
Fully recoverable rigid shape memory foam based on copper-catalyzed azide–alkyne cycloaddition (CuAAC) using a salt leaching technique†
Abeer A.
Alzahrani
a,
Mohand
Saed
 b,
Christopher M.
Yakacki
b,
Han Byul
Song
a,
Nancy
Sowan
c,
Joshua J.
Walston
a,
Parag K.
Shah
a,
Matthew K.
McBride
a,
Jeffrey W.
Stansbury
ad and
Christopher N.
Bowman
*ac
b,
Christopher M.
Yakacki
b,
Han Byul
Song
a,
Nancy
Sowan
c,
Joshua J.
Walston
a,
Parag K.
Shah
a,
Matthew K.
McBride
a,
Jeffrey W.
Stansbury
ad and
Christopher N.
Bowman
*ac
aDepartment of Chemical and Biological Engineering, University of Colorado Boulder, Boulder, CO, USA. E-mail: Christopher.bowman@colorado.edu
bDepartment of Mechanical Engineering, University of Colorado Denver, Denver, CO, USA
cMaterials Science and Engineering Program, University of Colorado Boulder, 596 UCB, Boulder, CO, USA
dDepartment of Craniofacial Biology, School of Dental Medicine, Anschutz Medical Campus, Aurora, CO, USA
First published on 29th November 2017
Abstract
This study is the first to employ the use of the copper-catalyzed azide–alkyne cycloaddition (CuAAC) polymerization to form a tough and stiff, porous material from a well-defined network possessing a high glass transition temperature. The effect of the network linkages formed as a product of the CuAAC reaction, i.e., the triazoles, on the mechanical behavior at high strain was evaluated by comparing the CuAAC foam to an epoxy-amine-based foam, which consisted of monomers with similar backbone structures and mechanical properties (i.e., Tg of 115 °C and a rubbery modulus of 1.0 MPa for the CuAAC foam, Tg of 125 °C and a rubbery modulus of 1.2 MPa for the epoxy-amine foam). When each foam was compressed uniformly to 80% strain at ambient temperature, the epoxy-amine foam was severely damaged after only reaching 70% strain in the first compression cycle with a toughness of 300 MJ m−3. In contrast, the CuAAC foam exhibited pronounced ductile behavior in the glassy state with three times higher toughness of 850 MJ m−3 after the first cycle of compression to 80% strain. Additionally, when the CuAAC foam was heated above Tg after each of five compression cycles to 80% strain at ambient temperature, the foam completely recovered its original shape while exhibiting a gradual decrease in mechanical performance over the multiple compression cycles. The foam demonstrated almost complete shape fixity and recovery ratios even through five successive cycles, indicative of “reversible plasticity”, making it highly desirable as a glassy shape memory foams.
Introduction
Shape-memory polymers (SMPs) are a class of smart polymeric materials that have the ability to store a temporary shape in a deformed state, and recover to a permanent shape upon exposure to a specific stimulus.1–3 SMP foams, in particular, possess several distinctive material capabilities that neat SMPs do not possess, such as lighter weight, greater volumetric expansion and dramatic permeability change upon actuation,4–6 as well as being able to compact to extremely small volumes compared to neat/regular SMPs.7 This enables the utilization of SMP foams in various applications such as deployable biomedical devices that facilitate minimally invasive vascular treatments8–10 and possesses a potential use for spacecraft structures where conservation of volume and weight is an extremely important concern including solar sails, solar arrays, and sunshields.11 In addition, SMP foams have been explored as self-repairing materials where a compressed secondary shape foam was utilized to partially close or fill damaged areas via its large volumetric expansion.6 All of these applications rely on large, on-demand shape deformations during programming of the shape-memory effect, which necessitates the design of tough foams to avoid failure typically associated with high strain loading.The majority of SMP foams utilize either polyurethane (PU),12–15 or epoxy16–20 -based materials. Although polyurethane SMP foams possess high-strain failure capability, the often low glass transition temperatures (Tg) of urethane-based foams limit their high temperature applications and modulus at high temperature. In contrast, thermosetting epoxy SMP foams have Tg readily available up to 90 °C, but the brittle nature of the epoxy-based foams at temperature below Tg restricts their use.16 Different techniques have been used to enhance the toughness of epoxies such as the addition of soft/thermoplastic particles,21 blending of copolymers,22 and utilization of composite materials.23,24 However, each one of these approaches have its own drawbacks, for example, incorporating soft particles in a glassy polymer compromises the mechanical properties of the composites,25 and the increase in particle content above an optimum level leads to aggregation and a further decrease in the composite strength.26 In addition, the mechanical properties of SMP foams are dictated by the nature of their chemical bonding and pore structure, i.e. porosity.27 Therefore, there is a significant variation in results depending on the monomer systems being used, making it difficult to generalize outcomes.16,28,29 Overall, it is desirable to design a tough, high Tg SMP foam that is repeatedly deformable to a highly compacted state and still demonstrates full recovery.
Moving away from the traditional chemistry of polyurethanes and epoxies, here, we introduce polymer foams fabricated for the first time using copper-catalyzed azide–alkyne cycloaddition (CuAAC) polymerization as a potential material for high strain SMP foams. CuAAC networks are formed through a step-growth mechanism, which leads to beneficial properties including delayed gelation, low polymerization shrinkage stress,30 and inherently uniform network structure31,32 as indicated by a narrow glass transition compared to conventional chain growth polymerization. These traits make this a good candidate for shape memory applications.33 Moreover, among the various reactions in the click chemistry toolbox, the CuAAC reaction is often considered to be the most capable reaction due to its ideal nature.34,35 For instance, this reaction proceeds efficiently and predictably under relatively facile reaction conditions to quantitative yields at ambient temperature and in an orthogonal manner. Therefore, implementing the CuAAC chemistry to design a polymer network allows a wide selection of azide and alkyne monomers and other orthogonal chemistries to be used to tune the physicochemical properties of the network.36,37
Additionally, the hetero-cyclic triazole ring, which forms in molar quantities during the CuAAC polymerization, significantly enhances the mechanical properties of the network due to its rigid nature.32,38–41 Hawker and co-workers demonstrated the potential of triazole linkages to improve the mechanical strength of a hydrogel using bifunctional alkyne-PEGs and tetrafunctional azide-PEGs that has 10 times greater strength than a hydrogel that was crosslinked by conventional chain growth polymerization.40 Although the CuAAC reaction has been studied extensively in solution,34,42–46 minimal work has been done to use it as a bulk polymerization reaction to form crosslinked networks with little done to understand the resulting mechanical properties.31,47–50 One of the few studies that evaluated CuAAC network was done by Baranek and coworkers who demonstrated that triazoles generated during the CuAAC polymerization would increase the Tg drastically, leading to glassy materials.32 Moreover, shape memory polymers formed via the CuAAC reaction exhibited outstanding attributes with shape fixity and recovery of 99%.31
The purpose of the study is to employ the “click” chemistry of CuAAC polymerizations to create glassy SMP foams that will exhibit (1) narrow glass transition temperature (Tg) significantly above ambient temperature, (2) enhanced stiffness and toughness of thermosetting foams based on these polymers, and (3) consistent shape-memory and recovery behavior over multiple compression cycles at high strain. Therefore, thermo-mechanical properties, shape-memory recovery behavior, and toughness of the CuAAC foam were evaluated in comparison with traditional epoxy-amine foams of similar Tg.
Experimental section
Materials
1,1,1-Tris(hydroxymethyl)propane, propargyl bromide solution (80 wt% in toluene), sodium hydroxide, sodium chloride, bisphenol A diglycidyl ether, lithium perchlorate, sodium azide, copper(II) chloride, N,N,N′,N′,N′′-pentamethyldiethylenetriamine (PMDETA), aniline, 1,6-diaminohexane, dimethyl sulfoxide, and acetonitrile were obtained from Sigma-Aldrich, and were used without further purification. All azides were synthesized according to the azide safety rules and handled with appropriate precaution when working with monomers, resins, and polymers in small quantities.51![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of diethyl ether:ethyl acetate (600 mL). The organic layer was washed three times with distilled water and brine, dried with anhydrous Na2SO4, and concentrated at reduced pressure. The concentrate was purified by flash chromatography (9:1 hexane:ethyl acetate) with 70% yield. 1H NMR (400 MHz, chloroform-d) δ 4.14 (d, J = 2.4 Hz, 6H), 3.42 (s, 6H), 2.42 (t, J = 2.4 Hz, 3H), 1.44 (q, J = 7.6 Hz, 2H), 0.90 (t, J = 7.6 Hz, 3H).
:1 mixture of diethyl ether:ethyl acetate (600 mL). The organic layer was washed three times with distilled water and brine, dried with anhydrous Na2SO4, and concentrated at reduced pressure. The concentrate was purified by flash chromatography (9:1 hexane:ethyl acetate) with 70% yield. 1H NMR (400 MHz, chloroform-d) δ 4.14 (d, J = 2.4 Hz, 6H), 3.42 (s, 6H), 2.42 (t, J = 2.4 Hz, 3H), 1.44 (q, J = 7.6 Hz, 2H), 0.90 (t, J = 7.6 Hz, 3H).
Methods
:1 monomers:methanol) and 80 wt% sodium chloride with respect to monomer. The ratio of 1.5:1 monomers to methanol was chosen to lower the resin viscosity, which improves the salt crystal distribution as needed to produce the foam. Sodium chloride was sifted through two different sized sieves with 350 μm mesh and 212 μm mesh. The average salt size was 290 ± 40 μm. The mold was rinsed with RainX to prevent polymer adhesion to the wall of the mold, and then sodium chloride was added and compacted in the mold with a Teflon rod. A slit was made in the bottom of the mold to allow air to escape when the reaction mixture was added. The reaction mixture was consisted of 50:50:1 azide:alkyne:CuCl2/PMDETA ratio based on the moles of functional groups. To the reaction mixture, 8 wt% of hexylamine with respect to monomer was added drop-wise to reduce Cu(II) to Cu(I). The reaction was allowed to proceed until the polymer was hard enough to remove from the mold without physical deformation. The polymer was heated overnight at 80 °C to ensure complete curing. Cured foams were soaked in 500 mL of DI water at 50 °C for 5 days with water changed once daily to leach out sodium chloride and hexylamine from the polymer. Foams were dried in vacuo at 100 °C for 24 h.
:1 monomers:acetonitrile) and 80 wt% sodium chloride with respect to monomer. Epoxy-amine polymer samples were formulated with a 1:0.5:0.5 bisphenol A diglycidyl ether:1,6-diaminohexane:aniline ratio based on the moles of functional groups. Bisphenol A diglycidyl ether was first dissolved in acetonitrile. 1,6-Diaminohexane and aniline were measured into a small vial and heated to 40 °C to melt. Sodium chloride was added to a mold that was previously treated with Rain-X. The amine mixture was added to the bisphenol A diglycidyl ether solution and mixed thoroughly before being added dropwise to sodium chloride in the mold. The mixture was allowed to cure overnight at ambient temperature. The following day, the polymers were removed from the mold and then left to cure at 80 °C for 48 h to ensure proper conversion. Cured foams were left in 500 mL of DI water at 50 °C for 5 days with water changed once daily to leach out sodium chloride from the polymer. Finally, foams were dried in vacuo at 100 °C for 24 h.
δ of the foam (1 mm thick × 6.5 mm wide × 10 mm long).
| Rf (%) = (εu/εm) × 100 | (1) |
| Rr = (εu − εp)/(εm − εp) × 100 | (2) |
Recovery tests were also performed using mechanical testing (Bose-ElectroForce 3200; Eden Prairie, MN) to monitor recovery as a function of temperature. After each cycle of deformation to 80% at ambient temperature, as described above, the recovery was measured as the sample was heated from 25 °C to 150 °C at a rate of 3 °C min−1 under constant preload force (0.15 N). Samples were held isothermal at 150 °C for 10 min to ensure equilibrium between cycles. The fixity and recovery ratio were calculated as in eqn (1) and (2) above.
Results and discussion
Fabrication of CuAAC and epoxy-amine foams
Here, we explore the shape-memory behavior, recovery and toughness of rigid CuAAC foams. The homogeneous, glassy CuAAC network was formed through copper-catalyzed azide–alkyne “click” reaction, see Scheme 1(a). The CuAAC foam was made via a salt-leaching technique52 in which the porosity is controlled by selecting the particle size and amount of the added salt particles as described in the Methods section. The CuAAC foam has 80% porosity and pore sizes range from approximately 200–350 μm with an average pore size of 290 ± 40 μm, see Fig. S1.† An epoxy-amine-based foam was used to compare with the CuAAC foam as a negative control, where monomers with similar backbone structures as in the CuAAC case were used to form the network as shown in Scheme 1(b). Specifically, a mixture of a flexible di-amine and a rigid mono-amine was used to formulate the epoxy-amine foam in order to achieve a glass transition temperature similar to the CuAAC foam. The epoxy-amine foam was also made by the same fabrication method as the CuAAC foam and presented 80% porosity. Additionally, in situ kinetic profiles of both the CuAAC and epoxy-amine polymerizations were investigated with respect to time via Fourier transform infrared spectroscopy (FT-IR) as presented in Fig. S2.† The Cu(II)–hexylamine system for the CuAAC polymerization proved to be an efficient catalyst to initiate the reaction, where over 90% conversion was obtained at ambient temperature in approximately 20 min. | ||
| Scheme 1 (a) The chemical structure of the monomers used to synthesize the CuAAC foam. (b) The chemical structure of the monomers used to synthesize the epoxy-amine foam. (c) A general scheme for the cyclization of an alkyne/azide system for CuAAC polymerization. | ||
Thermo-mechanical properties of CuAAC and epoxy-amine foams
The thermo-mechanical properties of this fully reacted CuAAC foam were evaluated by dynamic mechanical analysis (DMA) and compared with the epoxy-amine foam. As indicated in Fig. 1, the glass transition temperature (Tg) of the CuAAC foam was observed to be 115 °C with full width at half height of 10 ± 2 °C and a rubbery modulus of 1.0 MPa. The glass transition temperature (Tg) and the rubbery modulus of the epoxy-amine foam are observed to be 125 °C and 1.2 MPa, respectively, which are relatively analogous to the CuAAC foam. In addition, the mechanical properties of the CuAAC non-foam polymer and epoxy-amine non-foam polymer were evaluated using DMA and presented in Fig. S3.† While the Tg of each polymer was relatively equivalent to that of the foam, the rubbery modulus was significantly higher for both non-foam polymers than foam-based systems, as expected. The CuAAC and epoxy-amine non-foam polymers showed rubbery modulus values of 9.8 MPa and 32 MPa, respectively. It should be noted that molecular weight between crosslinks calculated from the rubbery modulus based on rubber elasticity is 540 g mol−1 for the bulk CuAAC polymer and 170 g mol−1 for the bulk epoxy-amine polymer. Thus, the crosslink density of the CuAAC polymer is 1.9 mol L−1, which is significantly lower than the epoxy-amine crosslink density of 6.1 mol L−1. This difference in crosslink density arises in order to achieve similar glass transition temperatures in the two polymers. The presence of the stiff triazole unit increases the relative Tg for the CuAAC polymers, necessitating a lower CuAAC crosslink density to achieve similar Tg values between the two polymers. Despite the equivalent Tg values of the two polymers, the lower crosslink density may enhance mobility and ductility of the polymer. However, as evidenced in Fig. 1, the effect of the different crosslink densities is minimized in the foams where the differences in rubbery modulus become minimal. | ||
| Fig. 1 Glass transition temperature (Tg) and storage modulus were measured for (a) the epoxy-amine foam and (b) the CuAAC foam. The epoxy-amine foam was formed with a mole ratio of 1:0.5:0.5 bisphenol A diglycidyl ether, 1,6-diaminohexane, and aniline with 80% porosity showing a glass transition peak with a Tg = 125 °C and a rubbery modulus = 1.2 MPa. The CuAAC foam was formed via a stoichiometric mole ratio of bisphenoldiazide, trialkyne-ether, 2 mol% CuCl2/PMDETA, and 8 wt% hexylamine with 80% porosity via click reaction showing a glass transition peak with a Tg = 115 °C and a rubbery modulus = 1.0 MPa. | ||
Compression and shape-memory recovery behavior of CuAAC and epoxy-amine foams
Rigid crosslinked polymers with a Tg much higher than ambient temperature most often exhibit brittle behavior when they are subjected to high strain since the immobility of the polymer chains limits their ability to dissipate energy through mechanical damping via bond rotation or by breaking secondary intermolecular interactions like hydrogen bonding or pi-stacking.53,54 The triazole linkage in the CuAAC foams affords the opportunity for hydrogen bonding and pi-stacking,55–57 each of which will lead to enhanced stiffness and toughness of the glassy CuAAC networks. Further, it is worth nothing that, while the triazole moieties are known to lead to these types of bimolecular, secondary interactions, the concentration of triazoles present in the networks here is significantly higher than those under which such interactions were identified previously. As such, one would expect a heightened level of such interactions. Here, we demonstrate the ductile behavior of the glassy CuAAC foam with high strain capability in compression compared to the brittle behavior of the glassy epoxy-amine foam as shown in Fig. 2. The CuAAC and epoxy-amine foams were compressed to 80% strain at a rate of 0.01 mm s−1 at ambient temperature (T ≪ Tg), and the shape-memory recovery behavior induced by temperature were evaluated over the multiple cycles of compression. It is noted that the epoxy-amine foam failed at 70% strain during the first compression cycle while the CuAAC foam was able to compress to 80% strain at ambient temperature over five successive cycles without failure, essentially eliminating the porous volume of the material in each cycle. The stress–strain response of the CuAAC foam to compression in Fig. 2(a) showed a typical behavior that is representative of the compression of a porous material. In addition, the homogeneous nature of the CuAAC network would help to distribute the stress equally through the network which is essential to minimize stress localization that would otherwise result in crack initiation. Over the course of each compression cycle, CuAAC foams experienced linear elastic and/or plastic deformation as well as densification. A decrease in the mechanical performance of the CuAAC foam after each cycle was also seen where the maximum stress decreased slightly for the first three cycles – 50, 48, 45 MPa respectively, from the first to the third cycle, while it decreased more significantly in the fourth cycle to 31 MPa. Similarly, the energy required to compress the CuAAC foam to 80% strain consequently decreased with compression subsequent cycles as presented in Fig. 2(b). This decline in maximum stress as well as energy required to compress 80% strain indicates that structural bonds in the foam are breaking after each cycle as a result of the applied strain. However, the foam dimensions were effectively the same even after a significant crack was initiated at the end of the fifth cycle of compression as shown in Fig. S4.† While the CuAAC foam exhibited toughness of 850 MJ m−3 when compressed to 80% strain without failure, the epoxy-amine foam presented almost three times lower toughness of 300 MJ m−3 and brittle behavior with failure at 70% strain during the first compression cycle as shown in Fig. 2(c). The image in Fig. 2(c) also illustrates the epoxy-amine foam was severely damaged after the first compression cycle. | ||
| Fig. 2 (a) A typical stress–strain response for 5 successive compression cycles performed on a single specimen of the CuAAC foams. The cylindrical CuAAC foam was compressed consecutively to 80% at ambient temperature with a strain rate of 0.01 mm s−1. The toughness or energy to 80% strain was calculated after the first cycle to be 850 MJ m−3. (b) Average toughness for 4 specimens of the CuAAC foams. Each specimen was compressed to 80% strain at ambient temperature (T ≪ Tg) with a strain rate of 0.01 mm s−1, followed by heating above Tg for 5 successive cycles. (c) Stress–strain curve obtained by compressing the epoxy-amine foam with a strain rate of 0.01 mm s−1 at ambient temperature. The energy to 70% strain for the epoxy-amine foam was calculated after the first cycle to be 300 MJ m−3. The image in the left corner shows the epoxy-amine foam severely damaged after the first cycle of compression. | ||
Despite having similar Tg values between these two glassy foams as demonstrated in Fig. 1, the significant difference in mechanical performance and shape memory-recovery behavior between the two foams arises due to the chemical composition differences between the networks. Specifically, the high concentration of triazole moieties present in the CuAAC foam is capable of inducing non-covalent, intermolecular interactions that lead to the observed high ductility achieved over multiple compression cycles as high as 80% strain in the glassy state. Previously, it has been demonstrated in triazole-containing small molecules that these groups are able to hydrogen bond, pi–pi stack, and undergo ring conformation changes.55–57 Furthermore, crosslinked networks made via CuAAC polymerization have demonstrated superior ductile behavior in comparison to methacrylate-based high Tg networks.37,50 To add to these findings, the comparison between the CuAAC and epoxy-amine based foams having equivalent Tg on the thermal reversibility and mechanical performance suggests that triazoles in the CuAAC foam contribute to the enhanced ductility.
Fixity and recovery ratios of the CuAAC foams upon heating above Tg after each compression cycle to 80% strain at ambient temperature were further evaluated and presented in Fig. 3. The foams maintained between 92 and 94% of the compressive strain when unloaded after each cycle. Fig. 3(a) demonstrated the network's ability to efficiently store elastic deformation at temperature well below Tg. The recovery ratios were then measured after reheating the samples above 115 °C. The CuAAC foam was able to recover fully through five successive cycles of deformation to 80% of its original height upon heating, presenting a significant volume change at ambient temperature with great resistance to fracture. This unique recovery behavior of the glassy CuAAC foam when reheated after such a significant plastic deformation at ambient temperature is known as “reversible plasticity”, a form of shape memory in which a material recovers fully when reheated after undergoing a significant plastic deformation at ambient temperature.39 The material was able to recover both the temporal elastic, linear region, and plastic region of deformation. The stress compressed the 10 mm length of SMP CuAAC foam uniformly in the axial direction to form a 2 mm tall disc that, when reheated, returns to its original shape as shown in Fig. 3(a) and the video in the ESI.† In addition, the tensile shape memory behavior of a CuAAC foam was also investigated and is presented in Fig. S5.† The foam was heated to 140 °C, and deformed to 12% strain followed by fixing the shape by cooling down to −10 °C. As a result, the CuAAC foam exhibited a dramatic shape change over a narrow range of temperature as dictated by a sharp glass transition temperature with repeatable shape memory cycles having both the free strain recovery (Rr) and shape fixity (Rf) around 99%. This was in consistent with the tensile shape memory behavior of a non-form CuAAC polymer investigated previously.31
 | ||
| Fig. 3 (a) The permanent shape (left) of a CuAAC foam, the 80% compressed foam (middle), and heating the foam above its Tg results in complete recovery of its original shape (right). The difference in color of the recovered foam is from thermal reduction of copper(II). (b) Average fixity and recovery for 4 specimens of foams. Each specimen was compressed to 80% strain for 5 successive cycles at ambient temperature (T ≪ Tg) with a strain rate of 0.01 mm s−1, followed by recovery of the compressed sample upon heating above Tg. The recovery and fixity were calculated after each cycle of compression. | ||
Since the 80% deformation corresponds closely to the maximum possible compression of the CuAAC foam of 80% porosity, the energy required for deformation of the CuAAC and epoxy-amine foams was also characterized at a relatively low strain (50%) over multiple successive cycles of compression at ambient temperature until failure occurs on each foam as shown in Fig. 4(a). Again the glassy CuAAC foam demonstrated great toughness and ability to recover plastic deformation upon heating above Tg for up to 13 cycles of compression while the epoxy-amine survived only for 2 compression cycles. As expected, each foam's mechanical performance declined along with successive cycles of compression, which indicates breaking of structural bonds occurred at temperature significantly lower than Tg under applied strain.
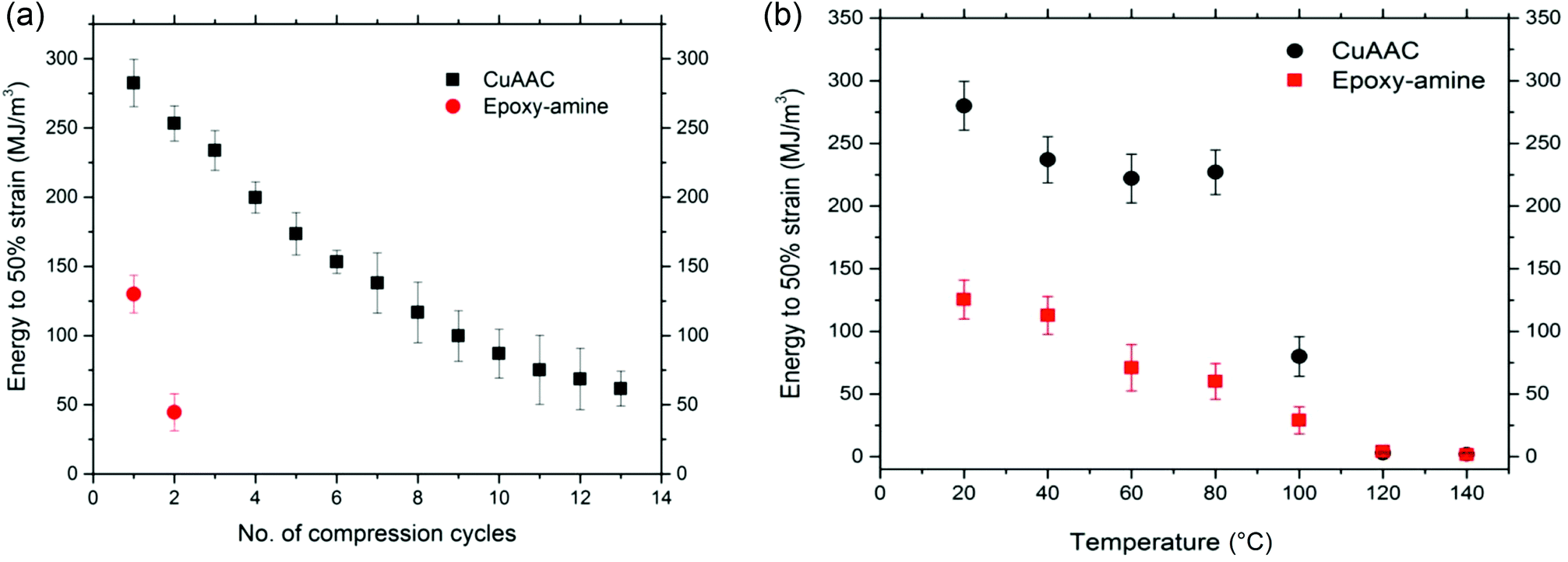 | ||
| Fig. 4 (a) Energy required to achieve 50% compression for CuAAC and epoxy-amine foams (n = 4). Each specimen was compressed repeatedly to 50% strain at ambient temperature (T ≪ Tg) with a strain rate of 0.01 mm s−1 until failure. The CuAAC foam was able to recover an average of 13 successive cycles of compressions upon heating above Tg while the epoxy-amine foam failed after only 2 cycles of compression. (b) The energy required for 50% strain plotted as a function of temperature for CuAAC foam (filled circle) and the epoxy-amine foam (filled square). Seven different foam samples for each system were compressed to 50% at different temperatures: 20, 40, 60, 80, 100, 120, and 140 °C. The energy values of CuAAC and epoxy-amine foams were calculated as the area under the stress–strain curve to 50% strain. | ||
The energy required for deformation of the CuAAC and epoxy-amine foam was also investigated as a function of temperature as shown in Fig. 4(b). Seven foams for each system were compressed to 50% strain at a rate of 0.01 mm s−1 at seven different temperatures: 20, 40, 60, 80, 100, 120, and 140 °C. To ensure uniform heating, each sample was held at a constant temperature for 10 min at each temperature point. Energy was measured as the area under the stress–strain curve. The energy required for both the CuAAC and epoxy-amine is the highest at ambient temperature; however, the CuAAC foam showed 2.5 times higher energy to 50% strain at ambient temperature than the energy of the epoxy-amine foam under the same conditions. As the temperature increases, the polymer becomes more ductile leading to reduction in the deformation resistance, and the energy declines for both the CuAAC and epoxy-amine foam. This behavior is particularly pronounced at temperatures close to the Tg in which both systems show a 97% reduction in energy just above the Tg regions.
Thermal free-strain recovery behavior of CuAAC foams
The thermal free-strain recovery behavior of compressed CuAAC foams is seen in Fig. 5. After being programmed to 80% compressive strain at ∼22 °C, the samples were allowed to freely recover while being heated to Tg + 40 °C at a rate of 3 °C min−1. The samples were then held at 150 °C for 10 min to ensure thermal equilibrium throughout the sample. In each cycle, the foams remained relatively stable up until the onset of glass transition (∼100 °C), as only 10% strain was recovered. After which, the foams rapidly expanded to at least 90% of its original length within the glass transition region. Full recovery was eventually achieved when the samples reached 150 °C and were held isothermally. While the overall recovery ratios after each compression cycle remained constant, the onset of recovery increased with each compression cycle. For example, the response of the 4th heating cycle was ∼20 °C higher compared to the 1st cycle. This delay in recovery is likely caused by the mechanical damage accumulated throughout the porous structure from cycle to cycle. | ||
| Fig. 5 Four sequential cycles of compression and recovery for a CuAAC foam. The foam was compressed to 80% strain at ambient temperature with a strain rate of 0.01 mm s−1, followed by recovery of the compressed sample upon heating to Tg + 40 °C with a heating rate of 3 °C min−1 under preload (0.15 N). Samples were held isothermal at 150 °C for 10 min between cycles. The same sample was compressed four times while monitoring the recovery as a function of temperature after each compression cycle. To observe thermal-recovery behavior of the foam more precisely, the sample was placed in contact with the compression plates used at both ends to monitor strain. | ||
Conclusions
This work has demonstrated a facile approach to fabricate foams based on CuAAC polymerizations with outstanding shape memory attributes that are formed from highly tough networks, particularly as compared to traditional epoxy-amine foams. The unique recovery with the capability for recoverable plastic deformation at ambient temperature under cyclic deformations as well as the stiffness and toughness of the glassy CuAAC network make these materials desirable as high-strain SMP foams. The CuAAC network also demonstrated a remarkable, reversible plasticity by presenting complete recovery of its original length through five consecutive compression cycles of 80% strain upon heating. The ability of the CuAAC foam to compact at ambient temperature to a small volume without fracture, having an easily tunable Tg with a wide range of monomer backbone structures combined with the unique chemistry of CuAAC such as chemoselectivity and orthogonally opens the door for porous CuAAC polymers to be widely employed in various smart material applications.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This research was funded by Saudi Arabia Culture Mission (SACM), Industry, University Cooperative Research Program (IUCRC), the National Science Foundation (Grant DMR 1310528), and the National Institutes of Health (NIH: 5U01DE023774).References
- A. Lendlein and S. Kelch, Angew. Chem., Int. Ed., 2002, 41, 2034–2057 CrossRef CAS.
- P. T. Mather, X. Luo and I. A. Rousseau, Annu. Rev. Mater. Res., 2009, 39, 445–471 CrossRef CAS.
- K. Gall, C. M. Yakacki, Y. Liu, R. Shandas, N. Willett and K. S. Anseth, J. Biomed. Mater. Res., Part A, 2005, 73, 339–348 CrossRef PubMed.
- D. Klempner, V. Sendijarevic and R. M. Aseeva, Handbook of Polymeric Foams and Foam Technology, Hanser, 2nd edn, 2004 Search PubMed.
- K. Hearon, P. Singhal, J. Horn, W. S. Iv, C. Olsovsky, K. C. Maitland, T. S. Wilson and D. J. Maitland, Polym. Rev., 2013, 53, 41–75 CrossRef CAS PubMed.
- G. Li and D. Nettles, Polymer, 2010, 51, 755–762 CrossRef CAS.
- D. Zhang, K. M. Petersen and M. A. Grunlan, ACS Appl. Mater. Interfaces, 2013, 5, 186–191 CAS.
- J. N. Rodriguez, M. W. Miller, A. Boyle, J. Horn, C.-K. Yang, T. S. Wilson, J. M. Ortega, W. Small, L. Nash, H. Skoog and D. J. Maitland, J. Mech. Behav. Biomed. Mater., 2014, 40, 102–114 CrossRef CAS PubMed.
- J. N. Rodriguez, F. J. Clubb, T. S. Wilson, M. W. Miller, T. W. Fossum, J. Hartman, E. Tuzun, P. Singhal and D. J. Maitland, J. Biomed. Mater. Res., Part A, 2014, 102, 1231–1242 CrossRef PubMed.
- C. M. Yakacki and K. Gall, in Advances in Polymer Science, 2010, vol. 226, pp. 147–175 Search PubMed.
- M. A. Di Prima, M. Lesniewski, K. Gall, D. L. McDowell, T. Sanderson and D. Campbell, Smart Mater. Struct., 2007, 16, 2330–2340 CrossRef CAS.
- A. Metcalfe, A.-C. Desfaits, I. Salazkin, L. H. Yahia, W. M. Sokolowski and J. Raymond, Biomaterials, 2003, 24, 491–497 CrossRef CAS PubMed.
- H. Tobushi, K. Okumura, M. Endo and S. Hayashi, J. Intell. Mater. Syst. Struct., 2001, 12, 283–287 CrossRef CAS.
- S. E. Chung and C. H. Park, J. Appl. Polym. Sci., 2010, 117, 2265–2271 CrossRef CAS.
- P. Singhal, J. N. Rodriguez, W. Small, S. Eagleston, J. Van de Water, D. J. Maitland and T. S. Wilson, J. Polym. Sci., Part B: Polym. Phys., 2012, 50, 724–737 CrossRef CAS PubMed.
- M. A. Di Prima, K. Gall, D. L. McDowell, R. Guldberg, A. Lin, T. Sanderson, D. Campbell and S. C. Arzberger, Mech. Mater., 2010, 42, 405–416 CrossRef.
- K. S. S. Kumar, R. Biju and C. P. R. Nair, React. Funct. Polym., 2013, 73, 421–430 CrossRef.
- E. A. Squeo and F. Quadrini, Smart Mater. Struct., 2010, 19, 105002 CrossRef.
- M. Di Prima, K. Gall, D. L. McDowell, R. Guldberg, A. Lin, T. Sanderson, D. Campbell and S. C. Arzberger, Mech. Mater., 2010, 42, 304–314 CrossRef.
- M. A. Di Prima, K. Gall, D. L. McDowell, R. Guldberg, A. Lin, T. Sanderson, D. Campbell and S. C. Arzberger, Mech. Mater., 2010, 42, 315–325 CrossRef.
- S. Kunz-Douglass, P. W. R. Beaumont and M. F. Ashby, J. Mater. Sci., 1980, 15, 1109–1123 CrossRef CAS.
- V. A. Beloshenko, Y. E. Beygelzimer, A. P. Borzenko and V. N. Varyukhin, Composites, Part A, 2002, 33, 1001–1006 CrossRef.
- Y. Liu, C. Han, H. Tan and X. Du, Polym. Adv. Technol., 2011, 22, 2017–2021 CrossRef CAS.
- B. J. Tiimob, V. K. Rangari and S. Jeelani, J. Appl. Polym. Sci., 2014, 131, 40867 CrossRef.
- J. Chen, A. J. Kinloch, S. Sprenger and A. C. Taylor, Polymer, 2013, 54, 4276–4289 CrossRef CAS.
- S. Fu, X. Feng, B. Lauke and Y. Mai, Composites, Part B, 2008, 39, 933–961 CrossRef.
- C. P. Frick, A. L. DiRienzo, A. J. Hoyt, D. L. Safranski, M. Saed, E. J. Losty and C. M. Yakacki, J. Biomed. Mater. Res., Part A, 2014, 102, 3122–3129 CrossRef PubMed.
- G. M. Odegard and A. Bandyopadhyay, J. Polym. Sci., Part B: Polym. Phys., 2011, 49, 1695–1716 CrossRef CAS.
- A. S. Argon and R. E. Cohen, Polymer, 2003, 44, 6013–6032 CrossRef CAS.
- H. B. Song, N. Sowan, P. K. Shah, A. Baranek, A. Flores, J. W. Stansbury and C. N. Bowman, Dent. Mater., 2016, 32, 1332–1342 CrossRef CAS PubMed.
- M. K. McBride, T. Gong, D. P. Nair and C. N. Bowman, Polymer, 2014, 55, 5880–5884 CrossRef CAS PubMed.
- A. Baranek, H. B. Song, M. McBride, P. Finnegan and C. N. Bowman, Macromolecules, 2016, 49, 1191–1200 CrossRef CAS PubMed.
- C. M. Yakacki, A. M. Ortega, C. P. Frick, N. Lakhera, R. Xiao and T. D. Nguyen, Macromol. Mater. Eng., 2012, 297, 1160–1166 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS PubMed.
- C. Barner-Kowollik, F. E. Du Prez, P. Espeel, C. J. Hawker, T. Junkers, H. Schlaad and W. Van Camp, Angew. Chem., Int. Ed., 2011, 50, 60–62 CrossRef CAS PubMed.
- A. A. Alzahrani, D. P. Nair, D. J. Smits, M. Saed, C. M. Yakacki and C. N. Bowman, Chem. Mater., 2014, 26, 5303–5309 CrossRef CAS.
- A. U. Shete and C. J. Kloxin, Polym. Chem., 2017, 8, 3668–3673 RSC.
- T. Gong, B. J. Adzima, N. H. Baker and C. N. Bowman, Adv. Mater., 2013, 25, 2024–2028 CrossRef CAS PubMed.
- D. D. Díaz, S. Punna, P. Holzer, A. K. Mcpherson, K. B. Sharpless, V. V. Fokin and M. G. Finn, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 4392–4403 CrossRef.
- M. Malkoch, R. Vestberg, N. Gupta, L. Mespouille, P. Dubois, A. F. Mason, J. L. Hedrick, Q. Liao, C. W. Frank, K. Kingsbury and C. J. Hawker, Chem. Commun., 2006, 2774–2776 RSC.
- B. Sandmann, B. Happ, M. D. Hager, J. Vitz, E. Rettler, P. Burtscher, N. Moszner and U. S. Schubert, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 239–247 CrossRef CAS.
- A. A. Alzahrani, A. H. Erbse and C. N. Bowman, Polym. Chem., 2014, 5, 1874–1882 RSC.
- B. J. Adzima, Y. Tao, C. J. Kloxin, C. A. DeForest, K. S. Anseth and C. N. Bowman, Nat. Chem., 2011, 3, 256–259 CrossRef CAS PubMed.
- Y. Liu, D. D. Díaz, A. A. Accurso, K. B. Sharpless, V. V. Fokin and M. G. Finn, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 5182–5189 CrossRef CAS.
- B. M. El-Zaatari, A. U. Shete, B. J. Adzima and C. J. Kloxin, Phys. Chem. Chem. Phys., 2016, 18, 25504–25511 RSC.
- M. A. Tasdelen and Y. Yagci, Tetrahedron Lett., 2010, 51, 6945–6947 CrossRef CAS.
- H. B. Song, A. Baranek and C. N. Bowman, Polym. Chem., 2016, 7, 603–612 RSC.
- B. Sandmann, B. Happ, J. Vitz, M. D. Hager, P. Burtscher, N. Moszner and U. S. Schubert, Polym. Chem., 2013, 4, 3938–3942 RSC.
- A. U. Shete, B. M. El-Zaatari, J. M. French and C. J. Kloxin, Chem. Commun., 2016, 52, 10574–10577 RSC.
- H. B. Song, X. Wang, J. R. Patton, J. W. Stansbury and C. N. Bowman, Dent. Mater., 2017, 33, 621–629 CrossRef CAS PubMed.
- S. Bräse, C. Gil, K. Knepper and V. Zimmermann, Angew. Chem., Int. Ed., 2005, 44, 5188–5240 CrossRef PubMed.
- W. L. Murphy, R. G. Dennis, J. L. Kileny and D. J. Mooney, Tissue Eng., 2002, 8, 43–52 CrossRef CAS PubMed.
- A. F. Senyurt, C. E. Hoyle, H. Wei, S. G. Piland and T. E. Gould, Macromolecules, 2007, 40, 3174–3182 CrossRef CAS.
- A. F. Senyurt, H. Wei, C. E. Hoyle, S. G. Piland and T. E. Gould, Macromolecules, 2007, 40, 4901–4909 CrossRef CAS.
- Y. Zhu, G. Wang, R. Wang and Z. Li, Cryst. Growth Des., 2009, 9, 4778–4783 CAS.
- D. Zornik, R. M. Meudtner, T. El Malah, C. M. Thiele and S. Hecht, Chem. – Eur. J., 2011, 17, 1473–1484 CrossRef CAS PubMed.
- Z. Karimov, I. Abdugafurov and B. Tashkhodjaev, (1-Phenyl-1H-1,2,3-triazol-4-yl)methyl pyridine-3-carboxylate, International Union of Crystallography, 2010, vol. 66 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7py01121k |
| This journal is © The Royal Society of Chemistry 2018 |