
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Biosynthesis and incorporation of an alkylproline-derivative (APD) precursor into complex natural products†
J.
Janata
 *,
Z.
Kamenik
,
R.
Gazak
,
S.
Kadlcik
and
L.
Najmanova
*,
Z.
Kamenik
,
R.
Gazak
,
S.
Kadlcik
and
L.
Najmanova
Institute of Microbiology, Czech Academy of Sciences, BIOCEV, Průmyslová 595, 252 50, Vestec, Czech Republic. E-mail: janata@biomed.cas.cz
First published on 8th March 2018
Abstract
Covering: up to 2017
This review covers the biosynthetic and evolutionary aspects of lincosamide antibiotics, antitumour pyrrolobenzodiazepines (PBDs) and the quorum-sensing molecule hormaomycin. These structurally and functionally diverse groups of complex natural products all incorporate rarely occurring 4-alkyl-L-proline derivatives (APDs) biosynthesized from L-tyrosine through an unusual specialized pathway catalysed by a common set of six proteins named Apd1–Apd6. We give an overview of APD formation, which involves unusual enzyme activities, and its incorporation, which is based either on nonribosomal peptide synthetase (PBDs, hormaomycin) or a unique hybrid ergothioneine-dependent condensation system followed by mycothiol-dependent sulphur atom incorporation (lincosamides). Furthermore, within the public databases, we identified 36 novel unannotated biosynthetic gene clusters that putatively encode the biosynthesis of APD compounds. Their products presumably include novel PBDs, but also novel classes of APD compounds, indicating an unprecedented potential for the diversity enhancement of these functionally versatile complex metabolites. In addition, phylogenetic analysis of known and novel gene clusters for the biosynthesis of APD compounds allowed us to infer novel evolutionary hypotheses: Apd3 methyltransferase originates from a duplication event in a hormaomycin biosynthetic gene cluster ancestor, while putative Apd5 isomerase is evolutionarily linked to PhzF protein from the biosynthesis of phenazines. Lastly, we summarize the achievements in preparing hybrid APD compounds by directing their biosynthesis, and we propose that the number of nature-like APD compounds could by multiplied by replacing L-proline residues in various groups of complex metabolites with APD, i.e. by imitating the natural process that occurs with lincosamides and PBDs, in which the replacement of L-proline for APD has proved to be an evolutionary successful concept.
 Z. Kamenik, R. Gazak, J. Janata, L. Najmanova and S. Kadlcik | Zdenek Kamenik acquired his PhD in analytical chemistry from Charles University in Prague in 2011 and then he spent a year as a postdoc in the group of Greg Challis at the University of Warwick, UK. Currently, he is a researcher in the group of Jiri Janata. Radek Gažák obtained his PhD in organic chemistry at the Institute of Chemical Technology in Prague, in 2006. He joined the group of Jiri Janata in 2012. Jiri Janata received his PhD in microbiology from the Institute of Microbiology, Academy of Sciences of the Czech Republic in 1995. He then moved to Yale University, School of Medicine as a post-doctoral fellow working with Wayne A. Fenton on inherited diseases caused by defects in amino acid metabolism. Since 2005, he has been a head of the laboratory for biology of secondary metabolism of the Institute of Microbiology AS CR in Prague. His research interests include biosynthesis of microbial natural products and resistance to antibiotics. Lucie Najmanova obtained a PhD in microbiology in 2002 from Charles University in Prague. She has been working with Jiri Janata focusing on the biosynthesis of microbial natural compounds since 2009. Stanislav Kadlcik received his PhD in microbiology from the University of Chemistry and Technology, Prague in 2014. He is a postdoctoral fellow in the laboratory of Jiri Janata focusing on the biosynthesis of natural products. |
1 Introduction
Most natural products synthesized in specialized secondary metabolism pathways are complex compounds consisting of several building blocks. These blocks can be regular intermediates supplied by primary metabolism, e.g. proteinogenic amino acids or acyl-CoA molecules. However, more often, several unusual precursors synthesized in specialized biosynthetic pathways are assembled into the final natural product. Additionally, each individual unusual precursor can be incorporated into several different structural contexts, i.e. it can be combined with distinct types of building blocks, resulting in complex natural products from structurally diverse families. Both the biosynthesis of specialized precursors and even more importantly their assembling systems, which are able to combine these specialized precursors, form the molecular evolution foundation for an enormous diversity of secondary metabolites. The overall prokaryotic biosynthetic potential has been partially uncovered over the last few years by global genome sequencing efforts and by the extensive analysis of known biosynthetic gene clusters (BGCs).1,2 BGCs of complex natural products exhibit sub-cluster mosaic patterns,3 where the biosynthesis of each specialized precursor is encoded by a specific gene sub-cluster, which represents an evolutionary independent subgroup of genes. These sub-clusters spread autonomously by horizontal gene transfer (HGT) among otherwise unrelated BGCs, resulting in the incorporation of encoded building blocks in a new structural context. Additionally, during evolution, sub-clusters pass through gene gain/loss changes, resulting in a variable tailoring of the complex compound moieties.4-Alkyl-L-proline derivatives (APDs) represent an example of a rarely occurring specialized building block. Although structurally similar to L-proline, it has been long known that APD is biosynthesized from another proteinogenic amino acid, L-tyrosine, through a specialized biosynthetic pathway unrelated to the biological formation of pyrroles.4 APD can be incorporated into at least three structurally and functionally diverse families of microbial complex natural compounds (Fig. 1), including the large group of antitumour agents pyrrolo[2,1-c][1,4]benzodiazepines (PBDs; for review see Gerratana5), the signalling molecule hormaomycin,6 and last but not least, the lincosamide antibiotic lincomycin.7 As expected, all sequenced BGCs coding for complex natural products with an APD moiety (hereinafter referred to as APD compounds) exhibit the following typical mosaic pattern: all of them share a set of five or six homologous genes, the APD biosynthetic gene sub-cluster (red arrows in Fig. 2), encoding a uniform APD moiety scaffold; whereas, the remaining part of BGCs is completely different for each group of APD compounds. Additionally, a variable set of genes, which encode post-condensation APD-tailoring enzymes, is present, especially in the BGCs of PBD compounds (orange arrows in Fig. 2), which results in a diversity of their APD moieties (Fig. 1).
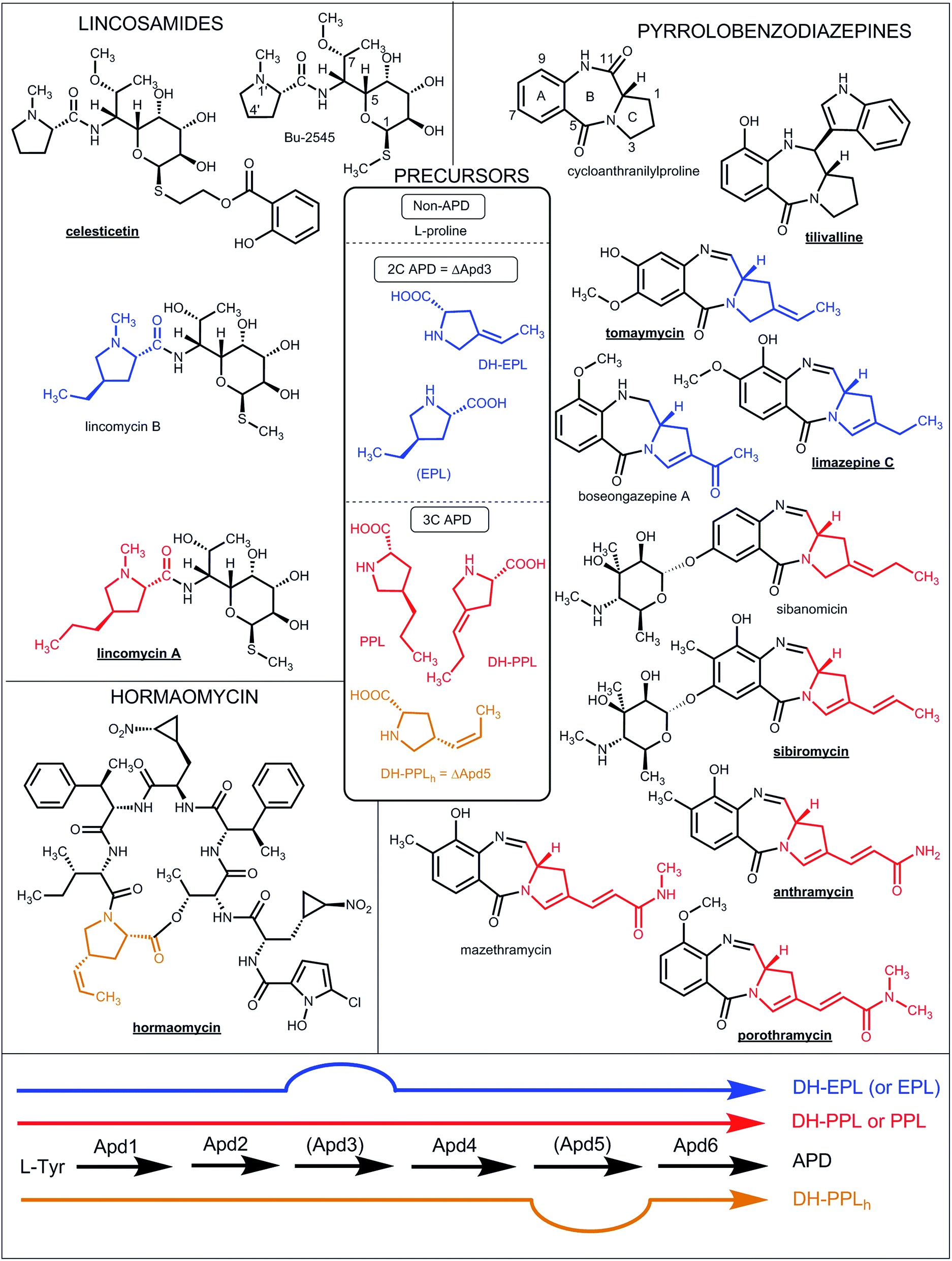 | ||
| Fig. 1 Natural lincosamides, pyrrolobenzodiazepines and hormaomycins incorporating L-proline or APDs. The names of compounds with already sequenced BGCs are in bold and underlined. The coloured lines at the bottom represent the APD biosynthetic pathways (for the detailed assignment of the APD biosynthetic proteins see Fig. 7 in Section 3.2) starting from L-tyrosine and resulting in the 2C APDs 4-ethylidene-L-proline (DH-EPL) and 4-ethyl-L-proline (EPL) (blue line; the Apd3 biosynthetic step is not involved), 3C APD 4-((Z)-propenyl)-L-proline (DH-PPLh; orange line; the Apd5 biosynthetic step is not involved) or 3C APDs 4-propyl-L-proline (PPL) and 4-propylidene-L-proline (DH-PPL) (red line). APD precursors (in the central frame) and the APD moieties in the final structures are coloured correspondingly. EPL in brackets indicates that it is only a side-product in PPL biosynthesis. | ||
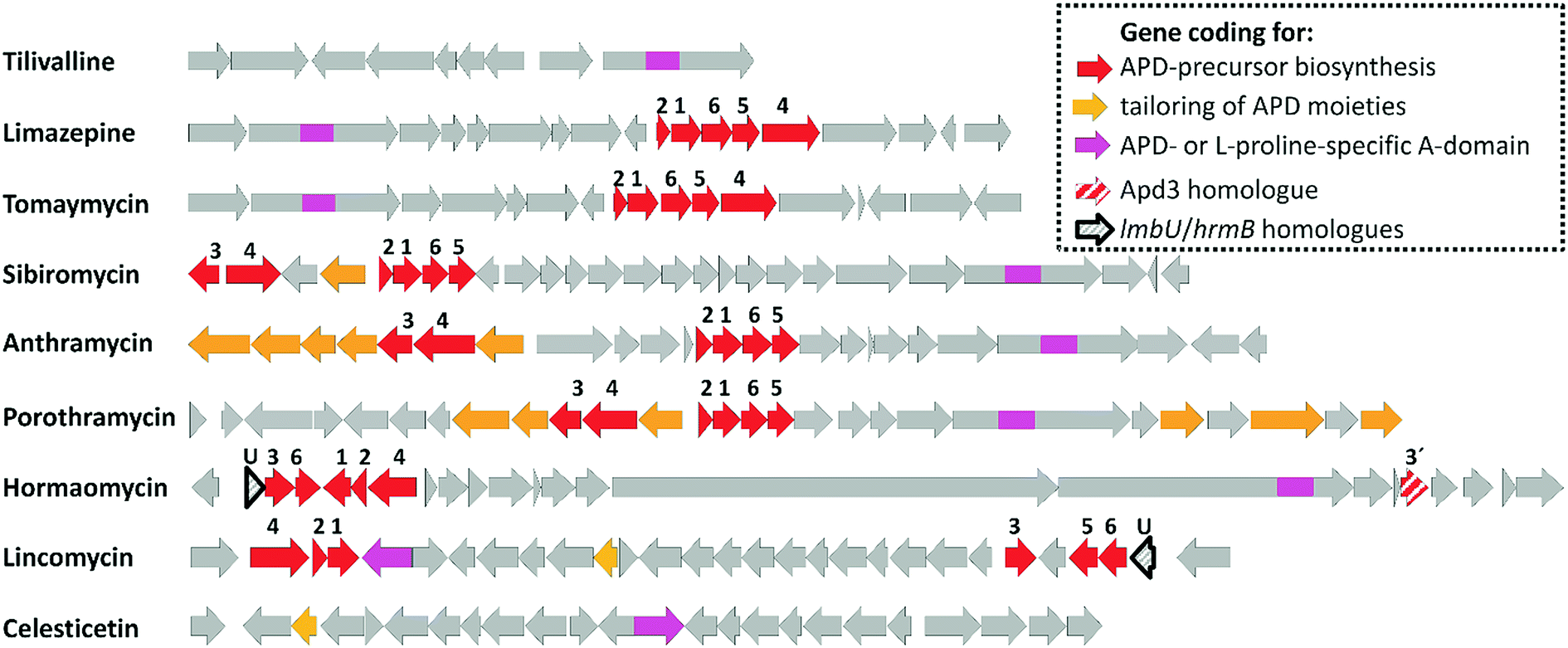 | ||
| Fig. 2 BGCs of APD compounds and two related non-APD compounds incorporating proteinogenic L-proline. The GenBank accession numbers for the respective BGCs are: HG425356 (tilivalline); KT381463 (limazepine); FJ768957 (tomaymycin); FJ768674 (sibiromycin); EU195114 (anthramycin); HQ872605 (porothramycin); HQ542230 (hormaomycin); EU124663 (lincomycin) and GQ844764 (celesticetin). The numbers above the red arrows correspond to the numbering of the apd genes (1 stands for apd1, 2 for apd2, etc.; for detailed assignment of the encoded Apd biosynthetic proteins see Fig. 7 in Section 3.2). The red striped arrows in the hormaomycin BGC represent a gene hrmS homologous to apd3, i.e. coding for the second C-methyltransferase; the black framed arrows “U” correspond to the putative regulatory gene lmbU in the lincomycin BGC and its homologue hrmB in the hormaomycin BGC. All of the remaining biosynthetic, resistance and regulatory or non-assigned genes are in grey. | ||
For the first time, this review covers the biosynthetic and evolutionary aspects of all three above-mentioned groups of APD compounds. We take advantage of several cutting edge papers published over the last few years on APD biosynthesis.8–10 We also draw on the recently discovered amazing model of APD incorporation into the truly “natural hybrid compound” lincomycin, where the amino acid APD is combined with amino-octose, another unusual precursor.7,11–17
Briefly, APD compounds represent a functionally beneficial alternative to related complex compounds with an integrated L-proline moiety (Chapter 2). Biosynthesis of the unified APD, which is incorporated into all APD compounds, is encoded by a nearly identical set of genes spread among BGCs by an HGT mechanism (Chapter 3). The APD precursor is then activated by the APD-specific adenylation domain (A-domain), an integral part of group-specific condensation systems. For all three groups of APD compounds, these APD-activating A-domains evolved independently by the adaptation of an ancestral L-proline-specific A-domain (Chapter 4). Finally, the activated APD precursor is integrated into different (in PDBs and hormaomycin; Chapter 5) or even radically different (in lincomycin; Chapter 6) structural contexts depending on the group-specific condensation system. In principle, even though APD biosynthetically originates from L-tyrosine, it structurally mimics L-proline and can therefore be incorporated into any complex compound instead of the L-proline precursor if the following two prerequisites are met: first, acceptance of the APD biosynthetic gene sub-cluster by the BGC of the L-proline-incorporating compound, and second, adaptation of the original L-proline-specific A-domain to use APD (Chapter 7).
2 Gene-structure-function basis of complex APD compounds
2.1 Overview of APD compounds and the encoding BGCs
So far, the APD moiety has been found in approximately two dozen complex natural products belonging to three groups of compounds; however, the distribution of APD compounds among these groups is very uneven. Except for hormaomycin and lincomycin, all the structurally characterized APD compounds belong to PBDs (Fig. 1 and 3). Within this group, a particularly high variability is typical among the APD moieties, including the length of the alkyl side-chain (two-carbon (2C APD) or three-carbon (3C APD)) and the further tailoring of the APD moiety.All hitherto structurally characterized APD natural compounds are microbial secondary metabolites produced specifically by Actinobacteria. To date, BGC sequences for seven structurally characterized APD compounds have been published (Fig. 2). Five of them encode PBDs: anthramycin produced by Streptomyces refuineus subsp. thermotolerans NRRL 3143,18 sibiromycin produced by Streptosporangium sibiricum ATCC 29053 (ref. 19) and porothramycin produced by Streptomyces albus subsp. albus ATCC 39897,20 all with 3C APD moieties, while tomaymycin produced by Streptomyces achromogenes var. tomaymyceticus21 and limazepine produced by Streptomyces sp. ICBB 8177,22 which have 2C APD moieties. The two remaining BGCs encode non-PBD APD compounds: hormaomycin produced by Streptomyces griseoflavus W-3846 and lincomycin produced by Streptomyces lincolnensis from the publicly unavailable industrial strain S. lincolnensis (78–11)23 or type strain S. lincolnensis ATCC 25466.24 Based on a comparison of the BGCs for APD compounds (Fig. 2), it is evident that a sub-cluster of five or six “red-arrow” genes, named apd1–apd6, shared across all BGCs for APD compounds should encode the common APD-precursor scaffold. When examining the evolutionary aspect, it is important to mention that there are PBDs as well as lincosamides in which L-proline is incorporated in place of an APD precursor (Fig. 1). Of these, BGCs for the PBD tilivalline produced by Klebsiella oxytoca25 and lincosamide celesticetin produced by Streptomyces caelestis13 are available (Fig. 2). As expected, the sub-cluster of apd1–apd6 genes is completely absent in both BGCs. Note that the production of non-APD PBDs is not limited to only Actinobacteria, as tilivalline is produced by γ-proteobacteria and cycloanthranilylproline, for example, by myxomycete Fuligo candida.26 It appears that the highly specialized APD biosynthetic pathway is the limiting factor maintaining the biosynthesis of APD complex compounds within Actinobacteria. Some groups of secondary metabolites of lower complexity, namely phenazines2 or related anthranilate derivatives,27 can be produced by a wide range of bacteria or filamentous fungi. The anthranilate derivative is the second precursor of PBD biosynthesis (see Section 2.2).
Comparative analysis of related BGCs was considered a useful tool for the identification of the APD biosynthetic gene sub-cluster, but failed to assign the functions to particular encoded biosynthetic proteins, postulate the order of biosynthetic steps in the APD pathway or even to elucidate APD precursor incorporation. An unexpected biochemistry, unusual enzymology and fascinating combinatorial potential in natural product synthesis (see the editorial in a recent special issue of Chemical Reviews)28 are behind the inspiration for the pathway-engineered approaches towards valuable bioactive compounds. However, the same challenging features seriously complicate the gene-to-molecule prediction, especially if an intermediate hydrolytic maturation step is involved, (i.e. a larger precursor is cleaved to form smaller constituents).29 Specifically, APD biosynthesis is based on the unprecedented reorganization of L-tyrosine into a structure mimicking the L-proline derivative.30,31 Additionally, APD incorporation into the lincomycin molecule is catalysed by a unique condensation system that involves hydrolytic maturation.7 The best illustration of these general difficulties is an example of lincosamide biosynthesis elucidated only in the last two to three years (see Chapter 6), i.e. twenty years after publication of lincomycin BGC.23 The functions of proteins participating in APD precursor biosynthesis are therefore based predominantly on the recent results of several groups and a combination of different experimental approaches (see the detail in Sections 3.1 and 3.2).
2.2 Pyrrolo[2,1-c][1,4]benzodiazepines (PBDs)
In the so far described natural PBDs, the A-ring can be modified at C-7, C-8 and/or C-9 by hydroxylation and/or methylation; the C-7 hydroxyl can then be further glycosylated or methylated. The additional glycosylation of the C-9 hydroxyl has also been published;33 however, in this case, the side-product (DH-sibiromycin diglycoside) structure has not been fully characterized. Modifications of the B-ring are exclusively targeted at C-11. The imine (N-10–C-11) is considered to be the main natural active form of PBDs.5 The imine forms can be easily solvated to carbinolamine or carbinolamine methyl ether forms depending on the purification and storage conditions. However, these forms are not considered as distinct derivatives of PBDs because the transition between imine, carbinolamine and carbinolamine ethers is reversible. Therefore, we propose that different forms of an individual PBD should not be given unique names. Accordingly, we follow the recommendation of Gerratana5 and refer to all PBDs only in their imine forms. A different situation is the irreversible oxidation at C-11, which affords stable cyclic dilactams, i.e. C-11-oxo-derivatives of PBDs.34–36 However, also in this case, we propose that these derivatives should not bear unique names, but instead herein their names are derived from the name of the corresponding non-oxidized PBD using a prefix “oxo“. Further modifications at C-11 are represented by the attachment of an indole residue in the structure of tilivalline, which was shown to proceed non-enzymatically,25,37–39 or by the reduction of the N-10–C-11 double bond to give PBDs with secondary amine groups, as in usabamycins and boseongazepines (Fig. 1 and 3).40,41 The C-ring modifications include hydroxyl and methoxy groups, endocyclic double bonds as well as 2C or 3C side chains at C-2 (for examples see Fig. 1 and 3).
 | ||
| Fig. 3 Additional representatives of natural PBDs. 2C APD moieties are in blue, while 3C APD moieties are in red. | ||
 | ||
| Fig. 4 Crystal structure of the anthramycin-DNA (CCAACGTTGG synthetic decamer) covalent adduct (PDB code 274D).55 DNA strands are coloured orange and yellow; the 3C chain of the APD moiety is red, while the tricyclic PBD scaffold is green. | ||
Seven natural PBDs with a 3C APD moiety in their final structures have been described, including anthramycin,42 sibiromycin,43 usabamycin A,40 mazethramycin,44 porothramycin,45 sibanomicin46 and one C11-oxo derivative oxoanthramycin (originally inappropriately named limazepine H).36 Another six PBDs incorporate a 2C APD moiety: tomaymycin and oxotomaymycin,47 prothracarcin (also named limazepine F),48 oxoprothracarcin,35 limazepine C49 and boseongazepine A.41 Additionally, besides the above-mentioned cycloanthranilylproline, also DC-81 and tilivalline incorporate unsubstituted L-proline.25,50 In the structures of chicamycin, abeymycin, RK-1441A and neothramycins. The proline moiety is further hydroxylated, while in RK-1441 this hydroxyl is additionally methylated.51–53
Once bound to DNA, PBDs have been shown to mediate a number of biological effects in cells, including DNA strand breakage,56 the inhibition of DNA processing enzymes57–59 or transcription factors60,61 and modulation of the signalling pathways.62,63 As many of these proteins and signalling pathways are upregulated in tumour cells, the described DNA binding effects could partially explain the molecular basis of the anticancer activities of PBDs.54 The DNA stabilizing effect has also been described for some PBD compounds incapable of forming the covalent linkage with the minor groove guanine residue. Antonow64 documented a library of synthetic C11-oxo-PBDs that displayed DNA helix-stabilizing activities through non-covalent interactions when the reactive C-11 was blocked. This could presumably explain the documented weak antitumour activity of usabamycins and boseongazepines,40,41 which have a secondary amine instead of an imine in their structures (Fig. 1 and 3).
Finally, besides DNA, PBDs can also target highly distinct biological structures. Natural C-11-oxo-PBD oxoanthramycin (limazepine H, Fig. 3) and its biosynthetically incomplete derivative limazepine G have been documented to inhibit neuraminidase, an enzyme that catalyses the release of progeny influenza viruses from infected host cells.36 Synthetic monomeric PBDs can inhibit human DNA ligase 1 by binding to its catalytic site.65
2.3 Hormaomycin
In contrast with PBDs, hormaomycin does not constitute a compound group with variable APD moieties; it does not even have a counterpart incorporating an L-proline instead of an APD, such as in lincosamides. Actually, a single hormaomycin side-product with a modified APD moiety was described containing a (2S,4R)-4-methylproline residue instead of a 4-propenyl residue.67 However, deuterium-labelled L-DOPA feeding experiments disproved its origin from L-Tyr.
In addition to hormaomycin, two other natural analogues, namely hormaomycin B and C, were isolated from a marine mudflat-derived Streptomyces strain SMN55 collected in Mohang, Korea.68 These analogues were accompanied by hormaomycin, which indicated that they were only side-products of hormaomycin biosynthesis arising from the omission of methylation at one phenylalanine residue and its incorporation instead of (β-Me)Phe.
2.4 Lincomycin (lincosamide antibiotics)
A common scaffold of lincosamide antibiotics apparently consists of two obligatory structural moieties: a specialized amino thio-octose unit condensed by an amide bond with a carboxyl group of an amino acid unit originating from proteinogenic L-proline (Bu-2545 and celesticetin) or APD (lincomycin). In celesticetin, an additional salicylate unit is connected via a two-carbon chain to a sulphur atom on the amino sugar moiety. Interestingly, recent results on lincosamide condensation7 uncovered an employment of another hidden biosynthetic participant: the sulphur atom in a lincomycin molecule (as well as sulphur atom and two-carbon linker of celesticetin) is the only remaining label of the L-cysteinyl from the mycothiol-conjugate precursor (for details see Section 6.2).14–17
In terms of the structural context of APD building block incorporation, the lincomycin molecule is the only known complex natural product where the amino acid APD moiety is attached to a biosynthetically different type of molecule, a specialized amino sugar. In lincosamide biosynthesis evolution, the unusual APD precursor “enters” into the already established complex biosynthetic system based on highly specialized sugar metabolism. This makes lincomycin biosynthesis an amazing model for the biosynthetic evolution of secondary metabolism (see Chapter 6).
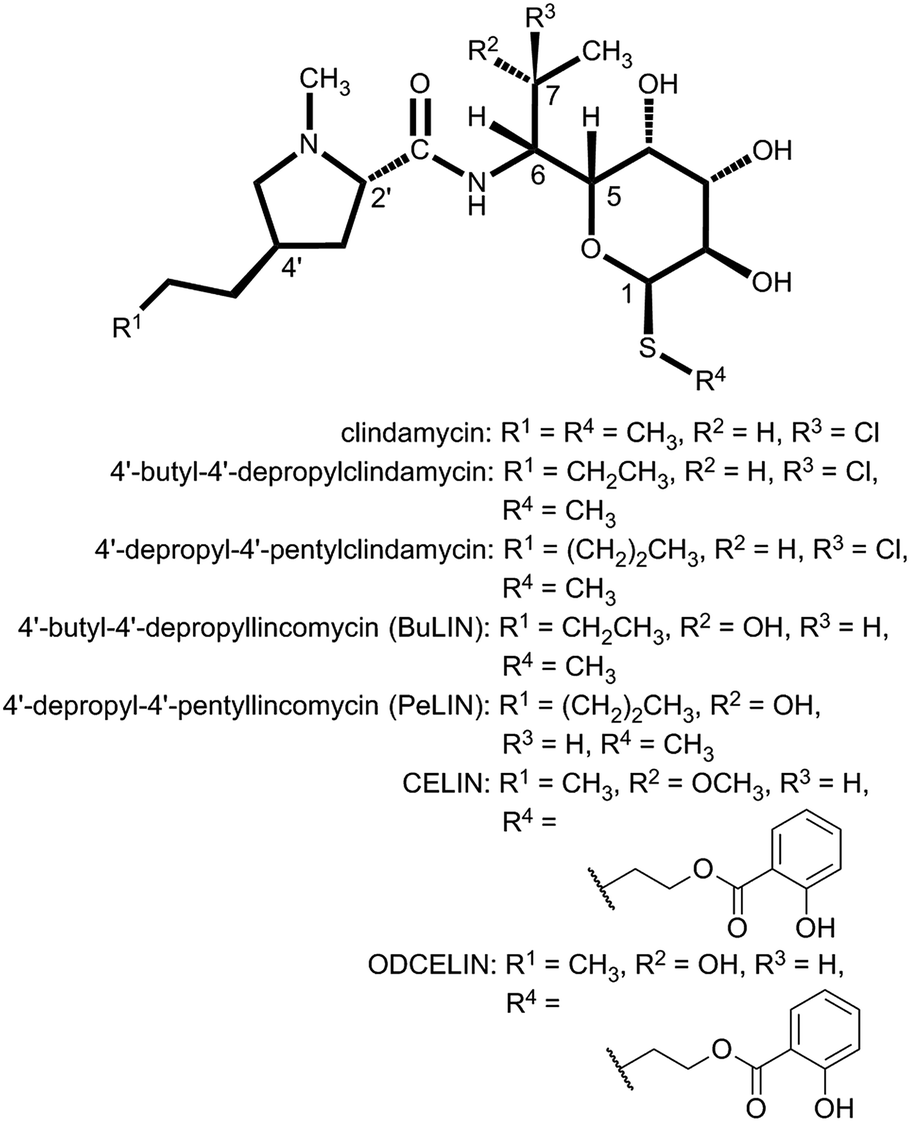 | ||
| Fig. 5 Structures of synthetic and biotechnologically prepared lincosamides. | ||
Their mode of action involves the inhibition of microbial protein synthesis by binding to the peptidyl transferase site of the 50S ribosome subunit and interference with the peptide chain initiation.80 A crystal structure of Deinococcus radiodurans and a later one of the Escherichia coli 50S ribosomal subunit complexed with lincosamide clindamycin documented this binding.81,82 Clindamycin has three hydroxyl groups in its sugar moiety (2-OH, 3-OH and 4-OH) that participate in the hydrogen-bond formation: 2-OH and 3-OH interact with N6 of nucleotide A2058 (E. coli numbering) of 23S rRNA. Dimethylation of the N6 group, which disrupts the hydrogen bonds, causes resistance to lincosamides83 as well as to macrolides occupying an overlapping binding site.84 Also the sulphur atom of the sugar moiety of clindamycin interacts with the 23S rRNA,81 indicating a crucial role for the whole structure of the unusual amino thio sugar moiety in the biological activity of lincosamides. The proline moiety is positioned close to the tyrosyl residue of the puromycin-binding site81 and its optional alkyl side-chain in the case of APD lincosamides (lincomycin, clindamycin) prolongs the lincosamide molecule towards the A-site t-RNA (Fig. 6).
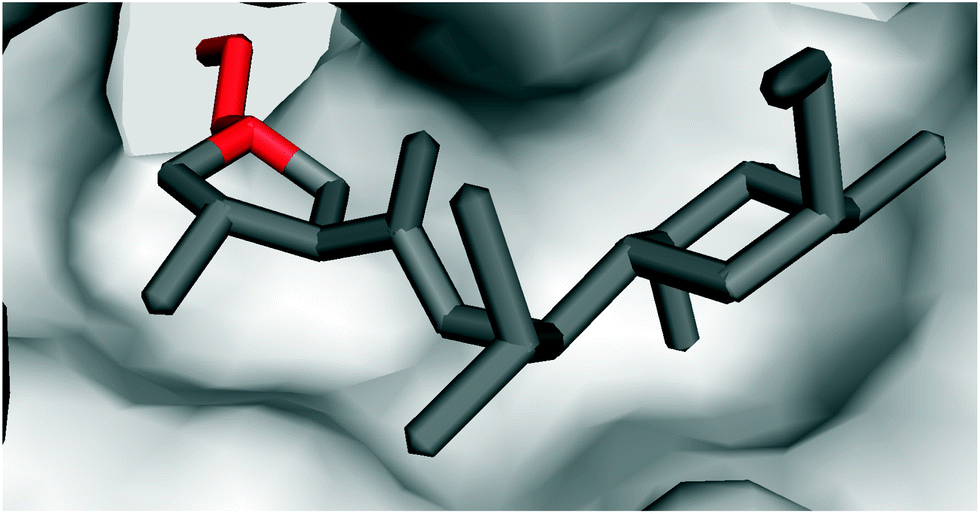 | ||
| Fig. 6 Crystal structure of clindamycin targeting the peptidyl transferase centre in the 50S ribosomal subunit of Deinococcus radiodurans (PDB code 1jzx);81 zoomed in view showing the blockade of the cavity oriented towards the A-site t-RNA. | ||
Moreover, clindamycin exhibits significant antiplasmodial activity85,86 by targeting protein synthesis in the specific plastid organelle, the apicoplast.87 Due to its low toxicity, clindamycin is recommended in combination with quinine for the treatment of Plasmodium falciparum malaria in patient risk groups and as a first-choice drug for women in the first trimester of pregnancy.88
2.5 Functional benefit of APD moieties
As described above, the biological targets and modes of action of PBDs and lincosamides are quite different. However, in both groups, the APD compounds seem to be evolutionary advantageous and exhibit better biological properties when compared to their counterparts that incorporate L-proline.This is more obvious for a small group of natural lincosamide antibiotics. The simplest natural product, Bu-2545 (Fig. 1), which consists of only L-proline- and amino-octose-derived moieties, exhibits antibacterial activity 1–2 order of magnitude lower than that of the relevant APD compound lincomycin.75 The third and last main natural lincosamide, celesticetin (Fig. 1), which also incorporates L-proline, exhibits moderate antibacterial activity (25–50% that of lincomycin).86 However, an unbiased assessment of the APD presence/absence impact on bioactivity can be obtained from a comparison of celesticetin and CELIN (the enzymatically prepared hybrid of lincomycin and celesticetin; Fig. 5), which differ exclusively in their L-proline/APD moiety. The minimal inhibition concentration for Kocuria rhizophila was 1600 nM for celesticetin, 400 nM for lincomycin and only 100 nM for CELIN.17 The impact of the APD alkyl side-chain length is evident from the comparison of lincomycin and lincomycin B (Fig. 1), a 2C APD derivative of lincomycin. Lincomycin B only exhibits approximately 25% of lincomycin activity.78 Finally, the same trend has been documented for synthetic derivatives of lincomycin and clindamycin with a prolonged alkyl side chain (Fig. 5), showing an increase in biological activity with the length of the APD alkyl side chain as 2C < 3C < 4C < 5C (maximum activity = 5C, 6C).86
It is evident that the ribosome binding activity of the lincosamide core structure (Bu-2545) can be increased by both the attachment of a salicylate moiety (celesticetin) at one side of the lincosamide core and by prolongation by the alkyl side chain at the other side (lincomycin). The increased biological activity of the prolonged lincosamide structures corresponds well with the known structure of clindamycin co-crystallized with the 50S ribosomal subunit81 (Fig. 6). This shows that the prolonged shape of the molecule by the APD alkyl side chain (red in Fig. 6) perfectly fits with the cavity oriented towards the A-site t-RNA.
The molecular basis for the positive effect of the APD side chain on the activity of the PBDs is different but analogous. The mechanism of action is based on the fitting of the PBD molecule into the DNA minor groove. The molecule has to fit into a defined space as well as possible, and the elongated APD side chain can be advantageous (see Fig. 4). The DNA binding activity of PBDs results from the sum of all the modifications to the molecule (for details see Chapter 5). However, the positive effect of the APD side chain length is evident from a comparison of the DNA affinity of several APDs, which showed the following trend from highest to lowest affinity: 3C APD sibiromycin > 3C APD anthramycin > 2C APD tomaymycin > non-APD DC-81 > non-APD neothramycin.58,89 Moreover, it was documented, that the non-covalent interactions between the DNA and PBD affect the recognition of the target sequence (i.e. finding the best low-energy configuration of DNA–PBD complex)57 and that the length and modifications of the APD side chain could (together with other modifications of the PBD scaffold) influence the sequence specificity of PBD binding and therefore qualitatively change the resulting biological effects.
In summary, the above-mentioned facts suggest that APD compounds and the APD biosynthetic pathway evolved as evolutionary more advantageous variants of their L-proline ancestors. Even though there are no described free APDs as final natural products, the establishment and evolution of the APD biosynthetic pathway was a crucial prerequisite for the molecular evolution of several groups of bioactive natural products.
3 APD precursor biosynthesis
3.1 Early labelling studies
The elucidation of the APD biosynthetic pathway began with lincomycin biosynthesis at the turn of 1960s' and 70's in the last millennium, a period characteristic of the extensive use of radiolabelled substrates. In 1969, Argoudelis et al. found out by fermentation of a culture of S. lincolnensis with 14C and deuterium-labelled substrates, followed by degradation of the formed lincomycin, that both the N–CH3 and terminal C–CH3 methyl groups of the lincomycin 4-n-propyl-L-hygric acid (N-methyl-4-propyl-L-proline) moiety originate from L-methionine (through biosynthetically formed S-adenosylmethionine, SAM).90 Later it was shown that 4-ethyl-L-proline (EPL) and 4-propyl-L-proline (PPL) accumulate in the culture broth of S. lincolnensis when the growth media was sulphur limited.91 The same authors proved using isotopically labelled substrates that the precursor of both EPL and PPL was L-tyrosine and that only seven carbon atoms out of the original nine carbons in L-tyrosine were incorporated into these precursors. Further contributions to APD biosynthesis were accomplished with PBDs based on double labelling and stable isotope experiments on anthramycin,92 tomaymycin34 and sibiromycin93 biosynthesis. These experiments indicated that an extradiol cleavage mechanism is involved in the pathway. Indeed, extradiol cleavage of L-DOPA is the key initial step in APD biosynthesis and was finally proven in an excellent study on lincomycin biosynthesis using deuterated- and 13C-labelled substrates in combination with 13C NMR and mass spectral analysis.4 The next fragment of knowledge was added to APD biosynthesis in 1992 with identification of an intermediate in the later phase of this pathway, here compound 6 (for structure see Scheme 1).94 This intermediate was isolated from the lincomycin non-producing strain of S. lincolnensis UC8292, which was incapable of synthesizing deazariboflavin, a reductase cofactor responsible for the reduction of 6 to PPL. Over these past three decades, several hypotheses regarding the APD biosynthetic pathway have been proposed and modified with the increase in knowledge.91,94,95 The proposal from 1992 was generally accepted until its revision in 2016, for which characterization of BGCs from natural APD compounds was a crucial prerequisite.8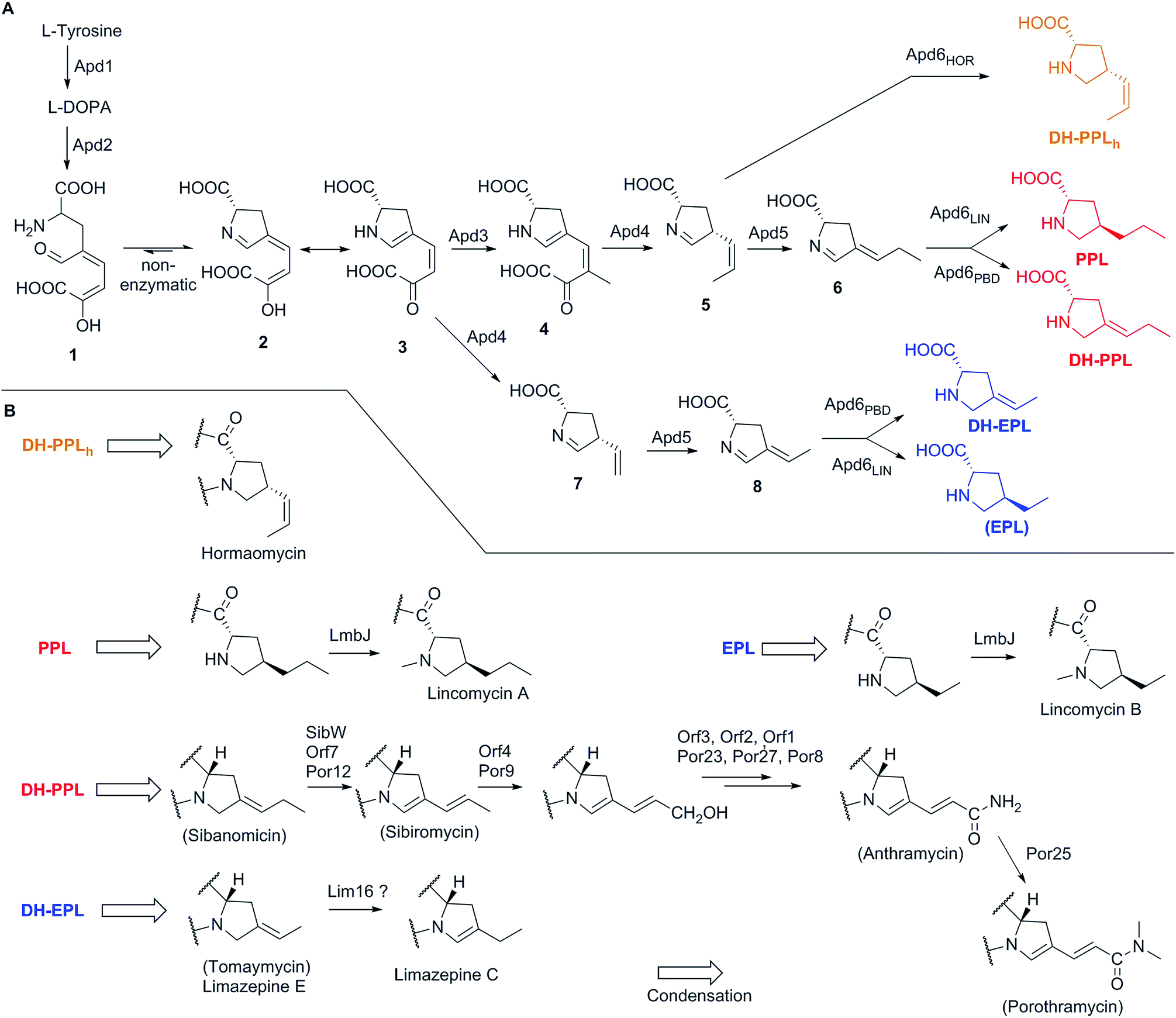 | ||
| Scheme 1 Biosynthesis of APDs and post-condensation tailoring of APD moieties. (A) APD biosynthesis. Final APDs are colour-coded in red (APDs biosynthesized through complete APD pathway), blue (incomplete APD pathway with the absence of Apd3 in the biosynthesis of DH-EPL or omission in the biosynthesis of EPL as a side-product shown in brackets) and orange (incomplete APD pathway – Apd5 absence) (B) post-condensation modifications of APD moieties. | ||
3.2 Current knowledge on APD biosynthesis
Knowledge of the BGCs encoding APD compounds (see Section 2.1 and Fig. 2) allowed for postulation of the six APD biosynthetic proteins. Given the history of individual BGC sequencing, the names of relevant homologous genes and encoded proteins are considerably heterogeneous. In the interest of clarity, we use the unified names apd1–apd6 or Apd1–Apd6 for these genes and proteins, respectively, throughout this review. The assignment of unified names is given in Fig. 7, which includes also the information on the level of the protein functional elucidation (indirect elucidation in vivo by gene inactivation and/or direct elucidation in vitro by testing with recombinant proteins). The overall biosynthetic machinery responsible for APD biosynthesis and modification of the APD moieties, which is consistent with all currently available data, is depicted in Scheme 1 and elaborated in more detail below. The full set of six APD biosynthetic proteins, i.e. the complete APD pathway, is required for the biosynthesis of 3C APDs PPL (precursor of lincomycin), and DH-PPL (precursor of PBDs with 3C APD moieties). In contrast, one of the six APD biosynthetic proteins is missing in the biosynthetic pathways of 3C APD DH-PPLh, a precursor of hormaomycin, and 2C APD DH-EPL, a precursor of PBDs with 2C APD moieties, respectively. Similarly in the complete APD biosynthetic machinery, one biosynthetic enzyme can be omitted resulting in an alternative precursor, e.g. 2C APD EPL precursor of lincomycin B, side-product of lincomycin.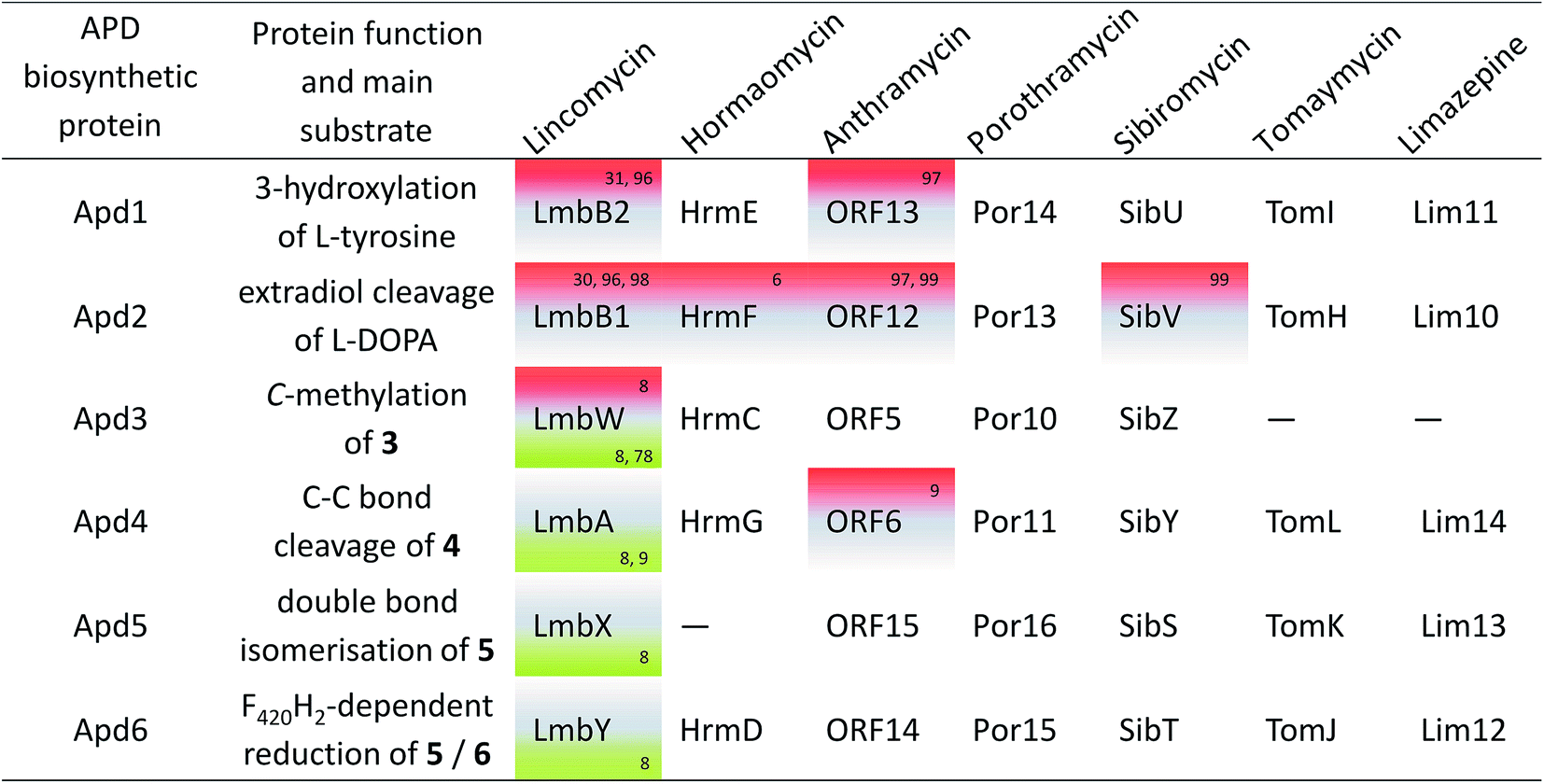 | ||
| Fig. 7 APD biosynthetic proteins. Proteins elucidated in vivo (gene inactivation experiments) are in green, proteins elucidated in vitro (tests with recombinant proteins) are in red. Corresponding references are included in the cell corners. The Apd enzyme numbers (left column) correspond to the proposed order of catalysed reactions in the APD biosynthetic pathway. | ||
L-DOPA subsequently underwent an extradiol cleavage by Apd2 L-DOPA-2,3-dioxygenase, resulting in intermediate 1, which was immediately subjected to a spontaneous intramolecular cyclization to form the yellow-coloured heterocyclic compound 3 (LmbB1,30,96,98 Orf12,97,99 SibV,99). Apd1 belongs to a single-domain type I extradiol dioxygenase of the vicinal oxygen chelate superfamily of enzymes, which use non-heme Fe(II) to bind and activate molecular oxygen for the subsequent insertion of two oxygen atoms via cleavage of the aromatic ring of L-DOPA.30 In a different biosynthetic pathway of the fungus Amanita muscaria, 1 produced by a 2,3-extradiol dioxygenase cyclizes into a different compound, namely muscaflavin.100 It is not clear what determines whether the APD pathway proceeds towards 2 and 3 or not otherwise.
In the subsequent course of reactions, an in vitro reaction with SAM-dependent Apd3 C-methyltransferase was employed to document the methylation of compound 3 to afford intermediate 4 (LmbW).8,78 However, it should be noted that 4 has not been fully structurally elucidated yet and that the conducted experiments did not unambiguously prove that the main native Apd3 substrate was 3 and not the later pathway intermediate 7 (or its enamine form 7a), which was previously proposed to be a substrate for this reaction.4,94 In contrast to 7, intermediate 3 as an 2-oxocarboxylic acid shares structural features with 5-guanidino-2-oxopentanoic and phenylpyruvic acids (Ppy), both of which are proven substrates of two functionally characterized C-methyltransferases homologous to Apd3, MrsA101 (26% identity to LmbW with a coverage of 80% according to blastp) and MppJ102 (26% identity, 84% coverage), respectively.103 Interestingly, in hormaomycin BGC, there are present genes encoding homologous C-methyltransferases for both similar substrates HrmS (for Ppy) and HrmC (Apd3 from hormaomycin biosynthesis).6 The schematic comparison of MppJ vs. HrmS based on the crystal structure of MppJ with bound Ppy (Fig. 8A) and the group of Apd3 proteins (Fig. 8B) reveals a high similarity in their active sites: featuring a mainly highly conserved motif formed by His243 and His295 residues, which together with Asp/Glu244 bind the central Fe3+ cation that, in cooperation with another highly conserved Arg127, fixes α-oxo-carboxylic moiety in both Ppy and 3 (Fig. 8). However, this group of Apd3 proteins contains an additional highly conserved motif, namely the Arg331 residue, which could be responsible for the fixing of the second carboxylic group in 3. Logically, this motif is missing in MppJ and HrmS (they instead contain Ser331 residue). Irrespective of the small differences in the active sites of both C-methyltransferase types, their high similarity indicates that the real native substrate of Apd3 indeed is 2-oxocarboxylic acid 3 and not an alternative substrate 7, as proposed earlier.
 | ||
| Fig. 8 Comparison of the active sites of two types of C-methyltransferases that methylate similar substrates, i.e. phenylpyruvate (A) and compound 3 (B). (A) Schematic active site of MppJ (Ser104; adopted from Zou103 and modified so that the deprotonation by Trp99 is, with respect to the generally low basicity of its indolic nitrogen, not required and enol form of Ppy is methylated) vs. HrmS (Cys104). (B) Schematic active sites of Apd3 (HrmC, LmbW, etc.). Violet colour indicates the variability of the proteins within each group, and the green colour depicts the variability in group (A) vs. group (B). Ppy, phenylpyruvate; SAM, S-adenosyl methionine. The numbering of residues corresponds to MppJ. | ||
The subsequent course of reactions requires the cleavage of an oxalyl residue from 4. The corresponding reaction was proposed to be performed by Apd4, affording the still hypothetical intermediate 5.8 Even though Apd4 is homologous to gamma-glutamyltransferases (it is an N-terminal nucleophile-hydrolase), the unprecedented ability of this protein family to cleave off a C–C bond was demonstrated recently by the in vitro cleavage of compound 3 (Orf6),9 which is a substrate of Apd4 in the case of 2C APDs (see Section 3.2.2). The proposed mechanism for Apd4 function is based on the release of a catalytic Thr residue (N-terminal nucleophile) by aspartate transfer, the generation of an alcoholate on the Thr residue hydroxyl group and the subsequent nucleophilic attack of this alcoholate on the carbonyl group of 3, which leads to the elimination of vinyl-dehydroproline 7 and the regeneration of the Thr residue by hydrolysis of the oxalate (Scheme 2A).9 However, the proposed mechanism does not explain the shift of the electrons into the sp2 C-atom (the authors solved this problem by protonating the electron-superfluous site), which differs from the usual β-elimination reactions, where the shift occurs into an sp3 C-atom. Therefore, a modification of the mechanism, involving the reorganization of the double bonds in 3 or 4 prior to the Thr-alcoholate nucleophilic attack or β-elimination reaction is necessary (our proposed modification is presented in Scheme 2B). In addition, this mechanism is applicable also for the main Apd4 substrate in the case of 3C APDs, i.e. intermediate 4, which should be further converted by this protein to 5, an intermediate with the same stereochemistry at C-4 and localization of the endocyclic double bond as DH-PPLh, for the final APD of hormaomycin biosynthesis.
 | ||
| Scheme 2 Proposed mechanism for Apd4 function. (A) Previously reported scheme for Orf6 and substrate 3 (adapted according to Zhong).9 B. Alternative mechanism common for both 3 and 4 that indicates the substrate double bond reorganization and more appropriately reflects a key β-elimination reaction. Compounds 5 and 7 are drawn in their enamine forms (compare with Scheme 1). | ||
Intermediate 5 requires the isomerization of its double bond to afford 6 (Scheme 1), i.e. the previously identified pathway intermediate.94 This step was proposed to be catalysed by the putative isomerase Apd5 based on in vivo experiments8 described in Section 3.2.2. Intermediate 6 is then proposed to be reduced by the putative F420H2-dependent reductase Apd6 to form the final product in the APD pathway.8 It was shown that lincomycin and lincomycin B incorporate the final APD as fully saturated PPL and EPL, respectively7,8,13 and that PBDs incorporate mono-unsaturated DH-PPL or DH-EPL.10 Comparison of the different degrees of saturation of these final APDs indicates that the double bonds reduction in 6 catalysed by Apd6 proceeds in these two groups of compounds in different ways (Scheme 1). We propose that Apd6 in lincomycin biosynthesis (Apd6LIN) catalyses the reduction of both the double bonds of 6, while Apd6 in PBD biosynthesis (Apd6PBD) reduces only the endocyclic double bond of the same intermediate. The function and particularly different reaction specificity of Apd6LIN and Apd6PBD have yet to be proven by in vitro experiments. However, our preliminary experimental results show that in contrast to Apd6PBD, Apd6LIN is able to reduce both the endo- and exocyclic double bonds of 6.
An additional protein, which was previously assigned to APD biosynthesis, is TomN encoded within tomaymycin BGC. TomN is a 4-oxalocrotonate tautomerase homologue with a solved protein structure, but its natural substrate has not been identified. However, TomN was shown to catalyze the efficient ketonization of dicarboxylic acid 2-hydroxymuconate, which is structurally reminiscent of 1. Therefore, 1 was proposed as a possible candidate of the TomN natural substrate.104 However, the role of TomN in APD biosynthesis (regardless of its exact natural substrate) is questionable because none of the remaining characterized BGCs of APD compounds encodes a homologue of this protein (including the BGC of 2C APD limazepine) and it is therefore not clear why TomN would be required exclusively in the biosynthesis of tomaymycin. One possible hypothesis is that TomN plays only an auxiliary function in the stabilization of a specific tautomer of an intermediate in APD biosynthesis and that this activity is not indispensable for APD biosynthesis.
3.3 Post-condensation modification of APD moieties
The APD biosynthesis is, to a substantial extent, conserved and results in a relatively narrow spectrum of four major APD precursors (Scheme 1). In contrast to the APD uniformity, the APD moieties of the final products are more structurally diverse, which is true for PBDs in particular. This diversity of the APD moieties is a result of post-condensation modifications, which occur in the biosynthesis of lincosamides and PBDs, but not hormaomycin. All the post-condensation steps are summarized in Scheme 1B.3.4 Genome mining and evolutionary aspects of APD compounds
 | ||
| Fig. 9 (A) Unrooted maximum-likelihood phylogenetic tree of Apd1 proteins represented by the name of the producing strain (for accession numbers of the respective proteins, see the ESI Table S1†). Bootstrap values (100 replicates) are indicated at the nodes. Apd1 from putative PBD BGCs are highlighted in shades of grey, and the lincomycin-producing strain S. lincolnensis is highlighted in blue; the hormaomycin producing strain S. griseoflavus is highlighted in violet. Microorganisms with characterized BGCs encoding the biosynthesis of known compounds are in bold. The highlighted Apd1 groups are marked with the abbreviations of APD compounds (putatively) encoded by the respective BGCs (the marker genes characteristic for the respective groups of BGCs are shown in brackets): LIN – lincomycin; HOR – hormaomycin; TOM – tomaymycins (homologues of tomP and tomD are present in the BGCs); LIM – limazepines (homologues of lim4, lim5); SIB – sibiromycins (homologues of sibC and sibH); ANT – anthramycins (homologues of orf2, orf3) and POR – porothramycins (homologues of por25, por26). The set of apd genes identified within the new BGCs obtained from genome mining is highlighted in red. The presence (+) or absence (−) of A-domains in the APD BGCs is marked in magenta. Where the present A-domain was additionally assessed to correspond with the overall sequence homology to L-proline-specific A-domains (Fig. 10), it is marked by ‘Pro’ in a magenta circle; the question mark means that the length of the available contig does not allow for determining the presence/absence of the A-domain in the BGC with sufficient certainty. (B) Proposed APD precursors. | ||
We found out that 19 out of the 36 newly identified BGCs contain, alongside the apd sub-cluster (complete or incomplete without apd3), also genes typical for the biosynthesis of PBDs, which allowed for a more detailed classification of the putative PBD products (Fig. 9 and Table S1†): (1) six BGCs encode ‘tomaymycins’; (2) one BGC for ‘limazepine’; (3) seven BGCs for ‘sibiromycins’; (4) four BGCs for ‘anthramycins’ and (5) one BGC for ‘porothramycin’. Without detailed analysis of the producing strains and production profiles, we cannot determine which of these gene clusters encode the new PBDs, but clear differences in the gene cluster composition suggest that at least three of them encode the biosynthesis of new sibiromycins, as follows. Nocardiopsis prasina contains a different composition of sugar biosynthetic genes, which suggests the production of a sibiromycin derivative with a different type of saccharide moiety. Actinomadura echinospora has a BGC without apd3, which most likely leads to the production of a novel type of sibiromycin derivative with a 2C APD moiety. The BGC of Dermacoccus sp. does not contain apd3 and apd6, and does not have sugar biosynthetic genes, but it contains a gene coding for a putative glycosyltransferase. The prediction of this product is more challenging, but the gene composition corresponds to a glycosylated sibiromycin derivative with a 2C APD moiety.
The remaining 17 out of the 36 newly identified BGCs do not contain (besides the apd genes) any additional marker genes required for the biosynthesis of PBDs, lincosamides or hormaomycin. This finding suggests that these BGCs encode novel classes of APD compounds, i.e. incorporating an APD moiety in a novel structural context. Moreover, 11 out of the 17 BGCs contain yet unidentified combinations of apd genes. The proposed products encoded by these APD sub-clusters are summarized in Fig. 9 and discussed as follows. None of the 17 BGCs encodes the complete apd sub-cluster, which means that lincomycin remains the only non-PBD compound with a full set of apd genes encoded within its BGC. Six BGCs contain apd genes encoding known incomplete APD pathways: the sub-cluster apd1, apd2, apd4–apd6 corresponding to the biosynthesis of DH-EPL or EPL, and the sub-cluster apd1–apd4, apd6 corresponding to the biosynthesis of DH-PPLh. Regarding the newly identified apd gene combinations, four types were revealed: (1) the sub-cluster apd1, apd2, apd4, apd6 corresponding to the biosynthesis of the 2C analogue of APD incorporated into hormaomycin, i.e. 4-((Z)-vinyl)-L-proline (I); this APD was incorporated into a lincosamide by a Δapd3Δapd5 deletion mutant of a lincomycin-producing strain;8 (2) the sub-cluster apd1–apd4 corresponding to the biosynthesis of 5, an intermediate of hormaomycin, 3C PBDs and lincomycin biosynthesis; (3) the sub-cluster apd1, apd2, apd6 corresponding to 2 and/or 3 (products of Apd1, Apd2), which can be reduced by Apd6 to give II or III; (4) the sub-cluster apd1, apd2 corresponding to a ‘minimal’ APD pathway, which would produce 2 and/or 3. The variability of new apd gene combinations suggests that at least four new APD building blocks can be expected in the yet unknown natural products.
The overall diversity of the new APD moieties will most likely be much higher depending on the structural context of the APD incorporation, which can be at least partially revealed by the presence of an A-domain activating the APD for a subsequent condensation reaction (see Chapter 4). In Fig. 9, we indicate if and what type of A-domain encoding sequence is present in the new BGCs. Specifically, 10 out of 17 BGCs for non-PBD APD compounds encode an A-domain sequence, which correspond to L-proline-specific A-domains by overall sequence homology. Considering the known APD-incorporating systems, these A-domains can be expected to have adapted for APD specificity (these A-domains are referred to as ‘L-proline-/APD-specific’). Four other new BGCs contain sequences corresponding to one or more A-domain(s), but distinct from those with L-proline/APD specificity; here, some of them could presumably activate a modified APD. A majority of the putative BGCs (13 out of 17) of the potential new non-PBD APD compounds bear at least three A-domain sequences regardless of their specificity (the number of the putative A-domains in each BGC is included in Table S1†), suggesting incorporation of the respective APDs or further modified APDs into a complex metabolite of a peptide type. For the remaining three BGCs, the incorporation context is more difficult to predict and a possible involvement of something other than the classical NRPS condensation system has to be taken into account.
It should be noted that some of the BGCs encoding the new combinations apd1, apd2, apd6 and apd1, apd2, apd4, apd6 also encode L-proline-/APD-specific A-domains, i.e. these new APD precursors are presumably incorporated by a condensation system, which is generally employed by one of the groups of characterized APD compounds. On the other hand, the process of APD incorporation will be particularly interesting in the case of APD sub-clusters without apd4. These pathways should produce APDs with a heavily substituted four carbon residue at C-4. However, the known APD condensing systems do not accept such precursors,8 and also, no natural compound with such a moiety have been reported. The condensing systems responsible for incorporating these APDs may thus represent novel enzymology.
Thorough analysis of all 36 new gene clusters and in-depth analysis of their possible products is beyond the scope of this review; however, we are convinced that the outlined BGCs may be a challenging inspiration for further extensive research in the field of natural products.
Origin of apd5 and evolution of PBD biosynthesis. PBDs are clearly the most abundant APD compounds with the highest structural diversity. The unconditional presence of apd5 in all 24 BGCs for PBD biosynthesis contrasts with its rare occurrence in the BGCs of non-PBD APD compounds (only 4 out of 19). This strongly suggests that PBDs were an entrance gate for Apd5 isomerase activity into APD biosynthetic pathway and that the apd5 ancestor gene was already involved in the acceptor BGC of an ancestral non-APD PBD. The acquisition of the yet incomplete sub-cluster apd1, apd2, apd4, apd6 resulted in the formation of a BGC for a 2C APD PBD similar to that of limazepines. Limazepines are the only known APD PBDs, which form an anthranilate precursor via trans-2,3-dihydro-3-hydroxyanthranilic acid (DHHA) through the chorismate/DHHA pathway, which is partially shared with the biosynthesis of phenazines (see Section 5.1). Specifically, the formation of a phenazine precursor is from the chorismate/DHHA pathway diverted by PhzF isomerase, homologous to Apd5 isomerase. It thus appears that Apd5 and PhzF evolved from a common ancestor and that limazepines represent the cradle of APD PBDs. Looking at the phylogenetic tree (Fig. 9), the establishment of APD PBDs (limazepine type) was presumably followed by simplification of the original chorismate/DHHA pathway to the chorismate/anthranilate pathway (tomaymycins) or by complete replacement for the kynurenine pathway, resulting in the establishment of another group of PBDs: 2C APD sibiromycins (e.g. newly found derivatives from Actinomadura echinospora or Dermacoccus sp. PE3). Subsequent acquisition of apd3 from a BGC of a non-PBD APD compound would then result in the predominant group of sibiromycins, i.e. sibiromycins with a 3C APD moiety. This event presumably caused the splitting of sibiromycins into the two evolutionary distinct groups in the Apd1 phylogenetic tree. The 3C APD sibiromycins from the lower branch (e.g. the sibiromycin derivative from Nocardiopsis prasina) appear to be evolutionary highly progressive because they evidently represent the origin for the evolution of anthramycins and porothramycins with heavily substituted APD moieties.
Origin of apd3 and evolution of hormaomycin/lincomycin biosynthesis. The evolution of non-PBD APD compounds is difficult to decode because the number of characterized compounds and BGCs is limited only to lincomycin and hormaomycin. However, the BGC of hormaomycin exhibits genetic features that give an indication regarding the evolutionary origin of apd3. During the evolution of the hormaomycin biosynthetic pathway, some of the genes encoding NRPS were duplicated,6 resulting also in the duplication of amino acid residues in its structure, including the unusual β-methyl-L-phenylalanine residue. The BGC of hormaomycin encodes two C-methyltransferase homologues: HrmC (i.e. Apd3 from hormaomycin biosynthesis; see Fig. 8), which is proposed to catalyse the methylation of 3 in APD biosynthesis, and HrmS (see Fig. 2, the red striped arrow), which is proposed to catalyse the C-methylation of benzylic position in phenylpyruvate (see Section 3.2.2). The structural similarity of HrmC and HrmS substrates and the sequence homology of these putative methyltransferases suggest that their ancestral gene was involved in the duplication event in the BGC of the hormaomycin ancestor; whereby, a gene encoding the attachment of β-methyl group in the β-methyl-L-phenylalanine residue was duplicated, with one of the encoded methyltransferases retaining the same substrate specificity (HrmS ancestor), while the other was adapted for the methylation of 3 in APD biosynthesis (HrmC ancestor). This new activity was subsequently incorporated into the biosynthesis of all 3C APD compounds, i.e. into more than a half the PBDs (13 out of 24) and five other non-PBD APD compounds, including lincomycin.
There are several clues indicating a common origin of the APD biosynthetic sub-cluster of hormaomycin and lincomycin BGCs: first, in the phylogenetic tree of Apd1 proteins, the lincomycin LmbB2 and hormaomycin HrmE were identified as the closest relatives. Second, the order of apd genes is conserved among PBD BGCs (at least in the sub-cluster apd2-apd1-apd6-apd5), but it differs in the BGCs of lincomycin and hormaomycin (apd4-apd2-apd1 in both; see Fig. 2). Finally, both lincomycin and hormaomycin BGCs comprise, in contrast to the BGCs of PBDs, an unusual putative regulatory gene (lmbU and hrmB, respectively) located immediately adjacent to the apd genes in both BGCs.108 These rare genes are homologous to the regulatory gene novE109 encoded within novobiocin BGC, while no homologues of this gene are present in the BGC of the non-APD lincosamide celesticetin, suggesting its relation to the apd sub-cluster. Therefore, we hypothesize that the lincomycin and hormaomycin APD sub-clusters originate from a common ancestor with the full set of apd genes and the adjacent putative regulatory gene homologous to lmbU. Two evolutionary events forming the current full APD pathway thus presumably met in this common ancestor: the duplication of the C-methyltransferase encoding gene resulting in apd3 and the acceptance of apd5 originating from an ancestral BGC of a PBD.
4 APD precursor activation
APDs are incorporated into the final complex natural compounds through two distinct machineries: NRPS systems are used to assemble PBDs and hormaomycins (see Chapter 5), and a hybrid condensation system is used to assemble lincosamides (see Chapter 6). Despite the difference in the condensation machineries, all known APD incorporation systems use the same key initial biosynthetic step. This comprises APD recognition and activation by an A-domain, which is either integrated into the NRPS polypeptide chain (modular A-domain) as in PBDs and hormaomycins or forms a discrete protein as in lincosamides (stand-alone A-domain). APD-specific A-domains evolved in a common principle by transformation of the ancestral L-proline-specific A-domains to use an unusual APD substrate, which was documented on the model of lincosamide biosynthesis.114.1 Lincosamides: transformation of L-proline-specific A-domain to use APD
In the lincosamide biosynthesis, L-proline or APD-precursor PPL is incorporated into celesticetin and lincomycin, respectively. CcbC and LmbC are the stand-alone A-domains, determining the overall substrate specificity of the respective condensation system. The high mutual sequence similarity of the pair of CcbC/LmbC A-domains (55.7% overall sequence identity) strongly suggests that they have a common L-proline-specific ancestor (see Section 4.2). Biochemical tests have shown significant differences in the substrate specificity of CcbC and LmbC: CcbC is strictly L-proline specific, whereas LmbC strongly (103 times) prefers PPL over L-proline.11 This reflects efficient adaptation to the new unusual substrate PPL and, at the same time, an effective rejection of the ancestral substrate L-proline for activation and the subsequent incorporation into lincosamide.The substrate specificity of A-domains can be predicted from the amino acid residues surrounding the substrate binding pocket (SBP).110–112 This so-called nonribosomal code consists of 10 amino acid residues. Two of them (lysine and glutamate), interacting with carboxyl- and amino-groups of the substrate, respectively, are conserved in the amino acid-activating A-domains. The remaining eight residues are variable, and their specific pattern is conserved in A-domains having the same substrate specificity. Even though the overall sequence homology of the biosynthetically related L-proline- and APD-specific A-domains CcbC and LmbC, respectively, is high, their nonribosomal codes differ dramatically at five of the eight variable residues, reflecting thus the APD-substrate specificity adaptation in SBP (see the ESI Fig. S1†).
Indeed, homology models of the SBP of both proteins, built according to the crystal structure of PheA, PDB ID 1AMU,110 visualised this dramatic reconstruction of the SBP during LmbC evolution.11 Specifically, spacious (phenylalanine, valine) or more hydrophilic (tyrosine) amino acid residues were substituted for smaller (alanine, glycine) or hydrophobic (leucine) residues, resulting in the formation of a hydrophobic channel essential for accommodation of the alkyl side chain of PPL. In addition to this, the homology models and biochemical tests showed that LmbC also efficiently activates synthetic APDs with prolonged 4C and 5C alkyl side chains.11 From a pharmaceutical point of view, this was a very important finding that enabled the preparation of more efficient lincosamide antibiotics by mutasynthesis (see Section 7.2).113
Recently, the proposed molecular mechanism of the A-domain SBP adaptation in LmbC evolution to prefer the APD substrate was experimentally confirmed.114 The above-mentioned three amino acid residues of the CcbC SBP were substituted to residues present at corresponding positions in LmbC by site-directed mutagenesis. The resulting substrate specificity of the triple-mutant CcbC protein was significantly shifted and mimicked the Km values of LmbC: first, PPL was significantly preferred over L-proline (Km differs by two orders of magnitude). Second, the Km values for synthetic APDs with prolonged alkyl side chain were even better than those for PPL, the natural substrate of LmbC.
4.2 Independent evolution of APD-specific A-domains
Previously performed phylogenetic analysis of APD-specific A-domains of published APD compounds showed that not only stand-alone A-domain LmbC, but also modular A-domains from the biosynthesis of PBDs and hormaomycin evolved from L-proline-specific ancestors.11 In addition, the adaptation for an APD occurred in parallel, i.e. independently for each of these three groups of APD compounds. We performed an analogous analysis updated for recently published L-proline- and APD-specific A-domains of known compounds and additionally for putative L-proline-/APD-specific A-domains from the BGCs of potential APD compounds uncovered in this review (see Section 3.4.1). The updated phylogenetic tree is depicted in Fig. 10, and nonribosomal codes of the (putative) A-domains are presented in the ESI Fig. S1.† This phylogenetic analysis fully complies with the previous results that the APD-specific A-domains evolved independently several times:11 all seven so far published APD-specific A-domains are spread among L-proline-specific A-domains (biochemically characterised or based on the known structure of the final product).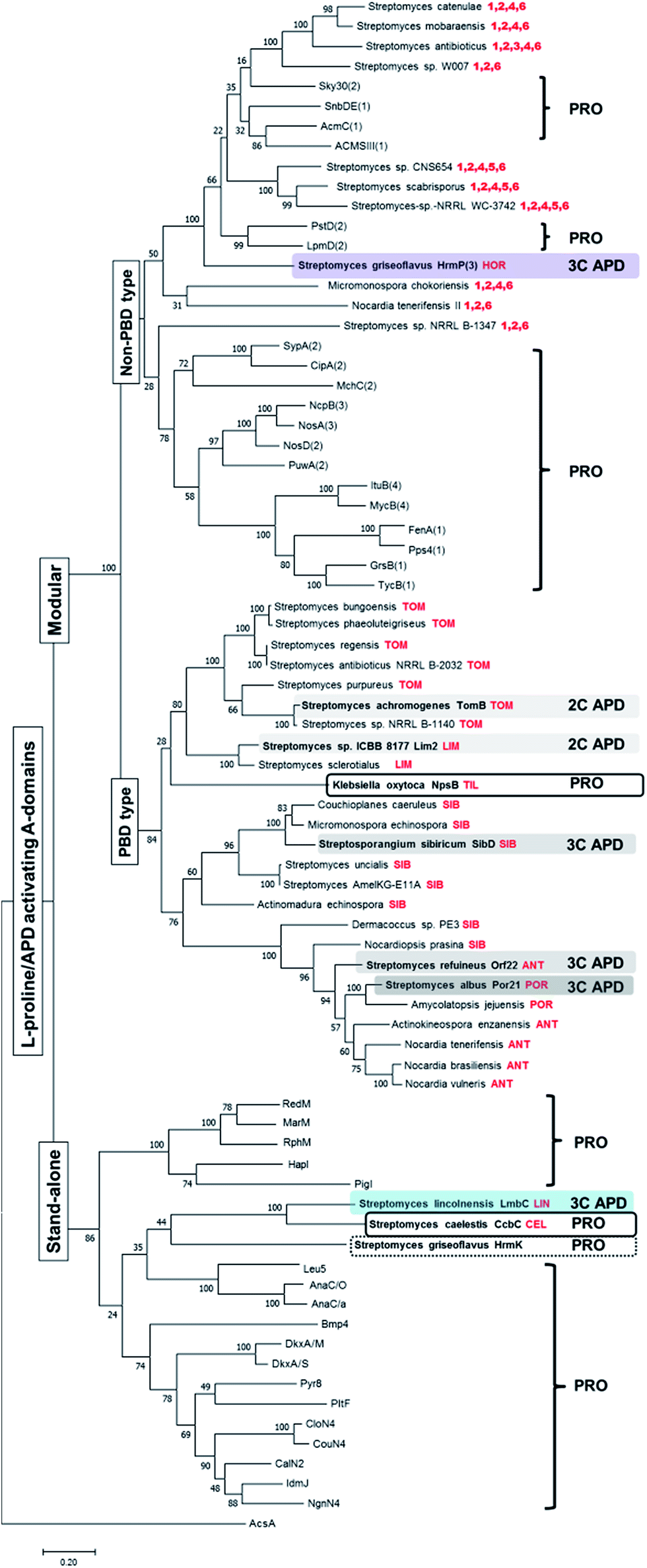 | ||
| Fig. 10 Rooted maximum-likelihood phylogenetic tree of L-proline/APD-specific A-domains. Bootstrap values (100 replicates) are indicated at the nodes. Acetyl-CoA synthetase AcsA (GenBank number WP_004399030.1) was used as the outgroup. The set of A-domains used for the phylogenetic tree construction comprised both the stand-alone and modular L-proline-specific A-domains with substrate specificity already experimentally verified or deduced from the metabolite structure and corresponding NRPS substrate specificity prediction (RedM),115 RphM,121 MarM,122 Pyr8,123 PigI,124 HapI,125 Bmp4,126 NgnN4,127 IdmJ,128 CalN2,129 CouN4,116 CloN4,130 AnaC/O,117 AnaC/a,131 DkxA/S,132 DkxA/M,132 Leu5,118 PltF,115 GrsB(1),133 TycB1(1),134 FenA(1),135 MycB(4),136 ItuB(4),137 Pps4(1),138 SypA(2),139 PuwA(2),140 MchC(2),141 NosD(2),142 NosA(3),142 NcpB(3),143 CipA(2),144 PstD(2)120 LpmD(2),119 ACMSIII(1),145 AcmC(1),146 Sky30(2)147 and SnbDE(1)148 together with APD-specific A-domains from the already published APD BGCs (highlighted in colours), two A-domains from the BGCs of related L-proline-incorporating compounds PBD tilivalline and lincosamide celesticetine and the proline-specific stand-alone domain HrmK from hormaomycin BGC (in frames). Finally the set was completed with putative APD-specific A-domains from the newly mined APD BGCs in the tree determined by the name of the producing microorganism and the respective putative APD compound, when predictable, or the numbers of encoded apd genes (in red colour). The number in parentheses behind the name of NRPS denotes the order of the A-domain in the NRPS protein chain, if relevant. The letter behind the slash denotes the source organism when the names of A-domains are the same. The names, substrates, organisms, nonribosomal codes and corresponding natural products are summarised in the ESI Fig. S1† The abbreviations stand for: TOM – tomaymycins; LIM – limazepines; SIB – sibiromycins; POR – porothramycins and ANT – anthramycins. | ||
Specifically, the stand-alone APD-specific A-domain LmbC from lincomycin biosynthesis clearly belongs to a clade together with stand-alone L-proline-specific A-domains, and its closest relative homologue is the L-proline-specific A-domain CcbC involved in the biosynthesis of the non-APD lincosamide celesticetin.11 The CcbC/LmbC pair, with a different substrate specificity, is related to other biochemically characterized stand-alone L-proline-specific A-domains employed in the biosynthesis of various pyrrole derivatives, including undecylprodigiosin (RedM),115 pyoluteorin (PltF),115 coumermycin A1 (CouN4),116 anatoxin-a (AnaC),117 leupyrrins (Leu5)118 and others (see Fig. 10 and the ESI Fig. S1†), indicating a common ancestral L-proline specificity. All modular L-proline-/APD-specific A-domains form distinct clades from the clade of stand-alone A-domains. Of these, those involved in PBD biosynthesis form a single distinct clade, including the confirmed APD-specific A-domains of tomaymycin (TomB), limazepines (Lim2), sibiromycin (SibD), anthramycin (Orf22) and porothramycin (Por21) as well as putative APD-specific domains from the 19 new putative BGCs, which presumably encode the biosynthesis of the PBDs. Note, that the modular L-proline-specific A-domain NpsB from the biosynthesis of non-APD PBD tilivalline belongs also in this “PBD” clade.
In contrast, the hormaomycin APD-specific modular A-domain, HrmP(3) and the 10 putative L-proline-/APD-specific A-domains from the novel BGCs of potential non-PBD APD compounds belong to several clades, which all are distinct from the “PBD” clade. Even though no L-proline-incorporating counterpart of hormaomycin has been identified, the sequentially closest homologues of APD-specific HrmP(3) are L-proline-specific modular A-domains LpmD(2) and PstD(2) from the biosynthesis of laspartomycin119 and friulimycin,120 respectively. Similarly, the 10 putative L-proline-/APD-specific A-domains from the new BGCs of the potential non-PBD APD compounds constitute a heterogeneous block interspersed among A-domains with confirmed L-proline specificity, suggesting that they evolved independently but in parallel from several different L-proline-specific ancestors.
From the overall viewpoint, we hypothesize that, in contrast to APD biosynthesis, which is spread by HGT of the apd sub-cluster, the condensation systems for the incorporation of APDs evolved independently by using the existing L-proline-incorporating systems and adapting their A-domain specificity.
5 NRPS-directed APD incorporation
5.1 PBD biosynthesis
In the biosynthesis of PBDs the APD precursor is condensed by bimodular NRPS to the activated anthranilic acid or its derivative (referred to also as an anthranilate precursor). Similarly to APD, this second PBD precursor is also synthesized in a specialized biosynthetic pathway.Anthranilate precursors of PBDs are biosynthesized through kynurenine or chorismate pathways, which are both derived from the primary metabolism. The kynurenine pathway starts with tryptophan, which is processed into variously modified 3-hydroxylated anthranilate precursors and incorporated into anthramycin,18,149 porothramycin20 and sibiromycin19,150 (Scheme 3A). The chorismate pathway can proceed via two distinct routes, both starting with seven steps of the shikimate pathway to yield chorismic acid. The first route (chorismate/anthranilate pathway, Scheme 3B) converts chorismic acid by anthranilate synthase to directly yield unsubstituted anthranilic acid, which is then incorporated into tomaymycin.21 The second route (chorismate/DHHA pathway, Scheme 3C), partially shared with the biosynthesis of phenazines and presumably the evolutionary oldest type of anthranilate biosynthesis in APD PBDs (see Section 3.4.2), involves the conversion of chorismic acid via DHHA and results in 3-hydroxylated anthranilate precursors. These precursors can be incorporated into APD PBD limazepines or non-APD PBD tilivalline.22,25 Consequently, the structure of the final PBD, particularly the presence/absence of the hydroxyl group at the C-9 of the PBD scaffold (corresponding to C-3 of the anthranilate precursor) does not clearly reflect the biosynthetic origin of its anthranilate moiety. This is true particularly for the limazepines22,25 and tilivalline25vs. anthramycin,18,149 sibiromycin19,150 and porothramycin,20 which incorporate 3-hydroxylated anthranilate precursors formed through distinct pathways (Scheme 3).
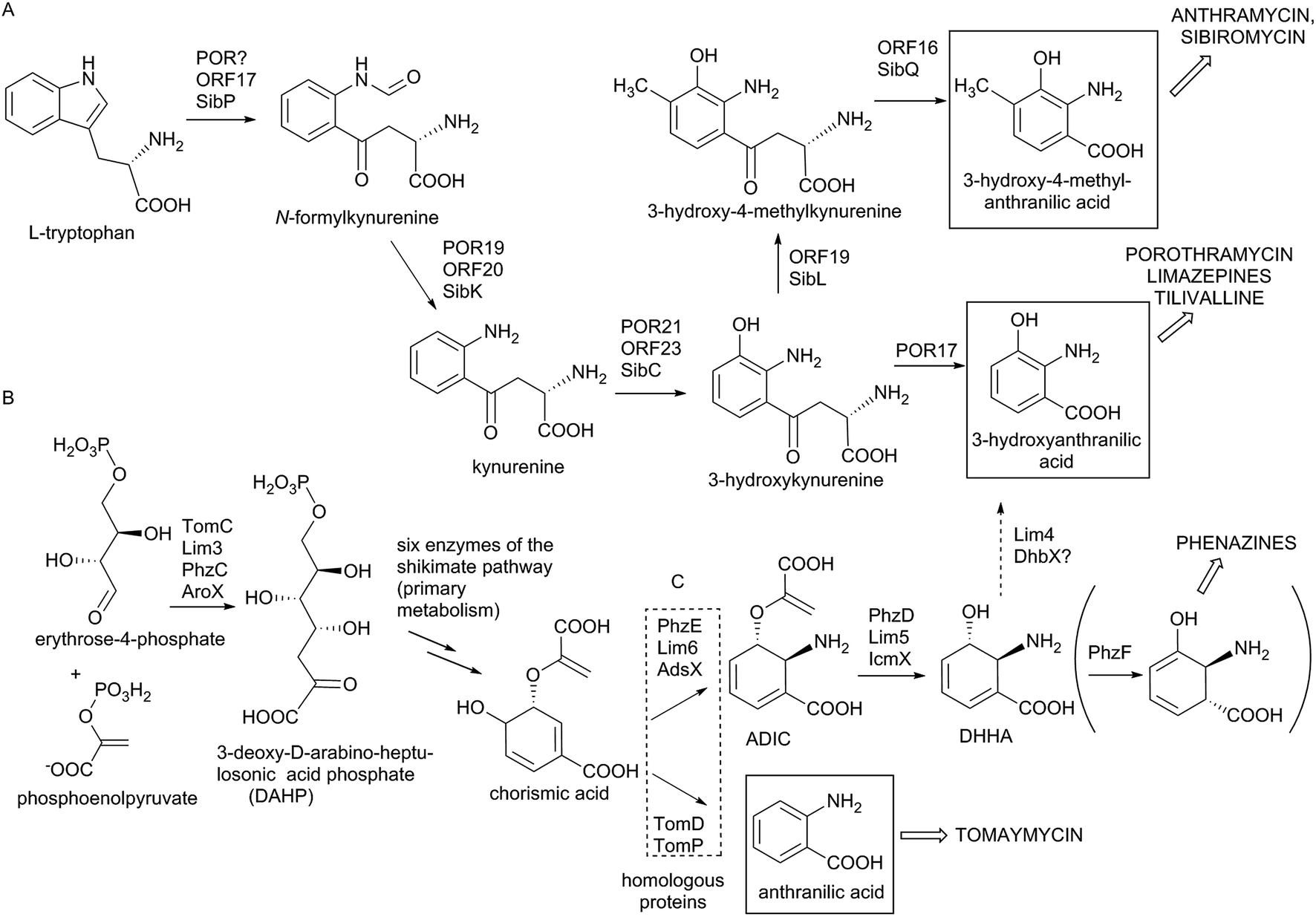 | ||
| Scheme 3 Biosynthesis of anthranilate precursors. (A) Kynurenine pathway resulting in anthranilate precursors of anthramycin, sibiromycin and porothramycin. (B) Shikimate-chorismate pathway resulting in the anthranilate precursor of tomaymycin. (C) Shikimate-chorismate pathway resulting in the anthranilate precursor of limazepines and tilivalline, partially shared with the biosynthesis of phenazines (Phz enzymes). Empty arrows indicate the entrance of the corresponding compounds to additional biosynthetic pathways (indicated by the names of the final compounds in capitals). | ||
Biosynthesis of anthranilate precursor through kynurenine pathway. In anthramycin, sibiromycin and porothramycin biosynthesis, tryptophan 2,3-dioxygenase (Orf17 or SibP) is proposed to cleave the pyrrole ring of L-tryptophan to yield N-formylkynurenine (Scheme 3A).18,19 This activity is not encoded within the BGC of porothramycin, but utilization of a homologue from the primary metabolic kynurenine pathway can be expected.20 Subsequently, N-formylkynurenine should be hydrolysed by aryl formamidase (Orf20, SibK, Por19) and oxidized by a kynurenine 3-monooxygenase (Orf23, SibC, Por21) to give 3-hydroxykynurenine (Scheme 3A). The subsequent steps have been confirmed biochemically only for sibiromycin biosynthetic proteins;150 however, the same strategy probably also applies to anthramycin biosynthesis.149In vitro tests showed that 3-hydroxykynurenine is C-methylated at C-4 (SibL, Orf19), and the resulting 3-hydroxy-4-methylkynurenine is cleaved by kynureninase (SibQ, Orf16) to yield 3-hydroxy-4-methylanthranilic acid (Scheme 3A), which is recognized and activated by the respective A-domain of NRPS. In porothramycin biosynthesis, the methylation step is omitted (even though the respective homologous methyltransferase Por18 is encoded in the BGC) and 3-hydroxykynurenine is presumably directly cleaved by kynureninase (Por17) to yield 3-hydroxyanthranilic acid (Scheme 3A).20 The methylation of the hydroxyl at the C-3 position of the anthranilate precursor was assigned to putative methyltransferase Por26. This step presumably also proceeds prior to condensation; however, it is not clear whether the main substrate is 3-hydroxykynurenine or 3-hydroxyanthranilic acid.
Biosynthesis of anthranilate precursors through the chorismate pathway. For tomaymycin, limazepine and tilivalline biosynthesis, the shikimate pathway is expected to deliver chorismic acid. All three BGCs contain an extra copy of a gene encoding the putative 3-deoxy-D-arabinose-heptulosonic-7-phosphate (DAHP) synthase (TomC, Lim3 and AroX), the key enzyme catalysing the first reaction of the shikimate pathway (Scheme 3B). The remaining six steps leading to chorismic acid are probably substituted by primary metabolic proteins. The fate of chorismic acid is different in the biosynthesis of tomaymycin and limazepines. In the case of tomaymycin, the chorismate/anthranilate pathway is employed; whereby, the chorismic acid is presumably converted by the putative anthranilate synthase TomD/TomP to form anthranilic acid (Scheme 3B), which is proposed to be activated by the respective A-domain. In the case of limazepines, the chorismic acid has been shown to enter the chorismate/DHHA pathway, whereby it is transformed by 2-amino-2-deoxy-isochorismate (ADIC) synthase (Lim6) and subsequently by isochorismatase (Lim5) to yield DHHA (Scheme 3C).22 Finally, DHHA is proposed to be converted by a putative 2,3-dihydro-2,3-dihydroxybenzoate dehydrogenase (Lim4) to form 3-hydroxyanthranilic acid. An identical pathway for the 3-hydroxylantranilic acid precursor is presumably also involved in tilivalline biosynthesis, as the homologues of Lim4-6 (AdsX, IcmX, DhbX, respectively) are encoded within its BGC.25
The anthranilate synthases, converting chorismic acid in tomaymycin (TomD/TomP), and ADIC synthase, converting the same substrate in limazepines (Lim6) (Scheme 3), are sequentially homologous, and unless they are biochemically characterized, their reaction specificity can be predicted only based on the presence/absence of genes coding for respective downstream biosynthetic reactions: ADIC processing DHHA synthase (Lim5) and 2,3-dihydro-2,3-dihydroxybenzoate dehydrogenase (Lim4). Accordingly, the 3-hydroxylated anthranilate precursor is formed through the chorismate/DHHA pathway also in the biosynthesis of paulomycins, diazepinomycin or benzoxazoles calcimycin, caboxamycin and A33853.129,151–154 In addition, Lim6 and Lim5 homologues are also involved in the biosynthesis of phenazines (PhzE and PhzD, respectively), where the resulting DHHA is transformed to 6-amino-5-oxocyclohex-2-ene-1-carboxylic acid (Scheme 3C) by the action of isomerase PhzF, which is highly homologous to Apd5 isomerases.155–157 We suppose that PhzF-like activity was an ancestor of the Apd5 isomerase in the APD biosynthesis (see Section 3.4.2).
Although the substrate specificity of the L-proline/APD A-domain is quite narrow (see Chapter 4), the anthranilate-specific A-domains seem to be quite promiscuous given the frequent identification of more than one side-product.32,149,150 Once recognized and bound to CP-domains, the precursors can be further modified both before and after the condensation step. However, current knowledge about the precise timing of the individual modifications is limited. Experimental evidence is available concerning anthramycin and sibiromycin biosynthesis, in which the A-domain of the condensing NRPS preferentially recognizes 3-hydroxy-4-methylanthranilic acid (Scheme 4).149,150 A further modification of the sibiromycin anthranilate moiety occurs during condensation at the SibE NRPS-bound precursor, which is hydroxylated by SibG at the C-5 position of the anthranilate precursor. After release from NRPS, this hydroxyl group is glycosylated by SibH, resulting in the attachment of a sugar moiety, sibirosamine.150
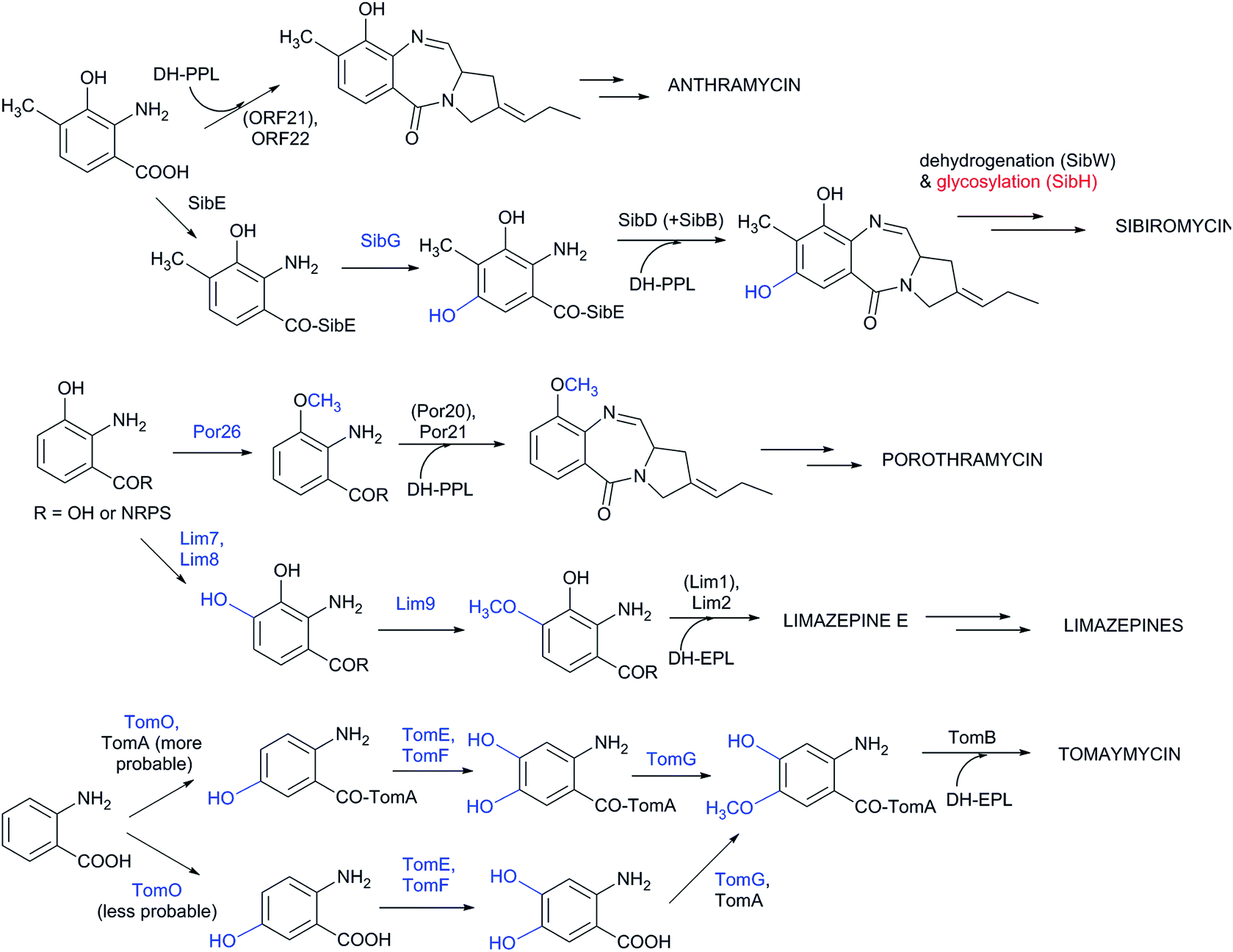 | ||
| Scheme 4 Modifications of anthranilates in PBDs. The protein-catalysing modifications of anthranilates prior to condensation and the resulting groups are in blue; post-condensation modifications are in red. Proteins in brackets indicate NRPS proteins, which activate anthranilates and are not drawn in the corresponding intermediates. | ||
The tomaymycin oxidoreductase, TomO, which is homologous to SibG, probably acts on the NRPS-bound substrate and, similarly to sibiromycin biosynthesis, hydroxylates the C-5 position of the anthranilate precursor (Scheme 4).150 Subsequently, while still bound to NRPS, hydroxylation at the C-4 position of the anthranilate precursor catalysed by TomE/TomF oxidoreductases and methylation of this hydroxyl group by TomG methyltransferase occurs to create the intermediate for condensation with DH-EPL (Scheme 4).32 The Lim7/Lim8 pair of enzymes, which are homologous to TomE/TomF, are involved in the biosynthesis of limazepines. Correspondingly, these putative oxidoreductases presumably catalyse the analogous hydroxylation at the C-4 position of the NRPS-bound anthranilate precursor (Scheme 4).22 The subsequent methylation of the C-4 hydroxyl was assigned to Lim9 based on its sequence similarity to tomaymycin methyltransferase TomG. However, these proteins would methylate hydroxyls in different positions (C-5 vs. C-4 of the anthranilate precursor; Scheme 4), suggesting that the putative homologous methyltransferases have a somewhat relaxed substrate specificity. With the exception of glycosylation in sibiromycin biosynthesis, the anthranilate precursors of PBDs are thus likely to enter the condensation reaction already in their final forms. The modifications at C-11 of the assembled PBD must occur post-condensationally; however, there is currently no information available concerning the biochemical origin of the C-11-oxo forms of PBDs and the secondary amines of usabamycins and boseongazepines.
5.2 Hormaomycin biosynthesis
In addition to the activated APD precursor, seven other amino acids enter the condensation step of hormaomycin biosynthesis. Of these, only L-isoleucine is proteinogenic. Together, five different types of specialized precursors participate in the condensation, as two unusual amino acids are incorporated twice in the resulting hormaomycin molecule. Beside genes coding for NRPS, a large portion of the hormaomycin BGC encodes enzymes involved in the biosynthesis of these specialized precursors.In the biosynthesis of 5-chloropyrrole 2-carboxylic acid (Chpca), L-proline is probably activated by a stand-alone A-domain protein, HrmK, and transferred to the CP HrmL to form a thioester-bound prolyl-CP.6 Following dehydrogenation of prolyl-CP to 2-pyrroloyl-CP, it is probably catalysed either by HrmM alone or HrmM with HrmN in cooperation, as both these proteins are similar to the known acyl-CoA dehydrogenases.6 Chlorination of the pyrrole is performed by HrmQ, as was shown in the combinatorial expression of hrmQ with genes of the clorobiocin BGC, which led to the production of hybrid aminocoumarins with an additional chlorine atom at position 5 of the pyrrole unit.162 In contrast, the protein responsible for the N-hydroxylation of the pyrrole ring is still unknown. It is believed that most of the biosynthetic steps occur on the HrmL-bound substrate. The final CP-bound product is then probably condensed with (3-Ncp)Ala attached to the CP of the first module of HrmO.6
Initial feeding studies indicated that the methyl group of β-methyl phenylalanine [(β-Me)Phe] is introduced by a SAM-dependent methyltransferase onto a suitable precursor generated from L-Phe.6 This methylation reaction is probably catalysed by the putative methyltransferase HrmS, which is homologous (52% identity, 97% coverage) to another described C-methyltransferase from Streptomyces hygroscopicus, MppJ.102 HrmS probably introduces a methyl group into α-keto acid phenylpyruvate, which has been demonstrated to be a natural substrate of MppJ (see also Section 3.2.1). The last step of (β-Me)Phe biosynthesis is the transamination reaction, which is probably catalysed by the corresponding enzyme from primary metabolism, due to the lack of a corresponding gene in the hormaomycin BGC. Note that hormaomycin BGC contains an additional gene encoding the C-methyltransferase, HrmC (Apd3), which is homologous to HrmS (22% identity; 90% coverage). HrmC is involved in 3C APD biosynthesis and its links to HrmS are discussed in Section 3.2.1 (regarding similarity of their active sites and substrates) and Section 3.4.2 (regarding the evolutionary aspects).
6 Lincosamide biosynthesis: natural hybrid system of APD incorporation
Lincosamides are defined by their central core being comprised of a naturally rare amino-octose, to which an amino acid (L-proline or an APD in natural lincosamides) is attached via an amide bond. Correspondingly, a large part of the lincosamide biosynthetic machinery proceeds through a sugar metabolic pathway, which is graphically illustrated by a comparison of the respective BGCs and by the proposed BGC for a hypothetical ‘minimal’ lincosamide in Fig. 11. Formation of the amide bond is catalysed by a unique hybrid condensation system, which apparently arose as a reflection of the need to connect an unusual combination of condensing partners, an amino sugar and an amino acid. The condensation system combines NRPS components that are responsible for amino acid activation (covered in Chapter 4) with NRPS-dissimilar ergothioneine-dependent activity, which is responsible for amino sugar conjugation/activation and subsequent amide bond formation.7,11,13 The condensation system subsequently determines the mycothiol-dependent post-condensation modifications, which result in sulphur atom incorporation into the lincosamide structure and which continue with a branch-point that directs whether the attachment of another building block, i.e. salicylic acid, will occur.7,14–17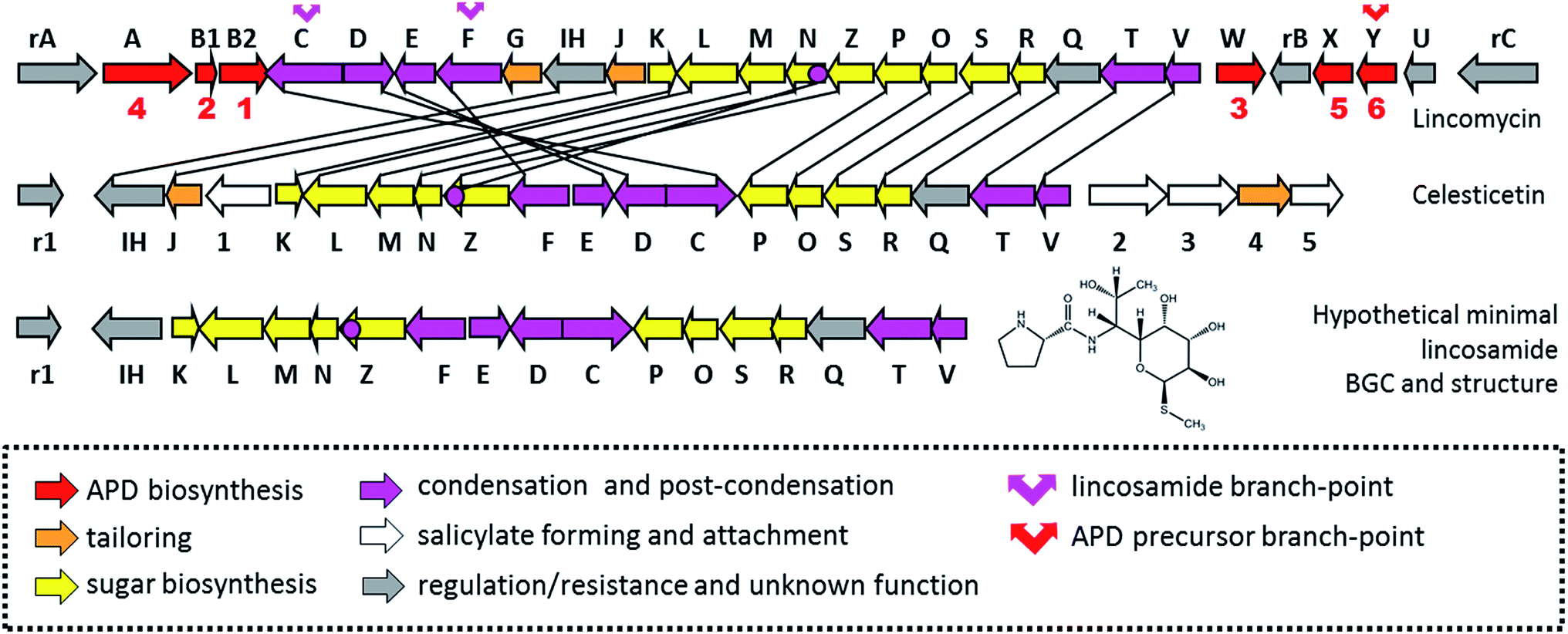 | ||
| Fig. 11 BGCs of lincomycin, celesticetin and hypothetical BGC coding for the biosynthesis of a ‘minimal’ lincosamide. The biosynthetic or regulatory genes are marked by corresponding capital letters or numbers; the resistance genes are marked by “r” and a corresponding capital letter or number. The red numbers bellow apd genes correspond to the number of APD biosynthetic steps catalysed by encoded proteins. The homologous genes are connected by black lines. | ||
6.1 Biosynthesis of the amino sugar precursor
Elucidation of the biosynthesis of the amino sugar precursor was conducted solely using a lincomycin model. However, the amino sugar precursors of the natural lincosamides celesticetin and Bu-2545 are identical to that of lincomycin; therefore, it can be assumed that its formation proceeds through the same machinery (this assumption is further supported by the comparison of lincomycin and celesticetin BGCs in Fig. 11). The first biosynthetic study of the amino sugar precursor was based on feeding experiments using 13C-labelled D-glucose,164 which showed that the amino-octose may be formed by a condensation reaction catalysed by a transaldolase from a pentose 5-phosphate (C5) and a C3 unit derived from the pentose phosphate pathway. Later, the first key intermediate of the amino sugar biosynthesis, D-erythro-D-gluco-octose 8-phosphate (13), was identified (Scheme 5).165 This led to the formulation of the following two initial enzymatic steps: a transaldol reaction catalysed by LmbR using D-fructose 6-phosphate (10) or D-sedoheptulose 7-phosphate (11) as the C3 donor and D-ribose 5-phosphate (9) as the C5 acceptor, followed by 1,2-isomerization catalysed by LmbN, which converts the resulting octulose 8-phosphate (12) to octose 8-phosphate (13) (Scheme 5).165 Further biosynthetic steps were elucidated using synthetic octose 1,8-bisphosphate (14), which was converted to octose 1-phosphate (15) in an in vitro reaction catalysed by LmbK phosphatase.12 It has been proposed that octose 8-phosphate (13) is converted to the regioisomeric octose 1-phosphate (15) through the 1,8-bisphosphate intermediate 14, where the second phosphorylation is presumably catalysed by the putative kinase LmbP.12 However, these authors claimed that direct confirmation of the LmbP function was unsuccessful due to difficulties in refolding the insoluble protein. Octose 1-phosphate (15) is then converted to nucleotide-activated octose 16 by a nucleotidylyltransferase LmbO.12 The last steps comprise epimerization of the C-4 hydroxyl group, the conversion of the C-6 hydroxyl group into an amino group and a formal reduction of the primary alcohol at C-8 into a methyl group. It is not clear which of these three alternative modifications proceeds first. Therefore, two alternative routes are proposed in this review (Scheme 5). Both routes are based on the assumption that the change of the spatial orientation of the C-4 hydroxyl group from equatorial to axial could cause better steric accessibility of the C-6 hydroxyl group for proteins participating in its transformation into an amine.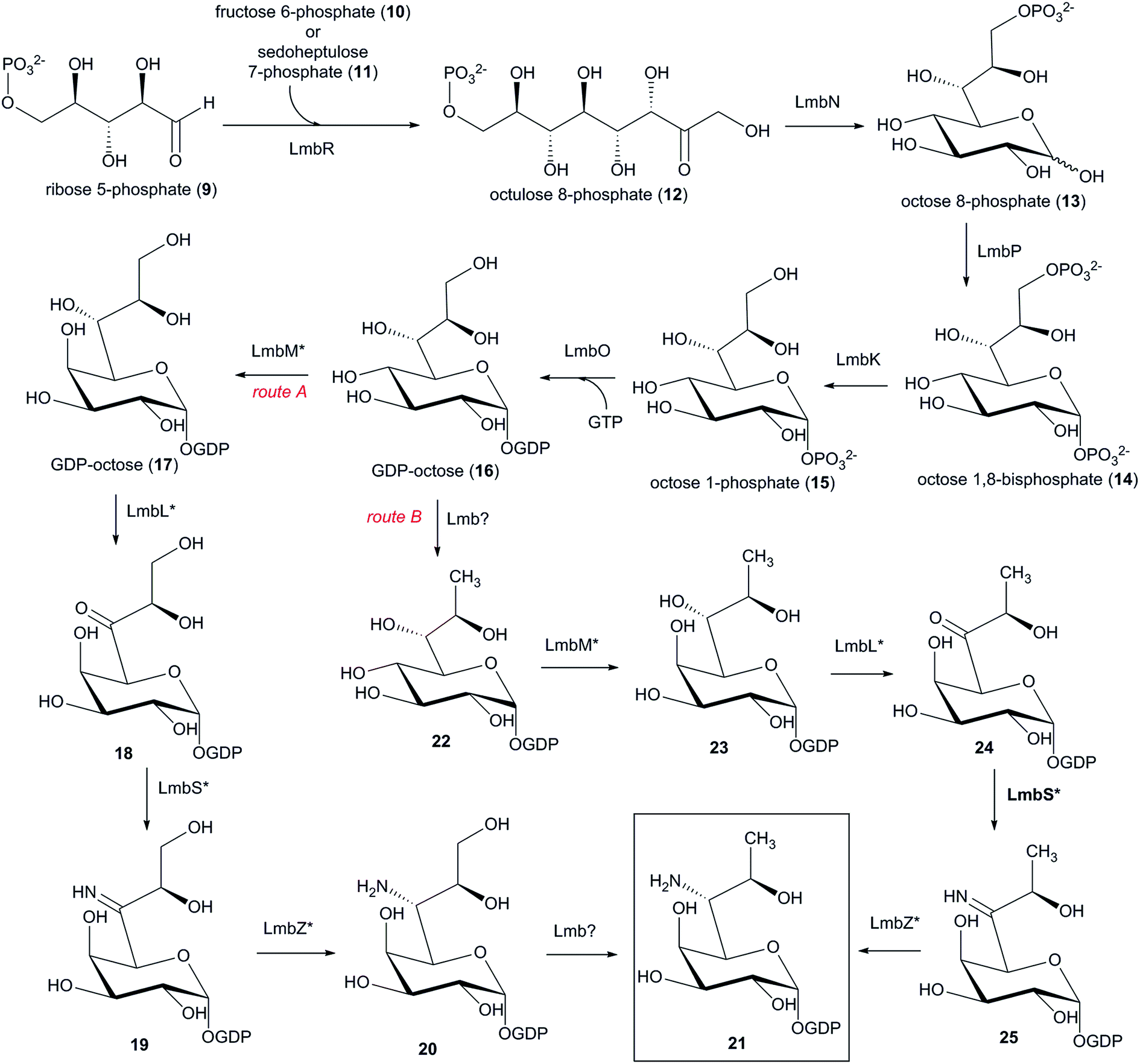 | ||
| Scheme 5 Biosynthesis of the amino-octose moiety of lincomycin. Functions of proteins marked with an asterisk are not proven. | ||
Accordingly, the epimerization should occur as the first reaction during the substitution 7-OH for 7-NH2. The number of the genes necessary for the first three steps of route A (Scheme 5) together with their predicted functions via BLAST analysis13 are in accordance with the following biosynthetic steps: a putative epimerase LmbM could epimerize the C-4 hydroxyl group of 16 affording D-galacto-octose 17 and the C-6-amino group may be introduced through three steps. LmbL (putative dehydrogenase) could catalyse the formation of ketone 18, which would give imine 19via the putative aminotransferase LmbS. The reduction of imine 19 by the putative oxidoreductase LmbZ could afford amine 20. The last step of route A represents the transformation of 20 to 21, which is, however, not a suitable candidate in neither lincomycin nor celesticetin BGC. An alternative route B starts by the formal reduction of GDP-octose 16 to 22 (Scheme 5). The remaining steps in route B are analogous to those of route A (epimerization of 22 to 23, followed by oxidation of 23 to 24, transamination of 24 to 25 and the final reduction of imine 25 to amine 21). Prior to linking amino sugar metabolism with amino acid metabolism (L-proline or APD), amino-octose 21 requires a transglycosylation step by ergothioneine, which is subsequently substituted by mycothiol to allow for the further maturation of the lincosamide skeleton (see Section 6.2).
6.2 Ergothioneine and mycothiol as hidden biosynthetic participants
Ergothioneine and mycothiol are low molecular weight thiols that are generally known for maintaining the redox potential in cells and therefore play an important role in cell detoxification processes. The major protective thiol in eukaryotes and most Gram-negative bacteria is glutathione. However, many taxonomically more specific thiols have been identified, including mycothiol and ergothioneine, gamma-glutamylcysteine, ovothiol, bacillithiol and others.166,167 Mycothiol is considered a glutathione surrogate and is the predominant thiol responsible for cell detoxification processes in most actinomycetes. Its general function lies in binding electrophiles (toxins, antibiotics) into S-conjugates, which are subsequently cleaved by Mca amidase. The resulting mercapturic acid derivatives, i.e. electrophiles bearing an N-acetylcysteine residue, are then excreted from the cell.168 Ergothioneine is biosynthesized by actinomycetes and fungi and is able to scavenge reactive oxygen species or reduce ferrylmyoglobin, which can be formed under oxidative stress.166 Aside from the protective role of the thiols, an intriguing precedent has been described for a constructive role for glutathione in the biosynthesis; in this case, it serves as a sulphur donor in the biosynthesis of gliotoxin.169 Interestingly, this sulphur-incorporation mechanism is reminiscent of the glutathione-dependent detoxification process.170 An analogous biosynthetic function was also revealed for coenzyme A. Although its function is not in cell protection, it was unexpected to reveal that it provides the cysteamine side chain in the biosynthesis of thienamycin.171 Concerning mycothiol and ergothioneine, a number of natural products have been identified as containing these thiols as part of their molecules; for instance, benzastatin JBIR-73, spithioneines A and B and clithioneine are ergothioneine S-conjugates;172–174 lusencimycins F and G are mycothiol S-conjugates; and lusencimycins D and E175 are mercapturic acid derivatives possibly converted from mycothiol S-conjugates by Mca amidase or its homologue. It is questionable whether these metabolites are simply thiol detoxification process products or whether the thiol residues have some significance for metabolite function (e.g. bioactivity), which would signify a biosynthetic relevance of the thiols. BGCs for these compounds, which could shed light on this aspect, have unfortunately not yet been published. Furthermore, even though it is a hidden to a large extent, a clearly biosynthetic role was revealed for ergothioneine and mycothiol, both of which have been shown to participate in the biosynthesis of lincosamides. Specifically, the formation of the lincosamide amide bond is dependent on the S-conjugation of ergothioneine with the amino sugar prior to the condensation reaction.7 The exact role of ergothioneine in this process remains elusive; however, it was proposed that it acts as an activator/carrier of the amino-octose precursor, similar to the role of a carrier protein or coenzyme A in amino acid activation and transfer.176 This function for ergothioneine, or protective low molecular weight thiols in general, would be unprecedented. Additionally, the involvement of ergothioneine mediates the incorporation of the sulphur atom so that it is bound in the final structure through an α-S-linkage. However, the sulphur atom originates from mycothiol in a process that is formally reminiscent of a mycothiol-dependent detoxification system; i.e. a mycothiol S-conjugate with lincosamide intermediate is formed. This intermediate is then transformed by a reaction catalysed by an Mca amidase homologue into a mercapturic acid derivative. This intermediate is not excreted from the cell as a detoxification waste product but is processed further so that only the sulphur atom label remains in the structure of lincomycin and Bu-2545 and in the ethanethiol residue in the structure of celesticetin.6.3 Unique amide bond formation and sulphur incorporation
Condensation and initial post-condensation reactions were elucidated using either lincomycin or celesticetin recombinant biosynthetic proteins, depending on the accessibility of their soluble forms. This portion of the biosynthesis is expected to proceed in the same manner in both compounds. NRPS-dissimilar condensation is initiated by the formation of a β-S-linkage between the amino-octose precursor 21 and ergothioneine in a reaction catalysed by LmbT/CcbT GTase (Scheme 6).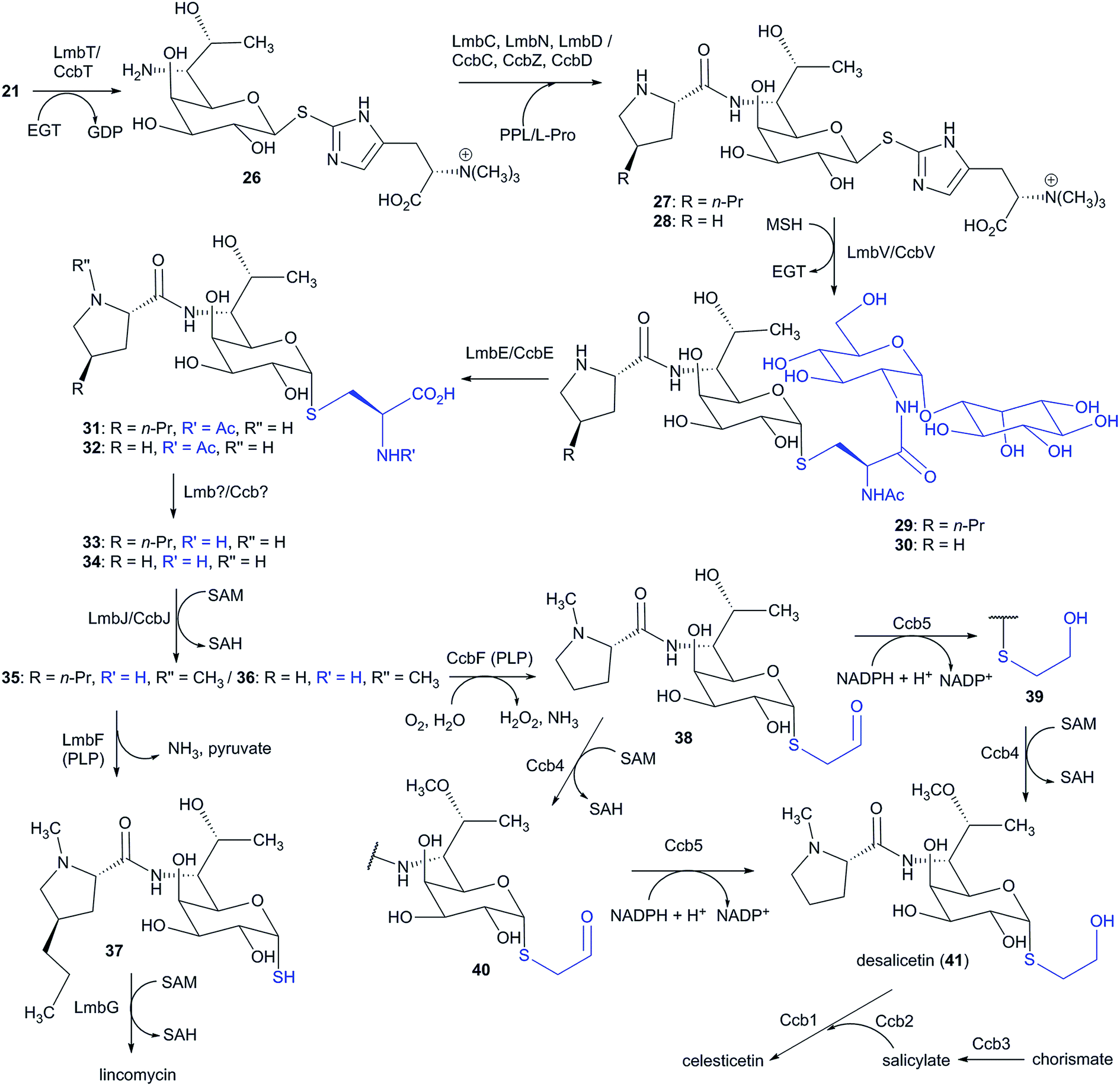 | ||
| Scheme 6 Condensation and post-condensation steps in the biosynthesis of the lincosamides lincomycin and celesticetin. Mycothiol residue and its step-by-step processing are highlighted in blue. EGT – ergothioneine, MSH – mycothiol. | ||
The resulting conjugate 26 then enters the condensation reaction with L-proline or an APD bound to the carrier protein domain of the LmbN/CcbZ bifunctional protein.13 Amide bond formation between the activated amino acid and ergothioneine-conjugated amino sugar is catalysed by the unique condensing enzyme LmbD/CcbD (Scheme 6), which exhibits no sequence similarity to any database of available proteins including other known condensing enzymes. It thus appears that LmbD/CcbD represents a condensing enzyme with a novel fold, suggesting that it evolved specifically for lincosamides. In the condensed lincosamide scaffold 27/28, the ergothioneine residue is replaced by mycothiol in a reaction catalyzed by LmbV/CcbV GTase with a conserved DinB-2 domain to afford the conjugate 29/30 with an α-S-linkage (Scheme 6), which is retained in the final lincosamide structure.7 The mycothiol residue of 29/30 is eliminated through a step-by-step sequence of post-condensation modifications starting with the removal of the 1-O-glucosamine-D-myoinositol pseudodisaccharide (Scheme 6). This biosynthetic step is catalysed by LmbE/CcbE amidase, which is homologous to the Mca amidase involved in the mycothiol detoxification process.7 The resulting mercapturic acid derivative 31/32 is unlocked for further biosynthetic steps by deacetylation of the N-acetylcysteine residue,14 for which no biosynthetic protein has been assigned. The resulting compound 33/34 is the major native substrate of SAM-dependent N-methyltransferase LmbJ/CcbJ, which attaches a methyl group at the amino acid moiety to give 35/36 (Scheme 6).14 This N-methyl ornamentation represents the last common step in the condensation and post-condensation biosynthetic steps of lincosamides (except for the incorporation of different amino acids). The N-methylation step catalysed by LmbJ can also occur after the reaction catalysed by LmbF (see Section 6.4), i.e. with the N-demethyl analogue of 37.14
6.4 Post-condensation diversification of lincosamide pathways
The unprecedented diversification of the lincosamide pathway is enabled by the homologous pyridoxal-5′-phosphate-dependent proteins LmbF and CcbF. These proteins process the intermediates 35 and 36 in different ways. LmbF catalyses β-elimination to form 37 with a sulphydryl group,14 which is subsequently methylated by LmbG S-methyltransferase to give the final pathway product lincomycin (Scheme 6).15,16 In contrast, CcbF catalyses an unusual decarboxylation-coupled oxidative deamination to preserve the two-carbon aldehyde attached at the sulphur atom, affording 38 (Scheme 6).14–16 The reactive aldehyde functional group in 38 is reduced by Ccb5 NADPH-dependent reductase to the alcohol 39 (Scheme 6). At this point, the hydroxyl group at the amino sugar moiety C-7 position is methylated by Ccb4 SAM-dependent O-methyltransferase to give desalicetin (41) (Scheme 6; an analogous step has to also occur in the biosynthesis of Bu-2545). To a lesser extent, the reactions catalysed by Ccb5 and Ccb4 can also proceed in the reverse order (Scheme 6).15,16 Subsequently, an ester bond is formed between the alcohol 41 and salicylic acid. The source of salicylic acid is presumably chorismic acid, from which it could be converted by a putative salicylate synthase Ccb3,13 which possesses a chorismate binding domain (as suggested by BLASTP). Prior to ester bond formation, salicylic acid is adenylated and transferred on CoA by Ccb2 salicylyl–CoA ligase. The salicylate–CoA conjugate and the lincosamide intermediate 41 are substrates for the unusual celesticetin-specific Ccb1 acyltransferase from the WS/DGAT-family of proteins, which catalyses the condensation of salicylic acid and 41, giving the final pathway metabolite celesticetin.17 Several previously identified natural lincosamides are products of the described biosynthesis with some alternations. Lincosamide Bu-2545, which does not have an identified biosynthetic gene cluster, is apparently formed by the same strategy as lincomycin except for the incorporation of an L-proline instead of an APD and for O-methylation of the hydroxyl group at C-7 of the sugar moiety, for which a homologue of Ccb4 O-methyltransferase should be responsible. O-Demethylcelesticetin arises as a result of Ccb4 methyltransferase omission, and celesticetin derivatives with incorporated anthranilic acid (formed in different pathways or in primary metabolism) are produced177 due to the relaxed substrate specificity by Ccb1 and Ccb2, which are responsible for the acid attachment.6.5 Evolutionary milestones in lincosamide biosynthesis
Considering the structural context of the APD incorporation, as the main topic of this review, lincomycin is an anomalous complex compound, combining rare amino acid APD with the unique amino thio-octose. However, regardless of the nature of the incorporated amino acid precursor (APD or L-proline), the lincosamide condensation system is a mysterious biosynthetic machine anyway; and is absolutely dissimilar to any other yet described system. In the evolution of lincosamide biosynthesis, we can detect two milestones: (I) set-up of the basic lincosamide condensation system, i.e. emergence of the lincosamide group, and (II) upgrade of the basic system to incorporate additional or more complex precursors.The basic lincosamide condensation system presumably produced a compound structurally close to simple Bu-2545, for which the BGC remains unknown too. Nevertheless, based on knowledge of lincosamide biosynthesis, we can easily predict all the indispensable genes. As is evident from the virtual BGC of ‘minimal’ lincosamide in Fig. 11, a majority of encoded proteins participate in the highly specialized sugar secondary metabolism: in biosynthesis of the special amino-octose or in the incorporation of a sulphur atom into its structure by unexpected metabolic coupling with ergothioneine and mycothiol metabolism.7 The clear prevalence of sugar metabolism resembling genes in the BGC reflects the core function of the unique amino thio-octose for the biological activity of lincosamides; whereby its structure is precisely “designed to fit” the ribosome target site (see Sections 2.4 and 2.5). When the proteinogenic L-proline is incorporated in the basic lincosamide structure (without the need for any biosynthetic genes in the BGC), three genes encode proteins directly involved in the amino acid moiety attachment and, only two of them resemble amino acid secondary metabolism: the stand-alone L-proline-specific A-domain and small CP-domain are general components of NRPS condensation systems in the secondary metabolism of peptides. The last but not least component of the sugar-amino acid condensing system is the most mysterious element of the whole lincosamide system; where, even though the condensation activity of the CcbD/LmbD protein has been experimentally demonstrated,7 the enzymatic reaction remains obscure. Although this protein participates in quite a common reaction, namely formation of the amide bond, it was identified exclusively in biosynthesis of lincosamides with no other homologues and no known structural motif or protein fold.
Altogether, this hybrid condensation system combining NRPS-like elements with unique reactions, including incorporation of the sulphur and formation of the amide bond, is dissimilar to any other yet described amino sugar incorporating system that employs only NRPS components, as in the biosynthesis of streptothricin178 and nourseothricin,179 or the pure NRPS-independent mechanism, as in the biosynthesis of small thiols mycothiol and bacillithiol180–183 or puromycin.184
The second milestone in lincosamide biosynthesis evolution was the upgrade to produce more efficient complex compounds by: first, the additional attachment of a salicylate moiety in celesticetin biosynthesis (when the reaction specificity of pyridoxal-5′-dependent F protein was modified); second and more interestingly, by the integration of specialized APD instead of L-proline (when the APD sub-cluster was accepted by HGT and the A-domain was adapted to activate the unusual substrate). The mysterious condensation system was thus even upgraded to connect two highly specialized secondary metabolites, instead of the combination of a secondary metabolite and primary metabolite.
In summary, a very small group of lincosamides offers an amazing list of strategies in molecular evolution, including the de novo establishment of a system for combination of precursors, acceptance of sub-clusters by HGT as well as the adaptation of substrate specificity and a shift of the reaction specificity of the involved enzymes.
7 Application potential inspired by Nature
The sub-cluster mosaic patterns of BGCs encoding complex natural products, combined with their unexpected biochemistry and unusual enzymology, can, on the one hand, seriously complicate the gene-to-molecule prediction, while on the other hand, once elucidated, it provides an amazing inspirational basis for pathway-engineered approaches towards the synthesis of valuable bioactive compounds. APD compounds as well as non-APD lincosamides represent specialized secondary metabolites of Actinobacteria formed through complex biosynthetic pathways. This is particularly true for lincomycin, where two specialized pathways, those for APD and amino sugar formation, meet one another. Recent achievements in the elucidation of APD biosynthesis and of the unique condensation system and post-condensation maturation of lincosamides have provided us with a great lesson on the mechanisms of the molecular evolution of complex secondary metabolites. Moreover, the newly acquired knowledge represents a blueprint from which we can mimic the ‘genetic engineering’ that has occurred in nature and can use this to prepare novel, unnatural, hybrid compounds.Two types of biosynthetic enzymes are essential from both an evolutionary and biotechnological point of view: (1) ‘pathway-forming’ proteins, which represent unusual proteins that are indispensable for the pathway (e.g. Apd2, LmbD/CcbD and Ccb1) and (2) ‘branch-forming’ proteins, which represent a pair of proteins responsible for pathway diversification achieved through a modified substrate or reaction specificity (e.g. Apd6LINvs. Apd6PBD, CcbC vs. LmbC and CcbF vs. LmbF). These enzymes represent the key tools for directing biosynthesis towards new hybrid compounds inspired by nature.
7.1 APD incorporation in a new context
4-Substitued prolines are considered as useful reagents in chemical synthesis, including in the design of new bioactive peptidomimetics,185 as these moieties occur frequently in biomolecules (proteins, peptides or even small molecules). The logical advantage of the C-4 position is the distance of the substituent from the residues participating in peptide bound formation, resulting in minimal steric hindrance and a potential to conjugate peptides to other chemical entities. In the case of small bioactive compounds consisting of two moieties only (like lincomycin or PBDs), formal 4-alkylation of the proline moiety offers the opportunity to extend molecule in the direction opposite to the other moiety and thus can yield the derivative with a prolonged shape. Besides APD compounds originating from L-tyrosine, exclusive for Actinobacteria, secondary metabolites with 4-methylproline moiety are produced in cyanobacteria from L-leucine.186 By screening 116 cyanobacteria strains from 8 genera, 11 new structurally diverse compounds (nonribosomal cyclic depsipeptides, nostoweipeptins and nostopeptolides) with 4-methyl-L-proline moiety were identified in two Nostoc strains.187 This suggests that the biosynthesis of natural products with 4-alkylated proline residue represents a successful natural biosynthetic concept that has evolved several times in parallel.8 Therefore, the knowledge regarding APDs and APD compound biosynthesis summarized in chapters 3 to 6 raises the question of whether APDs could replace 4-methyl-L-proline or even L-proline residues in a number of other bioactive molecules that contain these residues (published examples are listed in Section 4.2). The resulting compounds could benefit from APD incorporation in a similar way to lincosamides and PBDs (and possibly hormaomycin), which all exhibit different modes of action.7.2 Hybrid compounds using natural biosynthetic branching points
Evaluation of the antimicrobial and antiplasmodial activities of both natural and synthetic lincosamides revealed that the length of the alkyl side chain at C-4′ represents the molecule ‘hot spot’, which plays a crucial role in the efficiency of these compounds.86 Given the economic unfeasibility of preparing synthetic lincosamides, current knowledge regarding lincosamide biosynthesis has opened the door to preparing more efficient lincosamides without the need for their total synthesis. As an example, the relaxed substrate specificity of APD-specific A-domain LmbC and downstream proteins in lincomycin biosynthesis has enabled the preparation of lincomycin derivatives with an extended side chain at C-4′ by mutasynthesis. The mutasynthetic approach has resulted in more efficient 4′-butyl-4′-depropyl- and 4′-depropyl-4′-pentyl-lincomycin derivatives. Furthermore, the identification of different reaction specificity in LmbF/CcbF proteins (Scheme 6), which represent post-condensation ‘branch-forming’ points in the biosynthesis of lincomycin and celesticetin, has provided us with another opportunity to prepare more efficient lincosamides. Briefly, LmbF substrate produced by a deletion mutant strain of a lincomycin producer was purified and processed in vitro with CcbF and downstream celesticetin biosynthetic proteins Ccb5, (Ccb4), Ccb2, and Ccb1 to attach salicylic acid in the same manner as during the biosynthesis of celesticetin. The resulting compounds CELIN and ODCELIN (depending on whether celesticetin O-methyltransferase Ccb4 was employed; Fig. 5) exhibited more pronounced antibacterial activities than both celesticetin and lincomycin.17 These experiments not only produced more efficient hybrid lincosamides without chemical synthesis but also uncovered another ‘hot spot’ of the molecule: until then, the neglected salicylate moiety was shown to be of similar importance as the length of the alkyl side chain at C-4′. It seems that the salicylate moiety extends the lincosamide molecule towards the binding site of macrolides in the ribosome (see Section 2.4). Furthermore, it was shown that enzymes responsible for the attachment of salicylic acid in celesticetin biosynthesis, namely, Ccb2 salicylyl–CoA ligase and Ccb1 acyltransferase, have a relaxed substrate specificity, which allows for the incorporation of a number of benzoic acid derivatives into the lincosamide structure. Additionally, the authors proposed that using different biosynthetic systems, providing acids transferred to CoA, would possibly extend the spectrum of structures that could be attached to lincosamides beyond benzoic acid derivatives. The combination of the possible strategies to improve lincosamide efficiency has been summarized here, whereby construction of the respective engineered strains for their production and the application of simple synthetic steps, such as the efficiency improving chlorination at C-7 (i.e. the preparation of clindamycin from lincomycin), offer a broad spectrum of possibilities to prepare more efficient lincosamides, which could attract the attention of the pharmaceutical industry.The length and degree of saturation of the side chain of the APD moiety were also identified as one of several ‘hot spots’ for PBDs. Even though the preparation of antitumour agents based on PBDs has mainly focused on synthetically prepared compounds, the preparation of novel PBD scaffolds undoubtedly represents an interesting avenue of research. For instance, a suitable combination of modifications at the anthranilate and APD moieties could be explored.
8 Conclusion
The overall progress in the elucidation of APD biosynthesis has allowed us to postulate a sufficiently evidenced complete scheme of formation for this unusual precursor. However, several reactions still remain to be elucidated, and not all pathway intermediates have been fully characterized. In addition, despite the majority of Apd proteins catalysing their reactions through unusual and often unexpected enzymology, the structures of Apd proteins revealing the catalytic mechanisms in more detail have still not been solved. The so far seemingly limited distribution of APD compounds to only three groups of compounds was questioned by genome mining conducted for this review, which revealed that public databases contain a number of putative BGCs for novel APD compounds and showed that the non-PBD APD compounds are far more numerous than previously assumed. In other words, the already known trio of groups of APD compounds represents only a small portion of the existing variability.The APD compounds have never been described as independent biologically active compounds nor do they confer unique or specific functions on APD-incorporating compounds. So what is the driving force behind APD biosynthetic pathway evolution and distribution? The example of two more deeply studied groups of APD compounds (PBDs and lincosamides) could provide a possible answer. The APDs seem to be structurally and thus also functionally more advantageous variants of L-proline in many natural product structures, and moreover, the L-proline-incorporating biosynthetic systems are often almost ready to accept APDs as substitutes, requiring only a minor adaptation of key enzymes.
L-Proline, because of its specific structure, occupies a distinct position among the proteinogenic amino acids. Its incorporation confers specific properties to the final product (regardless of whether the product is a protein or a small molecule). The modification at C-4 of the L-proline molecule, especially formal C-4 alkylation, represents a structurally interesting alternative to the original L-proline, as documented by incorporation of the APD sub-cluster in the BGCs of more groups of natural compounds, which presumably originally incorporated only a simple L-proline. In each group of compounds, the final modification of the APD moiety has further evolved independently to efficiently improve the group-specific interaction with the target structure, which is reflected in the genetic composition of the respective sub-clusters. The genes in the PBD BGCs correspond to the formation of unsaturated APD moieties, which, due to their planar and slightly twisted shape, fit best into the minor groove of DNA. Additional functional groups of the APD moiety can improve the binding to DNA or shift the sequence specificity of the molecule. In the lincosamide structure, on the other hand, the fully saturated APD with a very long alkyl side-chain is needed to efficiently interfere with t-RNA binding in the A site of the ribosome.
APD sub-clusters are spread and integrated into the BGCs of variable groups of compounds relatively easily, resulting in a high diversity among the produced APD compounds. Therefore, genetic engineering employed to attain the combinatorial potential that could overcome processes in nature seems impossible. However, there are at least two examples showing that in some cases, we do have the opportunity to challenge nature. The first example is the enzymatic preparation of CELIN lincosamide, achieved through the combination of BGCs for the synthesis of highly specialized compounds, resulting in a chimera between natural lincomycin and celesticetin, i.e. an entirely nature-like hybrid compound. The failure of nature to come up with this combination may be explained by a complex metabolic coupling of lincosamide biosynthesis with other specialized cellular systems, such as the metabolism of low molecular weight thiols. It is possibly the reason for the extremely low natural frequency of this group of compounds, limiting also their natural combinatorial evolution. In such cases, the biological efficiency does not necessarily go hand in hand with their evolutionary success: lincosamides are medicinally important antibiotics and the antibacterial activity of CELIN is greater than that of any currently known natural lincosamides, including lincomycin. The other example of possibly successful genetic engineering by a combination of existing BGCs still remains hypothetical. This covers complex molecules with L-proline or 4-methyl-L-proline moieties, which could be replaced for APD moieties in order to prepare more efficient hybrid natural products. The possible target compounds form a broad spectrum of highly variable natural compounds often produced outside Actinobacteria, which could limit the combinatorial potential with Actinobacteria-specific APDs in nature by interspecies barriers. The combinatorial approaches of genetic engineering could in this case improve the natural ‘state of the art’.
9 Conflicts of interest
There are no conflicts to declare.10 Acknowledgements
The authors' laboratory work was supported by the project 17-13436Y from the Czech Science Foundation, the Ministry of Education, Youth and Sports of CR within the LQ1604 National Sustainability Program II (Project BIOCEV-FAR) and by the project “BIOCEV” (CZ.1.05/1.1.00/02.0109). The authors also thank their former colleague Tomas Kucera for his assistance with Fig. 5 and 7 and PhD students from their lab: Lucie Steiningerova, Simon Vobruba and Magdalena Pavlikova for their enormous effort to complete their experiments so that their relevant manuscripts could be already cited in this review.11 References
- P. Cimermancic, M. H. Medema, J. Claesen, K. Kurita, L. C. W. Brown, K. Mavrommatis, A. Pati, P. A. Godfrey, M. Koehrsen and J. Clardy, Cell, 2014, 158, 412–421 CrossRef PubMed.
- S. Mukherjee, R. Seshadri, N. J. Varghese, E. A. Eloe-Fadrosh, J. P. Meier-Kolthoff, M. Göker, R. C. Coates, M. Hadjithomas, G. A. Pavlopoulos and D. Paez-Espino, Nat. Biotechnol., 2017, 35, 676–683 CrossRef PubMed.
- M. H. Medema, P. Cimermancic, A. Sali, E. Takano and M. A. Fischbach, PLoS Comput. Biol., 2014, 10, e1004016 Search PubMed.
- N. M. Brahme, J. E. Gonzalez, J. P. Rolls, E. J. Hessler, S. Mizsak and L. H. Hurley, J. Am. Chem. Soc., 1984, 106, 7873–7878 CrossRef.
- B. Gerratana, Med. Res. Rev., 2012, 32, 254–293 CrossRef CAS PubMed.
- I. Höfer, M. Crüsemann, M. Radzom, B. Geers, D. Flachshaar, X. Cai, A. Zeeck and J. Piel, Chem. Biol., 2011, 18, 381–391 CrossRef PubMed.
- Q. Zhao, M. Wang, D. Xu, Q. Zhang and W. Liu, Nature, 2015, 518, 115–119 CrossRef CAS PubMed.
- P. Jiraskova, R. Gazak, Z. Kamenik, L. Steiningerova, L. Najmanova, S. Kadlcik, J. Novotna, M. Kuzma and J. Janata, Front. Microbiol., 2016, 7, 276 Search PubMed.
- G. Zhong, Q. Zhao, Q. Zhang and W. Liu, Nat. Commun., 2017, 8, 16109 CrossRef CAS PubMed.
- Z. Kamenik, S. Kadlcik, R. Gazak, S. Vobruba, L. Palanova, M. Kuzma and J. Janata, ACS Chem. Biol., 2017, 12, 1993–1998 CrossRef CAS PubMed.
- S. Kadlcik, T. Kucera, D. Chalupska, R. Gazak, M. Koberska, D. Ulanova, J. Kopecky, E. Kutejova, L. Najmanova and J. Janata, PLoS One, 2013, 8, e84902 Search PubMed.
- C. I. Lin, E. Sasaki, A. S. Zhong and H. W. Liu, J. Am. Chem. Soc., 2014, 136, 906–909 CrossRef CAS PubMed.
- J. Janata, S. Kadlcik, M. Koberska, D. Ulanova, Z. Kamenik, P. Novak, J. Kopecky, J. Novotna, B. Radojevic, K. Plhackova, R. Gazak and L. Najmanova, PLoS One, 2015, 10, e0118850 Search PubMed.
- Z. Kamenik, S. Kadlcik, B. Radojevic, P. Jiraskova, M. Kuzma, R. Gazak, L. Najmanova, J. Kopecky and J. Janata, Chem. Sci., 2016, 7, 430–435 RSC.
- M. Wang, Q. Zhao, Q. Zhang and W. Liu, J. Am. Chem. Soc., 2016, 138, 6348–6351 CrossRef CAS PubMed.
- R. Ushimaru, C.-I. Lin, E. Sasaki and H.-w. Liu, ChemBioChem, 2016, 17, 1606–1611 CrossRef CAS PubMed.
- S. Kadlcik, Z. Kamenik, D. Vasek, M. Nedved and J. Janata, Chem. Sci., 2017, 8, 3349–3355 RSC.
- Y. Hu, V. Phelan, I. Ntai, C. M. Farnet, E. Zazopoulos and B. O. Bachmann, Chem. Biol., 2007, 14, 691–701 CrossRef CAS PubMed.
- W. Li, A. Khullar, S. Chou, A. Sacramo and B. Gerratana, Appl. Environ. Microbiol., 2009, 75, 2869–2878 CrossRef CAS PubMed.
- L. Najmanova, D. Ulanova, M. Jelinkova, Z. Kamenik, E. Kettnerova, M. Koberska, R. Gazak, B. Radojevic and J. Janata, Folia Microbiol., 2014, 59, 543–552 CrossRef PubMed.
- W. Li, S. C. Chou, A. Khullar and B. Gerratana, Appl. Environ. Microbiol., 2009, 75, 2958–2963 CrossRef CAS PubMed.
- M. Pavlikova, Z. Kamenik, J. Janata, S. Kadlcik and L. Najmanova, Sci. Rep., 2018 Search PubMed , in press.
- U. Peschke, H. Schmidt, H. Z. Zhang and W. Piepersberg, Mol. Microbiol., 1995, 16, 1137–1156 CrossRef CAS PubMed.
- M. Koberska, J. Kopecky, J. Olsovska, M. Jelinkova, D. Ulanova, P. Man, M. Flieger and J. Janata, Folia Microbiol., 2008, 53, 395–401 CrossRef PubMed.
- G. Schneditz, J. Rentner, S. Roier, J. Pletz, K. A. Herzog, R. Bücker, H. Troeger, S. Schild, H. Weber and R. Breinbauer, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 13181–13186 CrossRef CAS PubMed.
- S. Nakatani, Y. Yamamoto, M. Hayashi, K. Komiyama and M. Ishibashi, Chem. Pharm. Bull., 2004, 52, 368–370 CrossRef CAS PubMed.
- C. T. Walsh, S. W. Haynes and B. D. Ames, Nat. Prod. Rep., 2012, 29, 37–59 RSC.
- W. A. van der Donk, Chem. Rev., 2017, 117, 5223–5225 CrossRef CAS PubMed.
- E. Trautman and J. Crawford, Curr. Top. Med. Chem., 2016, 16, 1705–1716 CrossRef CAS PubMed.
- K. L. Colabroy, W. T. Hackett, A. J. Markham, J. Rosenberg, D. E. Cohen and A. Jacobson, Arch. Biochem. Biophys., 2008, 479, 131–138 CrossRef CAS PubMed.
- J. Novotna, J. Olsovska, P. Novak, P. Mojzes, R. Chaloupkova, Z. Kamenik, J. Spizek, E. Kutejova, M. Mareckova, P. Tichy, J. Damborsky and J. Janata, PLoS One, 2013, 8, e79974 Search PubMed.
- A. von Tesmar, M. Hoffmann, J. Pippel, A. A. Fayad, S. Dausend-Werner, A. Bauer, W. Blankenfeldt and R. Müller, Cell Chem. Biol., 2017, 24(10), 1216–1227 CrossRef CAS PubMed.
- M. Šulc, I. Fadrhoncová, M. Jelínková, M. Chudomelová, J. Felsberg and J. Olšovská, J. Chromatogr. A, 2011, 1218, 83–91 CrossRef PubMed.
- L. H. Hurley, C. Gairola and N. V. Das, Biochemistry, 1976, 15, 3760–3769 CrossRef CAS PubMed.
- Y. Han, Y. Li, Y. Shen, J. Li, W. Li and Y. Shen, Drug Discoveries Ther., 2013, 7, 243–247 CAS.
- R. Jiao, H. Xu, J. Cui, H. Ge and R. Tan, J. Appl. Microbiol., 2013, 114, 1046–1053 CrossRef CAS PubMed.
- N. Mohr and H. Budzikiewicz, Tetrahedron, 1982, 38, 147–152 CrossRef CAS.
- H. Tse, Q. Gu, K. Sze, I. Chu, R. Kao, K. Lee, C. Lam, D. Yang, S. Tai, Y. Ke, E. Chan, W. Chan, J. Dai, S. Leung, S. Leung and K. Yuen, J. Biol. Chem., 2017, 292, 19503–19520 CrossRef CAS PubMed.
- E. Dornisch, J. Pletz, R. Glabonjat, F. Martin, C. Lembacher-Fadum, M. Neger, C. Hogenauer, K. Francesconi, W. Kroutil, K. Zangger, R. Breinbauer and E. Zechner, Angew. Chem., Int. Ed., 2017, 56, 14753–14757 CrossRef CAS PubMed.
- S. Sato, F. Iwata, S. Yamada, H. Kawahara and M. Katayama, Bioorg. Med. Chem. Lett., 2011, 21, 7099–7101 CrossRef CAS PubMed.
- M. Oh, J.-H. Jang, S.-J. Choo, S.-O. Kim, J. W. Kim, S.-K. Ko, N.-K. Soung, J.-S. Lee, C.-J. Kim and H. Oh, Bioorg. Med. Chem. Lett., 2014, 24, 1802–1804 CrossRef PubMed.
- W. Leimgruber, A. Batcho and F. Schenker, J. Am. Chem. Soc., 1965, 87, 5793–5795 CrossRef CAS PubMed.
- M. Brazhnikova, I. Kovsharova, N. Konstantinova, A. Mezentsev and V. Proshliakova, Antibiotiki, 1970, 15, 297–300 CAS.
- S. Kunimoto, T. Masuda, N. Kanbayashi, M. Hamada, H. Naganawa, M. Miyamoto, T. Takeuchi and H. Umezawa, J. Antibiot., 1980, 33, 665–667 CrossRef CAS PubMed.
- M. Tsunakawa, H. Kamei, M. Konishi, T. Miyaki, T. Oki and H. Kawaguchi, J. Antibiot., 1988, 41, 1366–1373 CrossRef CAS PubMed.
- J. Itoh, H.-O. Watanabe, S. Ishii, S. Gomi, M. Nagasawa, H. Yamamoto, T. Shomura, M. Sezaki and S. Kondo, J. Antibiot., 1988, 41, 1281–1284 CrossRef CAS PubMed.
- K. Kariyone, H. Yazawa and M. Kohsaka, Chem. Pharm. Bull., 1971, 19, 2289–2293 CrossRef CAS.
- K.-I. Shimizu, I. Kawamoto, F. Tomita, M. Morimoto and K. Fujimoto, J. Antibiot., 1982, 35, 972–978 CrossRef PubMed.
- S. Fotso, T. M. Zabriskie, P. J. Proteau, P. M. Flatt, D. A. Santosa and T. Mahmud, J. Nat. Prod., 2009, 72, 690–695 CrossRef PubMed.
- T. Fusao, K. Isao, T. Tatsuya, A. Kouzou, M. Makoto, I. Riyouji and F. Kazuhisa, Antibiotic DC-81 and its preparation, JPS58180487 (A), 1982.
- M. Konishi, M. Hatori, K. Tomita, M. Sugawara, C. Ikeda, Y. Nishiyama, H. Imanishi, T. Miyaki and H. Kawaguchi, J. Antibiot., 1984, 37, 191–199 CrossRef PubMed.
- J. E. Hochlowski, W. W. Andres, R. J. Theriault, M. Jackson and J. B. McAlpine, J. Antibiot., 1987, 40, 145–148 CrossRef PubMed.
- M. Miyamoto, S. Kondo, H. Naganawa, K. Maeda, M. Ohno and H. Umezawa, J. Antibiot., 1977, 30, 340–343 CrossRef PubMed.
- J. Mantaj, P. J. Jackson, K. M. Rahman and D. E. Thurston, Angew. Chem., Int. Ed., 2016, 56, 462–488 CrossRef PubMed.
- M. L. Kopka, D. S. Goodsell, I. Baikalov, K. Grzeskowiak, D. Cascio and R. E. Dickerson, Biochemistry, 1994, 33, 13593–13610 CrossRef CAS PubMed.
- J. M. Reid, S. A. Buhrow, M. J. Kuffel, L. Jia, V. J. Spanswick, J. A. Hartley, D. E. Thurston, J. E. Tomaszewski and M. M. Ames, Cancer Chemother. Pharmacol., 2011, 68, 777–786 CrossRef CAS PubMed.
- G. Wells, C. R. Martin, P. W. Howard, Z. A. Sands, C. A. Laughton, A. Tiberghien, C. K. Woo, L. A. Masterson, M. J. Stephenson and J. A. Hartley, J. Med. Chem., 2006, 49, 5442–5461 CrossRef CAS PubMed.
- M. S. Puvvada, J. A. Hartley, T. C. Jenkins and D. E. Thurston, Nucleic Acids Res., 1993, 21, 3671–3675 CrossRef CAS PubMed.
- M. S. Puvvada, S. A. Forrow, J. A. Hartley, P. Stephenson, I. Gibson, T. C. Jenkins and D. E. Thurston, Biochemistry, 1997, 36, 2478–2484 CrossRef CAS PubMed.
- P. Baraldi, B. Cacciarit, A. Guiotto, R. Romagnoli, G. Spalluto, A. Leoni, N. Bianchi, G. Feriotto, C. Rutigliano and C. Mischiati, Nucleosides, Nucleotides Nucleic Acids, 2000, 19, 1219–1229 CAS.
- M. Kotecha, J. Kluza, G. Wells, C. C. O'Hare, C. Forni, R. Mantovani, P. W. Howard, P. Morris, D. E. Thurston and J. A. Hartley, Mol. Cancer Ther., 2008, 7, 1319–1328 CrossRef CAS PubMed.
- Y.-W. Chou, G. C. Senadi, C.-Y. Chen, K.-K. Kuo, Y.-T. Lin, J.-J. Wang, J.-H. Lee, Y.-C. Wang and W.-P. Hu, Eur. J. Med. Chem., 2016, 109, 59–74 CrossRef CAS PubMed.
- M.-C. Hsieh, W.-P. Hu, H.-S. Yu, W.-C. Wu, L.-S. Chang, Y.-H. Kao and J.-J. Wang, Toxicol. Appl. Pharmacol., 2011, 255, 150–159 CrossRef CAS PubMed.
- D. Antonow, T. C. Jenkins, P. W. Howard and D. E. Thurston, Bioorg. Med. Chem., 2007, 15, 3041–3053 CrossRef CAS PubMed.
- M. Shameem, R. Kumar, S. Krishna, C. Kumar, M. I. Siddiqi, B. Kundu and D. Banerjee, Chem.-Biol. Interact., 2015, 237, 115–124 CrossRef CAS PubMed.
- S. Omura, H. Mamada, N.-J. Wang, N. Imamura, R. Oiwa, Y. Iwai and N. Muto, J. Antibiot., 1984, 37, 700–705 CrossRef CAS PubMed.
- X. Cai, R. Teta, C. Kohlhaas, M. Crüsemann, R. Ueoka, A. Mangoni, M. F. Freeman and J. Piel, Chem. Biol., 2013, 20, 839–846 CrossRef CAS PubMed.
- M. Bae, B. Chung, K.-B. Oh, J. Shin and D.-C. Oh, Mar. Drugs, 2015, 13, 5187–5200 CrossRef CAS PubMed.
- N. Andres, H. Wolf and H. Zähner, Z. Naturforsch., C: J. Biosci., 1990, 45, 851–855 CAS.
- K. Otoguro, H. Ui, A. Ishiyama, N. Arai, M. Kobayashi, Y. Takahashi, R. Masuma, K. Shiomi, H. Yamada and S. Omura, J. Antibiot., 2003, 56, 322–324 CrossRef PubMed.
- D. Mason, A. Dietz and C. DeBoer, Antimicrob. Agents Chemother., 1962, 2, 554–559 Search PubMed.
- C. DeBoer, A. Dietz, J. Wilkins, C. Lewis and G. Savage, Antibiot. Annu., 1954, 831–836 Search PubMed.
- H. Hoeksema, G. F. Crum and W. H. DeVries, Antibiot. Annu., 1955, 2, 837–841 Search PubMed.
- H. Hoeksema, J. Am. Chem. Soc., 1968, 90, 755–757 CrossRef CAS PubMed.
- M. Hanada, M. Tsunakawa, K. Tomita, H. Tsukiura and H. Kawaguchi, J. Antibiot., 1980, 33, 751–753 CrossRef CAS PubMed.
- J. Spizek, J. Novotna and T. Rezanka, Adv. Appl. Microbiol., 2004, 56, 121–154 CAS.
- A. Argoudelis, J. Fox, D. Mason and T. Eble, J. Am. Chem. Soc., 1964, 86, 5044–5045 CrossRef CAS.
- A. P. Pang, L. Du, C. Y. Lin, J. Qiao and G. R. Zhao, J. Appl. Microbiol., 2015, 119, 1064–1074 CrossRef CAS PubMed.
- D. Garrison, R. M. DeHaan and J. B. Lawson, Antimicrob. Agents Chemother., 1966, 7, 397–400 Search PubMed.
- T. Tenson and M. Ehrenberg, Cell, 2002, 108, 591–594 CrossRef CAS PubMed.
- F. Schlunzen, R. Zarivach, J. Harms and A. Bashan, Nature, 2001, 413, 814–821 CrossRef CAS PubMed.
- J. A. Dunkle, L. Xiong, A. S. Mankin and J. H. Cate, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 17152–17157 CrossRef CAS PubMed.
- J. I. Ross, E. A. Eady, J. H. Cove, C. E. Jones, A. H. Ratyal, Y. W. Miller, S. Vyakrnam and W. J. Cunliffe, Antimicrob. Agents Chemother., 1997, 41, 1162–1165 CAS.
- D. N. Wilson, Nat. Rev. Microbiol., 2014, 12, 35–48 CrossRef CAS PubMed.
- C. Lewis, J. Parasitol., 1968, 54, 169–170 CrossRef CAS PubMed.
- B. J. Magerlein, Adv. Appl. Microbiol., 1971, 14, 185–229 CAS.
- M. E. Fichera and D. S. Roos, Nature, 1997, 390, 407–409 CrossRef CAS PubMed.
- World Health Organization, Guidelines for the treatment of malaria, 2015 Search PubMed.
- D. E. Thurston, D. S. Bose, P. W. Howard, T. C. Jenkins, A. Leoni, P. G. Baraldi, A. Guiotto, B. Cacciari, L. R. Kelland and M.-P. Foloppe, J. Med. Chem., 1999, 42, 1951–1964 CrossRef CAS PubMed.
- A. D. Argoudelis, T. Eble, J. Fox and D. J. Mason, Biochemistry, 1969, 8, 3408–3411 CrossRef CAS PubMed.
- D. Witz, E. J. Hessler and T. L. Miller, Biochemistry, 1971, 10, 1128–1133 CrossRef CAS PubMed.
- L. H. Hurley, M. Zmijewski and C.-J. Chang, J. Am. Chem. Soc., 1975, 97, 4372–4378 CrossRef CAS PubMed.
- L. H. Hurley, W. L. Lasswell, R. K. Malhotra and N. V. Das, Biochemistry, 1979, 18, 4225–4229 CrossRef CAS PubMed.
- M. Kuo, D. Yurek, J. Coats, S. Chung and G. Li, J. Antibiot., 1992, 45, 1773–1777 CrossRef CAS PubMed.
- L. H. Hurley, Acc. Chem. Res., 1980, 13, 263–269 CrossRef CAS.
- D. Neusser, H. Schmidt, J. Spizek, J. Novotna, U. Peschke, S. Kaschabeck, P. Tichy and W. Piepersberg, Arch. Microbiol., 1998, 169, 322–332 CrossRef CAS PubMed.
- K. L. Connor, K. L. Colabroy and B. Gerratana, Biochemistry, 2011, 50, 8926–8936 CrossRef CAS PubMed.
- J. Novotna, A. Honzatko, P. Bednar, J. Kopecky, J. Janata and J. Spizek, Eur. J. Biochem., 2004, 271, 3678–3683 CrossRef CAS PubMed.
- S. Saha, W. Li, B. Gerratana and S. E. Rokita, Bioorg. Med. Chem., 2015, 23, 449–454 CrossRef CAS PubMed.
- F. Terradas and H. Wyler, 1991, 74, 124–140.
- S. D. Braun, J. Hofmann, A. Wensing, M. Ullrich, H. Weingart, B. Völksch and D. Spiteller, Appl. Environ. Microbiol., 2010, 76, 2500–2508 CrossRef CAS PubMed.
- Y. T. Huang, S. Y. Lyu, P. H. Chuang, N. S. Hsu, Y. S. Li, H. C. Chan, C. J. Huang, Y. C. Liu, C. J. Wu and W. B. Yang, ChemBioChem, 2009, 10, 2480–2487 CrossRef CAS PubMed.
- X.-W. Zou, Y.-C. Liu, N.-S. Hsu, C.-J. Huang, S.-Y. Lyu, H.-C. Chan, C.-Y. Chang, H.-W. Yeh, K.-H. Lin and C.-J. Wu, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2014, 70, 1549–1560 CAS.
- E. A. Burks, W. Yan, W. H. Johnson Jr, W. Li, G. K. Schroeder, C. Min, B. Gerratana, Y. Zhang and C. P. Whitman, Biochemistry, 2011, 50, 7600–7611 CrossRef CAS PubMed.
- L. Najmanova, E. Kutejova, J. Kadlec, M. Polan, J. Olsovska, O. Benada, J. Novotna, Z. Kamenik, P. Halada, J. Bauer and J. Janata, ChemBioChem, 2013, 14, 2259–2262 CrossRef CAS PubMed.
- J. Bauer, G. Ondrovicova, L. Najmanova, V. Pevala, Z. Kamenik, J. Kostan, J. Janata and E. Kutejova, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2014, 70, 943–957 CAS.
- A. Argoudelis, J. Coats, P. Lemaux and O. Sebek, J. Antibiot., 1972, 25, 445–455 CrossRef CAS PubMed.
- L. Chen, Z.-l. Wang, Q.-f. Zhao, S.-h. Gao and R.-f. Ye, Chem. Bioeng., 2011, 28, 37–41 CAS.
- V. Dangel, A. S. Eustaquio, B. Gust and L. Heide, Arch. Microbiol., 2008, 190, 509–519 CrossRef CAS PubMed.
- E. Conti, T. Stachelhaus, M. A. Marahiel and P. Brick, EMBO J., 1997, 16, 4174–4183 CrossRef CAS PubMed.
- T. Stachelhaus, H. D. Mootz and M. A. Marahiel, Chem. Biol., 1999, 6, 493–505 CrossRef CAS PubMed.
- G. L. Challis, J. Ravel and C. A. Townsend, Chem. Biol., 2000, 7, 211–224 CrossRef CAS PubMed.
- D. Ulanova, J. Novotna, Y. Smutna, Z. Kamenik, R. Gazak, M. Sulc, P. Sedmera, S. Kadlcik, K. Plhackova and J. Janata, Antimicrob. Agents Chemother., 2010, 54, 927–930 CrossRef CAS PubMed.
- S. Vobruba, S. Kadlcik, R. Gazak and J. Janata, PLoS One, 2017, 12, e0189684 Search PubMed.
- M. G. Thomas, M. D. Burkart and C. T. Walsh, Chem. Biol., 2002, 9, 171–184 CrossRef CAS PubMed.
- S. Garneau, P. C. Dorrestein, N. L. Kelleher and C. T. Walsh, Biochemistry, 2005, 44, 2770–2780 CrossRef CAS PubMed.
- A. Méjean, S. Mann, G. Vassiliadis, B. Lombard, D. Loew and O. Ploux, Biochemistry, 2010, 49, 103–113 CrossRef PubMed.
- M. Kopp, H. Irschik, K. Gemperlein, K. Buntin, P. Meiser, K. J. Weissman, H. B. Bode and R. Müller, Mol. BioSyst., 2011, 7, 1549–1563 RSC.
- Y. Wang, Y. Chen, Q. Shen and X. Yin, Gene, 2011, 483, 11–21 CrossRef CAS PubMed.
- C. Müller, S. Nolden, P. Gebhardt, E. Heinzelmann, C. Lange, O. Puk, K. Welzel, W. Wohlleben and D. Schwartz, Antimicrob. Agents Chemother., 2007, 51, 1028–1037 CrossRef PubMed.
- T. Kawasaki, F. Sakurai, S.-y. Nagatsuka and Y. Hayakawa, J. Antibiot., 2009, 62, 271 CrossRef CAS PubMed.
- S. M. Salem, P. Kancharla, G. Florova, S. Gupta, W. Lu and K. A. Reynolds, J. Am. Chem. Soc., 2014, 136, 4565–4574 CrossRef CAS PubMed.
- X. Zhang and R. J. Parry, Antimicrob. Agents Chemother., 2007, 51, 946–957 CrossRef CAS PubMed.
- A. K. Harris, N. R. Williamson, H. Slater, A. Cox, S. Abbasi, I. Foulds, H. T. Simonsen, F. J. Leeper and G. P. Salmond, Microbiology, 2004, 150, 3547–3560 CrossRef CAS PubMed.
- D. Kim, Y. K. Park, J. S. Lee, J. F. Kim, H. Jeong, B. S. Kim and C. H. Lee, J. Microbiol. Biotechnol., 2006, 16, 1912–1918 CAS.
- A. El Gamal, V. Agarwal, S. Diethelm, I. Rahman, M. A. Schorn, J. M. Sneed, G. V. Louie, K. E. Whalen, T. J. Mincer and J. P. Noel, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 3797–3802 CrossRef CAS PubMed.
- S. Maharjan, N. Aryal, S. Bhattarai, D. Koju, J. Lamichhane and J. K. Sohng, Appl. Microbiol. Biotechnol., 2012, 93, 687–696 CrossRef CAS PubMed.
- C. Li, K. E. Roege and W. L. Kelly, ChemBioChem, 2009, 10, 1064–1072 CrossRef CAS PubMed.
- Q. Wu, J. Liang, S. Lin, X. Zhou, L. Bai, Z. Deng and Z. Wang, Antimicrob. Agents Chemother., 2011, 55, 974–982 CrossRef CAS PubMed.
- F. Pojer, S.-M. Li and L. Heide, Microbiology, 2002, 148, 3901–3911 CrossRef CAS PubMed.
- A. Rantala-Ylinen, S. Känä, H. Wang, L. Rouhiainen, M. Wahlsten, E. Rizzi, K. Berg, M. Gugger and K. Sivonen, Appl. Environ. Microbiol., 2011, 77, 7271–7278 CrossRef CAS PubMed.
- P. Meiser, K. J. Weissman, H. B. Bode, D. Krug, J. S. Dickschat, A. Sandmann and R. Müller, Chem. Biol., 2008, 15, 771–781 CrossRef CAS PubMed.
- F. Saito, K. Hori, M. Kanda, T. Kurotsu and Y. Saito, J. Biochem., 1994, 116, 357–367 CrossRef CAS PubMed.
- H. D. Mootz and M. A. Marahiel, J. Bacteriol., 1997, 179, 6843–6850 CrossRef CAS PubMed.
- S. A. Samel, B. Wagner, M. A. Marahiel and L.-O. Essen, J. Mol. Biol., 2006, 359, 876–889 CrossRef CAS PubMed.
- E. H. Duitman, L. W. Hamoen, M. Rembold, G. Venema, H. Seitz, W. Saenger, F. Bernhard, R. Reinhardt, M. Schmidt and C. Ullrich, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 13294–13299 CrossRef CAS.
- K. Tsuge, T. Akiyama and M. Shoda, Journal of bacteriology, 2001, 183, 6265–6273 CrossRef CAS PubMed.
- K. Tsuge, T. Ano, M. Hirai, Y. Nakamura and M. Shoda, Antimicrob. Agents Chemother., 1999, 43, 2183–2192 CAS.
- B. K. Scholz-Schroeder, J. D. Soule and D. C. Gross, Mol. Plant-Microbe Interact., 2003, 16, 271–280 CrossRef CAS PubMed.
- J. Mareš, J. Hájek, P. Urajová, J. Kopecký and P. Hrouzek, PLoS One, 2014, 9, e111904 Search PubMed.
- S. C. Wenzel, B. Kunze, G. Höfle, B. Silakowski, M. Scharfe, H. Blöcker and R. Müller, ChemBioChem, 2005, 6, 375–385 CrossRef CAS PubMed.
- D. Hoffmann, J. M. Hevel, R. E. Moore and B. S. Moore, Gene, 2003, 311, 171–180 CrossRef CAS PubMed.
- H. Luesch, D. Hoffmann, J. M. Hevel, J. E. Becker, T. Golakoti and R. E. Moore, The Journal of organic chemistry, 2003, 68, 83–91 CrossRef CAS PubMed.
- C.-J. Huang, E. Pauwelyn, M. Ongena, D. Debois, V. Leclère, P. Jacques, P. Bleyaert and M. Höfte, Mol. Plant-Microbe Interact., 2015, 28, 1009–1022 CrossRef CAS PubMed.
- X. Wang, J. Tabudravu, M. E. Rateb, K. J. Annand, Z. Qin, M. Jaspars, Z. Deng, Y. Yu and H. Deng, Mol. BioSyst., 2013, 9, 1286–1289 RSC.
- F. Pfennig, F. Schauwecker and U. Keller, J. Biol. Chem., 1999, 274, 12508–12516 CrossRef CAS PubMed.
- S. Pohle, C. Appelt, M. Roux, H.-P. Fiedler and R. D. Süssmuth, J. Am. Chem. Soc., 2011, 133, 6194–6205 CrossRef CAS PubMed.
- V. Crecy-Lagard, V. Blanc, P. Gil, L. Naudin, S. Lorenzon, A. Famechon, N. Bamas-Jacques, J. Crouzet and D. Thibaut, J. Bacteriol., 1997, 179, 705–713 CrossRef PubMed.
- V. V. Phelan, Y. Du, J. A. McLean and B. O. Bachmann, Chem. Biol., 2009, 16, 473–478 CrossRef CAS PubMed.
- T. W. Giessen, F. I. Kraas and M. A. Marahiel, Biochemistry, 2011, 50, 5680–5692 CrossRef CAS PubMed.
- J. Li, Z. Xie, M. Wang, G. Ai and Y. Chen, PLoS One, 2015, 10, e0120542 Search PubMed.
- J. B. McAlpine, A. H. Banskota, R. D. Charan, G. Schlingmann, E. Zazopoulos, M. Piraee, J. Janso, V. S. Bernan, M. Aouidate and C. M. Farnet, J. Nat. Prod., 2008, 71, 1585–1590 CrossRef CAS PubMed.
- A. A. Losada, C. Méndez, J. A. Salas and C. Olano, Microb. Cell Fact., 2017, 16, 93 CrossRef PubMed.
- M. Lv, J. Zhao, Z. Deng and Y. Yu, Chem. Biol., 2015, 22, 1313–1324 CrossRef CAS PubMed.
- Q.-A. Li, D. V. Mavrodi, L. S. Thomashow, M. Roessle and W. Blankenfeldt, J. Biol. Chem., 2011, 286, 18213–18221 CrossRef CAS PubMed.
- J. F. Parsons, F. Song, L. Parsons, K. Calabrese, E. Eisenstein and J. E. Ladner, Biochemistry, 2004, 43, 12427–12435 CrossRef CAS PubMed.
- J. F. Parsons, K. Calabrese, E. Eisenstein and J. E. Ladner, Biochemistry, 2003, 42, 5684–5693 CrossRef CAS PubMed.
- S. Saha and S. E. Rokita, ChemBioChem, 2016, 17, 1818–1823 CrossRef CAS PubMed.
- M. Brandl, S. I. Kozhushkov, B. D. Zlatopolskiy, P. Alvermann, B. Geers, A. Zeeck and A. de Meijere, Eur. J. Org. Chem., 2005, 2005, 123–135 CrossRef.
- S. I. Kozhushkov, B. D. Zlatopolskiy, M. Brandl, P. Alvermann, M. Radzom, B. Geers, A. de Meijere and A. Zeeck, Eur. J. Org. Chem., 2005, 2005, 854–863 CrossRef.
- A. C. Jones, E. A. Monroe and W. H. Gerwick, Chem. Biol., 2011, 18, 281–283 CrossRef CAS PubMed.
- L. Heide, L. Westrich, C. Anderle, B. Gust, B. Kammerer and J. Piel, ChemBioChem, 2008, 9, 1992–1999 CrossRef CAS PubMed.
- M. Crüsemann, C. Kohlhaas and J. Piel, Chem. Sci., 2013, 4, 1041–1045 RSC.
- N. M. Brahme, J. Gonzalez, S. Mizsak, J. Rolls, E. Hessler and L. Hurley, J. Am. Chem. Soc., 1984, 106, 7878–7883 CrossRef CAS.
- E. Sasaki, C. I. Lin, K. Y. Lin and H. W. Liu, J. Am. Chem. Soc., 2012, 134, 17432–17435 CrossRef CAS PubMed.
- K. Van Laer, C. J. Hamilton and J. Messens, Antioxid. Redox Signaling, 2013, 18, 1642–1653 CrossRef CAS PubMed.
- R. C. Fahey, Biochim. Biophys. Acta, 2013, 1830, 3182–3198 CrossRef CAS PubMed.
- G. L. Newton, Y. Av-Gay and R. C. Fahey, Biochemistry, 2000, 39, 10739–10746 CrossRef CAS PubMed.
- D. H. Scharf, T. Heinekamp, N. Remme, P. Hortschansky, A. A. Brakhage and C. Hertweck, Appl. Microbiol. Biotechnol., 2012, 93, 467–472 CrossRef CAS PubMed.
- A. E. Salinas and M. G. Wong, Curr. Med. Chem., 1999, 6, 279–310 CAS.
- M. F. Freeman, K. A. Moshos, M. J. Bodner, R. Li and C. A. Townsend, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 11128–11133 CrossRef CAS PubMed.
- K. Motohashi, A. Nagai, M. Takagi and K. Shin-ya, J. Antibiot., 2011, 64, 281–283 CrossRef CAS PubMed.
- P. Fu and J. B. MacMillan, Org. Lett., 2015, 17, 3046–3049 CrossRef CAS PubMed.
- K. Konno, H. Shirahama and T. Matsumoto, Tetrahedron Lett., 1981, 22, 1617–1618 CrossRef CAS.
- S. B. Singh, D. L. Zink, K. Dorso, M. Motyl, O. Salazar, A. Basilio, F. Vicente, K. M. Byrne, S. Ha and O. Genilloud, J. Nat. Prod., 2009, 72, 345–352 CrossRef CAS PubMed.
- M. Wang, Q. Zhao and W. Liu, BioEssays, 2015, 37, 1262–1267 CrossRef PubMed.
- A. Argoudelis and T. Brodasky, J. Antibiot., 1972, 25, 194–196 CrossRef CAS PubMed.
- C. Maruyama, J. Toyoda, Y. Kato, M. Izumikawa, M. Takagi, K. Shin-ya, H. Katano, T. Utagawa and Y. Hamano, Nat. Chem. Biol., 2012, 8, 791–797 CrossRef CAS PubMed.
- N. Grammel, K. Pankevych, J. Demydchuk, K. Lambrecht, H. P. Saluz and H. Krügel, Eur. J. Biochem., 2002, 269, 347–357 CrossRef CAS PubMed.
- G. L. Newton, N. Buchmeier and R. C. Fahey, Microbiol. Mol. Biol. Rev., 2008, 72, 471–494 CrossRef CAS PubMed.
- V. K. Jothivasan and C. J. Hamilton, Nat. Prod. Rep., 2008, 25, 1091–1117 RSC.
- A. Gaballa, G. L. Newton, H. Antelmann, D. Parsonage, H. Upton, M. Rawat, A. Claiborne, R. C. Fahey and J. D. Helmann, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6482–6486 CrossRef CAS PubMed.
- D. Sareen, M. Steffek, G. L. Newton and R. C. Fahey, Biochemistry, 2002, 41, 6885–6890 CrossRef CAS PubMed.
- M. A. Rubio, P. Barrado, J. C. Espinosa, A. Jiménez and M. F. Lobato, FEBS Lett., 2004, 577, 371–375 CrossRef PubMed.
- A. Tolomelli, L. Ferrazzano and R. Amoroso, Aldrichimica Acta, 2017, 50, 3–15 Search PubMed.
- K. Fujii, K. Sivonen, K. Adachi, K. Noguchi, H. Sano, K. Hirayama, M. Suzuki and K.-i. Harada, Tetrahedron Lett., 1997, 38, 5525–5528 CrossRef CAS.
- L. Liu, J. Jokela, L. Herfindal, M. Wahlsten, J. Sinkkonen, P. Permi, D. P. Fewer, S. O. Døskeland and K. Sivonen, ACS Chem. Biol., 2014, 9, 2646–2655 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7np00047b |
| This journal is © The Royal Society of Chemistry 2018 |