
Cost-effective synthesis and solution processing of porous polymer networks through methanesulfonic acid-mediated aldol triple condensation†
Zi-Hao
Guo‡
 a,
Chenxu
Wang‡
b,
Qiang
Zhang
c,
Sai
Che
a,
Hong-Cai
Zhou
ab and
Lei
Fang
*ab
a,
Chenxu
Wang‡
b,
Qiang
Zhang
c,
Sai
Che
a,
Hong-Cai
Zhou
ab and
Lei
Fang
*ab
aDepartment of Chemistry, Texas A&M University, 3255 TAMU, College Station, Texas 77843, USA. E-mail: fang@chem.tamu.edu
bDepartment of Materials Science and Engineering, Texas A&M University, 3003 TAMU, College Station, Texas 77843, USA
cDepartment of Chemistry, Washington State University, Pullman, Washington, USA. E-mail: q.zhang@wsu.edu
First published on 27th December 2017
Abstract
A highly efficient aldol triple condensation method was developed for scalable synthesis of conjugated porous polymer networks. This bottom-up strategy features inexpensive starting materials and reagents as well as simple reaction procedures, which are ideal for mass production of functional organic porous materials. The resultant porous material demonstrated quick and selective adsorption of organic small molecules in aqueous solution. In addition, the pristine composition of the reaction mixture enabled solution processing of these cross-linked porous materials through a “soaking–heating–washing” procedure to form composite membranes that are essential for practical applications.
Introduction
Bottom-up synthesized microporous materials, such as metal–organic frameworks (MOFs),1,2 covalent organic frameworks (COFs)3,4 and porous polymer networks (PPNs),5–8 are promising candidates for gas storage,9 catalysis,10–12 sensing,13 environmental remediation,14,15 and molecular/ion separation.2,16–18 Among them, non-crystalline PPNs are usually constructed by irreversible cross-coupling of multi-functional monomers.7,19–22 The kinetically formed, conjugated rigid backbones of PPNs endow them with permanent porosity and extraordinary stability, which are in contrast to the crystalline frameworks (such as MOFs and COFs) that are built through dynamic bond formation.12,23 PPNs are therefore more suitable for processes and operations under harsh conditions. The large-scale production and applications of PPNs, however, still confront two major challenges: (i) most of the commonly used reactions for highly porous PPN syntheses require expensive metal catalysts/reagents and demanding operation-procedures, which add cost and risk to potential mass production. (ii) The insoluble and cross-linked nature of PPNs often prohibits feasible processing of these materials into forms relevant to practical applications, such as thin films and composites.24–26In this context, synthetic methods27–29 that allow for mass production and feasible solution-processing are in urgent demand for wider practical applications of PPNs. In order to achieve these objectives, several design principles should be followed: (1) the PPN backbone should be composed of rigid aromatic sp2 bonds, which give raise to persistent porous architecture as well as high chemical and thermal stability. (2) The starting materials, reagents/catalysts, and solvents should be of low cost and environmentally benign, while the reaction should tolerate the exposure to moisture and air. (3) Liquid-phase reaction with minimum number of reagents/catalysts is preferred, so that the reaction mixture could be used directly as the precursor for solution processing. Only a few literature examples could meet all the three requirements.30,31 We report herein a cost-effective, scalable synthesis of PPNs through aldol triple condensation reaction. It offers a green strategy for the mass production of highly stable PPNs that are able to adsorb organic molecules quickly and selectively. Moreover, the reaction enables solution-phase processing of these materials into microporous composite for advanced applications.
Experimental
General information
M2,32M3,33M434 and M535 were synthesized according to the procedure reported in the literature. M1 and other starting reagents were purchased from Aldrich or Alfa-Aesar and used without further purification. All dye molecules used in adsorption tests were purchased from Aldrich, TCI or The Science Company. The glass substrate was first rinsed with acetone. Subsequently, a thin layer of PTFE (purchased from Miller-Stephenson) was spray-coated on the substrate. Following this, the substrate was placed in an oven at 315 °C for 1 h. Carbon fiber paper (Toray 090) was purchased from Fuel Cell Store.Characterization
Thermogravimetric analysis (TGA) data were collected on Mettle-Toledo TGA-DSC-1 at a heating rate of 10 °C min−1 from 30 °C to 900 °C under N2 atmosphere. Solid state nuclear magnetic resonance (NMR) data were recorded on the Bruker Advance-400 Solids NMR spectrometer. N2 adsorption data were collected using the Micrometrics ASAP 2020 accelerated surface area and porosimetry system at 77 K. Samples were activated under vacuum at 120 °C for 12 h with the activation port equipped on ASAP 2020. Field-emission scanning electron microscopic images were collected using the FEI Quanta 600 FE-SEM. UV-vis absorption spectra were recorded on Shimadzu UV-2600 Spectrophotometer. The van der Waals diameters of dye molecules were calculated by Marvinsketch (version 17.23).Method
1,3,5-Triphenylbenzene
In a 25 mL-round-bottom flask, acetophenone (500 mg, 4.17 mmol) and methanesulfonic acid (80 mg, 0.83 mmol) were heated and stirred at 130 °C for 12 h. The mixture was subsequently neutralized by saturated NaHCO3 and extracted with CH2Cl2 (3 × 20 mL). The combined organic layers were dried by MgSO4, filtered, and concentrated in vacuum. The residue was purified by flash column chromatography (SiO2, hexane) to obtain the product as a white solid (364 mg, 1.19 mmol, 85.7%).General procedure of PPN synthesis by MSA-catalyzed ATC reaction
To a 20 mL-glass vial with cap, the monomer (1 equivalent) and methanesulfonic acid (10 to 20 equivalents) were added and heated at 110 °C for 12 h. A dark colored monolithic solid was obtained. After washing with water extensively, the solid was washed with ethanol for 24 h in a Soxhlet extractor. The product was dried under vacuum at 120 °C for 12 h.Organic molecules adsorption by PPN1 test
In each test, 10 mg PPN1 sample was added into 4 mL aqueous solution with organic solute. The concentration of solution was set to be 15 ppm except for that of bisphenol A solution, which was 22.8 ppm. The mixture was stirred at 150 rpm using a magnetic stir bar. At different time points, the solution was filtrated using a syringe filter for concentration test under UV-vis absorption spectroscopy. The concentration of solution was determined by recording UV-vis spectra at the following absorption wavelengths: 274 nm for BPA, 663 nm for MB, 432 nm for BthB, 550 nm for RdB, 550 nm for RB, and 495 nm for CR. The following equation was used to calculate the percentage of absorbed organic molecules:| Absorption percentage = (C0 − Ct)/C0 × 100% |
Each measurement was repeated twice to obtain the standard deviation.
Regeneration of PPN1 was achieved by sonication in acetone for 10 min. The regenerated material was then recovered by filtration for further dye removal test.
Thermodynamic parameters of the adsorption process were studied. Methylene blue solutions with different concentrations were treated by PPN1 samples. The relationship of the amount of dye absorbed at equilibrium, qe (mg g−1), and the residual dye concentration at equilibrium, ce (mg L−1), was plotted following the Langmuir isotherm model:
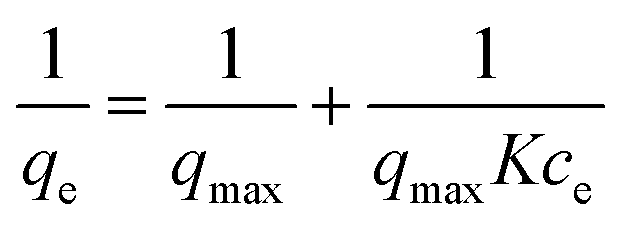
Carbon fiber paper/PPN1 composite membrane fabrication
A piece of carbon fiber paper (CFP) was soaked in monomer M1 solution in MSA (90 mg mL−1), which was then heated at 45 °C for 6 h to allow the formation of a gel. The soaked CFP was then taken out and the gel on the surface of CFP was wiped off.Subsequently, this CFP was further heated at 110 °C for 12 h to trigger the in situ polymerization of PPN inside the voids of CFP. After the reaction was completed, this sample was washed by DMF and ethanol. This sample was then soaked in the M1/MSA solution again and heated to 110 °C after 6 h of pre-treatment. Such “soaking–heating–washing” cycles were performed for 4 times to reach a reasonable high loading of PPN in the CFP matrix.
The methylene blue removal performance of CFP/PPN1 was evaluated by stirring 10 mg of CFP/PPN1 with 4 mL aqueous solution containing 10 ppm methylene blue for 12 h.
Result and discussion
It was reported in 1991![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 36 that tandem aldol triple condensation (ATC) reaction can convert 3 acetyl groups into a benzene ring under acidic condition through two aldol condensations, followed by [3+3] electrocyclic reaction and aromatization. Owing to its high efficiency and the C3 symmetry of the products, ATC has been widely used in the synthesis of star-shaped molecules37,38 and dendrimers.39 Recently, this reaction has also been employed in the preparation of organic microporous materials.30,40–42 In these reported reactions, however, either expensive reagents were used or the acids were easily decomposed under the reaction condition.36,40–42 Furthermore, most of these methods required air-tight procedures to protect the reaction from oxygen and moisture. Therefore, large-scale ATC synthesis of PPN as well as the desired feasible solution processing of composite membranes was still limited. We envisioned that methanesulfonic acid (MSA) could be an ideal liquid medium for the ATC reactions. MSA is non-toxic and thermally stable under 150 °C.43,44 Moreover, the inexpensive and environmentally benign nature of MSA allows it to serve not only as the reagent but also as the solvent for ATC reactions and solution-processing.
36 that tandem aldol triple condensation (ATC) reaction can convert 3 acetyl groups into a benzene ring under acidic condition through two aldol condensations, followed by [3+3] electrocyclic reaction and aromatization. Owing to its high efficiency and the C3 symmetry of the products, ATC has been widely used in the synthesis of star-shaped molecules37,38 and dendrimers.39 Recently, this reaction has also been employed in the preparation of organic microporous materials.30,40–42 In these reported reactions, however, either expensive reagents were used or the acids were easily decomposed under the reaction condition.36,40–42 Furthermore, most of these methods required air-tight procedures to protect the reaction from oxygen and moisture. Therefore, large-scale ATC synthesis of PPN as well as the desired feasible solution processing of composite membranes was still limited. We envisioned that methanesulfonic acid (MSA) could be an ideal liquid medium for the ATC reactions. MSA is non-toxic and thermally stable under 150 °C.43,44 Moreover, the inexpensive and environmentally benign nature of MSA allows it to serve not only as the reagent but also as the solvent for ATC reactions and solution-processing.
In a model reaction on acetophenone (ESI†), ATC reaction was conducted at 130 °C with catalytic amount of MSA (0.2 eq.) without additional solvent or reagent. The product 1,3,5-triphenylbenzene was isolated in 86% yield. Compared with previously reported ATC reactions and commonly used metal-catalyzed cross-coupling reactions, this method was highly efficient, easy to handle, and free of solvent. In this context, PPN syntheses were conducted using aromatic monomers (M1–M5, Fig. 1a) functionalized with multiple acetyl groups by this MSA-mediated ATC method.
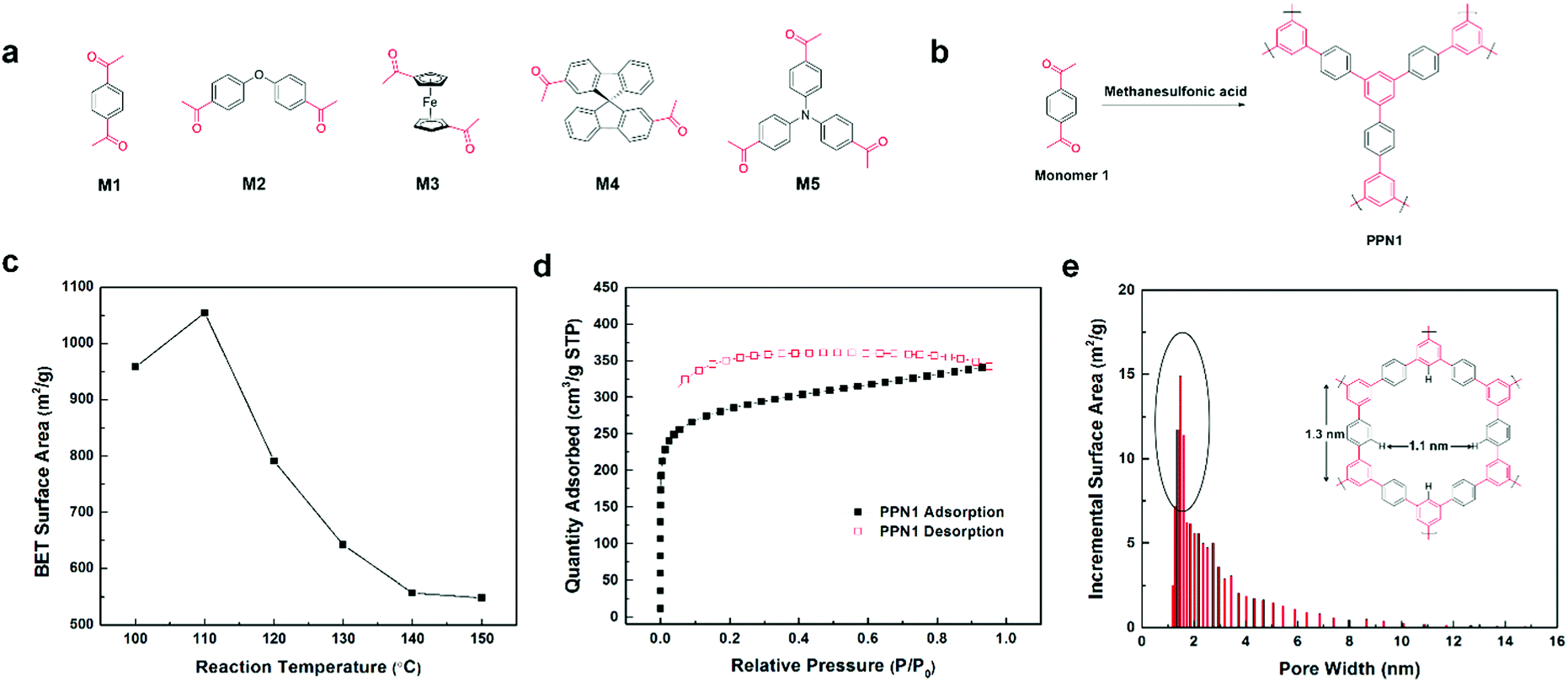 | ||
| Fig. 1 (a) Structural formula of the monomers M1–M5; (b) methanesulfonic acid-catalyzed ATC reaction for PPN1; (c) BET surface areas of PPN1 synthesized at different reaction temperatures; (d) 77 K N2 sorption isotherms and (e) pore size distribution of the PPN1 sample synthesized at 110 °C. | ||
The ATC polymerization of 1,4-diacetylbenzene (M1) was first carried out as a model (Fig. 1b). M1 was suspended in MSA (10 eq.) in an open reaction vessel. Upon heating for several minutes, a homogenous solution was obtained and the color gradually turned from yellow to orange (Fig. S1, ESI†), suggesting that the ATC reaction started to afford an extended conjugated π-system. After heating for 12 h and subsequent work-up, an insoluble red solid (PPN1) was isolated. The yields for PPN1, PPN3, PPN4 and PPN5 were almost quantitative, ranging from 90% to 99%. The yield of PPN2 was relatively low, ranging from 60% to 80% due to instability of the ether group in strong acid. It was important to note that no extra protective procedure was needed during the entire procedure, demonstrating the insensitive nature of MSA-mediated ATC reaction to oxygen or moisture.
A series of samples of PPN1 were produced at different reaction temperatures ranging from 100 °C to 150 °C. N2 adsorption–desorption isotherm measurements at 77 K and Brunauer–Emmet–Teller (BET) surface area analysis were conducted on these samples to screen the reaction conditions for high porosity. The BET surface area obtained at different reaction temperatures are shown in Fig. 1c. An optimized BET surface area of 1054 m2 g−1 was obtained at a reaction temperature of 110 °C, representing the highest value among PPNs synthesized by ATC methods.40,42,45 At lower temperatures, the reaction rate was slow and the solubility of reaction intermediates was poor, leading to a lower conversion and hence lower porosity in the product. Above 110 °C, a monotonous decrease in BET surface areas of the products was attributed to the formation of defects: under higher temperatures, the microporous network grew too quickly so that more defects were formed to lower the microporosity.7 With the optimized reaction temperature of 110 °C, the pore volume of the product was 0.42 cm3 g−1. Pore size distribution analysis (Fig. 1e) showed that the majority of the pores were with diameters in the range of 1–2 nm, matching the theoretically calculated diameter of the smallest repeating cyclic structure. A small amount of larger pores (<10 nm) were also present, probably because of the kinetically trapped defects in this sample.
The structure of PPN1 was also investigated with FT-IR spectroscopy (Fig. 2). There were two peaks around 1700 cm−1 corresponding to the carbonyl stretching of unreacted acetyl (1718 cm−1) and α,β-unsaturated ketone (1683 cm−1). For the PPN1 formed at a lower temperature (100 °C), the stronger peak at 1718 cm−1 indicated that a larger fraction of the acetyl groups were unreacted due to the relatively lower conversion. At higher temperatures, the signal of acetyl stretching was weakened significantly, while the intensity of the α,β-unsaturated ketone peak increased, giving rise to a broad peak centered at around 1700 cm−1. The relative intensity of the benzene stretching peak (1507 cm−1), compared with that of the broad peak at 1700 cm−1, first increased from 100 °C to 110 °C and then decreased from 110 °C to 150 °C, indicating that 110 °C was the optimized temperature for the highest conversion and lowest defect level. These results agreed well with the BET surface area measurements (Fig. 1c) and the corresponding hypothesis. In addition, solid-state 13C CP/MAS NMR spectroscopy (Fig. S15, ESI†) revealed two major signals (138.7 and 124.1 ppm) corresponding to unsubstituted aromatic carbon atoms and substituted benzene carbon atoms, respectively, agreeing with the proposed backbone constitution of PPN1. Due to the low sensitivity of this method, however, the expected peaks corresponding to defects, such as acetyl group and α,β-unsaturated ketone, were not identifiable. In contrast, elementary analysis (Table S1, ESI†) showed trace amounts of oxygen left over in PPN1, corresponding to the unreacted defects.
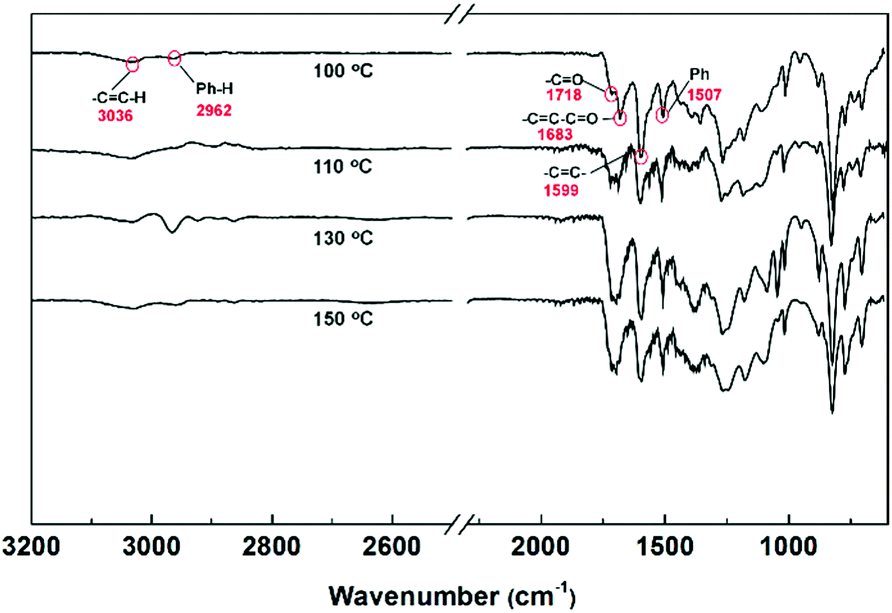 | ||
| Fig. 2 FT-IR spectroscopy of PPN1 samples obtained from different reaction temperatures (100 °C, 110 °C, 130 °C and 150 °C). | ||
PPN2 to PPN5 were synthesized from monomers M2–M5 under the optimized condition (Table 1). The BET surface areas of PPN2, PPN3 and PPN5 were lower than that of PPN1. The decrease in porosity was attributed to (i) the higher flexibility of these monomer, and (ii) network interpenetration due to their longer molecular lengths.7PPN4 showed extremely low porosity, probably because of the bulkiness of the spirofluorene that filled up the pores. N2 sorption isotherms for PPN1, 2, 3 and 5 at 77 K (Fig. 1c and Fig. S2–S5, ESI†) showed high gas uptake at low relative pressures and a flat course in the intermediate section, representing typical Type I adsorption–desorption isotherms. Thermogravimetric analyses (TGA) (Fig. S7, ESI†) demonstrated good thermal stability of PPN1, PPN2, PPN4, and PPN5 with decomposition temperatures over 400 °C owing to the robust nature of their rigid sp2-rich backbones. The only exception, PPN3, showed a distinctive weight loss before 200 °C, because of the lower intrinsic thermal stability of the ferrocene unit.46
| Microporous polymer | Monomer | S BET (m2 g−1) | V Micro (cm3 g−1) | V total (cm3 g−1) |
|---|---|---|---|---|
| a Surface area calculated from N2 adsorption–desorption isotherm at 77 K using the BET method. b Micropore volume calculated from N2 adsorption isotherm using the t-plot method. c Total pore volume at P/P0 = 0.97. | ||||
| PPN1 | M1 | 1054 | 0.28 | 0.42 |
| PPN2 | M2 | 515 | 0.04 | 0.20 |
| PPN3 | M3 | 699 | 0.04 | 0.25 |
| PPN5 | M5 | 729 | 0.17 | 0.31 |
| CFP/PPN1 | M1 | 216 | N/A | N/A |
Despite the amorphous nature of PPN1, the large pore volume and narrow pore size distribution promises its application in size-selective adsorption of molecular solutes in the solution.18,47,48 To test the efficiency and selectivity of PPN1 in adsorbing organic molecules, aqueous solutions of organic compounds (15–23 ppm in 4 mL water) with different van der Waals size, ranging from 0.8 nm to 2.5 nm (Fig. 3c and Fig. S10, ESI†), were treated with 10 mg PPN1 powder. These organic samples are either dyes or UV-absorbers, so that UV-visible absorption spectroscopy can be employed to test the adsorption efficiency (Fig. 3 and Fig. S8, ESI†). After adding PPN1 to the solution, dye molecules with small van der Waals diameters, such as bisphenol A (BPA) and methylene blue (MB), were fully adsorbed in short time (5 minutes for MB and 15 minutes for BPA), leading to diminished UV-vis absorption in the solution. In contrast, for rhodamine B (RdB), bromothymol blue (BB), Congo red (CR) and Rose Bengal (RB) that possess van der Waals diameters matching or larger than the maximum pore size of PPN1, the solution concentration remained high even after 3 h. The selective adsorption of BPA and MB by PPN1 in mild agitation shows its potential for rapid and selective removal of small organic molecules in water.15 Adsorption selectivity of PPN1 is comparable with those porous materials containing specific supramolecular receptors15,49 and outperformed prevalent commercial porous materials, such as active carbon and zeolite.18 These high performances can be attributed to the strong hydrophobicity and the narrow size distribution of the pores in PPN1.50 In addition, the fact that the size-selectivity was not sensitive to the charge of the molecules indicated that the selectivity was primarily a result of the size matching effect and not electrostatic interactions. Furthermore, through the Langmuir isotherm plot, a high value of maximum adsorption capacity (139 mg g−1) of PPN1 to MB solution was obtained (Fig. S11, ESI†). The adsorption capability of PPN1 can be feasibly regenerated for at least 3 times after simply bath-sonicating the PPN1 in acetone for 10 min at room temperature.
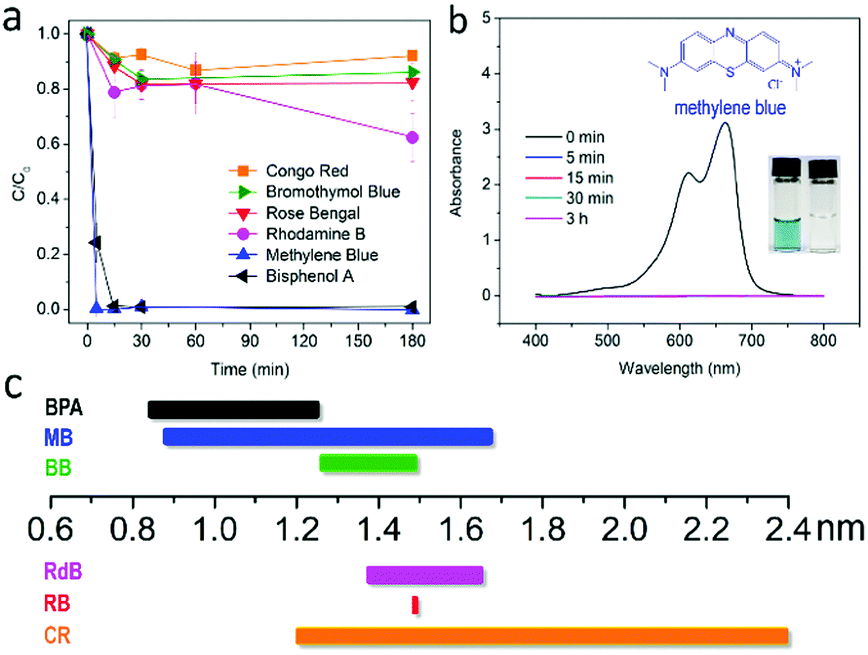 | ||
| Fig. 3 (a) Plot of relative concentration changes of different dye solutions versus time upon treating with PPN1; (b) example of UV-vis spectra of MB solution at different time after the addition of PPN1, inset shows the photo of MB solution before and 5 min after the adsorption process started and (c) molecular sizes of the tested dye molecules, the bars range from the minimal projection diameters to maximal projection diameters of the solutes. | ||
Although the rigid and cross-linked nature endow PPNs with excellent stability and solvent resistance, the processing of such materials could be challenging.31,51,52 Using MSA-mediated ATC synthesis, however, this problem can be addressed by taking advantage of the pristine nature of the reaction mixture30,31 because no other reagent was added into the reaction other than MSA. Using this method, it was possible to produce PPN composites with a supporting matrix to integrate important properties related to practical applications, such as mechanical robustness and electrical conductivity. For example, in situ ATC reaction of M1 solution in MSA can be performed in the presence of carbon fiber paper (CFP) to afford porous PPN1/CFP composite. In this composite, the voids and holes in CFP matrix were filled up with PPN1. In order to fill up all the space in CFP paper, 4 cycles of “soaking–heating–washing” procedure were performed (Fig. 4a), leading to a 55% weight-gain in the composite after the incorporation of PPN (Fig. S13, ESI†). Top and cross-section view SEM images of CFP and CFP/PPN1 (Fig. 4b, c and Fig. S14, ESI†) shows that the PPN infiltrated deep into the CFP. N2 sorption isotherm measurement demonstrated the porous nature of the composite (BET surface area 216 m2 g−1) despite the large composition of carbon fiber matrix53 (Fig. S6, ESI†). The CFP/PPN1 composite also possessed excellent methylene blue removal efficiency (97.7%) (Fig. S19, ESI†) although its adsorption kinetics was slower than that of PPN1 powder. Overall, this method enables the fabrication of functional PPN products mechanically supported by a strong fiber matrix.
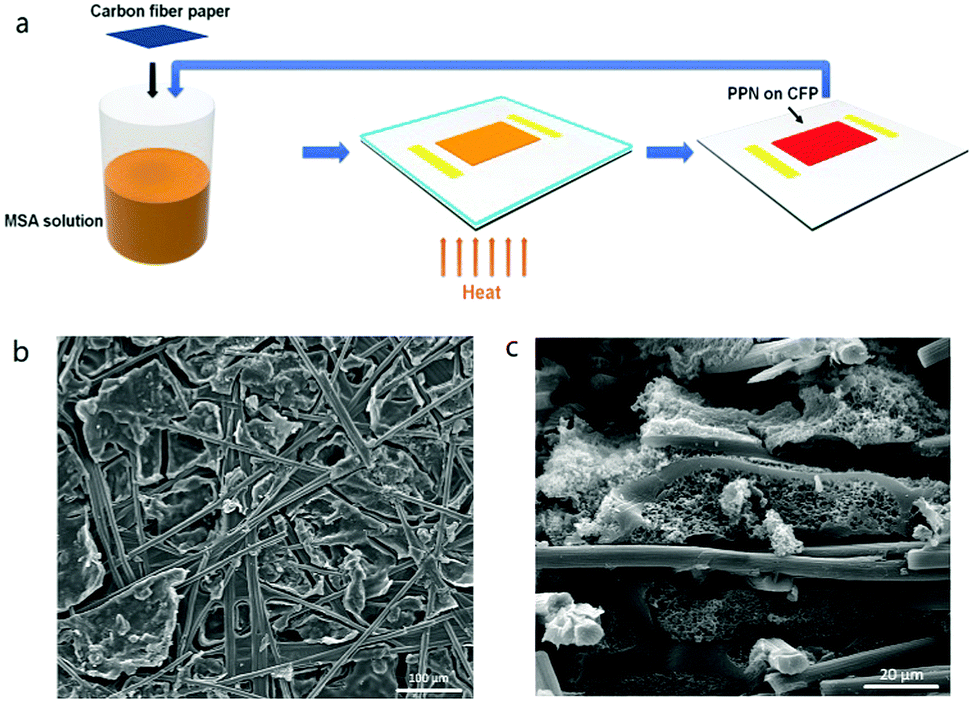 | ||
| Fig. 4 (a) “Soaking–heating–washing” cycles for the fabrication of CFP/PPN1 composite membrane. Scanning electron microscopy (SEM): (b) top view and (c) cross-section view of CFP/PPN1 composite membrane. | ||
Conclusions
In summary, cost-effective bottom-up syntheses of organic microporous polymer networks were achieved using methanesulfonic acid-mediated aldol triple condensation reaction. This method features inexpensive starting materials and reagents as well as simple reaction procedure. The hydrophobic nature and narrow size distribution of the pores of the resultant material enabled rapid and selective adsorption of organic molecules in aqueous environment. Pristine composition of the reaction precursor allowed solution processing of porous composite membranes of these insoluble PPNs through a “soaking–heating–washing” strategy, enabling the application of composite PPN materials with supporting materials. Overall, this study paves the way for large-scale, practical applications of PPN materials for purification, filtration, catalysis and sensing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by Start-up Funds from Texas A&M University, two Welch Foundation Grants (A-1898 and A-1725), and a Welch Foundation Endowed Chair Grant (HJZ A-0030). C. W. acknowledges the support from a China Scholarship Council scholarship (201507040025). The authors would like to thank Dr Vladimir Bakhmoutov for his meaningful discussion regarding the solid state NMR in this work.Notes and references
- O. M. Yaghi, M. O’Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705–714 CrossRef CAS PubMed.
- J. R. Li, J. Sculley and H. C. Zhou, Chem. Rev., 2012, 112, 869–932 CrossRef CAS PubMed.
- P. J. Waller, F. Gandara and O. M. Yaghi, Acc. Chem. Res., 2015, 48, 3053–3063 CrossRef CAS PubMed.
- X. Feng, X. Ding and D. Jiang, Chem. Soc. Rev., 2012, 41, 6010–6022 RSC.
- Y. Xu, S. Jin, H. Xu, A. Nagai and D. Jiang, Chem. Soc. Rev., 2013, 42, 8012–8031 RSC.
- A. I. Cooper, Adv. Mater., 2009, 21, 1291–1295 CrossRef CAS.
- D. Yuan, W. Lu, D. Zhao and H.-C. Zhou, Adv. Mater., 2011, 23, 3723–3725 CrossRef CAS PubMed.
- W. Lu, D. Yuan, D. Zhao, C. I. Schilling, O. Plietzsch, T. Muller, S. Bräse, J. Guenther, J. Blümel, R. Krishna, Z. Li and H.-C. Zhou, Chem. Mater., 2010, 22, 5964–5972 CrossRef CAS.
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T. H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724–781 CrossRef CAS PubMed.
- J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC.
- M. Yoon, R. Srirambalaji and K. Kim, Chem. Rev., 2012, 112, 1196–1231 CrossRef CAS PubMed.
- H. Xu, J. Gao and D. Jiang, Nat. Chem., 2015, 7, 905–912 CrossRef CAS PubMed.
- J. W. Colson and W. R. Dichtel, Nat. Chem., 2013, 5, 453–465 CrossRef CAS PubMed.
- B. Aguila, Q. Sun, J. A. Perman, L. D. Earl, C. W. Abney, R. Elzein, R. Schlaf and S. Ma, Adv. Mater., 2017, 29, 1700665 CrossRef PubMed.
- A. Alsbaiee, B. J. Smith, L. Xiao, Y. Ling, D. E. Helbling and W. R. Dichtel, Nature, 2016, 529, 190–194 CrossRef CAS PubMed.
- J. R. Li, R. J. Kuppler and H.-C. Zhou, Chem. Soc. Rev., 2009, 38, 1477–1504 RSC.
- Y. Zhang, S. Yuan, X. Feng, H. Li, J. Zhou and B. Wang, J. Am. Chem. Soc., 2016, 138, 5785–5788 CrossRef CAS PubMed.
- J. Byun, H. A. Patel, D. Thirion and C. T. Yavuz, Nat. Commun., 2016, 7, 13377 CrossRef CAS PubMed.
- J. Weber and A. Thomas, J. Am. Chem. Soc., 2008, 130, 6334–6335 CrossRef CAS PubMed.
- L. Chen, Y. Honsho, S. Seki and D. Jiang, J. Am. Chem. Soc., 2010, 132, 6742–6748 CrossRef CAS PubMed.
- J. X. Jiang, F. Su, A. Trewin, C. D. Wood, N. L. Campbell, H. Niu, C. Dickinson, A. Y. Ganin, M. J. Rosseinsky, Y. Z. Khimyak and A. I. Cooper, Angew. Chem., Int. Ed., 2007, 46, 8574–8578 CrossRef CAS PubMed.
- J.-X. Jiang, F. Su, A. Trewin, C. D. Wood, H. Niu, J. T. A. Jones, Y. Z. Khimyak and A. I. Cooper, J. Am. Chem. Soc., 2008, 130, 7710–7720 CrossRef CAS PubMed.
- L. Huang, H. Wang, J. Chen, Z. Wang, J. Sun, D. Zhao and Y. Yan, Microporous Mesoporous Mater., 2003, 58, 105–114 CrossRef CAS.
- C. Zhu and L. Fang, Curr. Org. Chem., 2014, 18, 1957–1964 CrossRef CAS.
- O. Knopfmacher, M. L. Hammock, A. L. Appleton, G. Schwartz, J. Mei, T. Lei, J. Pei and Z. Bao, Nat. Commun., 2014, 5, 2954 Search PubMed.
- R. P. Bisbey, C. R. DeBlase, B. J. Smith and W. R. Dichtel, J. Am. Chem. Soc., 2016, 138, 11433–11436 CrossRef CAS PubMed.
- S. H. Je, O. Buyukcakir, D. Kim and A. Coskun, Chem, 2016, 1, 482–493 CAS.
- S. Wang, C. Zhang, Y. Shu, S. Jiang, Q. Xia, L. Chen, S. Jin, I. Hussain, A. I. Cooper and B. Tan, Sci. Adv., 2017, 3, e1602610 CrossRef PubMed.
- E. Troschke, S. Grätz, T. Lübken and L. Borchardt, Angew. Chem., Int. Ed., 2017, 56, 6859–6863 CrossRef CAS PubMed.
- X. Zhu, C. Tian, S. Chai, K. Nelson, K. S. Han, E. W. Hagaman, G. M. Veith, S. M. Mahurin, H. Liu and S. Dai, Adv. Mater., 2013, 25, 4152–4158 CrossRef CAS PubMed.
- X. Zhu, C. Tian, S. M. Mahurin, S. Chai, C. M. Wang, S. Brown, G. M. Veith, H. Luo, H. Liu and S. Dai, J. Am. Chem. Soc., 2012, 134, 10478–10484 CrossRef CAS PubMed.
- J. K. Ray, S. Gupta, D. Pan and G. K. Kar, Tetrahedron, 2001, 57, 7213–7219 CrossRef CAS.
- Y. Li and Y. Zheng, J. Appl. Polym. Sci., 2016, 133, 43217 Search PubMed.
- C. Stobe, R. Seto, A. Schneider and A. Lützen, Eur. J. Org. Chem., 2014, 6513–6518 CrossRef CAS.
- T.-S. Hsiao, T.-L. Chen, W.-L. Chien and J.-L. Hong, Dyes Pigm., 2014, 103, 161–167 CrossRef CAS.
- S. S. Elmorsy, A. Pelter and K. Smith, Tetrahedron Lett., 1991, 32, 4175–4176 CrossRef CAS.
- C. Huang, W. Fu, C.-Z. Li, Z. Zhang, W. Qiu, M. Shi, P. Heremans, A. K.-Y. Jen and H. Chen, J. Am. Chem. Soc., 2016, 138, 2528–2531 CrossRef CAS PubMed.
- F. Cherioux and L. Guyard, Adv. Funct. Mater., 2001, 11, 305–309 CrossRef CAS.
- X.-Y. Cao, W.-B. Zhang, J.-L. Wang, X.-H. Zhou, H. Lu and J. Pei, J. Am. Chem. Soc., 2003, 125, 12430–12431 CrossRef CAS PubMed.
- M. Rose, N. Klein, I. Senkovska, C. Schrage, P. Wollmann, W. Böhlmann, B. Böhringer, S. Fichtner and S. Kaskel, J. Mater. Chem., 2011, 21, 711–716 RSC.
- F. M. Wisser, K. Eckhardt, D. Wisser, W. Böhlmann, J. Grothe, E. Brunner and S. Kaskel, Macromolecules, 2014, 47, 4210–4216 CrossRef CAS.
- Y.-C. Zhao, D. Zhou, Q. Chen, X.-J. Zhang, N. Bian, A.-D. Qi and B.-H. Han, Macromolecules, 2011, 44, 6382–6388 CrossRef CAS.
- Y. Zou, T. Yuan, H. Yao, D. J. Frazier, D. J. Stanton, H.-J. Sue and L. Fang, Org. Lett., 2015, 17, 3146–3149 CrossRef CAS PubMed.
- M. D. Gernon, M. Wu, T. Buszta and P. Janney, Green Chem., 1999, 1, 127–140 RSC.
- S. Yuan, B. Dorney, D. White, S. Kirklin, P. Zapol, L. Yu and D.-J. Liu, Chem. Commun., 2010, 46, 4547–4549 RSC.
- A. C. de Souza, A. T. N. Pires and V. Soldi, J. Therm. Anal. Calorim., 2002, 70, 405–414 CrossRef CAS.
- S. Kitagawa, Angew. Chem., Int. Ed., 2015, 54, 10686–10687 CrossRef CAS PubMed.
- S. M. Kuznicki, V. A. Bell, S. Nair, H. W. Hillhouse, R. M. Jacubinas, C. M. Braunbarth, B. H. Toby and M. Tsapatsis, Nature, 2001, 412, 720–724 CrossRef CAS PubMed.
- D. Shetty, I. Jahovic, J. Raya, F. Ravaux, M. Jouiad, J.-C. Olsen and A. Trabolsi, J. Mater. Chem. A, 2017, 5, 62–66 CAS.
- R.-X. Yang, T.-T. Wang and W.-Q. Deng, Sci. Rep., 2015, 5, 10155 CrossRef CAS PubMed.
- C. Gu, N. Huang, Y. C. Chen, L. Qin, H. Xu, S. Zhang, F. Li, Y. Ma and D. Jiang, Angew. Chem., Int. Ed., 2015, 54, 13594–13598 CrossRef CAS PubMed.
- M. F. Jimenez-Solomon, Q. L. Song, K. E. Jelfs, M. Munoz-Ibanez and A. G. Livingston, Nat. Mater., 2016, 15, 760 CrossRef CAS PubMed.
- Y. Chen, S. Li, X. Pei, J. Zhou, X. Feng, S. Zhang, Y. Cheng, H. Li, R. Han and B. Wang, Angew. Chem., Int. Ed., 2016, 55, 3419–3423 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7qm00485k |
| ‡ These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2018 |