
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Degradable high Tg sugar-derived polycarbonates from isosorbide and dihydroxyacetone†
Daniel
Hult
 ,
Sandra
García-Gallego
,
Tobias
Ingverud
,
Oliver C. J.
Andrén
and
Michael
Malkoch
*
,
Sandra
García-Gallego
,
Tobias
Ingverud
,
Oliver C. J.
Andrén
and
Michael
Malkoch
*
KTH Royal Institute of Technology, Department of Fibre and Polymer Technology, Teknikringen 56-58, SE-100 44, Stockholm, Sweden. E-mail: malkoch@kth.se
First published on 14th March 2018
Abstract
Polycarbonates from isosorbide and dihydroxyacetone (DHA) have been synthesised using organocatalytic step-growth polymerization of their corresponding diols and bis-carbonylimidazolide monomers. By the choice of the feed ratio and monomer activation, either isosorbide or ketal protected DHA, random and alternating poly(Iso-co-DHA) carbonates have been formed. Thermal properties by DSC and TGA were herein strongly correlated to the monomer composition. Dilution studies using 1H-NMR spectroscopy of a model compound DHA-diethyl carbonate in CD3CN and D2O highlighted the influence of α-substituents on the keto/hydrate equilibrium of DHA. Further kinetics studies in the pH* range of 4.7 to 9.6 serve to show the hydrolytic pH-profile of DHA-carbonates. The hydrolytic degradation of deprotected polymer pellets shows an increased degradation with increasing DHA content. Pellets with a random or alternating configuration show different characteristics in terms of mass loss and molecular weight loss profile over time.
Introduction
Aliphatic poly(carbonate)s (APCs) are still considered as an unexplored family of polymers. This has mainly been due to their slow degradability and low glass-transition temperatures (Tg). In comparison with their aromatic analogues, the inherent properties of APCs are today seen as attractive features for biomedical applications.1,2 In an effort to expand the uses of APCs, researchers are currently focusing on the sustainable development of poly(carbonate)s through the synthesis of renewable monomers and polymers with degradable features.3,4 From a biomedical point of view, poly(carbonate)s exhibit appealing degradation properties. Unlike poly(ester)s, APCs do not cause a local lowering of pH, which can be problematic for pH-sensitive drugs and the surrounding tissue.2 The degradation of APCs occurs by surface erosion in vivo rather than the bulk degradation behaviour exhibited by poly(ester)s.2In another context, isosorbide is an attractive building-block being a rigid, chiral, non-toxic and renewable replacement for petrochemical monomers such as bis-phenol A.5–7 It is a sugar-based diol derived from the hydrolysis of starch, sucrose or cellulose, and has an annual production in tens of thousand tons.5 Isosorbide produces polymers with a high Tg and great mechanical and optical properties.6 Efforts to make poly(isosorbide) carbonates degradable have been made through the incorporation of di-acidic co-monomers to produce poly(ester-carbonate)s capable of enzymatic degradation when used with a poly(ester) content higher than 50%.6 An alternative approach to increase the degradability of various co-polymers is reported in a pioneering study by Putnam and co-workers,8 using the sugar-derivative dihydroxyacetone (DHA), a metabolic intermediate in the glycolysis cycle.4 DHA can be synthesized by the selective oxidation of glycerol, an abundant waste product from biodiesel production.9 The multiple forms of DHA in solution, together with the high reactivity of its ketone, limit the number of available polymerization conditions. Nevertheless, successful synthetic procedures have been reported towards poly(carbonate)s and poly(carbonate-ester)s.4 Among them, the semi-crystalline poly(DHA) carbonate has proven difficult to process given its high melting point coinciding with the degradation temperature and it required co-monomers such as caprolactone and lactide (LA) to decrease the melting temperature.8,10 In terms of biodegradability, the presence of DHA in several materials – including random co-polymers of LA-DHA, alternating aliphatic poly(ester)s, block co-polymers with DHA and poly(ethylene)glycol (PEG) –can be related to their degradation profile.8,11–13 Surprisingly, to date little is known in regard to the mechanism behind the improved hydrolytic degradation exhibited by DHA.3
Our group recently reported on a procedure to generate and tailor the final configuration of APCs from bis-carbonylimidazolide monomers of 1,3- and higher diols.14 The monomers were attained on multi-gram scales through the activation of diols by 1,1′-carbonyldiimidazole (CDI), which readily precipitate out of solution and can be isolated by simple filtration. The resulting monomers were then polymerized through Fluoride Promoted Carbonylation (FPC) step-growth polymerization to afford a library of functional APCs. During the development of FPC polymerization, a competing ring-closing depolymerization mechanism was observed, which resulted in scrambling and in some cases an under-representation of 1,3-diols in the final polymer. This phenomenon was especially prevalent for the less nucleophilic diol isosorbide, with a final composition of 76/24 isosorbide to 2,2-dimethylpropane-1,3-diol. Considering the scrambling phenomenon as an advantage, it is apparent that random co-polymers can be achieved with the similarity of what is achieved with ring-opening polymerization (ROP), using step-growth polymerization from simpler and more easily scalable monomers.
In the present study, we tailor the degradability of isosorbide poly(carbonate)s through their copolymerization with DHA. High Tg amorphous poly(carbonate)s are accomplished that can generate high-modulus materials that degrade in neutral to weakly alkaline solutions. A thorough analysis on the pH-profile of model compound degradation by 1H-NMR spectroscopy, together with a polymer degradation study by SEC and gravimetric measurements, serves to shed some light on the behaviour of this class of copolymers.
Experimental
Materials
1,1′-Carbonyldiimidazole (CDI), isosorbide (Iso) and dihydroxyacetone (DHA) were purchased from Carbosynth (Berkshire, UK). Isosorbide (98%) was purified by re-crystallization from acetone prior to use in polymerizations. All other chemicals were purchased from Sigma-Aldrich (St Louis, MO, USA) and used without further purification. HPLC grade solvents were used for all synthetic procedures, and dried over vacuum/heat-activated molecular sieves (3 Å) prior to use. Diazabicycloundecene (DBU) was dried and stored over 3 Å molecular sieves. All glassware and magnets were flame dried under vacuum and filled with argon prior to use. Dihydroxyacetone dimethylacetal ((MeO)2DHA) (1)15 and isosorbide-biscarbonylimidazolide (Iso-bis-Im) (2)14 were prepared according to published protocols, and complete structural characterization is reported in the ESI.†Methods
NMR ( 1 H and 13 C) experiments were performed on a Bruker Avance 400 (100) MHz NMR instrument. 1H-NMR spectra were referenced to the residual solvent peak of (CD3)2SO δ 2.50, CDCl3δ 7.26 and D2O δ 4.79, and 13C-NMR spectra to (CD3)2SO δ 39.52 and CDCl3δ 77.16. Quantitative 13C-NMR spectroscopy was performed with 200 mg sample in 0.8 mL (CD3)2SO with 20 mM chromium(III) acetylacetonate and a relaxation time of 10 s. 2D-NMR techniques including COSY, HSQC, HMBC, DEPT and NOESY were used to assign 1H and 13C shifts.Size exclusion chromatography (SEC) was carried out on a TOSOH EcoSEC HLC-8320 GPC system equipped with an EcoSES RI detector and three columns from PSS GmbH (PSS PFG 5 μm; Microguard, 100 Å and 300 Å). A mobile phase of DMF with 0.01 M LiBr (0.2 mL min−1) at 50 °C was used together with a linear polymethylmethacrylate (PMMA) calibration method.
Differential scanning calorimetry (DSC) was performed on a Mettler Toledo DSC820, at a heating/cooling rate of 10 °C min−1 under a nitrogen flow (50 mL min−1). The glass transition temperature (Tg) was selected as the midpoint of transition on the second heating cycle (20 °C to 200 °C).
Thermogravimetric analysis (TGA) was performed on a Mettler Toledo TGA/DSC1, using the STARe software to process the data. A sample of 5 mg was placed in a 70 μL ceramic crucible and heated from 30 °C to 500 °C at a rate of 10 °C min−1 under a nitrogen flow (50 mL min−1).
Synthesis of compounds
Polymerization of poly(Iso-co-(MeO)2DHA)
All polymerizations were performed in 50–100 mL round-bottom flasks, fitted with magnetic stir bars and flame dried under vacuum. Varying feed ratios of (MeO)2DHA (1), bis-carbonylimidazolide (2 or 4) and isosorbide (3) were added in powder form (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 eq. diol to bis-carbonylimidazolide), sealed and cycled with vacuum/argon three times. Stock solutions of DBU and solvent (CHCl3 or CH3CN) were dried overnight on vacuum/heat-activated 3 Å molecular sieves, and then added to the reaction vessel using a syringe. Final catalytic loading was 1 mol% DBU with regard to bis-carbonylimidazolide. Polymerisations in solvent were either conducted at room temperature or at 50 °C under magnetic stirring for 5 h to 16 h. Neat polymerization was conducted at 100 °C to 120 °C, by adding the catalyst directly to the reaction vessel and reacting for 3 h to 4 h. The DBU was subsequently quenched with 5 eq. of acetic acid and precipitated from CHCl3 into methanol, forming a white powder precipitate, in scales of 2 to 10 g and yields of 87–97%. δH (400 MHz; CDCl3) 5.05–5.11 (2H, br m, H7 and H10), 4.89 (1H, br t, H9), 4.53 (1H, br d, H8), 4.29–4.17 (4H, br m, CH2 1 and 3), 4.10–3.97 (2H, br m, CH2 6), 3.96–3.85 (2H, br m, CH2 11), 3.27 (6H, br s, CH3 4). δC (100 MHz; CDCl3) 154.1–153.2 (m, O(CO)O), 98.3 (C2), 85.8 (C8), 81.6 (C7), 81.1 (C9), 77.3 (C10), 73.2 (C6), 70.6 (C11), 63.3 (CH2(CO) CH2 1 and 3), 48.6 (CH3 4).
:1 eq. diol to bis-carbonylimidazolide), sealed and cycled with vacuum/argon three times. Stock solutions of DBU and solvent (CHCl3 or CH3CN) were dried overnight on vacuum/heat-activated 3 Å molecular sieves, and then added to the reaction vessel using a syringe. Final catalytic loading was 1 mol% DBU with regard to bis-carbonylimidazolide. Polymerisations in solvent were either conducted at room temperature or at 50 °C under magnetic stirring for 5 h to 16 h. Neat polymerization was conducted at 100 °C to 120 °C, by adding the catalyst directly to the reaction vessel and reacting for 3 h to 4 h. The DBU was subsequently quenched with 5 eq. of acetic acid and precipitated from CHCl3 into methanol, forming a white powder precipitate, in scales of 2 to 10 g and yields of 87–97%. δH (400 MHz; CDCl3) 5.05–5.11 (2H, br m, H7 and H10), 4.89 (1H, br t, H9), 4.53 (1H, br d, H8), 4.29–4.17 (4H, br m, CH2 1 and 3), 4.10–3.97 (2H, br m, CH2 6), 3.96–3.85 (2H, br m, CH2 11), 3.27 (6H, br s, CH3 4). δC (100 MHz; CDCl3) 154.1–153.2 (m, O(CO)O), 98.3 (C2), 85.8 (C8), 81.6 (C7), 81.1 (C9), 77.3 (C10), 73.2 (C6), 70.6 (C11), 63.3 (CH2(CO) CH2 1 and 3), 48.6 (CH3 4).
Deprotection of poly(Iso-co-(MeO)2DHA)
Deprotection of dimethyl ketal was performed using trityl tetrafluoroborate Ph3CBF4 (1 eq.) in CH2Cl2.10,17 To a round-bottom flask fitted with a magnetic stir bar, 300 mL of CH2Cl2, poly(Iso-co-(MeO)2DHA) (3.00 g, 8.98 mmol ketal units), Ph3CBF4 (2.96 g, 8.98 mmol, 1 eq. per (MeO)2DHA repeating unit) and H2O (180 μL, 8.98 mmol) were added. The reaction was allowed to be stirred at room temperature for 1 h. The solvent was subsequently removed in vacuo and purified by precipitation into methanol, affording poly(Iso-co-DHA) as a white powder (2.1 g, 82%). δH (400 MHz; (CD3)2SO) 5.07 (1H, br m, H6), 5.01 (1H, br m, H9), 4.97 (4H, br m, CH2 1 and 3), 4.85 (1H, br m, H7), 4.78 (1H, br m, H8), 3.99 (1H, br m, H10), 3.85 (3H, br m, H5, H5′ and H10′). δC (100 MHz; (CD3)2SO) 197.6 (C2), 153.5–153.2 (C4, C4′), 85.2 (C8), 81.2 (C9), 80.6 (C7), 77.3 (C6), 72.3 (C10), 70.3 (C5), 68.8 (C1 and C3).pH-Titration of the model compound (A)
For the hydrolysis study of the model compound DHA-diethyl carbonate (A), acetate and phosphate buffers were prepared in D2O between pH* 3.8 and 9.0, where the pH* notation refers to the pH value recorded using a standard glass electrode pH-probe in D2O. This is called the “cancel-out” approach, whereby the constant term (pD = pH* + 0.4) used to convert the pH to pD is assumed to cancel out the increase in the pKa of acids in D2O.18,19 Due to the limited solubility of A in D2O, experiments were carried out in a mixture of D2O and CD3CN. As a consequence of this, a second assumption was made, where it has been shown that pH scales linearly with the volume fraction of MeCN (ϕMeCN) added to an aqueous buffer through the function mpH (eqn (1), see the ESI†).20| swpH − wwpH = mpHϕMeCN | (1) |
Degradation study
For each material, 100 mg of a finely ground polymer was pressed into a pellet and cut into four equal pieces of approximately 25 mg. The polymer samples were then submerged into 50 mM phosphate buffer (pH 8.0) and incubated at 37 °C for a week. Duplicates of the samples were isolated at different time intervals, lyophilized and analysed both gravimetrically and by SEC.Results and discussion
Synthesis of poly(carbonate)s
With the aim of evaluating poly(Iso-co-DHA) carbonates and the effect the final architecture has on their properties, we relied on the step-growth polymerization of bis-carbonylimidazolides and diols to afford a library of rigid polycarbonates (Scheme 1 and Table 1). While the former study unfolded FPC chemistry using cesium fluoride (CsF) as an inorganic polymerization catalyst,14 herein we evaluate the highly potent organocatalyst DBU commonly used for ring-opening polymerization.21 In this regard, DBU was chosen in an effort to maximize the incorporation of DHA 1,3-diol into the final polymers. Kinetic experiments for the polymerization of bis-carbonylimidazolide and diols showed a satisfactory rate increase with DBU concentration, indicating that it is a viable catalyst (Fig. S1†). Polymerization reactions were carried out with a catalytic loading of 1 mol% with respect to diol, in either solvent (1.0 M monomer) or under neat conditions. Compared to CsF used in the previous study, polymerizations with DBU behaved in a similar manner, as seen for polymers I and II (Table 1). DHA's propensity to cyclize was most apparent in polar solvents such as acetone and acetonitrile (Fig. S2†). A similar behaviour was also observed in the case of high isosorbide content polymer VI at an elevated temperature (50 °C) in chloroform.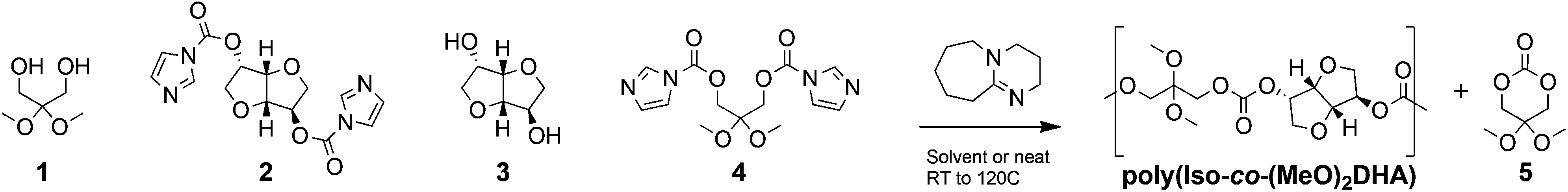 | ||
| Scheme 1 Step-growth polymerization using monomers 1 to 4 to form poly(Iso-co-(MeO)2DHA) carbonates, with the formation of cyclic monomer 5. | ||
| Polymer | Solventa | Monomers | Time (h) | Cyclization (5)e (%) | DHA feed/finalf (%) | M n (g mol−1) | Đ | Yield (%) |
|---|---|---|---|---|---|---|---|---|
|
a Unless otherwise stated, reactions were carried out at room temperature using DBU (1 mol%) as a catalyst.
b Reaction was carried out at 120 °C using DBU as a catalyst.
c Reaction was carried out at 100 °C using CsF as a catalyst.
d Reaction was conducted at 50 °C.
e 1H-NMR integration of δ 4.28 and 3.31 ppm.
f 1H-NMR integration of the Iso-segment at δ 4.88 and the DHA-segment at 4.21 ppm.
|
||||||||
| I | Neatb | 1 + 2 | 4 | 6 | 50/48 | 9356 | 1.78 | 87 |
| II | Neatc | 1 + 2 | 3 | 8 | 50/47 | 7292 | 1.76 | 91 |
| III | (CH3)2CO | 1 + 2 | 16 | 11 | 50/45 | 4556 | 1.53 | 87 |
| IV | CH3CN | 1 + 2 | 16 | 10 | 50/45 | 9231 | 1.99 | 90 |
| V | CHCl3 | 1 + 2 | 16 | 4 | 50/48 | 7857 | 1.83 | 97 |
| VI | CHCl3d |
1 + 2 + 3 | 4 | 10 | 25/21 | 5378 | 1.84 | 94 |
| VII | CHCl3 | 3 + 4 | 16 | 2 | 50/50 | 7153 | 1.65 | 92 |
| VIII | CHCl3 | 1 + 3 + 4 | 16 | 4 | 75/74 | 5790 | 1.49 | 88 |
A closer look at the configuration of polymers using 13C-NMR spectroscopy reveals significant scrambling when using monomer 1 (Fig. 1). Dyad analysis of carbonyls with 13C-NMR spectra indicates that the reaction of monomers 3 and 4 to generate polymer VII shows a strict alternating sequence with two observed shifts at δ 154.0 and 153.8 ppm, corresponding to the endo- and exo-configuration of isosorbide. However, the integration of carbonyl shifts using the inverse-gated decoupling 13C-NMR spectra of polymers V and VI, accomplished from monomers 1, 2 and 3, indicates statistically random co-polymers (Table S1†). This competing depolymerization mechanism was observed for both catalysts when a terminal 1,3-diol was present in the system.14 The highest degree of incorporation of residual six-membered monomer 5 was observed when using chloroform. With this in mind, the final properties of polymers can be tailored to fit the specific needs of the application.
 | ||
| Fig. 1 13C-NMR spectra of the carbonyl region for poly(Iso-co-(MeO)2DHA) showing the different dyad combinations based on the monomer feed. Reported values for poly(isosorbide) (■).14 | ||
Deprotection of methyl-ketal in poly(Iso-co-(OMe)2DHA) was evaluated through the previously reported acetal deprotection protocols.10,15 Mixtures of TFA and water led to significant degrees of deprotection with concomitant hydrolysis as reported by Putnam and co-workers.15 The most satisfactory results were achieved with trityl tetrafluoroborate Ph3CBF4 and water (1:1) in dichloromethane.10,17 Trityl-deprotection limited the acid-catalyzed hydrolysis of carbonates and showed the visual precipitation of the polymer with increasing DHA-content. This procedure afforded 80–90% deprotection, confirmed by 1H-NMR and 13C-NMR spectra. After precipitation into cold methanol, poly(Iso-co-DHA) carbonates were isolated as glassy white powders with Mn in the range of 4500–9350 and PDI values in the range of 1.49–1.99.
Thermal properties
Thermal analysis of the co-polymers revealed the stark effect each sugar-derived monomer has on the final performance of the material. The keto-functional DHA and the bicyclic isosorbide provide a roughly a 100 °C difference in Tg for the two homo-polymers, 68 °C and 169 °C respectively.8,14 Differential scanning calorimetry revealed that all co-polymers were amorphous with a single glass transition. Deprotection of poly(Iso-co-(OMe)2DHA) led to an increase in Tg. For the deprotected poly(Iso-co-DHA) carbonates, an increasing trend in Tg was observed in the range from 80 °C to 127 °C, going from 25% (VIII-d) to 75% (VI-d) isosorbide content (Fig. 2 and Table 2). Deprotected polymers showed some instability at temperatures above the Tg which is consistent with thermal degradation. This was further verified by thermogravimetric analysis.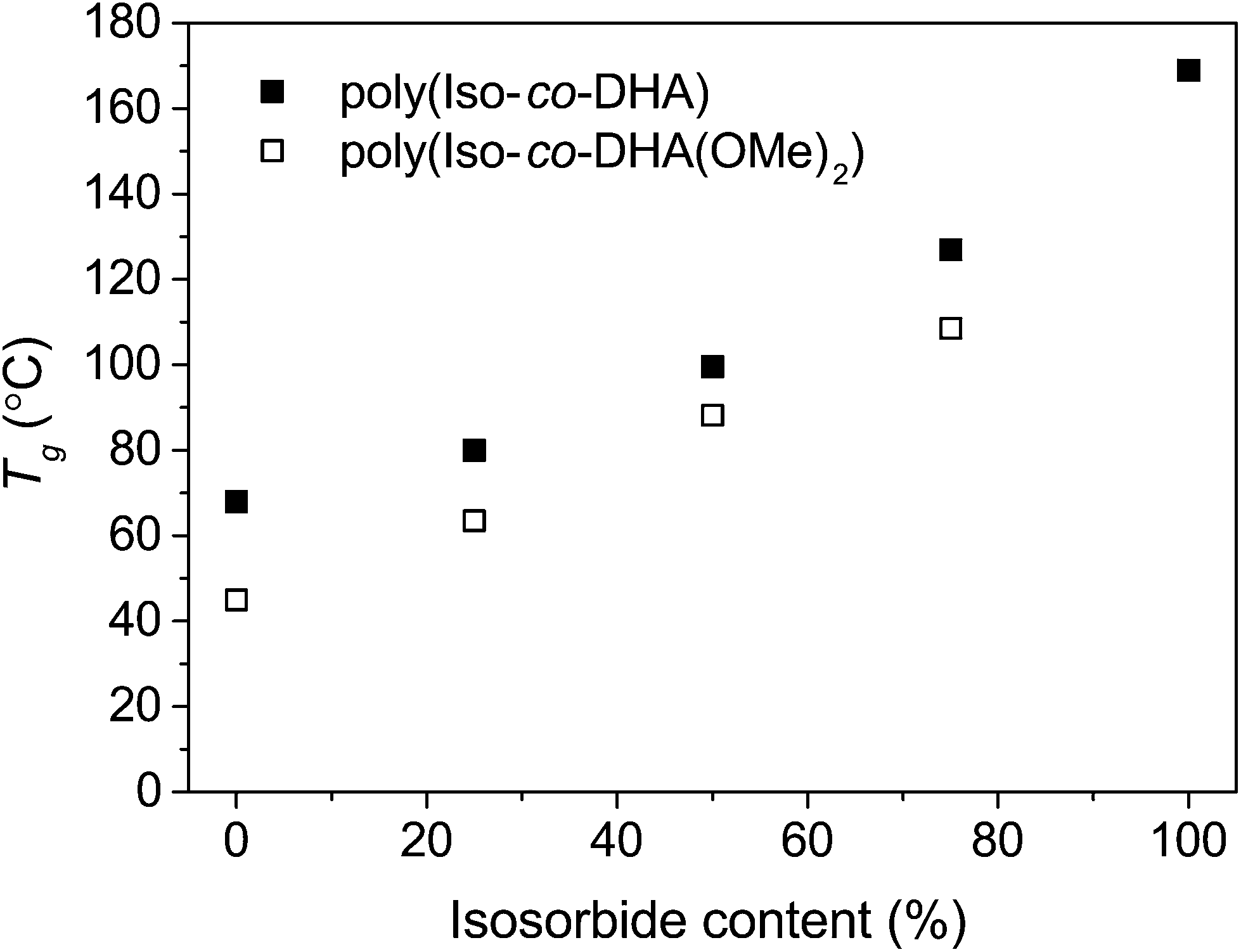 | ||
| Fig. 2 Glass-transition temperatures (Tg) as a function of isosorbide content for ketal protected (□) and deprotected polymers (■). Reported values for homo-polymers poly(DHA)8 and poly(isosorbide).14 | ||
| Polymera | Iso/DHA | M n (kg mol−1) | Đ | T g (°C) | T d-5% (°C) | T d-max (°C) |
|---|---|---|---|---|---|---|
| a “d” indicates deprotected polymer. b Referenced values for poly(isosorbide).14 | ||||||
| IX | 100/0 | 20.4 | 1.9 | 169 | 336 | 379 |
| VI-d | 75/25 | 7.6 | 1.8 | 127 | 268 | 380 |
| V-d | 50/50 | 4.4 | 1.6 | 100 | 194 | 288/368 |
| VII-d | 50/50 | 4.5 | 1.7 | 101 | 251 | 237 |
| VIII-d | 25/75 | 2.0 | 1.5 | 80 | 191 | 285 |
In terms of thermal degradation, TGA revealed an increasing trend in the onset of degradation, Td-5% values (corresponding to 5% mass loss), with increasing isosorbide content (Fig. 3). Looking at the 75% DHA polymer (VIII-d) a sharper decrease was observed at lower temperatures leading to a maximum rate of degradation (Td-max) of 237 °C from the more thermally labile blocky DHA configuration. This is consistent with the results from Simon et al. for low crystallinity copolymers with ε-caprolactone and DHA, having lower Td-5% values for higher DHA content.10 For the 50/50 random co-polymer (V-d) a bimodal degradation behaviour was observed with two Td-max values at 288 °C and 368 °C (Fig. 3). This behaviour is presumably due to its random configuration yielding short blocks of the two monomers. A comparison between the random (V-d) and alternating (VII-d) (50/50) co-polymers indicated that opting for an alternating chain configuration increases Td-5% from 194 °C to 251 °C, which is consistent with a more thermally stable polymer (Fig. 3). The alternating co-polymer showed a similar onset to the isosorbide-rich polymer (VI-d) with a Td-5% at 268 °C, having a broad transition and a Td-max value similar to the homo-polymer of isosorbide (Table 2). The homo-polymer of isosorbide showed a single-step thermal decomposition process with a Td-5% at 336 °C, which is in good accordance with the literature values.6 With the intended goal of forming degradable and rigid poly(carbonate)s, the chiral isosorbide proves to be an excellent co-monomer to afford high Tg amorphous materials.
 | ||
| Fig. 3 TGA-thermograms for poly(isosorbide) and poly(Iso-co-DHA). | ||
Hydrolytic degradation study
Thermal data clearly showed a decrease in poly(carbonate) stability with the keto-functional DHA building-block. In terms of hydrolytic instability, ketones can be elusive as they often exist in more than one form in solution, be it with their enol-tautomer or in equilibrium with their hydrated form (gem-diol). Based on the previous findings on DHA-containing poly(ester)s and poly(carbonate)s, it is apparent that these polymers show a rate higher than the expected rate of hydrolysis.8,12 Neighboring group participation of β-carbonyls for the hydrolysis of esters has been discussed to a great extent in the literature.22–26 Several studies have shown that the intramolecular attack of hydrated β-carbonyls can significantly increase the rate of hydrolysis.26Hydrolysis of the model-compound DHA(EC)2
To better understand the behavior of DHA-containing poly(carbonate)s, the diethyl carbonate analogue of DHA (A) was studied using 1H-NMR spectroscopy in D2O and CD3CN (Fig. 4a). DHA is known to co-exist in its keto, hydrate and enol forms.27 When mixed in D2O/CD3CN both DHA (E) and DHA-(EC)2 (A) instantly form an equilibrium between the keto- and hydrate forms. The CH2 α-protons of the various DHA-forms in the 1H-NMR spectra show a downward shift of about δ 0.8 ppm between their keto- and hydrate forms.27 This is also observed in the case of diethyl carbonate A (δ 4.82 ppm) to B (δ 4.04 ppm), and DHA E (δ 4.26 ppm) to F (δ 3.42 ppm) (see the ESI† for assignments). No enol-form could be observed through 1H-NMR spectroscopy and this was further confirmed by HSQC and HMBC 2D-NMR analysis (assignments of NMR spectra can be found in the ESI†). | ||
| Fig. 4 (a) Keto/hydrate equilibria of the model compound (A/B) and degradation products monoethyl carbonate (C/D) and DHA (E/F). (b) Amount of hydrate as a function of CD3CN fraction in D2O (lines are included as a visual guide). Equilibrium constants in D2O/CD3CN (øCD3CN = 0.4, 17 mM) were found to be 0.80 (KAB), 0.26 (KCD) and 0.11 (KEF). | ||
In order to investigate the influence that the overall polarity of the medium has on the keto/hydrate equilibrium, a series of dilution tests were performed using D2O and CD3CN. Fig. 4b indicates that DHA (E), being predominantly water soluble, decreases its concentration upon dilution with CD3CN in D2O, going from a ratio of 4:1 as reported by Davis27 to a ratio of 9:1, affording the equilibrium constant KEF = [F]/[E] = 0.11, in D2O/CD3CN (øCD3CN = 0.4). The predominantly acetonitrile soluble DHA-(EC)2 (A) increases its concentration upon dilution with D2O in CD3CN, from the 100% keto-form in CD3CN to the 44% hydrate form in D2O/CD3CN (øCD3CN = 0.4), KAB = [B]/[A] = 0.80. The major difference between the two equilibria is the presence of two electron-withdrawing carbonate groups in diethyl carbonate (A), while DHA (E) contains two electron-donating hydroxyl groups, meaning that the carbons in A are significantly more electron-poor than those in E. The keto/hydrate equilibrium was not affected by alterations in the pH* range studied from 4.7 to 9.6, using 50 mM acetate- and phosphate-buffer solutions in D2O/CD3CN (øCD3CN = 0.4).
Degradation studies by 1H-NMR spectroscopy in D2O/CD3CN (øCD3CN = 0.4) revealed a third equilibrium constant between mono-ethyl carbonates C and D, KCD = [D]/[C] = 0.26. Under the conditions used, adding one carbonate to DHA doubles the concentration of hydrate in solution. On the other hand, having two electron-withdrawing carbonates (A) will quadruple the amount of hydrate formed in solution, as compared to DHA (E).
To investigate further which form of DHA causes rapid hydrolysis, kinetic studies were performed on the diethyl carbonate analogue (A) of DHA in the pH* range of 4.7 to 9.6. Kinetic data for the formation of ethanol fit well with a pseudo first-order reaction with increasing rates going from neutral to alkaline conditions (Fig. 5a), with a first order dependence on [OD−] (Fig. 5b). If the carbonate hydrolysis were to go through an intermediate enol-form, significant deuteration of CH2 protons in the α-position would be observed.28 This was not the case, suggesting that the seemingly hydrolytically unstable nature of these compounds likely stems from the hydrated form.
 | ||
| Fig. 5 pH-Profile for the hydrolysis of DHA-(EC)2 (A) in D2O/CD3CN (øCD3CN = 0.4). (a) Observed pseudo first-order rate constants vs. pH*, (b) log–log plot of observed pseudo first-order rate constants, showing a second-order overall rate constant kEtOH = 0.82 × 105 (M−1 h−1) for the formation of ethanol. | ||
For pH* in the range of 5.0 to 8.0, reactions conform well to an equilibrium approximation, meaning that the rates of the irreversible steps (k2 and k4, eqn (2)) can be seen as significantly slower than those of the reversible hydration reactions. This means that the kinetics can be seen as a two-step consecutive reaction (eqn (3)), where the concentration at any given point in time is given by [A]0 = [A + B]t + [C + D]t + [E + F]t, using the equilibrium constants (KAB, KCD and KEF) to solve for each individual component. At pH* values above 8.0, reactions deviated from equilibrium over time.
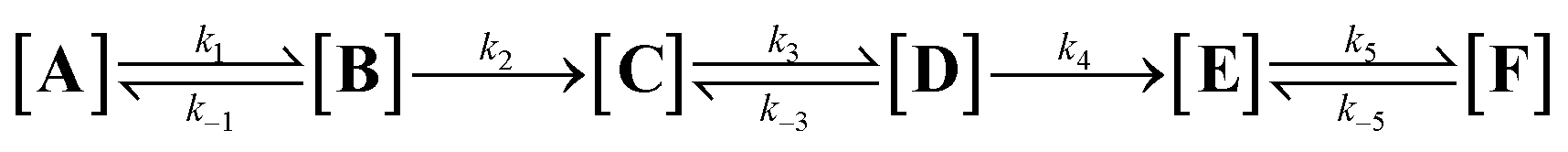 | (2) |
 | (3) |
The rate of the first irreversible step (k′) is taken from the rate consumption of diethyl carbonates A and B, with known initial concentrations [A]0 (see the ESI†). Using the observed rate constants (kobs for A and B), a second-order overall rate constant can be determined with respect to [OD−] and [A + B], k′ = 1.31 × 105 (M−1 h−1) (Fig. S3†). The second irreversible step was evaluated by fitting the integrated rate law to the experimental data (Fig. S4†). The best fit to the data suggests that the second step (k′′) has a slower rate than the first step (k′). This is consistent with the equilibrium of the mono-ethyl-carbonate being shifted towards the keto-form, making k′′ the rate limiting step in the formation of E.
Overall, these results would suggest that the hydrolytic instability is highly dependent on the ability of substrates to become hydrated. Neighbouring group participation has been showcased for poly(glycerol) carbonates, where the poly(1,2-glycerol) carbonate with a pendant primary hydroxyl group increases the degradation rates in solution.29,30 In a recent study by Ricapito et al. on poly(ethylene glycol) (PEG) and poly(DHA) carbonate hydrogels, it was shown that having an extra methylene group on each side, in the case of 1,5-dihydroxy-pentan-3-one, provided significantly slower degradation compared to hydrogels formed with PEG and DHA-carbonates.12 As the dilution study suggested, decreasing the polarity of the surrounding medium and decreasing the electron-withdrawing effect on the α-carbons limit the ability to form hydrates, which subsequently affects the hydrolytic degradation of the compound. In the polymer case, being confined to a hydrophobic micro-environment together with limited conformational mobility, chemical degradation rates are expected to be less pronounced. The hydrolytic degradation of the polymers is assessed in the following section.
Hydrolytic degradation of poly(Iso-co-DHA) polycarbonates
Based on the insights gained from model compound studies, further degradation investigations were carried out on polymers in their solid state. Previous studies on LA-DHA co-poly(ester-carbonate)s have indicated an increased degradation behaviour with increasing DHA content, with more surface-like erosion characteristics.8 Pellets of co-polymers poly(Iso-ran-DHA) with varying DHA contents from 25% to 75% (V-d, VI-d and VIII-d) and the alternating version of poly(Iso-alt-DHA) containing 50% DHA (VII-d) were exposed to an accelerated aging study in 50 mM phosphate buffer at pH 8.0 and 37 °C. Being first order in [OH−], going from physiological pH 7.4 to 8.0, the chemical degradation rates are expected to increase by a factor of 4.Within 1 day the 75% DHA containing (VIII-d) poly(Iso-co-DHA) had lost most of its mass (Fig. 6a). After 5 hours of immersion in aqueous buffer VIII-d became insoluble in DMF, affording swollen transparent gel-like pieces. The random poly(Iso-ran-DHA) 50% (V-d) and 25% (VI-d) DHA containing pellets remained physically intact with low mass loss over the first 2 days. The 50% co-polymer V-d showed a faster trend in terms of molecular weight loss compared to its 25% (VI-d) counterpart, indicating a traditional surface erosion type of degradation.2,6
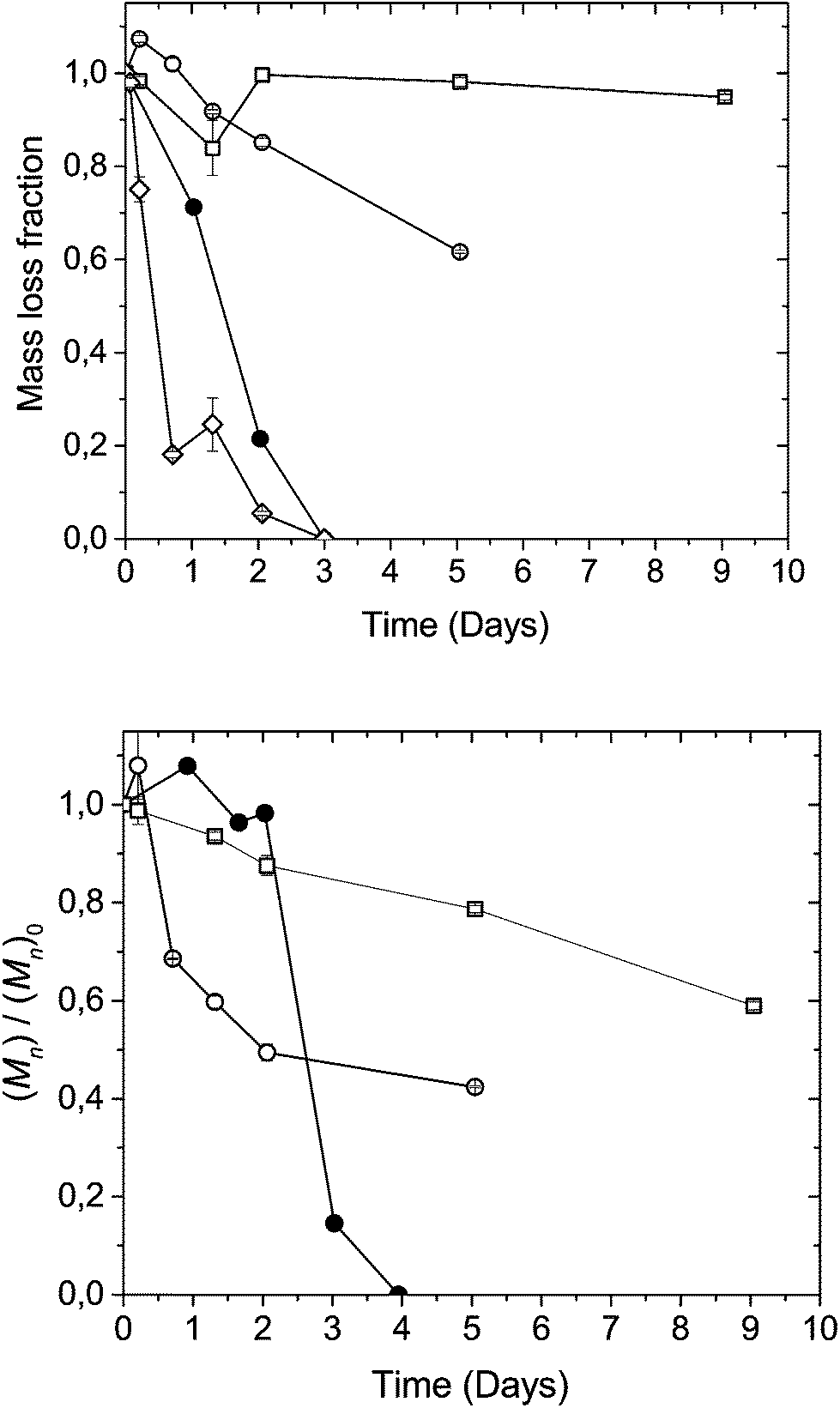 | ||
| Fig. 6 Fractional mass loss (a) and Mn loss by SEC (b) for the hydrolytic degradation of poly(Iso-co-DHA); VI-d 75–25 (□), V-d 50–50 (○), VIII-d 25–75 (◊), and the alternating VII-d 50–50 (●), in 50 mM phosphate buffer at pH 8 and 37 °C. | ||
The alternating poly(Iso-alt-DHA) (VII-d) displayed a different type of degradation behaviour. Pellets of VII-d showed little molecular weight loss in the first 2 days (Fig. 6a). However, pellets showed continuous mass loss, followed by rapid degradation upon swelling in the buffer solution. After 2 days the pellets had completely disintegrated, presumably due to increases in stress and crack formation due to water uptake, leading to increased plasticization and rapid hydrolysis. Endo and co-workers have reported radical differences in the solubility of polystyrene with pendant tri-carbonyls upon the formation of its hydrate form.31 The alternating analogue (VII-d) consists of one type of carbonate linkage, namely from DHA to isosorbide, with a more limited conformational mobility compared to its blocky random counterpart (V-d). The alternating analogue should thus be less prone to an intramolecular nucleophilic attack, given that each DHA-unit is anchored to the more rigid isosorbide. The random co-polymers contain three types of carbonates: DHA–DHA, DHA–isosorbide and isosorbide–isosorbide (Fig. 1). These three types of linkages are expected to have different probabilities of hydrolysis by chemical degradation. The DHA–DHA carbonate linkages should be more prone to random chain-scission, having greater conformational mobility and two adjacent β-carbonyls per carbonate, according to what was observed related to thermal stability in TGA measurements (Fig. 3). Previous studies have shown that DHA polycarbonates pose high surface energy, with an especially high polar contribution, presumably from the ketone.15 These findings would suggest that a higher DHA content provides a more polar microenvironment. Comparing the high isosorbide content polymer VI-d (25% DHA) with the alternating VII-d (50% DHA), with little to no repeating DHA–DHA carbonates, it would seem that the higher isosorbide content co-polymer is less prone to water uptake, as can be seen in Fig. 6, both in terms of mass loss and molecular weight changes over time. The results suggest that not only does the amount of the labile DHA monomer affect the degradation behavior of the final polymer construct, but also that the distribution along the polymer chain will greatly affect water-uptake as a consequence. Furthermore, scrambled polymers with dyad sequences similar to those achieved by ROP protocols show a greater stability over time in a weakly alkaline buffer.8,32
Conclusions
Rigid, amorphous and degradable poly(carbonate)s have been produced from the sugar-based isosorbide and the sugar metabolic derivative dihydroxyacetone (DHA). In order to tailor the final properties, the bi-cyclic chiral isosorbide diol proved to be excellent for achieving high Tg amorphous co-polymers. The keto-functional DHA was also shown to provide the hydrolytic instability necessary to make the poly(carbonate)s degradable. Hydrolytic instability presented by the DHA co-monomer likely stems from carbonates having β-carbonyl neighbouring groups, capable of forming hydrates. Model compound studies indicate that ketones with two adjacent carbonates show a large degree of hydration, with the loss of one carbonate leading to the equilibrium being shifted towards the keto form. The pH-profile of the hydrolysis reaction indicates that it is first order in [OH−] affording substantial hydrolysis going from neutral to weakly alkaline conditions. In terms of polymer degradation of solid pellets, the rate of random chain-scission increases with DHA content, providing degradation rates greater than conventional poly(carbonate)s and even poly(ester)s. Changing the monomer configuration had a marked effect on the degradation profiles, as the alternating configuration indicated little initial chain scission, with greater initial water uptake. This behaviour may be attributed to the fact that chains have limited mobility, with an even distribution of the polar DHA, capable of forming hydrates along the polymer. Rapid hydrolysis was only observed after the pellets had swollen and disintegrated. The co-polymers containing more blocky regions of isosorbide showed less initial mass loss due to water uptake followed by a gradual molecular weight loss over time. These rigid, amorphous and degradable poly(carbonate)s are seen as good candidates for the bone restoration field. In this field, it is necessary to provide initial mechanical support and ideally with high-modulus properties that match the surrounding mineral tissue followed by induced macro-porosity for potential tissue ingrowth.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was generously supported by the Swedish Research Council VR (2011-5358, 2010-435 and 2015-04779) and Knut and Alice Wallenberg Foundation KAW (2012-0196). This project has received funding for Sandra García-Gallego from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie Grant Agreement No. 655649.Notes and references
- J. Xu, E. Feng and J. Song, J. Appl. Polym. Sci., 2014, 131, 39822 CrossRef.
- S. Tempelaar, L. Mespouille, O. Coulembier, P. Dubois and A. P. Dove, Chem. Soc. Rev., 2013, 42, 1312–1336 RSC.
- D. K. Schneiderman and M. A. Hillmyer, Macromolecules, 2017, 50, 3733–3749 CrossRef CAS.
- N. G. Ricapito, C. Ghobril, H. Zhang, M. W. Grinstaff and D. Putnam, Chem. Rev., 2016, 116, 2664–2704 CrossRef CAS PubMed.
- C. Dussenne, T. Delaunay, V. Wiatz, H. Wyart, I. Suisse and M. Sauthier, Green Chem., 2017, 19, 5332–5344 RSC.
- F. Fenouillot, A. Rousseau, G. Colomines, R. Saint-Loup and J. P. Pascault, Prog. Polym. Sci., 2010, 35, 578–622 CrossRef CAS.
- M. Rose and R. Palkovits, ChemSusChem, 2012, 5, 167–176 CrossRef CAS PubMed.
- J. R. Weiser, P. N. Zawaneh and D. Putnam, Biomacromolecules, 2011, 12, 977–986 CrossRef CAS PubMed.
- R. M. Painter, D. M. Pearson and R. M. Waymouth, Angew. Chem., Int. Ed., 2010, 49, 9456–9459 CrossRef CAS PubMed.
- J. Simon, J. V. Olsson, H. Kim, I. F. Tenney and R. M. Waymouth, Macromolecules, 2012, 45, 9275–9281 CrossRef CAS.
- J. N. Korley, S. Yazdi, K. McHugh, J. Kirk, J. Anderson and D. Putnam, Biomaterials, 2016, 98, 41–52 CrossRef CAS PubMed.
- N. G. Ricapito, J. Mares, D. Petralia and D. Putnam, Macromol. Chem. Phys., 2016, 217, 1917–1925 CrossRef CAS.
- P. N. Zawaneh, S. P. Singh, R. F. Padera, P. W. Henderson, J. A. Spector and D. Putnam, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 11014–11019 CrossRef CAS PubMed.
- J. V. Olsson, D. Hult, S. García-Gallego and M. Malkoch, Chem. Sci., 2017, 8, 4853–4857 RSC.
- A. N. Zelikin, P. N. Zawaneh and D. Putnam, Biomacromolecules, 2006, 7, 3239–3244 CrossRef CAS PubMed.
- M. Lecouvey, C. Dufau, D. El Manouni and Y. Leroux, Nucleosides Nucleotides, 1999, 18, 2109–2120 CAS.
- D. H. R. Barton, P. D. Magnus, G. Smith, G. Streckert and D. Zurr, J. Chem. Soc., Perkin Trans. 1, 1972, 542–552 RSC.
- K. H. Scheller, V. Scheller-Krattiger and R. B. Martin, J. Am. Chem. Soc., 1981, 103, 6833–6839 CrossRef CAS.
- A. Krȩżel and W. Bal, J. Inorg. Biochem., 2004, 98, 161–166 CrossRef.
- X. Subirats, M. Rosés and E. Bosch, Sep. Purif. Rev., 2007, 36, 231–255 CrossRef CAS.
- N. E. Kamber, W. Jeong, R. M. Waymouth, R. C. Pratt, B. G. G. Lohmeijer and J. L. Hedrick, Chem. Rev., 2007, 107, 5813–5840 CrossRef CAS PubMed.
- J. A. Walder, R. S. Johnson and I. M. Klotz, J. Am. Chem. Soc., 1978, 100, 5156–5159 CrossRef CAS.
- W. N. Washburn and E. R. Cook, J. Am. Chem. Soc., 1986, 108, 5962–5964 CrossRef CAS PubMed.
- M. L. Bender and M. S. Silver, J. Am. Chem. Soc., 1962, 84, 4589–4590 CrossRef CAS.
- F. Ramirez, B. Hansen and N. B. Desai, J. Am. Chem. Soc., 1962, 84, 4588–4588 CrossRef CAS.
- K. Bowden, Chem. Soc. Rev., 1995, 24, 431–435 RSC.
- L. Davis, Bioorg. Chem., 1973, 2, 197–201 CrossRef CAS.
- S. J. Reynolds, D. W. Yates and C. I. Pogson, Biochem. J., 1971, 122, 285–297 CrossRef CAS PubMed.
- H. Zhang and M. W. Grinstaff, J. Am. Chem. Soc., 2013, 135, 6806–6809 CrossRef CAS PubMed.
- J. Geschwind and H. Frey, Macromolecules, 2013, 46, 3280–3287 CrossRef CAS.
- T. Dei, K. Morino, A. Sudo and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 2245–2251 CrossRef CAS.
- J. R. Weiser, A. Yueh and D. Putnam, Acta Biomater., 2013, 9, 8245–8253 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8py00256h |
| This journal is © The Royal Society of Chemistry 2018 |