
Effective macrophage delivery using RAFT copolymer derived nanoparticles†
K. S.
Montgomery
ab,
R. W. M.
Davidson
a,
B.
Cao
bc,
B.
Williams
bc,
G. W.
Simpson
b,
S. K.
Nilsson
 bc,
J.
Chiefari
b and
M. J.
Fuchter
*a
bc,
J.
Chiefari
b and
M. J.
Fuchter
*a
aChemistry Department, Imperial College London, South Kensington Campus, SW7 2AZ, UK. E-mail: m.fuchter@imperial.ac.uk
bCSIRO Manufacturing, Bag 10, Clayton South, Victoria 3169, Australia
cARMI, Monash University, Clayton, Victoria 3800, Australia
First published on 28th November 2017
Abstract
Reversible addition fragmentation chain transfer (RAFT) polymerisation provides a highly controlled means to assemble copolymers of different architectures for a variety of applications, including drug delivery. Polymers consisting of a butyl methacrylate-co-methacrylic acid p(BMA-co-MAA) hydrophobic block and a poly(ethylene glycol) methyl ether methacrylate p(PEGMA-475) hydrophilic block were synthesised via RAFT polymerisation and self-assembled into micelles. A range of micelle particles of different sizes were obtained by varying the composition of the block copolymers. The micelles were crosslinked to form nanoparticles and fluorescently labelled to study cellular internalisation. The prepared nanoparticles were extensively taken up by primary murine macrophages and a promising candidate was identified. To demonstrate effective delivery of a cell impenetrable cargo a fluorescent dye, 4′,6-diamidino-2-phenylindole (DAPI), was encapsulated inside the nanoparticles and successfully delivered to macrophages. The nanoparticles’ stability at increased temperatures and at low concentrations, the tunability of their synthesis and their extensive internalisation by macrophages and performance makes them highly promising delivery vehicles for a range of therapeutics and imaging agents.
Introduction
The use of nanoparticles for cellular delivery of drugs and other agents is currently an extremely popular area of research, with targets ranging from tumour cells to inflamed tissue.1 The use of these vehicles enables the delivery of hydrophobic, insoluble, cell impenetrable, or toxic entities in a specific and controlled manner.2–4 Delivery to macrophages is of particular interest due to their role in the innate immune response, as well as their involvement in the development of cancerous tumours and many other diseases, including arthritis and AIDS.5–9 A number of different vehicles have been investigated for delivery to macrophages including liposomes, vesicles, dendrimers and micelle-based nanoparticles.10–14 Of note, as reported by Nardin et al. and Vyas et al., delivery vehicles have the ability to deliver drugs to macrophages with a much higher efficacy than when the drugs are administered alone.10,15,16Polymeric micelles, formed from amphiphilic block copolymers which self-assemble under aqueous conditions, can be crosslinked to give stable nanoparticles for drug delivery applications.14,17,18 To enable self-assembly in an aqueous environment block polymers need to have distinct hydrophilic and hydrophobic portions: the hydrophobic block forms the micelle core while the hydrophilic block forms the corona, resulting in a particle which is able to encapsulate lipophilic agents such as drugs or dyes.19–21 The required block copolymers are commonly synthesised via controlled radical polymerisation techniques, such as atom-transfer radical-polymerisation (ATRP), nitrox-mediated radical polymerisation (NMP) and RAFT, which give high control over polymer composition and therefore allow for the formation of micelles tailored to suit a wide range of applications.22–28
The ease of use and broad versatility of RAFT polymerisation, in terms of both reaction conditions and monomer choice, allows for the controlled synthesis of a large range of block copolymers with low dispersity.27,29,30 Since the development of the technique in 1998, RAFT has been used to make a vast variety of polymeric structures, in part due to the functional group tolerance of this method, which permits the use of a wide array of compatible functionalised monomers.31 Furthermore, RAFT is amenable to the use of automated parallel synthesisers which allow for the construction of large polymer libraries and the use of one-pot sequential polymerisation techniques to obtain quasi-block copolymers.32–35 Indeed, in many cases quasi-block copolymers are suitably similar in properties and applications to their pure block copolymer counterparts.34 For example, Guerrero-Sanchez et al. synthesised a library of quasi-block copolymers and found them to be very similar to equivalent ‘pure’ block copolymers when analysed using differential scanning calorimetry.34 Hinton et al. showed that quasi-triblock copolymers were able to deliver siRNA to cells more efficiently than pure triblock copolymers.36
Herein we report the design and synthesis of amphiphilic RAFT block copolymers, which can subsequently be self-assembled in water to give micelle particles. We detail the use of quasi-block polymerisation via RAFT to access polymers suitable for such self-assembly and crosslinking of the resultant micelles to give stable nanoparticles that exhibited extensive cellular internalisation by primary macrophages. As proof of principle, we show that these polymeric nanoparticles can be used to encapsulate a dye, which cannot normally penetrate living cells, and deliver it to macrophages.
Results and discussion
The design of the target copolymers is shown in Fig. 1. The aim of this work was to access block copolymers suitable for the development of nanoparticles for cellular delivery. The hydrophobic portion of the polymer consisted of a mixture of methacrylic acid (MAA) and butyl methacrylate (BMA), which are known to polymerise at a similar rate.37 The MAA within this block was used to endow acidic functionality to the nanoparticle core to promote the encapsulation of basic cargoes. The hydrophilic portion of the polymer was poly(ethylene glycol) methacrylate (PEGMA-475). Poly(ethylene glycol) (PEG) is commonly used in drug delivery systems as it is safe for in vivo use and the PEGMA-475 monomer was compatible with the RAFT agent used in this work (Fig. 1).1,3 Additional functionality was incorporated into the PEGMA-475 block to allow for (i) crosslinking of the nanoparticle's shell, to stabilise the particle once assembled, and (ii) fluorescent labelling, which enabled tracking of the nanoparticles in cell based studies.38–40 Specifically, an N-acryloxysuccinimide (NAS) monomer was selected for crosslinking – neighboring succinimide units were crosslinked using a diamine linker – and methacryloxyethyl thiocarbamoyl rhodamine B (MRhB) was selected as a suitable fluorescent label (Fig. 1).41 We note that upon internalisation of the nanoparticles, the diamine crosslinker may be susceptible to degradation by peptidases that are found in high levels in macrophages, thereby facilitating the efficient release of the cargo.42–44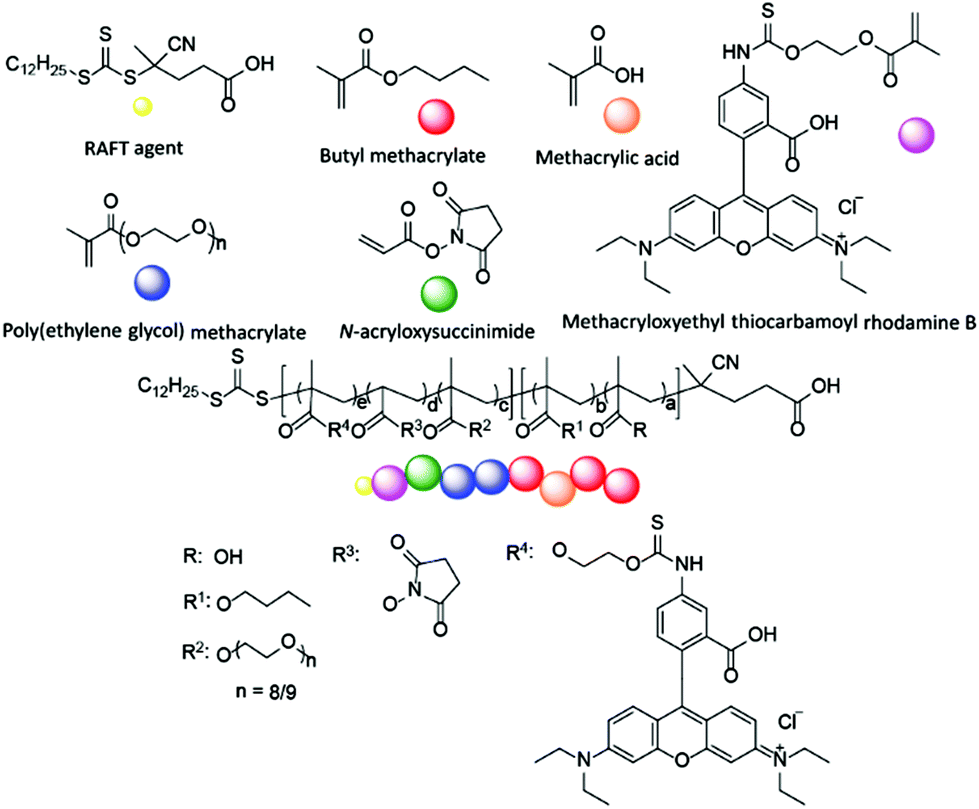 | ||
| Fig. 1 Design of block copolymers. a = 4–81, b = 27–198, c = 11–32, d = 3–10, e = 0.05. Square parentheses represent copolymer blocks. | ||
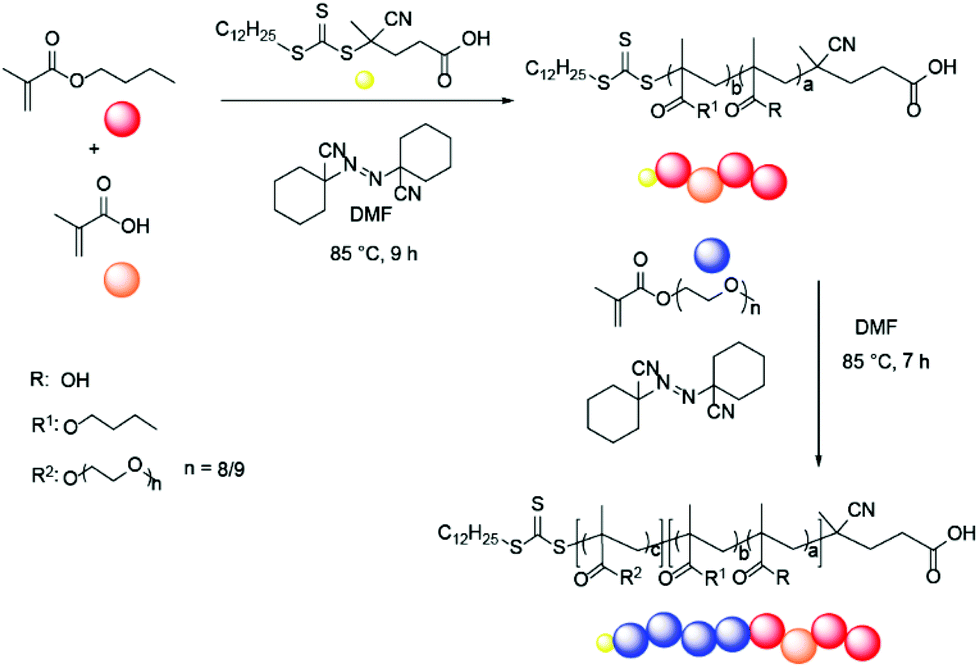
Polymer synthesis commenced with BMA and MAA copolymerisation, using varying ratios (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 4:1, 2:1) of BMA and MAA (Scheme 1 and Table 1). A range of initiators and RAFT agents were surveyed for their ability to access the target molecular weight copolymers with low dispersity. It was found that the methacrylate-compatible RAFT agent 4-cyano-4[(dodecylsulfanylthiocarbonyl)sulfanyl] pentanoic acid in combination with 1,1′-azobis(cyclohexanecarbonitrile) (ACHN) as an initiator was the optimal system (Scheme 1).45 Notably, the use of relatively short (9 h) reaction times, ACHN as the initiator and low MAA concentrations helped reduce the formation of dead polymer chains as evidenced by the dispersity index (Tables 1 and 2). The final BMA:MAA molar ratio in the copolymer was controlled by the comonomer feed ratio in the reaction solution. MAA conversion was generally found to be slightly lower than BMA conversion; by approximately 5–10%. The target molecular weight of the copolymer was determined by the stoichiometry of the RAFT agent and monomers in the reaction mixture.
:1, 4:1, 2:1) of BMA and MAA (Scheme 1 and Table 1). A range of initiators and RAFT agents were surveyed for their ability to access the target molecular weight copolymers with low dispersity. It was found that the methacrylate-compatible RAFT agent 4-cyano-4[(dodecylsulfanylthiocarbonyl)sulfanyl] pentanoic acid in combination with 1,1′-azobis(cyclohexanecarbonitrile) (ACHN) as an initiator was the optimal system (Scheme 1).45 Notably, the use of relatively short (9 h) reaction times, ACHN as the initiator and low MAA concentrations helped reduce the formation of dead polymer chains as evidenced by the dispersity index (Tables 1 and 2). The final BMA:MAA molar ratio in the copolymer was controlled by the comonomer feed ratio in the reaction solution. MAA conversion was generally found to be slightly lower than BMA conversion; by approximately 5–10%. The target molecular weight of the copolymer was determined by the stoichiometry of the RAFT agent and monomers in the reaction mixture.
 | ||
| Scheme 1 Quasi-block copolymerisation reaction, a = 2–10, b = 16–25, c = 6–9. Square parentheses represent copolymer blocks. | ||
| BMA:MAA ratio |
QB total Mn (ĐM) | M n of BMA/MAA block | M n of PEGMA block | Micelle diameter (nm) | PB total Mn (ĐM) | M n of BMA/MAA block | M n of PEGMA block | Micelle diameter (nm) |
|---|---|---|---|---|---|---|---|---|
| Molecular weight (Mn) and micelle size of quasi-block (QB) copolymers (black) compared to molecular weight and micelle size of pure block (PB) copolymers (blue). Mn values were calculated using end group analysis by 1H-NMR. ĐM values were determined via GPC (THF SEC). | ||||||||
| 9:1 |
5900 (1.12) | 3100 | 2800 | 16 | 7000 (1.12) | 3200 | 3800 | 42 |
| 9:1 |
8000 (1.17) | 3700 | 4300 | 26 | 7600 (1.18) | 3800 | 3800 | 40 |
| 4:1 |
6300 (1.12) | 2900 | 3400 | 25 | 6300 (1.19) | 3000 | 3300 | 27 |
| 2:1 |
6900 (1.23) | 3600 | 3300 | 24 | 5900 (1.17) | 3000 | 2900 | 25 |
| Polymers synthesised were either pure block (PB) or quasi-block (QB). Four different polymer block structures were investigated (Fig. 3). Polymer Mn and ĐM values were determined via GPC (THF SEC). Nanoparticle diameters were measured in PBS using DLS at 25 °C. Internalisation was measured by mean channel fluorescence (MCF) of macrophages. MCF values were normalised to allow a direct comparison between samples. Nanoparticle internalisation was also normalised with regards to sample rhodamine fluorescence (displayed value, details in ESI, Fig. S6 and Table S9).a Samples had 2 DLS peaks, indicating two different particle sizes, the main peak is displayed. Highlighted samples (green) were selected for further studies. |
|---|

|
Following synthesis of the p(BMA-co-MAA) block, the resultant copolymer was used as a macroRAFT agent and chain extended in the second polymerisation step to add the PEGMA-475 block (Scheme 1). ACHN was once again used as the initiator and the target molecular weight of the PEGMA-475 block was controlled by the ratio of macroRAFT agent to the monomer. Pure block and quasi-block polymerisation methods were surveyed for the formation of the target block copolymers, which are summarised in Table 1.
Micelles were generated from the target block copolymers, through self-assembly in water. Dynamic light scattering (DLS) and the pendant drop method were used to determine the critical micelle concentration (CMC) and therefore select the concentrations to be used for subsequent characterisation and cross linking of the nanoparticles (ESI pages S12–S15†). Dynamic light scattering (DLS) was subsequently used to determine whether self-assembled micelles generated from pure block copolymers or their quasi-block counterparts (Table 1) influenced micelle formation. Despite the less precise nature of block composition for quasi-block polymerisation it was found that stable micelles were still formed. Since quasi-block polymerisation is efficient and applicable to parallel synthesis, this was the primary method of choice to generate target polymers for cellular internalisation studies.
A larger library of p(BMA-co-MAA)-b-p(PEGMA-475) block copolymers with a range of molecular weights (5900–14700 Da) and BMA:MAA ratios (9:1, 4:1 and 2:1) was synthesised to compare polymer composition and molecular weight to the size of the resultant micelles (ESI Table S6†). These copolymers formed micelles ranging in size from 16 to 143 nm. Analysis of the data showed a weak positive trend between the total molecular weight of the polymer chain and the size of the micelle particle formed; higher molecular weight polymers led to larger micelles (Fig. 2). The hydrophobic p(BMA-co-MAA) block Mn had a more significant effect on micelle size than the hydrophilic PEGMA-475 block Mn. The RAFT-end group was removed from a number of polymers to determine if it affected micelle formation; the size of the micelles formed did not change once the end group had been removed (ESI pages S17–18†).
 | ||
| Fig. 2 Total polymer molecular weight vs. micelle size (top) for the library of synthesised micelles, p(BMA-co-MAA) copolymer block molecular weight vs. micelle size (middle) and PEGMA-475 block molecular weight vs. micelle size (bottom). Molecular weights were calculated from 1H-NMR spectra, data displayed in ESI Table S6.† | ||
Following this, the incorporation of an N-acryloxysuccinimide (NAS) monomer and a rhodamine B monomer, methacryloxyethyl thiocarbamoyl rhodamine B (MRhB), were investigated in order to design nanoparticles suitable for cellular internalisation studies. NAS was incorporated to allow for crosslinking of the micelles with a diamine linker to form stable nanoparticles, while MRhB was incorporated to enable tracking of the nanoparticles in vitro. To investigate whether the order of the monomer units in the polymer influenced micelle formation, four different variations in polymer composition were investigated when incorporating NAS and MRhB into the hydrophilic section of the polymers. All nanoparticles were shell cross linked, with four different polymer compositions, A, B, C and D, explored. Compositions are graphically depicted in Fig. 3.
 | ||
| Fig. 3 Different polymer block structures. | ||
Polymers categorised as A were p(BMA-co-MAA)-b-p(PEGMA-475)-b-p(NAS-co-MRhB) triblock copolymers where the NAS and MRhB were distributed in the outer core. B polymers were p(BMA-co-MAA)-b-p(PEGMA-475)-b-p(NAS)-b-p(MRhB) tetrablock copolymers where MRhB is on the surface of the nanoparticle. C polymers were p(BMA-co-MAA)-b-p(PEGMA-475-co-NAS)-b-p(MRhB) triblock copolymers where the cross linker NAS was in the middle part of the shell. D polymers were p(BMA-co-MAA)-b-p(PEGMA-475-co-NAS-co-MRhB) diblock copolymers where the NAS cross linker and MRhB was distributed throughout the shell of the nanoparticle. As the method to make category A polymers was the most efficient route to incorporating NAS and MRhB monomers into the existing library of p(BMA-co-MAA)-b-p(PEGMA-475) block copolymers, the majority of the polymers were chain extended using this method. Polymers were chain extended using the same polymerisation conditions described earlier to make the initial block copolymers (85 °C in DMF, using ACHN as the initiator), for 6 or 7 h (see ESI for full experimental details, pages S7–S8†). The resultant polymers were then self-assembled and crosslinked (Scheme 2) to create stable nanoparticles; chain extended block copolymers were dissolved in water and reacted with the polyetheramine JEFFAMINE D230 (see ESI pages S7–8 for full experimental details†). Successful crosslinking was demonstrated by the improved stability of crosslinked nanoparticles in THF and isopropanol when compared to the non-crosslinked analogues (ESI Table S8†).
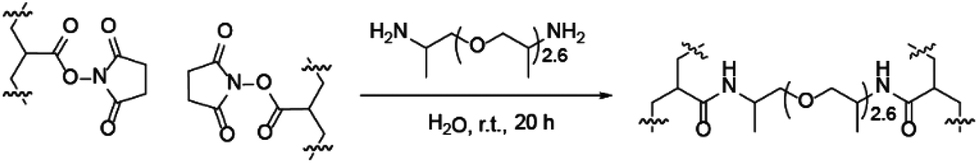 | ||
| Scheme 2 Micelles were crosslinked to form stable nanoparticles using a diamine linker. | ||
Cellular internalisation studies using murine macrophages were performed on the library of synthesised nanoparticles (Table 2). Purified activated murine macrophages (Gr1−Mac1+F4/80+ cells) were harvested and sorted from bone marrow or the peritoneal cavity. Following immunolabelling for the cellular markers Gr-1, Mac-1 and F4/80, cells were sorted via fluorescence activated cell sorting (FACS) (details of labelling and sorting strategies are outlined in the ESI, pages S8–S11†).46 The internalisation of all twenty-two rhodamine-labelled nanoparticles was investigated using flow cytometric analysis (Table 2). Specifically, the rhodamine-labelled nanoparticles were incubated with macrophages and internalisation initially characterised based on an increased fluorescence intensity of rhodamine in the gated Gr1−Mac1+F4/80+ macrophage population.
Mean channel fluorescence (MCF) values were normalised using one sample, tested on multiple days. To compensate for any variability in nanoparticle fluorescence (i.e. if different samples contained different amounts of rhodamine) nanoparticle internalisation was normalised by dividing MCF values by the total fluorescence of each sample; these results are displayed as ‘rhodamine normalised internalisation’ in Table 2. The MCF of untreated cells, used as a negative control in all experiments, was <150. MCF values for all samples were well above 150, indicating that all nanoparticles were successfully internalised by macrophages. Internalisation of representative nanoparticles was also cross-checked by fluorescence microscopy (see ESI Fig. S5†).
While all twenty-two fluorescent nanoparticles studied were taken up by macrophages, some nanoparticles appeared to be internalised to a higher extent than others (Table 2). Although it has been reported in the literature that particle size has a significant effect on internalisation by macrophages, no clear correlation was observed between nanoparticle size (or polymer molecular weight) and higher levels of internalisation in this case (ESI Fig. S6†).47–53 Despite this, some copolymer compositions clearly displayed higher levels of internalisation and therefore five polymers (3, 4, 10, 11 and 15, coloured green in Table 2) were selected for further validation and study. Firstly, further replicates were carried out for the five selected nanoparticles. Nanoparticle internalisation varied slightly between experiments but trends were consistent across the replicates: (4) > (15) > (3) > (11) ≈ (10) (Fig. 4).
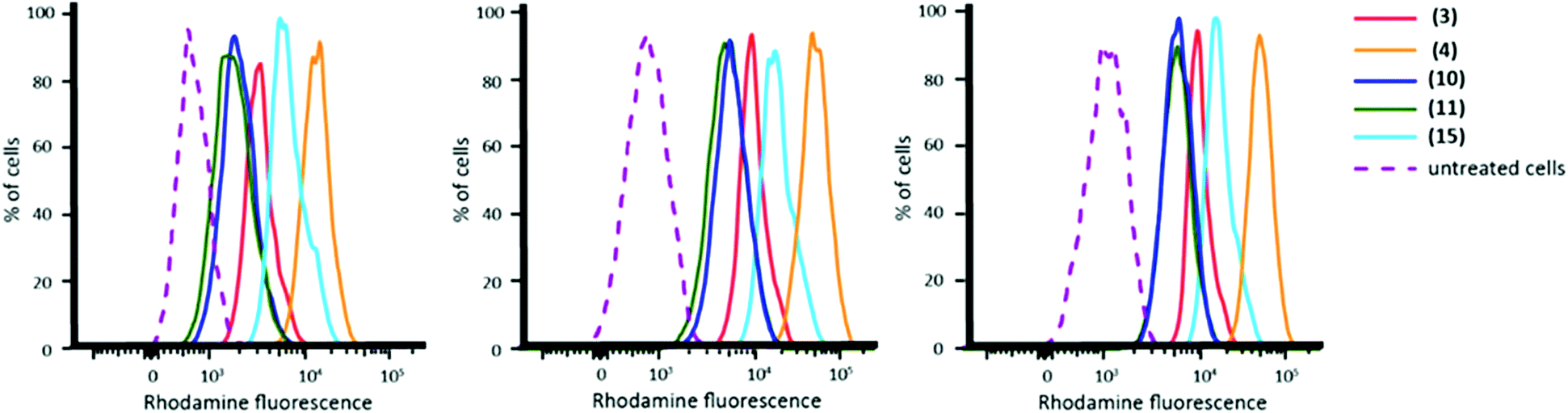 | ||
| Fig. 4 FACS plots showing the fluorescence intensity of cells incubated with rhodamine-labelled nanoparticles (3), (4), (10), (11) and (15) in three separate experiments. NB: Plots show rhodamine fluorescence prior to normalisation. | ||
It is prudent to note that rhodamine fluorescence as measured by flow cytometry is not a direct measurement of nanoparticle internalisation as, owing to the varying numbers of rhodamine functionalised polymer chains that constitute a nanoparticle, equimolar quantities of nanoparticle may exhibit different degrees of fluorescence intensity. Consequently, to quantify nanoparticle internalisation we normalised the fluorescence intensity of each nanoparticle according to the number of polymer chains. The micelle aggregation number was determined using multi-angle light scattering, which showed a range from 18 (nanoparticle (11)) to 89 (nanoparticle (15)); the full results can be found in ESI Table S10.† Once rhodamine fluorescence per nanoparticle was taken into account, the trend for nanoparticle internalisation was (11) > (4) > (3) ≈ (15) ≈ (10) (ESI Table S10†).
Given the extensive cellular internalisation of the polymeric nanoparticles, efforts then turned to the use of these systems to deliver a suitable cargo to macrophages. A fluorescent blue dye, 4′,6-diamidino-2-phenylindole (DAPI), was selected for encapsulation since: (i) it has basic functionality, making it more likely to be successfully retained inside the acidic nanoparticle core. (ii) DAPI permeates live cell membranes poorly unless used at high concentration (≥25 μg ml−1) so the efficient delivery of any DAPI to macrophages had to be due to successful encapsulation and transport of the dye within the nanoparticles. (iii) The fluorescence emission profiles of rhodamine and DAPI do not overlap and thus the two dyes can be used simultaneously as detection of their respective fluorescence can be unambiguously distinguished.54,55
Nanoparticle (11) was selected for DAPI encapsulation as it had the highest level of internalisation out of the particles tested (see ESI Table S10†) and was therefore identified to be the most promising candidate for drug and dye delivery. DAPI was encapsulated in nanoparticles via passive diffusion by stirring a solution of particles and DAPI for 24 h (full details can be found in the ESI, page S8†).
Nanoparticles containing encapsulated DAPI were extensively dialysed to remove unencapsulated DAPI. After dialysis, the DAPI fluorescence of micellar solutions was measured to confirm successful encapsulation; 2.0 μg ml−1 was determined to be present in the nanoparticle solution (ESI Fig. S7†). DAPI-encapsulated nanoparticles successfully delivered DAPI to macrophages (Fig. 5) and a linear correlation was found between number of nanoparticles internalised and the amount of DAPI delivered (ESI Fig. S10†).
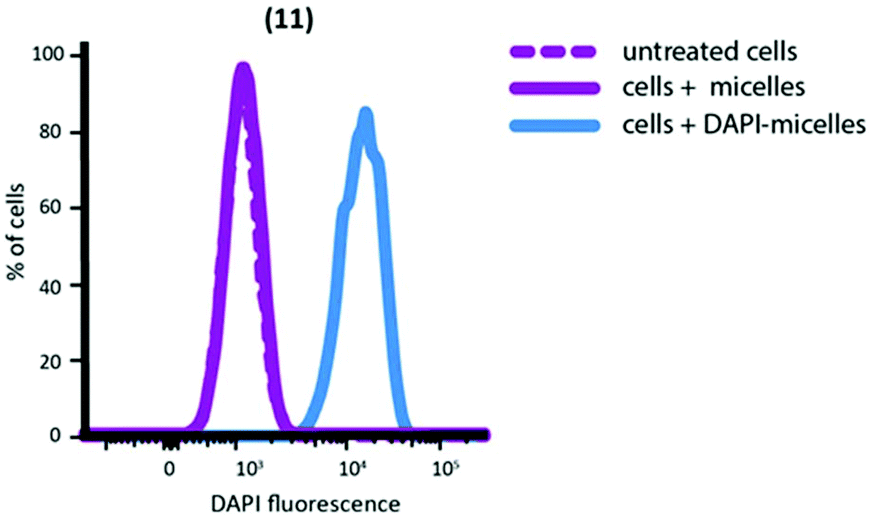 | ||
| Fig. 5 FACS plot showing delivery of DAPI to peritoneal macrophages by nanoparticle (11). Untreated cells and nanoparticles not containing DAPI are shown for comparison. | ||
In contrast, no DAPI internalisation was observed when cells were treated with DAPI without the aid of nanoparticle assisted delivery (ESI Fig. S9†). Collectively, these results confirm the ability of our nanoparticles to encapsulate and efficiently deliver molecules that would otherwise have poor cell-penetration.
Conclusions
A library of quasi-block copolymers was synthesised by varying the chain lengths and composition of BMA-co-MAA and PEGMA blocks, which successfully self-assembled into a series of micelles. Block copolymers were fluorescently labelled using a rhodamine B monomer and a NAS-diamine crosslinking system was used to form stable nanoparticles for in vitro use. Cellular studies using primary macrophages showed that the nanoparticles were successfully internalised and that they were able to deliver an encapsulated, non cell-penetrating, dye to cells. Together, our data provide evidence that quasi-block copolymer synthesis can be used in place of pure-block copolymer synthesis for drug delivery applications (providing suitable monomers are employed). The crosslinked nanoparticles have proven to be promising delivery vehicles due to their stability at low concentrations, their ability to encapsulate basic cargo and their high level of cellular internalisation.Live subject statement
All experiments were performed in compliance with the National Health and Medical Research Council of Australia's guidelines and were approved by the Monash Animal Research Platform ethics committee.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank Carlos Guerrero-Sanchez for his help with ChemSpeed experiments, Jess Hatwell-Humble and Dani Cardozo for animal assistance, Andrea Reitsma, Chad Heazlewood and Shen Heazlewood for technical assistance. We would also like to thank Huntsman Corporation for the donation of the polyamine, JEFFAMINE D230, which was used as a crosslinker in this work.Funding for this work was provided by the Commonwealth Scientific and Industrial Research Organization (CSIRO), Cancer Research UK (grant C21484A6944) and the Engineering and Physical Sciences Research Council (EPSRC).
Notes and references
- G. Gaucher, M.-H. Dufresne, V. P. Sant, N. Kang, D. Maysinger and J.-C. Leroux, J. Controlled Release, 2005, 109, 169–188, DOI:10.1016/j.jconrel.2005.09.034.
- L. Y. Lin, N. S. Lee, J. Zhu, A. M. Nyström, D. J. Pochan, R. B. Dorshow and K. L. Wooley, J. Controlled Release, 2011, 152, 37–48, DOI:10.1016/j.jconrel.2011.01.009.
- N. Nasongkla, E. Bey, J. Ren, H. Ai, C. Khemtong, J. S. Guthi, S.-F. Chin, A. D. Sherry, D. A. Boothman and J. Gao, Nano Lett., 2006, 6, 2427–2430, DOI:10.1021/nl061412u.
- Y. Kim, M. H. Pourgholami, D. L. Morris and M. H. Stenzel, J. Mater. Chem., 2011, 21, 12777–12783, 10.1039/c1jm11062d.
- S. Gordon and S. Rabinowitz, Adv. Drug Delivery Rev., 1989, 27–47, DOI:10.1016/0169-409X(89)90036-7.
- J. Condeelis and J. W. Pollard, Cell, 2006, 124, 263–266, DOI:10.1016/j.cell.2006.01.007.
- Y. Luo, H. Zhou, J. Krueger, C. Kaplan, S.-H. Lee, C. Dolman, D. Markowitz, W. Wu, C. Liu, R. A. Reisfeld and R. Xiang, J. Clin. Invest., 2006, 116, 2132–2141, DOI:10.1172/JCI27648.
- N. Maruotti, F. P. Cantatore, E. Crivellato, A. Vacca and D. Ribatti, Histol. Histopathol., 2007, 22, 581–586, DOI:10.14670/HH-22.581.
- S. M. Bol, V. Cobos-Jiménez, N. A. Kootstra and A. B. Van't Wout, Future Virol., 2011, 6, 187–208, DOI:10.2217/fvl.10.93.
- C. Kelly, C. Jefferies and S.-A. Cryan, J. Drug Delivery, 2011, 1–11, DOI:10.1155/2011/727241.
- P. Broz, N. Ben-Haim, M. Grzelakowski, S. Marsch, W. Meier and P. Hunziker, J. Cardiovasc. Pharmacol., 2008, 51, 246–252, DOI:10.1097/FJC.0b013e3181624aed.
- P. V. Kumar, A. Asthana, T. Dutta and N. K. Jain, J. Drug Targeting, 2008, 14, 546–556, DOI:10.1080/10611860600825159.
- S. S. Yu, C. M. Lau, W. J. Barham, H. M. Onishko, C. E. Nelson, H. Li, C. A. Smith, F. E. Yull, C. L. Duvall and T. D. Giorgio, Mol. Pharm., 2013, 10, 975–987, DOI:10.1021/mp300434e.
- A. M. Jhaveri and V. P. Torchilin, Front. Pharmacol., 2014, 5, 77, DOI:10.3389/fphar.2014.00077.
- A. Nardin, M.-L. Lefebvre, K. Labroquère, O. Faure and J.-P. Abastado, Curr. Cancer Drug Targets, 2006, 6, 123–133, DOI:10.2174/156800906776056473.
- S. P. Vyas, M. E. Kannan, S. Jain, V. Mishra and P. Singh, Int. J. Pharm., 2004, 269, 37–49, DOI:10.1016/j.ijpharm.2003.08.017.
- A. Blanazs, S. P. Armes and A. J. Ryan, Macromol. Rapid Commun., 2009, 30, 267–277, DOI:10.1002/marc.200800713.
- P. V. Messina, J. M. Besada-Porto and J. M. Ruso, Curr. Top. Med. Chem., 2014, 14, 555–571, DOI:10.2174/1568026614666140121112118.
- T. Miller, G. Van Colen, B. Sander, M. M. Golas, S. Uezguen, M. Weigandt and A. Goepferich, Pharm. Res., 2013, 30, 584–595, DOI:10.1007/s11095-012-0903-5.
- A. S. Narang, D. Delmarre and D. Gao, Int. J. Pharm., 2007, 345, 9–25, DOI:10.1016/j.ijpharm.2007.08.057.
- S. Kim, Y. Shi, J. Y. Kim, K. Park and J.-X. Cheng, Expert Opin. Drug Delivery, 2010, 7, 49–62, DOI:10.1517/17425240903380446.
- J.-S. Wang and K. Matyjaszewski, Macromolecules, 1995, 28, 7572–7573, DOI:10.1021/ma00126a041.
- M. Sawamoto and M. Kamigaito, Trends Polym. Sci., 1996, 4, 371–376 CAS.
- D. H. Solomon, E. Rizzardo and P. Cacioli, US4581429 Search PubMed; Chem. Abstr. 1985 102 221335q Search PubMed.
- G. Moad and D. H. Solomon, The Chemistry of Free Radical Polymerization, Pergamon Press, London, 1995 Search PubMed.
- K. Matyjaszewski and J. Spanswick, Mater. Today, 2005, 8, 26–33, DOI:10.1016/S1369-7021(05)00745-5.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562, DOI:10.1021/ma9804951.
- M. H. Stenzel, Chem. Commun., 2008, 3486–3503, 10.1039/b805464a.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2006, 59, 669–692, DOI:10.1071/CH06250.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2009, 62, 1402–1472, DOI:10.1071/CH09311.
- G. Moad, E. Rizzardo and S. H. Thang, Polymer, 2008, 49, 1079–1131, DOI:10.1016/j.polymer.2007.11.020.
- A. Akinc, D. M. Lynn, D. G. Anderson and R. Langer, J. Am. Chem. Soc., 2003, 125, 5316–5323, DOI:10.1021/ja034429c.
- J. J. Haven, C. Guerrero-Sanchez, D. J. Keddie and G. Moad, Macromol. Rapid Commun., 2014, 35, 492–497, DOI:10.1002/marc.201300459.
- C. Guerrero-Sanchez, L. O'Brien, C. Brackley, D. J. Keddie, S. Saubern and J. Chiefari, Polym. Chem., 2013, 4, 1857–1862, 10.1039/c3py21135e.
- J. J. Haven, C. Guerrero-Sanchez, D. J. Keddie, G. Moad, S. H. Thang and U. S. Schubert, Polym. Chem., 2014, 5, 5236–5246, 10.1039/C4PY00496E.
- T. M. Hinton, C. Guerrero-Sanchez, J. E. Graham, T. Le, B. W. Muir, S. Shi, M. L. V. Tizard, P. A. Gunatillake, K. M. McLean and S. H. Thang, Biomaterials, 2012, 33, 7631–7642, DOI:10.1016/j.biomaterials.2012.06.090.
- D. J. Keddie, Chem. Soc. Rev., 2014, 43, 496–505, 10.1039/C3CS60290G.
- K. B. Thurmond, T. Kowalewski and K. L. Wooley, J. Am. Chem. Soc., 1996, 118, 7239–7240, DOI:10.1021/ja961299h.
- K. B. Thurmond, T. Kowalewski and K. L. Wooley, J. Am. Chem. Soc., 1997, 119, 6656–6665, DOI:10.1021/ja9710520.
- Y. Li, B. S. Lokitz, S. P. Armes and C. L. McCormick, Macromolecules, 2006, 39, 2726–2728, DOI:10.1021/ma0604035.
- S. Pascual and M. J. Monteiro, Eur. Polym. J., 2009, 45, 2513–2519, DOI:10.1016/j.eurpolymj.2009.06.009.
- L. Juillerat-Jeanneret, J.-D. Aubert and P. Leuenberger, J. Lab. Clin. Med., 1997, 130, 603–614, DOI:10.1016/S0022-2143(97)90110-4.
- H. L. Jackman, F. Tan, D. Schraufnagel, T. Dragović, B. Dezsö, R. P. Becker and E. G. Erdös, Am. J. Respir. Cell Mol. Biol., 1995, 13, 196–204, DOI:10.1165/ajrcmb.13.2.7626287.
- Y. Waumans, G. Vliegen, L. Maes, M. Rombouts, K. Declerck, P. Van Der Veken, W. Vanden Berghe, G. R. Y. De Meyer, D. Schrijvers and I. De Meester, Inflammation, 2016, 39, 413–424, DOI:10.1007/s10753-015-0263-5.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2005, 58, 379–410, DOI:10.1071/CH05072.
- A. A. Cassado, M. R. D'Império Lima and K. R. Bortoluci, Front. Immunol., 2015, 6, 225, DOI:10.3389/fimmu.2015.00225.
- H. Yue, W. Wei, Z. Yue, P. Lv, L. Wang, G. Ma and Z. Su, Eur. J. Pharm. Sci., 2010, 41, 650–657, DOI:10.1016/j.ejps.2010.09.006.
- S. S. Yu, C. M. Lau, S. N. Thomas, W. G. Jerome, D. J. Maron, J. H. Dickerson, J. A. Hubbell and T. D. Giorgio, Int. J. Nanomed., 2012, 7, 799–813, DOI:10.2147/IJN.S28531.
- J. A. Champion, A. Walker and S. Mitragotri, Pharm. Res., 2008, 25, 1815–1821, DOI:10.1007/s11095-008-9562-y.
- Y. Tabata and Y. Ikada, Biomaterials, 1988, 9, 356–362, DOI:10.1016/0142-9612(88)90033-6.
- M. K. Pratten and J. B. Lloyd, Biochim. Biophys. Acta, 1986, 881, 307–313, DOI:10.1016/0304-4165(86)90020-6.
- D. Paul, S. Achouri, Y.-Z. Yoon, J. Herre, C. E. Bryant and P. Cicuta, Biophys. J., 2013, 105, 1143–1150, DOI:10.1016/j.bpj.2013.07.036.
- Y. Yeo, Nanoparticulate Drug Delivery Systems: Strategies, Technologies, and Applications, John Wiley & Sons, 2013 Search PubMed.
- D. Zink, N. Sadoni and E. Stelzer, Methods, 2003, 29, 42–50, DOI:10.1016/S1046-2023(02)00289-X.
- B. Chazotte, Cold Spring Harb. Protoc., 2011, 6, 80–83, DOI:10.1101/pdb.prot5556.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7py01363a |
| This journal is © The Royal Society of Chemistry 2018 |