- F. Fages, Angew. Chem., Int. Ed., 2006, 45, 1680 CrossRef CAS PubMed.
- A. Ajayaghosh and V. K. Praveen, Acc. Chem. Res., 2007, 40, 644 CrossRef CAS PubMed.
- D. K. Smith, Molecular Gels-Nanostructured Soft Materials, in Organic Nanostructures, ed. J. L. Atwood and J. W. Steed, Wiley-VCH, Weinheim, 2008 Search PubMed.
- N. M. Sangeetha and U. Maitra, Chem. Soc. Rev., 2005, 34, 821 RSC.
- S. S. Babu, V. K. Praveen and A. Ajayaghosh, Chem. Rev., 2014, 114, 1973 CrossRef CAS PubMed.
- R. G. Weiss and P. Terech, Molecular Gels, Springer, Dordrecht, 2006, p. 978 Search PubMed.
- G. O. Lloyd and J. W. Steed, Nat. Chem., 2009, 1, 437 CrossRef CAS PubMed.
- M.-O. Piepenbrock, M. N. Clarke and J. W. Steed, Langmuir, 2009, 25, 8451 CrossRef CAS PubMed.
- J. W. Steed, Chem. Soc. Rev., 2009, 38, 506 RSC.
- J. Boekhoven and S. I. Stupp, Adv. Mater., 2014, 26, 1642 CrossRef CAS PubMed.
- P. K. Vemula, N. Wiradharma, J. A. Ankrum, O. R. Miranda, G. John and J. M. Karp, Curr. Opin. Biotechnol., 2013, 24, 1174 CrossRef CAS PubMed.
- V. S. Balachandran, S. R. Jadhav, P. K. Vemula and G. John, Chem. Soc. Rev., 2013, 42, 427–438 RSC.
- N. Dey, S. K. Samanta and S. Bhattacharya, ACS Appl. Mater. Interfaces, 2013, 5, 8394 CAS.
- A. Marsavelski, V. Smrecki, R. Vianello, M. Zinic, A. M. Milankovic and A. Santic, Chem. – Eur. J., 2015, 21, 12121 CrossRef CAS PubMed.
- H. Maeda, Chem. – Eur. J., 2008, 14, 11274 CrossRef CAS PubMed.
- P. Xing, X. Chu, M. Ma, S. Li and A. Hao, Phys. Chem. Chem. Phys., 2014, 16, 8346 RSC.
- P. A. Gale, N. Busschaert, C. J. E. Haynes, L. E. Karagiannidis and I. L. Kirby, Chem. Soc. Rev., 2014, 43, 205 RSC.
- J. Feng and H. Zhang, Chem. Soc. Rev., 2013, 42, 387 RSC.
- A. Ajayaghosh, V. K. Praveen and C. Vijayakumar, Chem. Soc. Rev., 2008, 37, 109 RSC.
- D. D. Dıaz, D. Kuhbeck and R. J. Koopmans, Chem. Soc. Rev., 2011, 40, 427 RSC.
- A. Mishra and R. Gupta, Dalton Trans., 2014, 43, 7668 RSC.
- J. L. Sessler, P. A. Gale and W. S. Cho, Anion Receptor Chemistry, The Royel Society of Chemistry, Cambridge, UK, 2006 Search PubMed.
- K. Liu and J. W. Steed, Soft Matter, 2013, 9, 11699 RSC.
- D. Ghosh, I. Lebedytė, D. S. Yufit, K. K. Damodaran and J. W. Steed, CrystEngComm, 2015, 17, 8130 RSC.
- M. Paul, K. Sarkar and P. Dastidar, Chem. – Eur. J., 2015, 21, 255 CrossRef CAS PubMed.
- N. N. Adarsh and P. Dastidar, Chem. Soc. Rev., 2012, 41, 3039 RSC.
- T. Tu, X. Feng, Z. Wang and X. Liu, Dalton Trans., 2010, 39, 10598 RSC.
- A. Dey, S. K. Mandal and K. Biradha, CrystEngComm, 2013, 15, 9769 RSC.
- S. Samai and K. Biradha, Chem. Mater., 2012, 24, 1165 CrossRef CAS.
- K. Ghosh, D. Kar, S. Panja and S. Bhattacharya, RSC Adv., 2014, 4, 3798 CAS.
- K. Ghosh, S. Panja and S. Bhattacharya, RSC Adv., 2015, 5, 72772 RSC.
- K. Ghosh and S. Panja, RSC Adv., 2015, 5, 12094 RSC.
- S. Panja, S. Bhattacharya and K. Ghosh, Langmuir, 2017, 33, 8277 CrossRef CAS PubMed.
- J. Mann, A. Baron, Y. O. Boahen, E. Johansson, G. Parkinson, L. R. Kelland and S. Neidle, J. Med. Chem., 2001, 44, 138 CrossRef CAS PubMed.
- S. Bhattacharya and P. Chaudhuri, Chem. – Asian J., 2007, 2, 648 CrossRef CAS PubMed.
- P. Chaudhuri, B. Ganguly and S. Bhattacharya, J. Org. Chem., 2007, 72, 1912 CrossRef CAS PubMed.
- U. E. Spichiger-Keller, Chemical Sensors and biosensors for Medicinal and Biological Applications, Wiley-VCH, Weinheim, Germany, 1998 Search PubMed.
- Z. Xu, S. Zheng, Y. Yoon and D. R. Spring, Analyst, 2010, 135, 2554 RSC.
- X. L. Ni, X. Zeng, C. Redshaw and T. Yamato, Tetrahedron, 2011, 67, 3248 CrossRef CAS.
- M. C. Linder and M. H. Azam, Am. J. Clin. Nutr., 1996, 69, 797S Search PubMed.
- B. Sarkar, in Metal Ions in Biological Systems, ed. H. Siegel and A. Siegel, Marcel Dekker, New York, 1981, vol. 12, pp. 233–282 Search PubMed.
- Z. Zhang, D. Wu, X. Guo, X. Qian, Z. Lu, Q. Zu, Y. Yang, L. Duan, Y. He and Z. Feng, Res. Toxicol., 2005, 18, 1814 CrossRef CAS PubMed.
- V. Amendola, L. Fabbrizzi, M. Licchelli, C. Mangano and P. Pallavicini, Acc. Chem. Res., 2001, 34, 488 CrossRef CAS PubMed.
- G. E. Mckeown-Eyssen, J. Ruedy and A. Neims, Am. J. Epidemiol., 1983, 118, 470 CrossRef CAS PubMed.
- L. Wang, H. Zhang, C. Wang and T. Ma, ACS Sustainable Chem. Eng., 2013, 1, 205 CrossRef CAS.
- L. Cui, J. Wu and H. Ju, ACS Appl. Mater. Interfaces, 2014, 6, 16210 CAS.
- K. Ghosh, S. Panja and S. Bhattacharya, ChemistrySelect, 2017, 2, 959 CrossRef CAS.
- K. Ghosh, A. R. Sarkar and A. Patra, Tetrahedron Lett., 2009, 50, 6557 CrossRef CAS.
- J. Makarevic, Z. Stefanic, L. Horvat and M. Zinic, Chem. Commun., 2012, 48, 7407 RSC.
- Z. Zhao, J. W. Y. Lamb and B. Z. Tang, Soft Matter, 2013, 9, 4564 RSC.
- X. Ma, J. Zhang, N. Tang and J. Wu, Dalton Trans., 2014, 43, 17236 RSC.
- H. Y. Liu, H. Wu, J. F. MaY., Y. Liu, B. Liu and J. Yanger, Cryst. Growth Des., 2010, 10, 4795 CAS.
- K. Ghosh and S. Panja, ChemistrySelect, 2016, 1, 3667 CrossRef CAS.
- S. Panja, S. Debnath and K. Ghosh, J. Photochem. Photobiol., A, 2017, 348, 110 CrossRef CAS.
- A. Y.-Y. Tam and V. W.-W. Yam, Chem. Soc. Rev., 2013, 42, 1540 RSC.
- Q. Sun and B. Yan, Bioorg. Med. Chem. Lett., 1998, 8, 361 CrossRef CAS PubMed.
- MMX calculation was performed using PC model, serena software, version 9.200.
- A. Yoshimura, K. Nozaki, N. Ikeda and T. Ohno, J. Phys. Chem., 1996, 100, 1630 CrossRef CAS.
- M. Haga, M. M. Ali, S. Koseki, K. Fujimoto, A. Yoshimura, K. Nozaki, T. Ohno, K. Nakajima and D. J. Stufkens, Inorg. Chem., 1996, 35, 3335 CrossRef CAS.
- R. Ramachandran, G. Prakash, S. Selvamurugan, P. Viswanathamurthi, J. G. Malecki and V. Ramkumar, Dalton Trans., 2014, 43, 7889 RSC.
- P. T. Chou, G. R. Wu, C. Y. Wei, C. C. Cheng, C. P. Chang and F. T. Hung, J. Phys. Chem. B, 2000, 104, 7818 CrossRef CAS.
- P. Job, Ann. Chim., 1928, 9, 113 CAS.
- J. Lnczedy, T. Lengyel and A. M. Ure, IUPAC Compendium of Analytical Nomenclature, Definitive Rules, 1997, web edition, IUPAC, 2002.
- J. R. Macdonald, Ann. Biomed. Eng., 1992, 20, 289 CrossRef CAS PubMed.
- F. Kremer and A. Schönhals, Broadband dielectric spectroscopy, Springer, Berlin, New York, 2003 Search PubMed.
- R. J. Kortschot, A. P. Philipse and B. H. Erné, J. Phys. Chem. C, 2014, 118, 11584 CAS.
- S. Kitajima, F. Bertasi, K. Vezzu, E. Negro, Y. Tominaga and V. D. Noto, Phys. Chem. Chem. Phys., 2013, 15, 16626 RSC.
- B. K. Money, K. Hariharan and J. Swenson, J. Phys. Chem. B, 2012, 116, 7762 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Gelation results, emission, absorption, FTIR and 1H NMR spectra, Job plots, binding curves, detection limit and copies of 1H, 13C NMR and HRMS. See DOI: 10.1039/c7qm00505a |
| This journal is © the Partner Organisations 2018 |