
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceClassic fungal natural products in the genomic age: the molecular legacy of Harold Raistrick†
Raissa
Schor
and
Russell
Cox
 *
*
Institut für Organische Chemie, BMWZ, Leibniz Universität Hannover, Germany. E-mail: russell.cox@oci.uni-hannover.de
First published on 14th March 2018
Abstract
Covering: 1893 to 2017
Harold Raistrick was involved in the discovery of many of the most important classes of fungal metabolites during the 20th century. This review focusses on how these discoveries led to developments in isotopic labelling, biomimetic chemistry and the discovery, analysis and exploitation of biosynthetic gene clusters for major classes of fungal metabolites including: alternariol; geodin and metabolites of the emodin pathway; maleidrides; citrinin and the azaphilones; dehydrocurvularin; mycophenolic acid; and the tropolones. Key recent advances in the molecular understanding of these important pathways, including the discovery of biosynthetic gene clusters, the investigation of the molecular and chemical aspects of key biosynthetic steps, and the reengineering of key components of the pathways are reviewed and compared. Finally, discussion of key relationships between metabolites and pathways and the most important recent advances and opportunities for future research directions are given.
 Raissa Schor | Raissa Schor received a B. Sc. in Chemistry and Management from the University of Ulm in 2010 and a M. Sc. in Chemistry of the Julius-Maximilians-Universität Würzburg in 2013. She joined the Cox group as a PhD student in 2013, where she worked on the elucidation of the biosynthesis of fungal meroterpenoids. |
 Russell Cox | Russell Cox was born in 1967 in the New Forest in the UK where he grew up. He studied chemistry at the University of Durham, and then worked with Prof. David O'Hagan at the same institution for his PhD, studying the biosynthesis of fungal metabolites. Post-doctoral periods with Professor John Vederas FRS in Edmonton Alberta, and Professors David Hopwood FRS and Tom Simpson FRS at Norwich and Bristol in the UK were followed by his appointment as a lecturer in the School of Chemistry at the University of Bristol where he rose to become full Professor of Organic and Biological Chemistry. He moved to become Professor of Microbiological Chemistry at the Leibniz Universität Hannover in Germany in 2013. He has served as an editorial board member of Natural Product Reports until 2012, is currently an editorial board member of RSC Advances, and has been past chair of the Directing Biosynthesis series of scientific conferences. |
1. Harold Raistrick 1890–1971 and fungal natural products
In July 1929, at the age of 38, Harold Raistrick was appointed to his first academic post as University Chair of Biochemistry at the University of London.1 By the end of 1934 he had published 42 scientific papers in the fledgling area of natural products chemistry and been elected to the Royal Society, publishing 20 papers during 1931 alone. This remarkable period of scientific productivity was built on an equally remarkable 8 year period when Raistrick was the head of biochemical research at the Nobel Explosives Company based in Ardeer in Scotland. Raistrick's work at that time had focussed on the production of crucial feedstocks for the production of explosives by fermentation processes. However Raistrick realised that microorganisms, and fungi in particular, were adept at chemical transformations unavailable to industry at that time, and in the production of natural products with interesting and useful properties in other spheres of life. It was the results of these investigations which catalysed the start of his academic career in London, and which also helped to found today's science of fungal biotechnology and engineered biosynthesis (Fig. 1).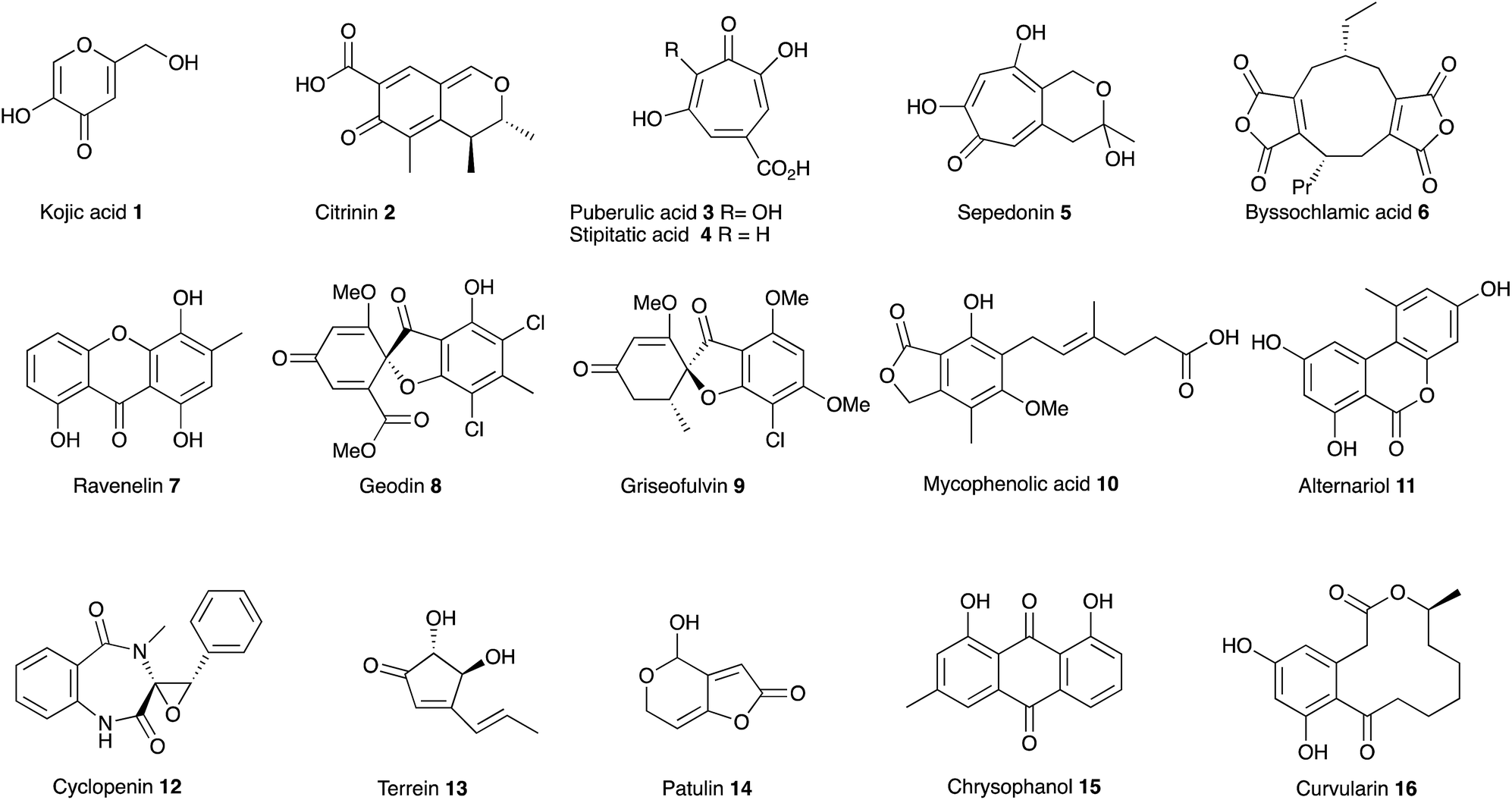 | ||
| Fig. 1 Structures of key compounds. | ||
Raistrick described the discovery and investigation of compounds still of key interest today. These included kojic acid 1,2 citrinin 2 (ref. 3) the tropolones puberulic acid 3 (antimalarial), stipitatic acid 4 and sepedonin 5, and the maleidride byssochlamic acid 6. Compounds such as the xanthone ravenelin 7 were the first examples of compounds in their class. Others such as geodin 8 and griseofulvin 9 had unprecedented structures, and griseofulvin 9 and mycophenolic acid 10 are still clinically important compounds today. He focussed on the key orders of fungi which still make up the majority of the productive organisms exploited in industry and academia today including varied species of Aspergillus, Penicillium and Fusarium. Other compounds such as alternariol 11, cyclopenin 12, terrein 13 and patulin 14 remain of importance today as mycotoxins involved in crop and food spoilage. Raistrick also isolated and characterised numerous anthraquinones such as chrysophanol 15 which is a key intermediate in the biosynthesis of many anthraquinones and xanthones, and curvularin 16 which later had a role in development of ideas about the processive nature of fungal polyketide biosynthesis. Raistrick also had a role in the penicillin story where he was an early collaborator of Fleming.4
In many cases these compounds were among the first examples of classes of compounds now taken for granted as natural products of fungi. Their structures became the inspiration for generations of chemists involved in analysis, synthesis, biosynthesis, and more latterly genetic and genomic studies. Compounds such as citrinin 2 have served as key models at all stages of the development of the science of fungal natural products – from early isolation and structure elucidation (e.g. Cram and Whalley 1948),5 its first synthesis in 1949, through the earliest speculation and experiments on biosynthesis (for example Birch and coworkers in 1958),6 to the development of sophisticated isotopic labelling and NMR strategies (e.g. Staunton and coworkers7 and Sankawa and coworkers8 in the 1980s) to the more recent understanding of the biosynthetic gene cluster (e.g. Nihira and coworkers, 2007).9 Similarly compounds reported by Raistrick such as the tropolones (e.g. puberulic acid 3 in 1931) have stimulated important developments by others in the field of bonding theory (e.g. Dewar, 1945),10 and new ideas about synthesis (e.g. Todd) and biosynthesis (e.g. Robinson) from major figures in the field of organic chemistry. Other compounds such as the nonadrides (Section 4) also drove biosynthetic speculation, the development and application of new analytical methods to natural products chemistry, and also contributed to new ideas about biomimetic synthesis (e.g. Barton, Baldwin, Sutherland and coworkers). Raistrick's example also no-doubt contributed to the explosion in fungal natural product prospecting in the years after 1945.
Many of Raistrick's compounds have remained of key interest to microbiological chemists up to today because of their unusual structures, their useful biological properties, and the fascinating questions underpinning their biosynthesis. Modern investigations of many of the compounds have revealed the secrets of their biosynthesis, and illuminated the hidden relationships between the different classes of fungal natural products – particularly within the polyketides and meroterpenoids which form a rich vein of Raistrick's compounds. It seems highly likely that interest will continue in understanding and exploiting these compounds as the science moves towards rational manipulation of fungal biosynthetic pathways and production of new compounds. It seems timely, therefore, to review the most important work by briefly illustrating the historical context and most up-to-date discoveries for the main classes of Raistrick's compounds (Sections 2–9). Furthermore, despite the undoubted significant advances in the field, some key biosynthetic questions are still unanswered and some of the remaining challenges will be briefly explored in Section 10.
2. Alternariol
Raistrick and Thomas first reported alternariol 11 in 1953 as a crystalline solid isolated from Alternaria tenuis and its structure was determined by chemical means. Alternariol is a potent mycotoxin, and as many Alternaria species both produce 11 and infect food crops and stored feed, contamination with this compound presents a serious problem. Given its important ecological role, it is remarkable that relatively few studies of the biosynthesis of alternariol 11 have been reported. Early work by Thomas proved the compound was a typical non-reduced polyketide, biosynthesised in the expected way from [14C]-acetate.11 More sophisticated experiments reported by Staunton, Abell, Garson and Leeper used [2-2H3]-acetate and 2H NMR as part of a study designed to probe the enolization of intermediates prior to ring formation. The results showed that significant 2H wash-out was observed at position 4 (relative to position 6 and other deuterated positions) consistent with the idea that a 4,5-enol 18a is a key intermediate, supporting the idea that a cis-intermediate must be formed prior to ring formation (Scheme 1).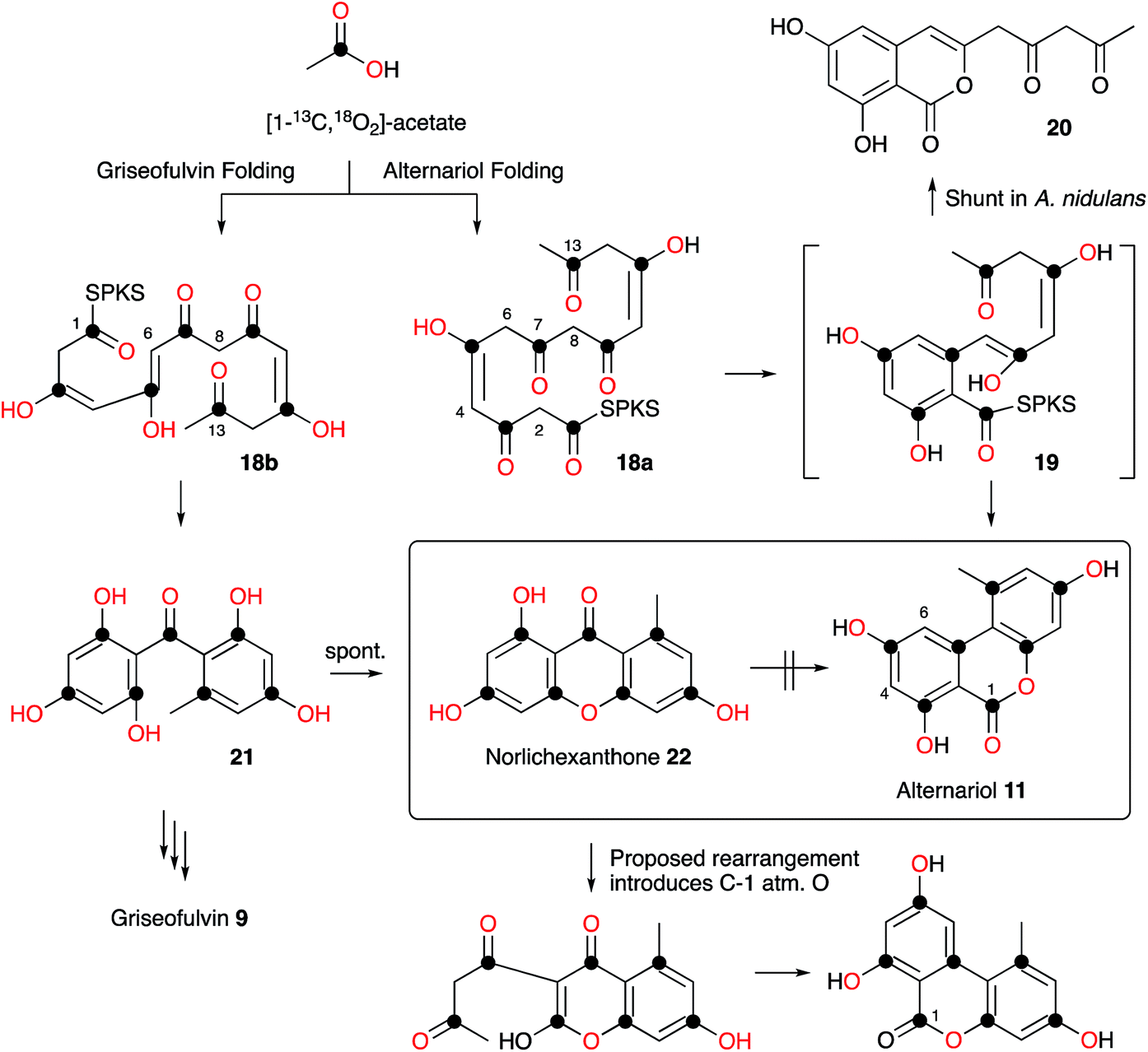 | ||
| Scheme 1 Labelling studies and folding patterns in alternariol 11 and norlichexanthone 22. | ||
Wang, Oakley and coworkers reported a novel method for the discovery of the function of many of the cryptic non-reducing polyketide synthase (nrPKS) genes of Aspergillus nidulans. They systematically replaced the native promoters of nrPKS genes with constitutive promoters and isolated new compounds produced by the engineered strains.12 In the case of the alternariol nrPKS, the lack of an obvious chain-releasing domain in the PKS itself meant that an adjacent gene encoding a β-lactamase-type thiolesterase also had to be induced. This led to production of alternariol 11 as well as the shunt dehydrocitreoisocoumarin 20. Contemporaneous work by Fischer and coworkers identified a highly-reducing PKS (PKSJ) in Alternaria alternata as a potential alternariol synthase, but direct (e.g. knockout or heterologous expression) evidence for its involvement is lacking.13 More recently, Chooi and coworkers have isolated a true alternariol synthase gene (SnPKS19) from the plant pathogen Parastagonospora nodorum and proven its role by heterologous expression. SnPKS19 encodes the expected nr-PKS. Intriguingly SnPKS19 is a close homolog of the Penicillium aethiopicum norlichexanthone (NLX) synthase gene gsfA which is involved in the biosynthesis of griseofulvin (see Section 7): GfsA produces norlichexanthone 22 as a shunt metabolite. Previous labelling studies by Stinson and coworkers14 had suggested that norlichexanthone 22 was a precursor of alternariol, but labelling studies by Simpson using [1-13C, 18O2]-acetate disproved this hypothesis (Scheme 1).15 The differences between the nrPKS gsfA and SnPKS19 must control the regiochemistry of the first ring formation in each heptaketide which are 1–6 in norlichexanthone 22 and 2–7 in alternariol 11 biosynthesis.
Alternariol remains of key interest as a mycotoxin contaminant of food production, particularly soft fruits, berries and in viniculture. The current detailed level of understanding of its biosynthesis, coupled with the development of new CRISPR/Cas9 methods in the main producing host A. alternata16 may lead to methods for the production of A. alternata strains deficient in its biosynthesis, although the release of such strains to the wild would require significant regulatory work. The discovery of the close biochemical link between the alternariol and griseofulvin pathways suggests a possible common evolutionary history which may allow discovery of other related compounds and pathways, and future in vitro investigations could focus on the origins of the different ring-folding selectivities in the two closely related PKS, giving further details of how the intermediate polyketo intermediates are stabilised and selectively cyclised.
3. Geodin, ravenelin and the emodin pathway
The central role of the anthraquinone emodin 23 in fungal secondary metabolism is reflected in its very early discovery as a fungal metabolite and its structural proof by synthesis in 1924.17 Raistrick made many contributions to this area, notably through the first report of the xanthone ravenelin 7 (ref. 18) from Helminthosporium ravenelii and geodin 8 (ref. 19) from Aspergillus terreus, coincidentally both in 1936. It would not be until many years later that the biosynthetic links between the seemingly very different anthraquinones, xanthones and geodins would be elucidated, but these classes of compounds now form a key and widespread family of fungal metabolites, for which the molecular aspects of biosynthesis are only just becoming clear.In the case of geodin 8, Raistrick proposed possible structures, with the assistance of Cedric Hassall, in 1947 based on extensive chemical degradation and derivatisation,20 although it was not until 1958 when Ian Scott and Derek Barton provided the now-accepted structure, using IR spectroscopy as a key plank of evidence.21 The structure of ravenelin 7 was solved rapidly by Raistrick, and this proved to be the first fungal xanthone. Birch, Simpson and Westerman reported the results of the first important biosynthetic study of xanthones in 1975. Feeding [1-13C]- and [1,2-13C2]-acetate to H. ravenelii resulted in incorporation of intact acetates into the methylated aromatic C-ring, but scrambling of acetate in the A-ring, clearly indicating the intermediacy of a symmetrical benzophenone intermediate such as 25.22 Earlier work by Gatenbeck23 and Hassall24 had already shown that questin (1-O-methyl emodin) 26 is efficiently converted to the benzophenone sulochrin 27 (Scheme 3) in Aspergillus terreus and it seemed likely that a similar process in H. ravenelii could convert a simple non-reduced polyketide such as emodin 23 to a benzophenone, such as monodictyphenone 25, and then on to a xanthone such as ravenelin 7. However, further experiments would be needed to confirm this hypothesis. Vederas and coworkers provided some of the required evidence through the use of [1-13C, 18O2]-acetate feeding experiments (Scheme 2).25 The results showed that the 4-oxygen and either the 8-oxygen or the 10a-oxygen are not derived from acetate, and are probably introduced from atmospheric oxygen. This supports an oxidative removal of the emodin 10-carbon, most likely by a biological Baeyer–Villiger-like reaction which would thereby introduce the atmospheric 8/10a oxygen prior to ring closure to the xanthone skeleton. The intact bond between 13C-10a and 18O-10a in 7 also confirms the mode of ring closure.
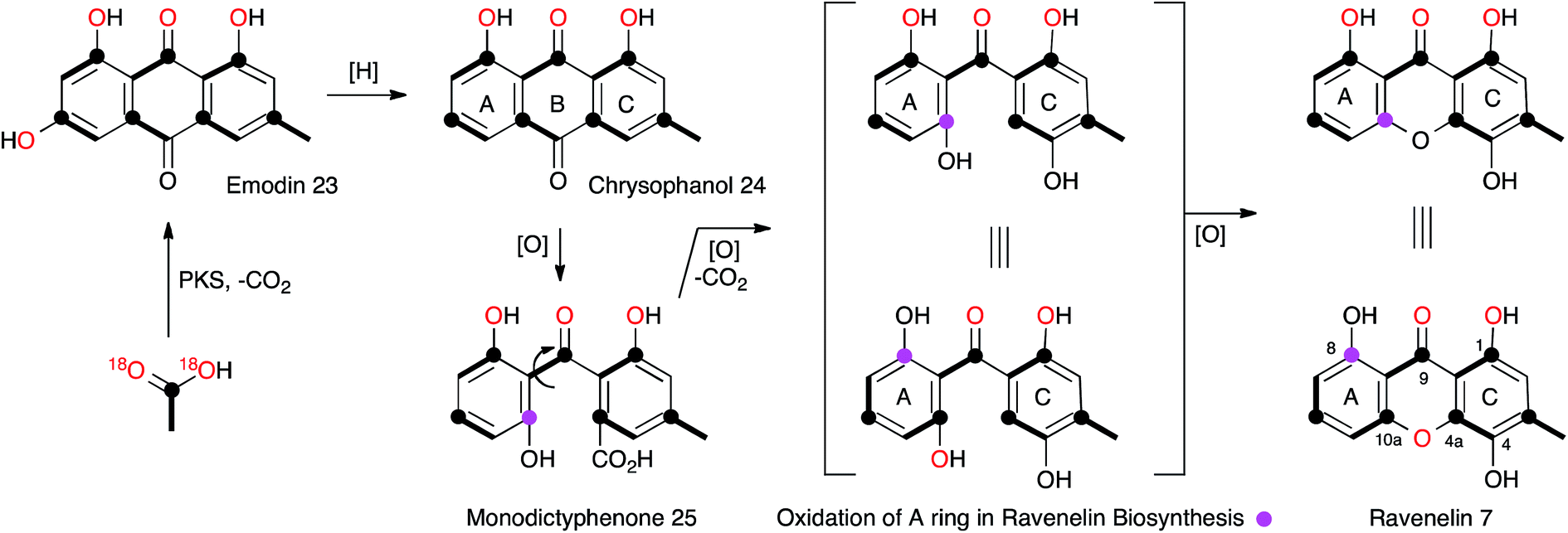 | ||
| Scheme 2 Labelling pattern in ravenelin 7 reaveled from isotopic feeding experiments. | ||
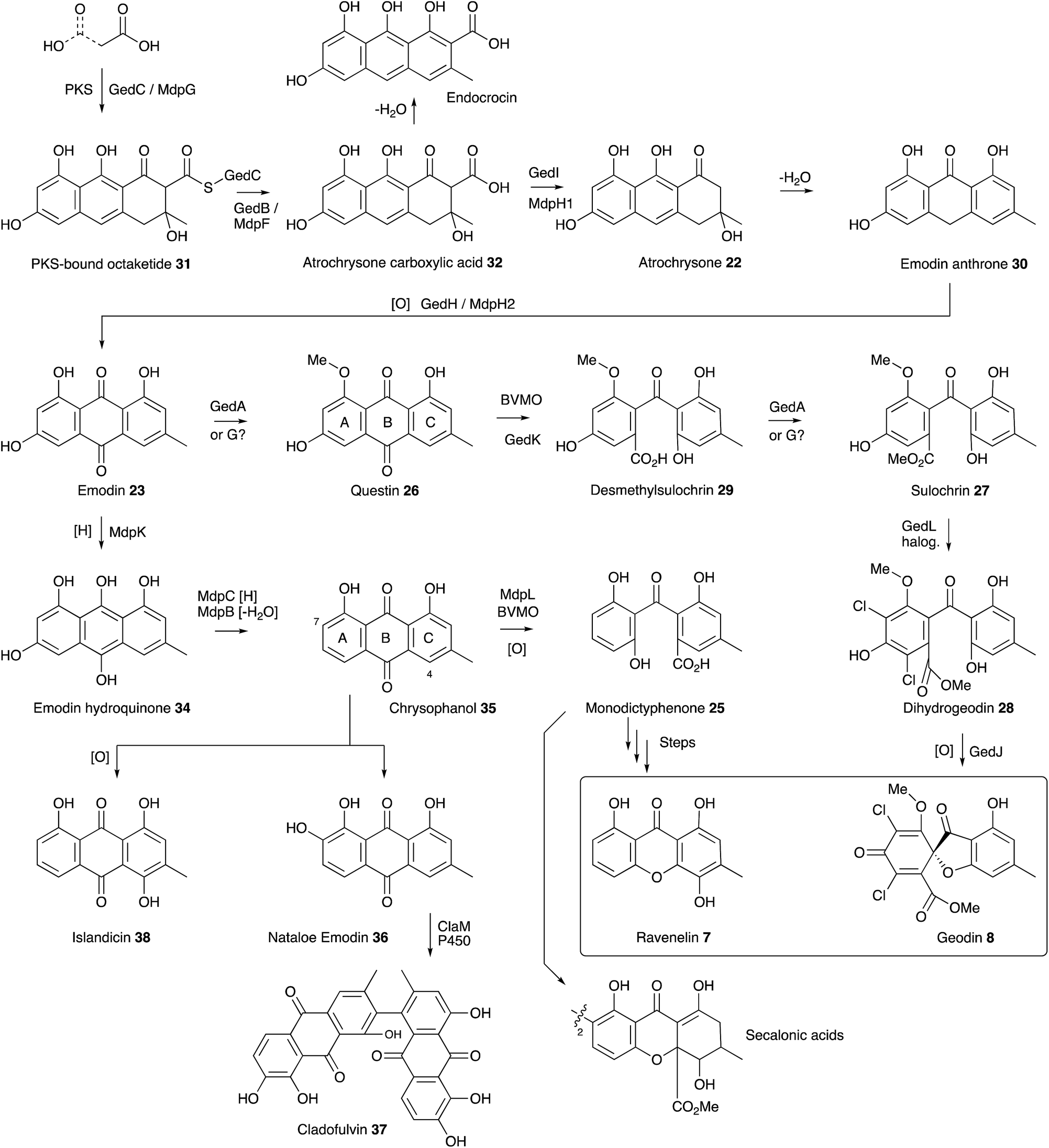 | ||
| Scheme 3 Summary of the emodin pathway to geodin 8, ravenelin 7 and related compounds. | ||
Meanwhile a direct link between emodin 23 and geodin 8 was proven by Fujimoto and coworkers who fed [UL-14C]-emodin to surface cultures of A. terreus and showed incorporation into geodin 8 and dihydrogeodin 28.26 This led to a different strategy to understand the biosynthetic pathway by Japanese groups who purified active enzymes from cell-free extracts of A. terreus. The first success came in the isolation of an emodin 1-O-methyl transferase (e.g. a questin 26 synthase) by Sankawa, Ebizuka and Fujii.27 Next came evidence for the final step of the pathway when dihydrogeodin oxidase (DHGO) was purified from A. terreus and shown to effectively stereoselectively convert dihydrogeodin 28 to geodin 8 itself.28 The enzyme is an unusual blue-copper protein, and quite different in sequence to the P450 enzyme which catalyses the parallel step in griseofulvin biosynthesis (Section 7). The DHGO gene was cloned through a process involving expression of an A. terreus cDNA library and probing it with a DHGO antibody. This led to identification of a partial DHGO encoding gene which could be used to capture a full-length genomic clone.29
The enzymatic system which catalyses the key anthraquinone ring cleavage was also purified from WT A. terreus by the Sankawa group. The purified protein effectively converts questin 26 to desmethylsulochrin 29in vitro, presumably via a Baeyer–Villiger/hydrolysis sequence, and requires O2 and NADPH.30 Fractionation of this preparation into two less active fractions which regained full activity on recombination suggested the active system consists of an oxygenase and an associated electron transfer protein. Another oxidase, putatively involved in the route, was isolated from A. terreus by the same group.31 Emodin anthrone oxygenase is a non-heme iron protein which converts emodin anthrone 30 to emodin 23. Finally, the sulochrin PKS was identified by degenerate PCR32 using primers specific for non-reducing PKS which identified a gene fragment which could be used for targeted knockout.33
Availability of the A. terreus genome, combined with the sequence information obtained from the experiments described above, allowed Mortensen, Larsen and coworkers to identify the geodin BGC (ged) and reconstitute it in A. nidulans.34 Thirteen open reading frames were transferred from A. terreus to A. nidulans, along with their native promoters and terminators and this converted A. nidulans to a geodin 8 producer. These experiments also unambiguously identified the required chlorinase protein (GedL), but did not dissect the precise timings of all steps.
The ubiquity of fungal xanthones has stimulated very many biosynthetic studies over the past half century, and there is no space here for a comprehensive discussion. However recent molecular advances, particularly into the biosynthesis of monodictyphenone 25 (mdp),35 the prenylated xanthones,36 and the secalonic acids (otherwise known as the ergochromes)37 have shed significant light onto the early steps of ravenelin 7 and geodin 8 biosynthesis and highlighted the key intermediate as emodin 23. It is now certain that the non-reducing PKS MdpG (homologous to GedC) produces a non-reduced PKS-bound octaketide 31 which is hydrolytically released by MdpF (GedB) to produce atrochrysone carboxylic acid 32, which in turn gives atrochrysone 33 and emodin anthrone 30 after decarboxylation and hydrolysis (GedB/MdpF). Conversion to emodin 23 itself is catalysed by MdpH2 (GedH), although Müller and coworkers have convincingly argued that emodin 23 is probably present as the corresponding reduced emodin hydroquinone 34in vivo.38 In in vitro experiments MdpC and MdpB respectively provide the key intermediate chrysophanol 35 (Raistrick, 1950)39via a reduction–elimination sequence. Simpson has convincingly argued that chrysophanol 35 is subjected to a Baeyer–Villiger oxidation catalysed by MdpL to give monodictyphenone 25 which would be a precursor of ravenelin 7 and the secalonic acids.40 Information concerning these later molecular steps is so far missing. Chrysophanol 35 is also the substrate for hydroxylation, e.g. at C-7 to provide nataloe emodin 36, the precursor of cladofulvin 37,41 or at C-4 to provide the isomeric islandicin 38 (Raistrick 1949).42
The geodin pathway branches from emodin 23 and proceeds via questin 26 to desmethylsulochrin 29. Labelling studies show that the regioselectivity of this BVMO must be opposite to that operating in the mdp pathway, i.e. C-ring oxidation. Finally, methyl ester formation, chlorination and oxidative ring closure give geodin 8. Although the geodin pathway is now well understood, key questions regarding the later-stage steps and mechanisms of many other anthraquinone and xanthone pathways, such as those to the secalonic acids, remain to be solved. One recent highlight has been the discovery of the P450 system responsible for the oxidative dimerisation steps which form the bis-anthraquinones such as cladofulvin 37.40
Genomic studies are beginning to show that the mdp pathway is very widespread in fungi and it appears that early evolution of the steps to emodin 23 have formed the foundations for later diversification to many different bioactive structures involved in key processes of pathogenicity, particularly with respect to plants and other fungi. It seems likely that genomic investigations of these pathways using an evolutionary rationale will probably lead to discoveries of many other related compounds and pathways. Of the pathways which are now known to flow from emodin 23, for example to the large class of secalonic acids, key molecular questions remain regarding the (often) oxidative rearrangements leading to the complex final structures.
4. The maleidrides
Glauconic acid 39 (Penicillium purpurogenum), reported by Nadine Wijkman in 1931,43 was the first member of a family of carbocyclic maleic anhydrides which have become known as maleidrides. Raistrick and Smith reported byssochlamic acid 6 from Byssochlamys fulva soon after in 1933.44 As in the cases of many other compounds isolated and purified during this period, the structure elucidation proved very difficult, and it was not until the early 1960s that definitive structures were elucidated by Baldwin, Barton, Sutherland and coworkers (chemical degradation)45 and Sim and Robertson (Crystallography)46 for both compounds. The related glaucanic acid 40 (Fig. 2) is isomeric to byssochlamic acid 6 and Barton and Sutherland speculated that such compounds could be derived by dimerisation of C9 monomers – coining the term nonadride for this class47 – with different regiochemical dimerisations explaining the structural differences. Heaveadride 41 (Helminthosporium heveae) isolated by MacMillan and coworkers in the early 1970s falls into the same C9-derived family,48 but other compounds such as rubratoxin B 42 (ref. 49) and the structurally more complex phomoidrides 43 (ref. 50) must be derived from larger precursors. Most family members contain two maleic anhydride moieties, but the selective herbicide cornexistin 44 (ref. 51) contains only one. Despite the increasing numbers of larger compounds discovered the name nonadrides later became associated with the central 9-membered ring. Later related octadrides such as zopfiellin 45 (ref. 52) and viburspiran 46,53 and heptadrides such as agnestadride A 47 (ref. 54) have been discovered with 8- and 7-membered central rings respectively, leading to the general term maleidrides (Fig. 2).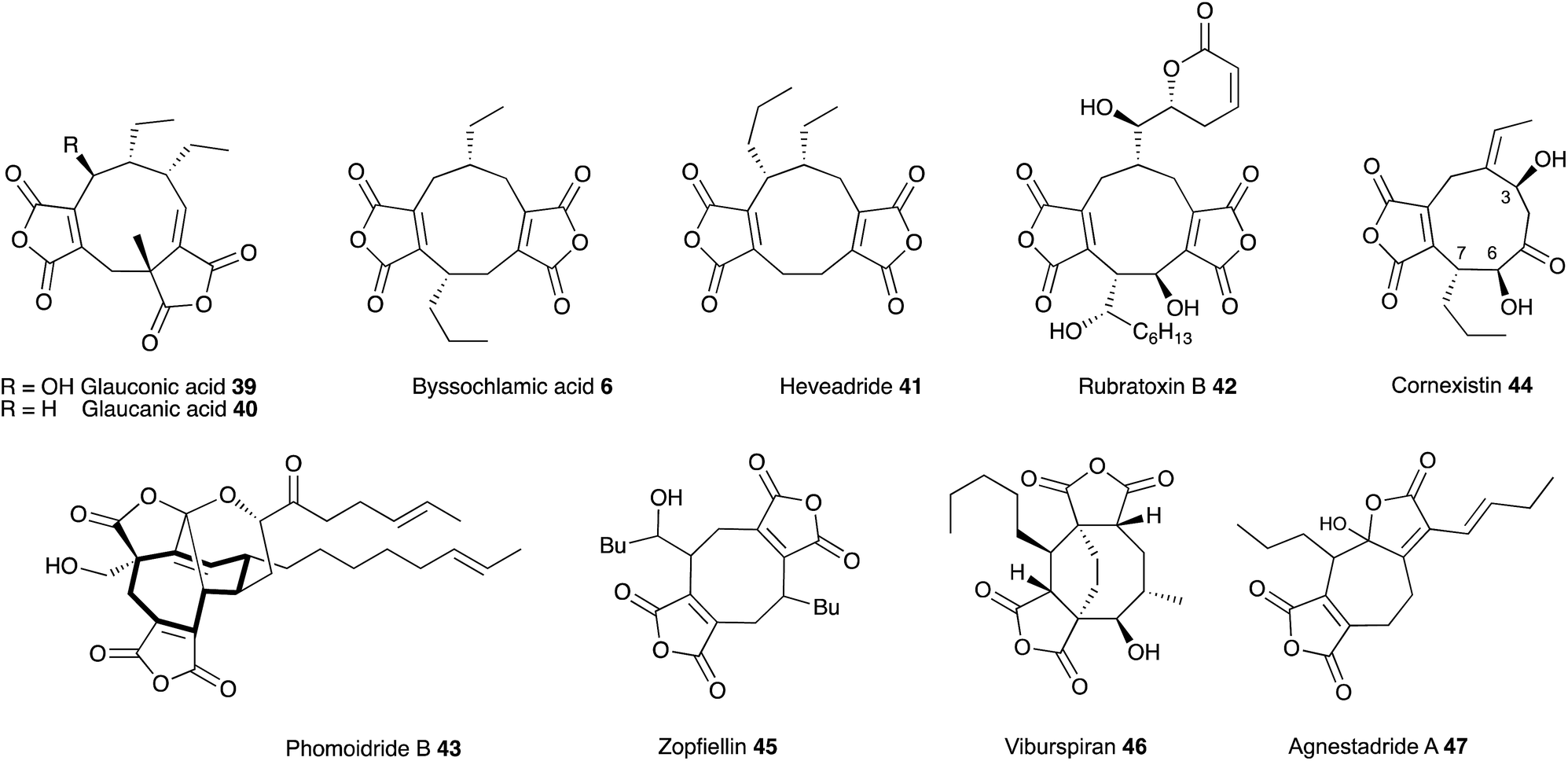 | ||
| Fig. 2 Structures of key maleidrides. | ||
As soon as the structures of 6 and 39 were elucidated their biosynthetic origins were of interest. Sutherland and coworkers reported the results of [1-14C]- and [2-14C]-acetate feeds in 1968 which showed the incorporation of three acetates head-to-tail in each C9 monomer during biosynthesis of glauconic acid 39 in Penicillium purpurogenum. Scrambling of label from acetate at the other three carbons suggested the use of a Krebs cycle intermediate for the remaining C3 unit and this was confirmed by feeding [2,3-14C2]-succinate which suggested that oxaloacetate was a more likely precursor than pyruvate.55 The results supported a previous suggestion that a polyketide or fatty acid is condensed with oxaloacetate to make a citrate-like intermediate which dehydrates to the observed maleic anhydride 48 (Scheme 4).46 The symmetrical incorporation of the labels strongly supported the idea that dimerisation of such an intermediate formed the nine-membered ring in 39etc. This hypothesis was confirmed by feeding 2-(1-3H]-n-butyl)-3-methylmaleic anhydride (i.e. dihydro-48) to P. purpurogenum and observation of symmetrical tritium incorporation into 39, albeit at low levels of incorporation (<0.5%). Later results showed that the unsaturated anhydride 48 is incorporated much more efficiently (ca. 50%).56
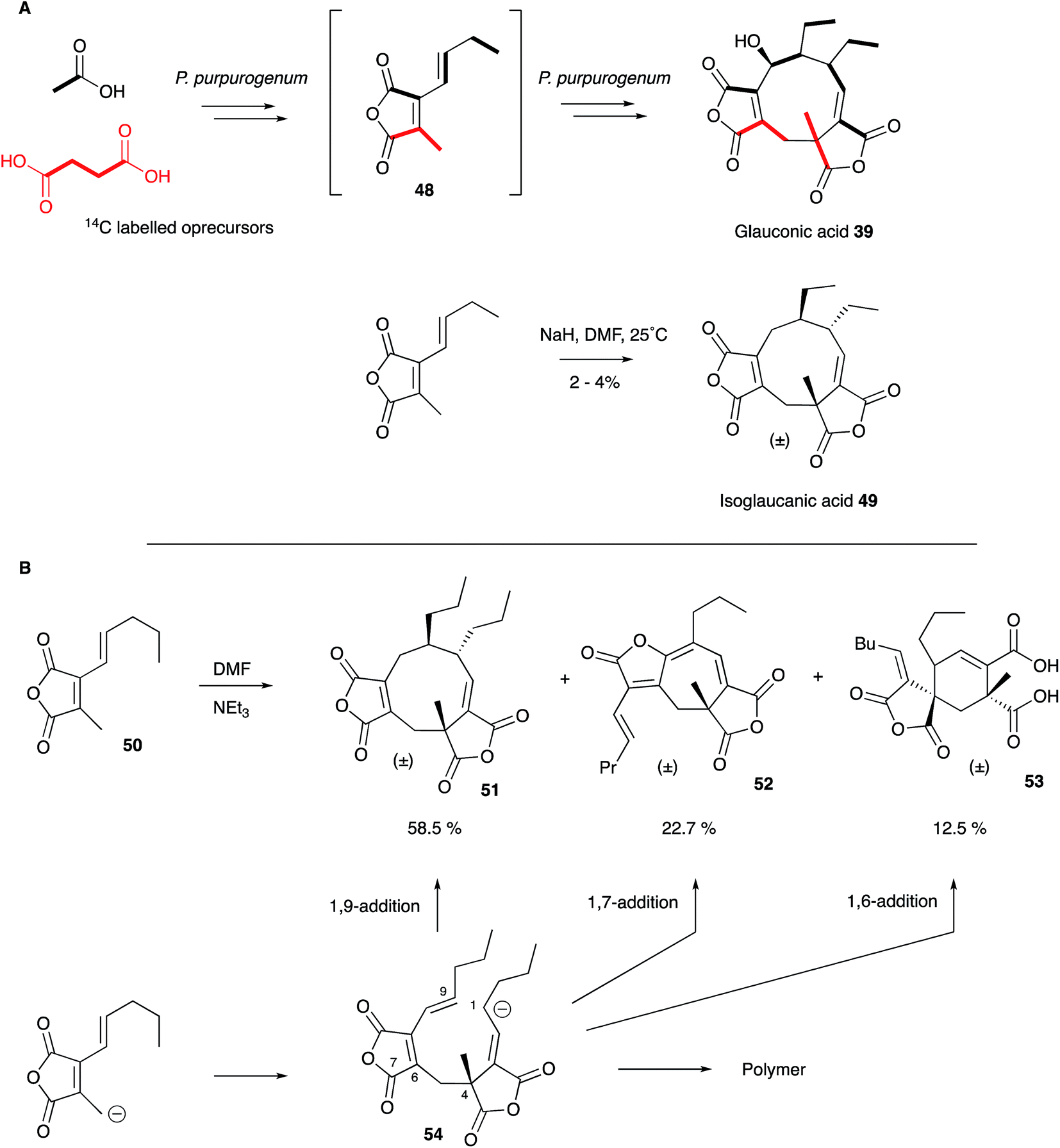 | ||
| Scheme 4 Biosynthetic and Biomimetic routes to maleidrides: (A) Sutherland's work; (B) Baldwin's work. | ||
These results encouraged Sutherland and coworkers to attempt a biomimetic synthesis of glauconic acid 39, reported in 1972. Treatment of the anhydride precursor 48 with either NaH or Et3N produced a low yield (2–4%) of the nonadride isoglaucanic acid 49. This observation supported the proposed mechanism of formation of the nonadrides as either a concerted 6π + 4π cyclodimerisation, or a stepwise polar process (Scheme 4A).55 Baldwin and coworkers extensively reinvestigated this biomimetic process using an n-pentenyl maleic anhydride 50 (rather than the n-butenyl analogue 48 used by Sutherland) during the late 1990s. Although still low, optimisation of reaction conditions resulted in yield improvements (up to 8.5% for 51), and the isolation of isomeric compounds with six- (53) and seven-membered rings (52) in similar amounts.57 The structural relationships between these compounds suggest an initial step in which an anion attacks at C-4 to form the common intermediate 54 (Scheme 4B). Three different modes of cyclisation are then possible to afford the observed 9-, 7- and 6-membered products, and this favours the stepwise mechanism over the concerted 6π + 4π addition. The use of polar solvents improved overall yields through stabilisation of the required anionic starting material, and the addition of Mg2+ ions favoured formation of cyclised products over polymers, presumably via coordination and activation of the maleic anhydride electrophile.
Further progress in understanding the biosynthesis of maleidrides did not come for another 15 years and had to await the development of full genome sequencing and effective molecular methods in fungi. A thorough reinvestigation of the secondary metabolite profile of the original B. fulva strain investigated by Raistrick in the 1930s by Cox, Simpson and coworkers revealed the presence of several related compounds.54 Two heptadrides (47 and its dehydration product) were isolated and these appear to derive from the same pathway as byssochlamic acid 6 itself. The carboxymethyl maleic anhydride 55 was also isolated from a fungus for the first time, and was shown to be unstable to spontaneous decarboxylation in solution to give the methyl maleic anhydride 48 shown by Sutherland and Baldwin to be an in vitro precursor of the nonadrides. This compound appears to be derived from condensation of a polyketide and oxaloacetate in a citrate synthase-like process in agreement with the original suggestions of Sutherland and others. Full genome sequencing of B. fulva was then achieved. A search for a biosynthetic gene cluster (BGC) encoding a fungal highly reducing polyketide synthase (hr-PKS) and a citrate synthase (CS) enzyme revealed a candidate cluster which also encoded a hydrolase, a 2-methylcitrate dehydratase (2MCDH) and several proteins of unknown function.58 Transcriptomic analysis linked activity of the cluster with metabolite production and defined possible limits of the BGC. Knockout of the PKS gene then gave definitive evidence for its involvement in the biosynthesis of the maleic anhydride 55 and both the nonadride 6 and heptadride 47 components (Scheme 5).
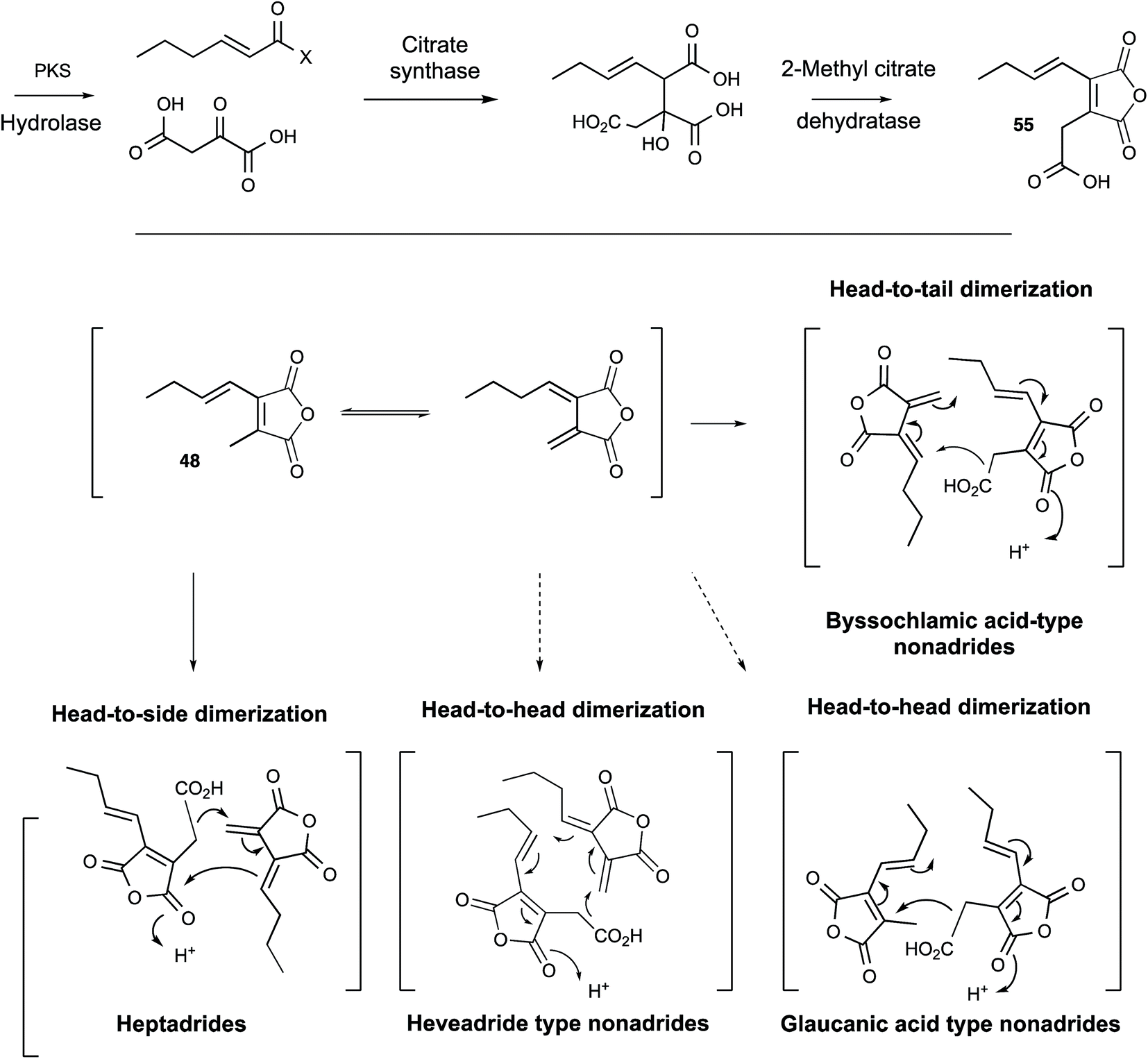 | ||
| Scheme 5 Biosynthesis of maleidrides. | ||
Stepwise reconstruction of the gene cluster in the fungal host Aspergillus oryzae was then used to probe the function of the biosynthetic components. Coexpression of the PKS, hydrolase, CS and 2MCDH encoding genes led to production of the carboxymethyl maleic anhydride 55, as expected, but not the nonadrides or heptadrides.57 Parallel work by the group of Oikawa, focussing on the biosynthesis of the phomoidrides 43, came to a similar conclusion that a hr-PKS, CS and 2MCDH are involved in the production of the monomer.59 The byssochlamic acid BGC contains several genes of unknown function. Two of these encode proteins with homology to ketosteroid isomerases (KSI), and two more show homology to phosphatidylethanolamine binding proteins (PEBP). Coexpression of the core genes encoding the biosynthesis of 55 with the two KSI-encoding genes increases the titre of 55 and produces byssochlamic acid 6. Addition of the two PEBP genes increases the titre again, giving significantly more 6 and also produces the heptadride 47.
Two recent studies have revealed the complex series of oxidative steps which convert the nonadride skeleton into more complex and biologically more interesting compounds (Scheme 6A). Genome sequencing strategies have revealed the BGC responsible for the biosynthesis of cornexistin 44 which is a potent and selective herbicide.60 Once again the core genes encode an hr-PKS, hydrolase, CS and 2MCDH. However the cornexistin BGC differs from that responsible for byssochlamic acid 6 biosynthesis in including only single copies of the KSI and PEBP genes. Unlike the BGC for 6, the cornexistin cluster encodes numerous oxidases and other genes of unknown function. While the oxidases were shown to be involved in late-stage decoration of the cornexistin skeleton, the processes involved in removal of one of the maleic anhydrides could not be elucidated by knockout experiments and remain cryptic.
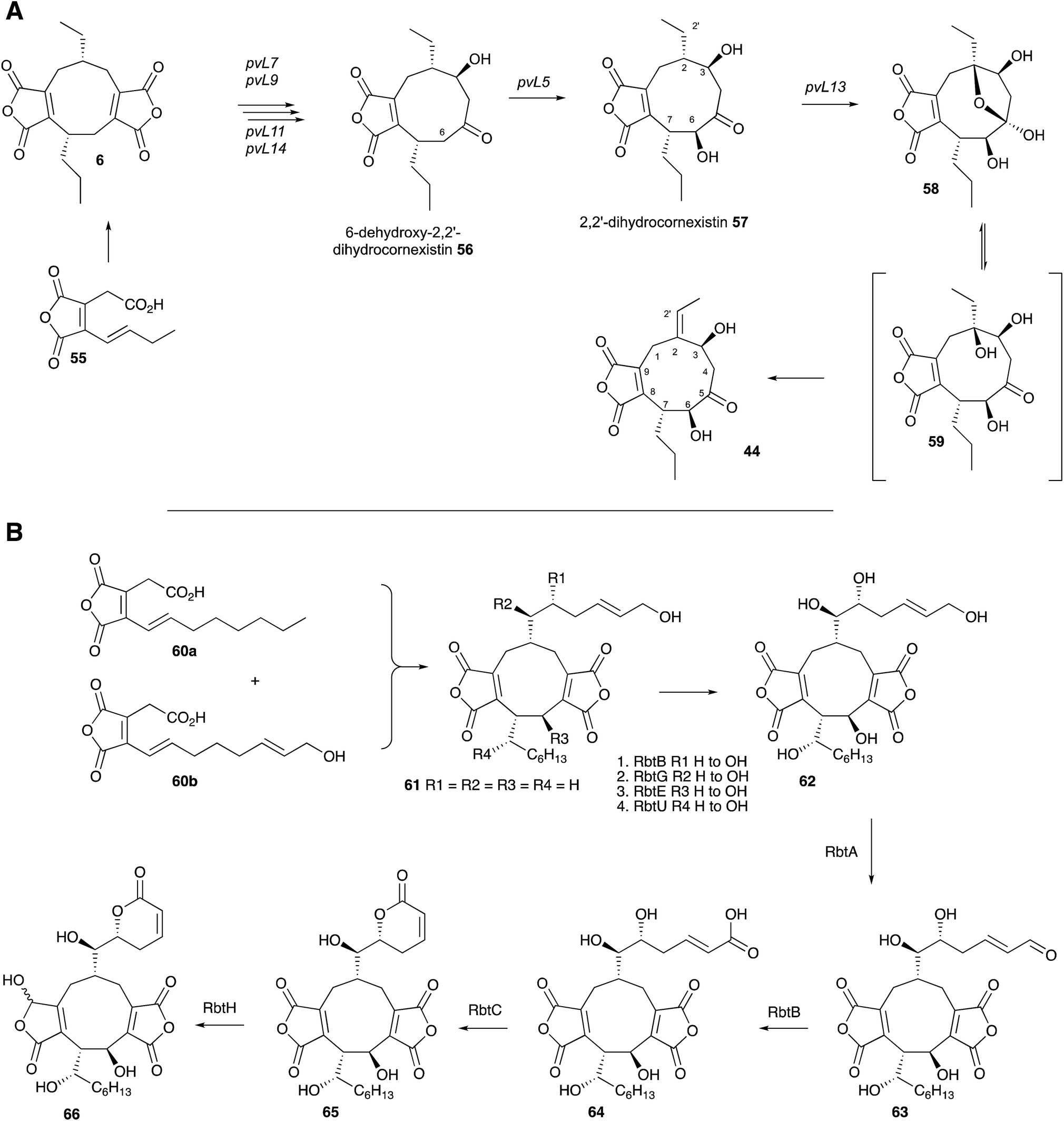 | ||
| Scheme 6 Biosynthesis of cornexistin 44 and rubratoxin B 66. | ||
In the case of the biosynthesis of rubratoxin B 65, a potent and specific inhibitor of protein phosphatase 2, Yu, Tang, Hu and coworkers revealed the BGC by full genome sequencing of Penicillium dangeardii and genome mining using the byssochlamic acid biosynthetic genes as targets.61 This revealed a BGC containing the expected PKS, CS, 2MCDH, KI and PEBP genes, as well as genes encoding seven further redox enzymes. Knockout experiments supported by re-feeding, and in vitro assays of isolated recombinant enzymes elucidated the oxidative steps, and also implicated an interesting possibility of an early hydroxylation of one maleic anhydride precursor 60 and an asymmetric dimerisation to form the initial nonadride 61 (Scheme 6B). The pathway also features a dual functional non-heme iron dependent oxygenase (RbtB) which appears to act at two unrelated points in the pathway. Finally, an interesting new reductase (RbtH) which can convert one of the maleic anhydrides to the corresponding γ-hydroxybutenolide of rubratoxin A 66 was discovered.
Thus the early predictions of Sutherland and others regarding the likely biosynthesis have been proven correct with combination of polyketide and citrate-processing steps. The use of PKS, hydrolase and CS also features in the biosynthesis of other fungal metabolites such as the squalestatins62 and these genes form a functional motif which appears to be widely distributed in fungi. Once again it seems that evolution of a particular biosynthetic pattern leads to an intermediate structure which can be diversified to many other bio-active compounds by addition of genes encoding new functions.
It seems clear that the KSI component of the synthetic machinery can form the nine-membered ring by a dimerisation process, but the precise chemical steps remain to be elucidated – for example it is not yet clear if the decarboxylation of the initial maleic anhydride (e.g.55) is coupled to the dimerisation or not. In the case of byssochlamic acid 6 biosynthesis the KSI and PEBP proteins can produce both 9- and 7-membered rings, but as yet no explanation has been found for the biosynthesis of the related 8-membered octadrides such as viburspiran 46 and zopfiellin 45. Baldwin's biomimetic work shows that the chemistry to form the various regioisomers is feasible, even without enzyme catalysis, but selectivity-controlling features of the biological processes remain elusive. There are also numerous examples of linear family members such as cordyanhydrides63 A and B 67 and 68 (Fig. 3) and the biosynthesis of these has not yet been investigated. It seems likely that future advances will focus on in vitro studies of these mysterious enzymes to provide a better level of understanding, particularly of their mechanism and selectivity.
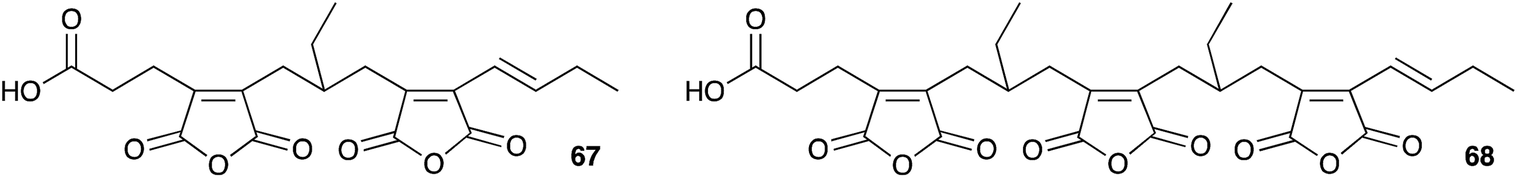 | ||
| Fig. 3 Cordyanhydrides. | ||
5. Citrinin and the azaphilones
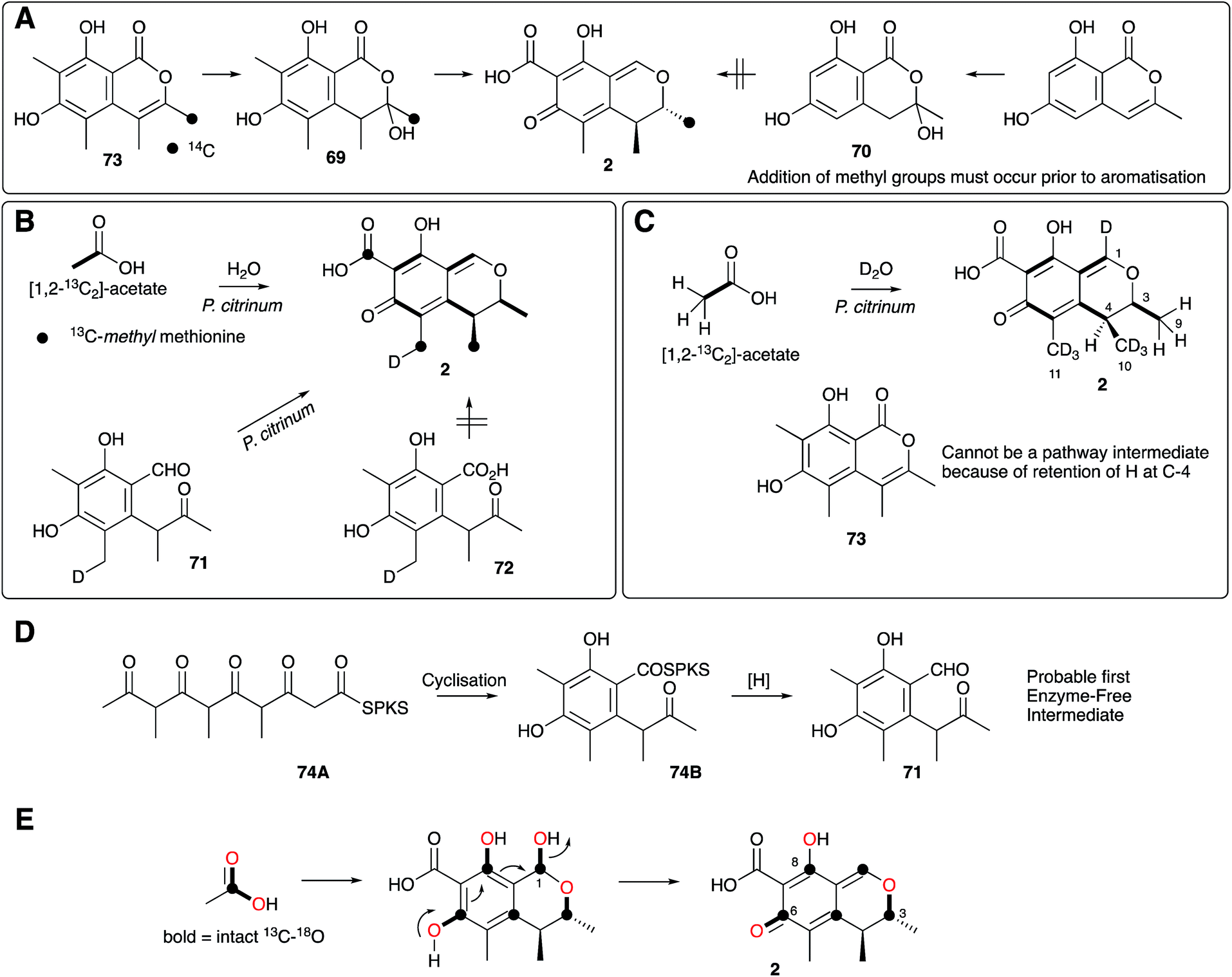
Citrinin 2 was first reported by Raistrick and Hetherington in 1931 as a yellow component of the organic extract of Penicillium citrinum.64 Lack of useful spectroscopic methods meant that Raistrick and Robert Robinson's early studies to elucidate its structure relied exclusively on chemical and physical methods.65 The deduced structures were unsatisfactory and many groups struggled with this problem through the 1940s during which a lively series of papers disputed each other's claims.66 Further efforts by Cram refined, but did not solve the structure67 and it was not until 1948 that Whalley and coworkers elucidated the now accepted quinomethide structure.68 Early researches focussed on the potential antibacterial properties of citrinin, no doubt stimulated by the emerging work with penicillin from another Penicillium species, but it was soon clear that citrinin was too toxic for medical use.Schwenk and coworkers69 and Birch and coworkers70 published almost contemporaneous reports of the incorporation of 14C-labelled acetate into citrinin which confirmed its polyketide origin. The results of Birch showed that the three branching carbons derived from the C1 pool (formic acid), but Schwenk showed that the S-methyl of methionine is incorporated more efficiently and these were the first demonstrations of the incorporation of methyl branches into fungal polyketides, confirming an earlier idea of R. B. Woodward.71 Staunton and coworkers concluded that these branching groups must be added prior to aromatisation because 14C-sclerotinin 69 was weakly incorporated into citrinin by P. citrinum, but its des-methyl analogue 70 was not (Scheme 7A).72
 | ||
| Scheme 7 Summary of key isotopic labelling experiments which elucidated various steps of citrinin 2 biosynthesis. | ||
Investigations of citrinin biosynthesis served as the basis for the development of a number of innovative isotopic labelling experiments by the Staunton group using stable isotopes during the late 1970s and early 1980s. Development of procedures for the synthesis of specifically isotopically labelled late-stage precursors were developed by Staunton, and independently by Scolastico and coworkers, to show that the ketoaldehyde 71 is the likely first enzyme-free intermediate (Scheme 7B);73 and inverse feeding experiments using [1,2-13C2-2,2,2-1H3]-acetate in media made from D2O provided a sensitive way to trace acetate protons during biosynthesis. For example, observation of 1H at C-4 of 2 after feeding [1,2-13C2, 1H3]-acetate to P. citrinum grown in D2O showed that 73 cannot be a direct precursor (Scheme 7C).74 The previously observed conversion of 69 to 2 in low yield (Scheme 7A) therefore probably arises from adventitious reactions in P. citrinum. Together, results from these experiments showed that the polyketide is most likely methylated as the chain is assembled (processive biosynthesis) to give an enzyme-bound intermediate such as 74A, which then cyclises and aromatises to 74B before reductive release to give 71 (Scheme 7D). Subsequent complex tailoring reactions must then give 2.
Meanwhile, Sankawa and coworkers used citrinin as a test-bed for early experiments using 17O- and 18O-labelled acetates, dual labelled with 13C in Aspergillus terreus.75 Their work clearly showed that both 18O and 17O can be effective tracers in biosynthetic studies. 18O is most conveniently detected by its isotope shift on attached 13C nuclei in 13C NMR, while 17O is NMR active itself and can be directly observed. Retention of intact 13C–18O bonds from [1-13C, 18O2]-acetate at C-6 suggested that the quinomethide is formed by a simple mechanism involving elimination of water from a hemiacetal at C-1 (Scheme 7E).
Despite the extensive work of many groups on the topic of citrinin biosynthesis reported in the literature over 30 years, other poorly interpreted work has also appeared and been highly cited. In particular the work of Francois, Hajjaj and coworkers into the biosynthesis of citrinin in Monascus ruber is widely referenced despite an incorrect interpretation of the labelling pattern from [1,2-13C2]-acetate leading to a biosynthetic hypothesis which is starkly different from the previously deduced pathways in A. terreus and P. patulum.76 This route is proposed to involve the cleavage of an acetate despite the labelling pattern from their own data clearly showing that all acetates are, in fact, incorporated intact. This report has misled several subsequent researchers,77 and is unfortunately not the only misinterpretation of data in the citrinin story.
A gene encoding the citrinin 2 PKS (citS, also known as pksct) was first discovered in Monascus purpureus by Nihira and coworkers, reported in 2005, by using a PCR screening approach.78 This led to the gradual discovery of a larger genomic fragment containing the complete citrinin BGC and an activator gene ctnA.79 In a key experiment Nihira and coworkers transferred the entire genomic fragment from M. purpureus to the fungal host Aspergillus oryzae with the aim of heterologously producing citrinin 2. Citrinin 2 could be detected in this experiment, but in vanishingly low titre. Titres could be increased to ca. 1.5 mg L−1 by over-expression of ctnA.80 However no significant analysis of the clustered genes, or accompanying biochemical investigations were reported.
Significant progress has been made more recently in understanding citrinin biosynthesis at the molecular level. Cox and He reported a series of experiments in which the citrinin BGC from Monascus ruber was systematically reconstructed in A. oryzae by using strong promoters for each structural gene.81 The citrinin synthase gene citS encodes an iterative non-reducing PKS. Expression of citS alone led to the production of the keto-aldehyde 71 in agreement with the earlier hypotheses of Staunton and others. This is evidently released from the PKS by its terminal reductive release domain, so the PKS requires no other release mechanism. However, if citA encoding a serine hydrolase is coexpressed with citS a much higher titre of 71 was observed. Townsend and coworkers recently showed that the hydrolase appears to act as an ‘unblocking’ system, releasing incorrectly synthesised intermediates from the PKS and restoring its processivity.82 Systematic co-expression with each of the remaining citrinin BGC components then elucidated the post-PKS pathway in full. The first step is oxidation of C-12 by a non-heme iron dioxygenase encoded by citB to form the 12-alcohol 75. This is then oxidised to the C-12 aldehyde 76 by citC, a nicotinamide dependent oxidoreductase, and then again to the 12-carboxylic acid 77 by citD which is a typical aldehyde dehydrogenase. The final step, catalysed by citE, is stereoselective reduction of the 3-ketone of 77, closure of the dihydropyran ring and formation of the quinomethide 2 by dehydration. The pathway is in full agreement with the previous labelling studies, and negates the pathways suggested by Hajjaj and others. A further advantage of the A. oryzae expression system described here is the high titres obtained-up to 50 mg L−1. The pathway proceeds via unstable aldehyde intermediates 76 and 77 and these give rise to numerous shunt pathways in which probable spontaneous reactions diversify the citrinin skeleton to 78–81 (Scheme 8).
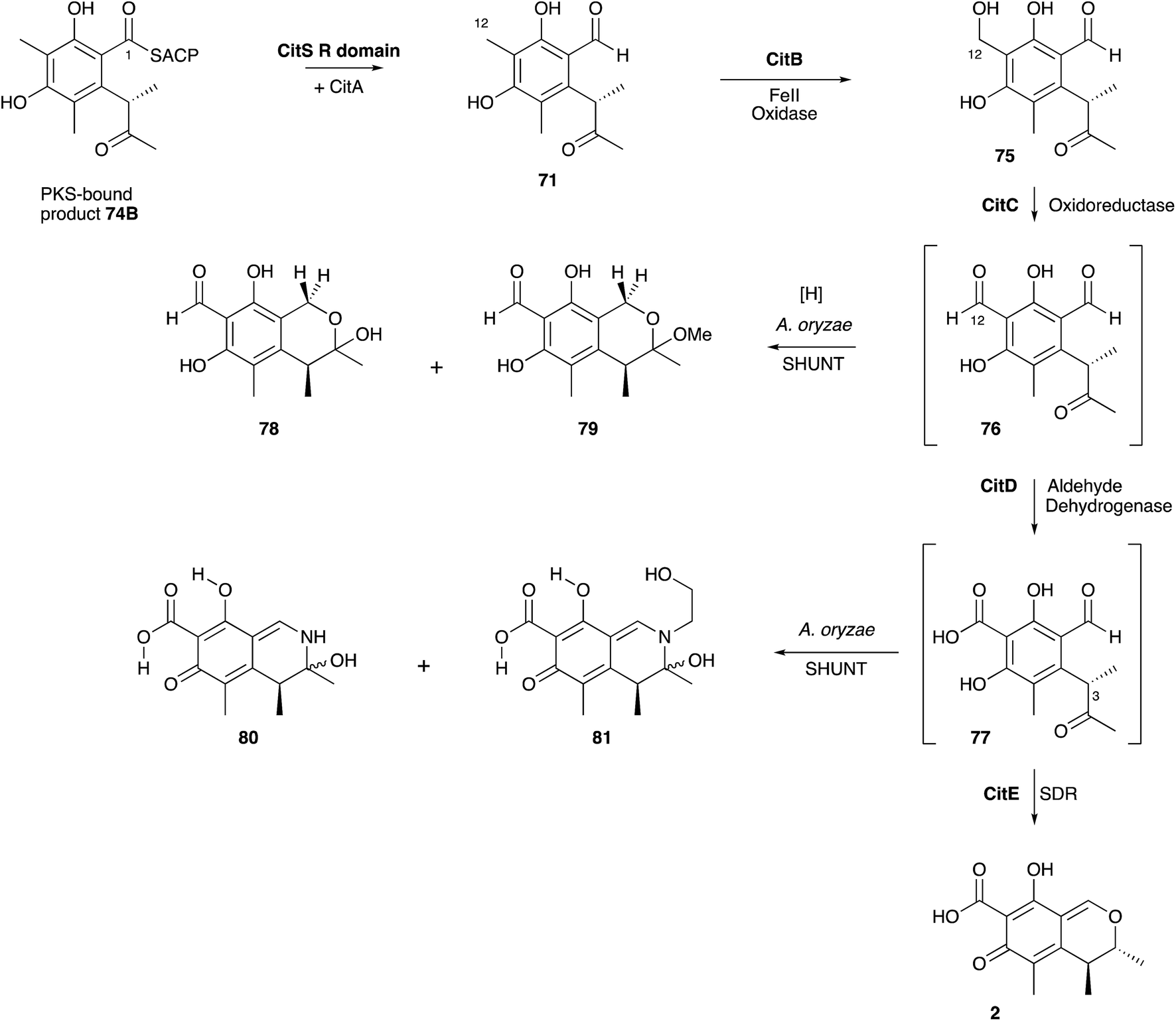 | ||
| Scheme 8 Biosynthetic pathway to citrinin 2 and related shunts. | ||
Citrinin is closely related to the azaphilone class of fungal metabolites. These compounds are so-named because of their propensity to react with amines non-enzymatically. This is illustrated by the fact that aldehyde intermediates such as 76 and 77 in the citrinin pathway are rapidly shunted to chromanes and dihydroisoquinolines such as 78-79 and 80-81 respectively. In A. oryzae ethanolamine appears to act as the nucleophilic amine. Citrinin itself is resistant to amination. The biosynthesis of the closely related azaphilones is now well understood in the cases of chaetoviridin and related compounds such as the Monascus pigments and proceeds via a pathway parallel to the citrinin route in which reductive release of a non-reduced polyketide is followed by successive oxidations.83
Full understanding of the citrinin pathway at the molecular level allows other parallels to be drawn with known fungal pathways. For example citB, a non-heme Fe-II oxygenase, is functionally equivalent to TropC in the tropolone pathway (Section 9). However during tropolone biosynthesis the aromatic ring has been oxidised (by the FAD-dependent TropB) and this allows oxidative ring expansion to form the tropolone nucleus. In the case of citrinin 2 the more reduced aromatic nucleus is stable and does not rearrange. Compounds such as sepedonin 5,84 another of the Raistraick compounds, feature structural motifs common to both the tropolones and citrinin and it seems likely that the sepedonin BGC will feature both a citB-type and a TropC-type oxidase.
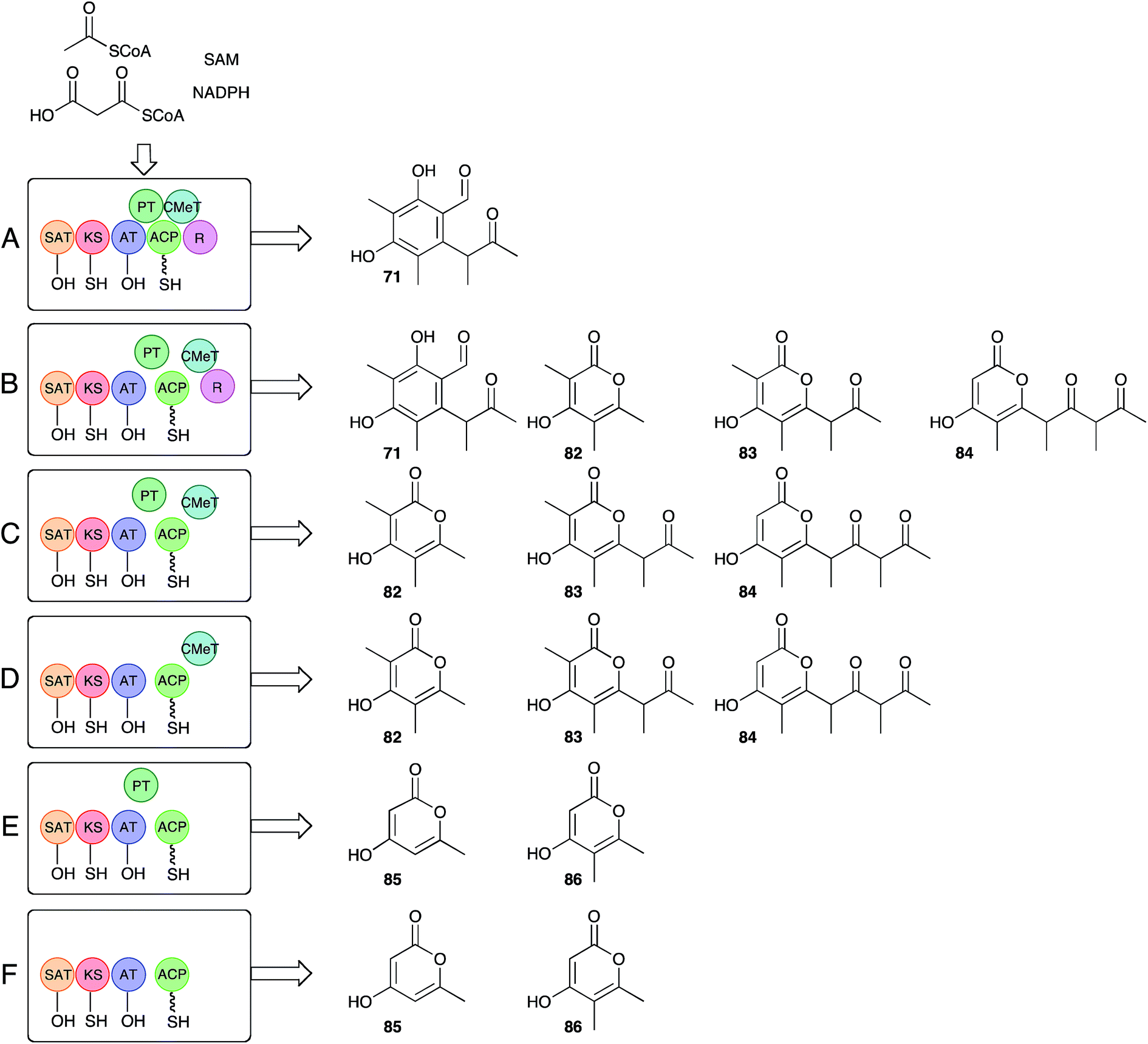
Recent work by Townsend and coworkers has set about answering long-standing questions regarding the precise function of the citrinin PKS itself.85 Bioinformatic analysis of the citrinin PKS shows it belongs to the non-reducing class of iterative fungal PKS, and consists of (N to C) starter unit acyl transferase (SAT), ketosynthase (KS), acyl transferase (AT), product template (PT), acyl carrier protein (ACP), C-methyltransferase (C-MeT) and reductive release (R) catalytic domains. Much is already known about this class of PKS. For example the SAT domain loads the starter unit (acetate in this case) from CoA to KS, while AT loads a malonyl unit to the ACP. KS catalyses chain extension to create acetoacetyl ACP. In the absence of other reactions these processes are iterative (4 extensions in the case of citrinin), with the growing β-ketoacyl chain being stabilised and cyclised by the PT domain prior to reductive release giving 2 (Scheme 9A). In the case of citS, however, the CMeT domain shows a programmed role and methylates β-carbons after extensions 1, 2 and 3, but not after extension 4.
 | ||
| Scheme 9 Townsend's deconstruct/reconstruct experiments with citS. | ||
The approach of Townsend and coworkers was to take a ‘deconstruction–reconstruction’ approach. They expressed fragments of citS in E. coli to obtain separate SAT-KS-AT, PT, ACP, CMeT and R domains which were recombined in vitro in the presence of substrates and cofactors. Remarkably the dissected and reassembled synthase retains the ability to synthesise 71, although pyrones 82–84, indicative of spontaneous release, were also produced (Scheme 9B). In the absence of the reductive release domain a range of pyrones were also made (Scheme 9C), and further removal of the PT domain gave much the same result (Scheme 9D). Removal of the CMeT domain provided only triketides showing that methylation must occur before chain extension (Scheme 9E and F). Observation of very low levels of 86 were rationalised by copurification of methylacetoacetyl CoA which could act as a starter unit. These results clearly show that the interplay between the C-MeT and KS selectivities is crucial for correct product production in this type of PKS, and support other recent observations of kinetic competition between catalytic domains for ACP-bound substrates as a likely key factor controlling the programming of iterative fungal PKS more generally.86–88
While citrinin itself is a mycotoxin, other azaphilones and related compounds are of major interest as safe food colourants and preservatives, especially in oriental cuisine. Many efforts to understand and exploit their biosynthesis derive from attempts to produce the dyestuffs in higher titre and more selectively. The recent successes in using full and partial pathway expression in the case of the monascus pigments have begun to link the molecular and chemical features required for the production of particular colours of pigments and it seems likely that this work will be extended to include rational efforts to produce new compounds to-order. Such engineering efforts could obviously focus on both the PKS itself, building on the knowledge gained by Townsend and coworkers of the citrinin PKS, but parallel work by Molnar (see Section 6 below) shows that rational domain-swaps can also be deployed. Increased knowledge of the activities and selectivities of the numerous tailoring redox transformations in varied pathways should also lead to productive mix and match experiments in which recombinant PKS and tailoring genes can be combined.
6. Curvularin and the dihydroxyphenylacetic acid lactones
Raistrick and Rice discovered curvularin 16, a metabolite of Penicillium gilmanii, in 1952. The paper communicating its discovery was not published until 1971 shortly after Raistrick's death.89 This was the first example of a widespread class of fungal metabolites now known as the dihydroxyphenylacetic acid lactones (DAL), which are in-turn related to the resorcylic acid lactones (RAL). Musgrave, coincidentally working at Raistrick's old laboratories at the ICI Nobel division at Ardeer, made the first reports of curvularin in 1956,90 and the first biosynthetic studies and structure were reported by Arthur Birch, Herchel Smith and coworkers in 1959.91 The structure was one of the first solved using Birch's acetate hypothesis. Dehydrocurvularin 87 was reported by Musgrave in 1967,92 and this compound has proved to be an important testbed for developing ideas about the biosynthesis of highly reduced polyketides in fungi.Vederas fed Alternaria cinerariae, a producer of 87, with a wide range of 13C, 2H and 18O-labelled acetates and used 13C-NMR to determine the origin of all atoms and bonds in 87. In particular [2-13C, 2H3]-acetate labelled intact C–D units (Scheme 10), and chemical conversion to 88 provided a system in which the stereochemistry at C-12 could be assessed by NMR. Oleic acid 89 was isolated from the same fermentations, and degradation to 90 also allowed stereochemical assignment. The results of these studies showed clearly that the remaining 2H, at C-2-derived carbons, is incorporated with opposite stereoselectivity in the polyketide and fatty acid (Scheme 10). This indicates that fungal fatty acid synthase (FAS) and the dehydrocurvularin hrPKS must possess enoyl reductase (ER) components with opposite stereoselectivity for the reprotonation step, forming 91 and 92 respectively as intermediates.93
 | ||
| Scheme 10 Isotopic feeding experiments in Alternaria cinerariae. | ||
A key early question regarding the mechanism of fungal hrPKS was whether β-processing occurs processively (after each chain extension reaction) or after construction of a complete poly-β-keto carbon skeleton such as occurs during the construction of non-reduced polyketides. Dehydrocurvularin 87 is an interesting case in which the initial tetraketide appears highly reduced, while the final tetraketide appears non-reduced. Vederas and coworkers neatly addressed this point by synthesising and feeding isotopically labelled di-, tri- and tetra-ketides 93–98 to A. cinerariae as their N-acetyl cysteamine thiolesters (SNAC). To prevent degradation of these advanced precursors they also supplemented β-oxidation inhibitors. In addition, to detect incorporation of intact precursors, the compounds were labelled with 13C2 units placed between polyketide acetate positions. 13C NMR of isolated 87 showed intact incorporation of 93, 94 and 97, strongly supporting the processive mechanism of highly reduced polyketide formation.94 The tetraketide 97 was incorporated with surprisingly high efficiency (ca. 70%) (Scheme 11).
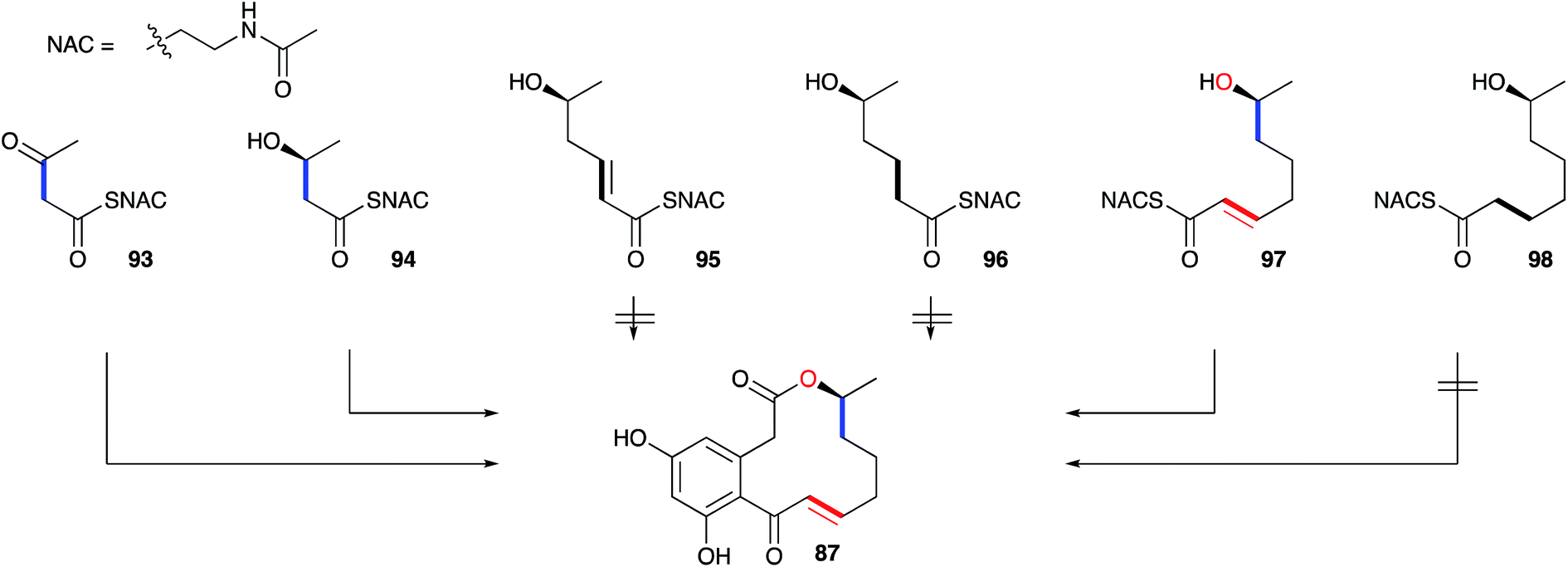 | ||
| Scheme 11 Incorporation of advanced labelled precursors into dehydrocurvularin 87 in Alternaria cinerariae. | ||
Molecular information was supplied by Molnar's group who isolated the dehydrocurvularin BGC from Aspergillus terreus. Two PKS are required for synthesis, a hrPKS (AtCURS1) and a nrPKS (AtCURS2), and this was proven by heterologous expression of these two components in yeast. The hrPKS provides a tetraketide starter unit for the nrPKS, and experiments in yeast in which only the nrPKS was expressed allowed DAL formation when synthetic tetraketides (i.e.97) were fed as SNAC thiolesters (Scheme 12A).95 This neatly explains the fact that the tetraketide 97 is so efficiently incorporated into 87 – it is the starter unit for the nrPKS. The nrPKS extends 97 by four more units and catalyses the aromatic cyclisation. Final off-loading by the nrPKS TE domain forms the macrocycle. The use of two PKS, where a hrPKS supplies the starter unit for a subsequent nrPKS is the same as observed to be involved in the biosynthesis of resorcylic acid lactones (RAL), which are another large class of fungal metabolites.
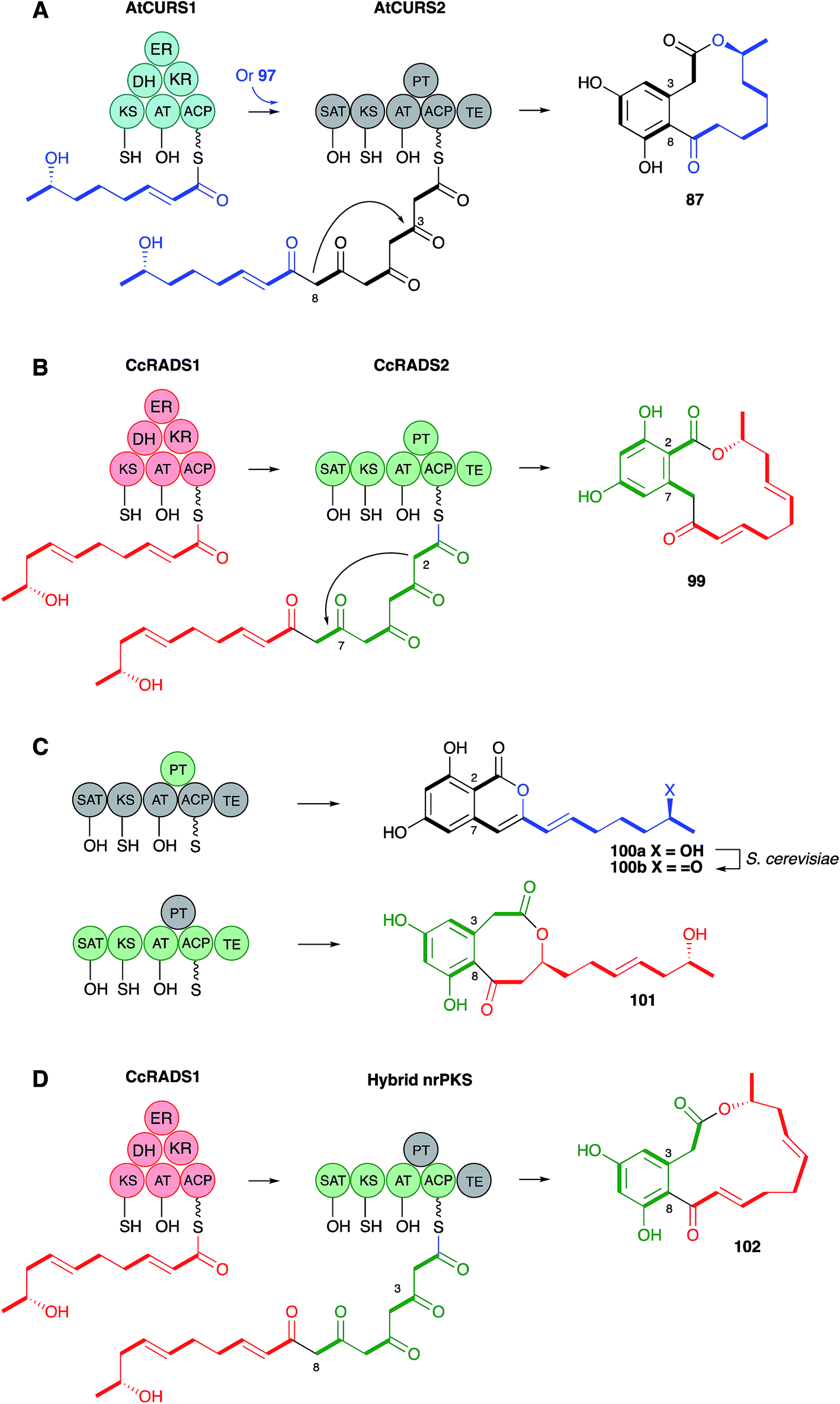 | ||
| Scheme 12 Engineering of DAL and RAL systems to control ring cyclisation and TE regioselectivity. | ||
The Molnar group also reported significant progress in understanding and engineering the cyclisation events in the nrPKS component.96 During biosynthesis of the DAL class of metabolites nrPKS ring formation occurs at the 3–8 positions (Scheme 12A), while in the RAL class, for example monocillin 99, the ring formation is 2–7 (Scheme 12B). Ring formation is catalysed by the product template (PT) domain of the nrPKS, and this evidently also controls the regiochemistry since swapping the PT domains led to the formation of the reprogrammed compounds (Scheme 12C). Homology modelling of the PT domains themselves led to the identification of key residues involved in folding and cyclisation and a triple mutation of the AtCURS2 PT domain led to clean conversion from a synthase which made 87 to one which makes 100b.
The AtCURS2 nrPKS releases its normal product, 87, using its thiolesterase (TE) domain. Similarly the monocillin nrPKS CcRADS2 releases 99. Molnar and coworkers used a similar domain swap strategy to probe the intrinsic selectivities of these TE domains.97 All 8 permutations of TE (AtCURS2 or CcRADS2), aromatic substitution pattern (3–8 or 2–7) and side chain structure (tetraketide or pentaketide) were assessed by building chimeric PKS systems in vivo. For example, to assess a pentaketide starter unit with a 3,8 cyclisation pattern and the dehydrocurvularin TE, CcRADS1 was paired with CcRADS2 in which the PT and TE domains were swapped-in from ArCURS2. This provided the new compound radilarin 102 (Scheme 12D). Further work to increase the range of functional DAL/RAL hybrids showed that combinations of the hrPKS and nrPKS can be made with PKS systems which make saturated and unsaturated starter units and nrPKS which make only three extensions.98 Methylated compounds such as 104 can be created if the hrPKS component has a CMeT domain, such as AfoG, the starter unit synthase for preasperfuranone 103 which is an early intermediate in azaphilone biosynthesis.99 In this case the AtCURS2 PKS had to be engineered to contain the AfoE SAT domain (Scheme 13). These experiments clearly show the opportunities for rational production of defined compounds within the DAL/RAL family and are among the most advanced to date in the sphere of natural product biosynthetic engineering in general.
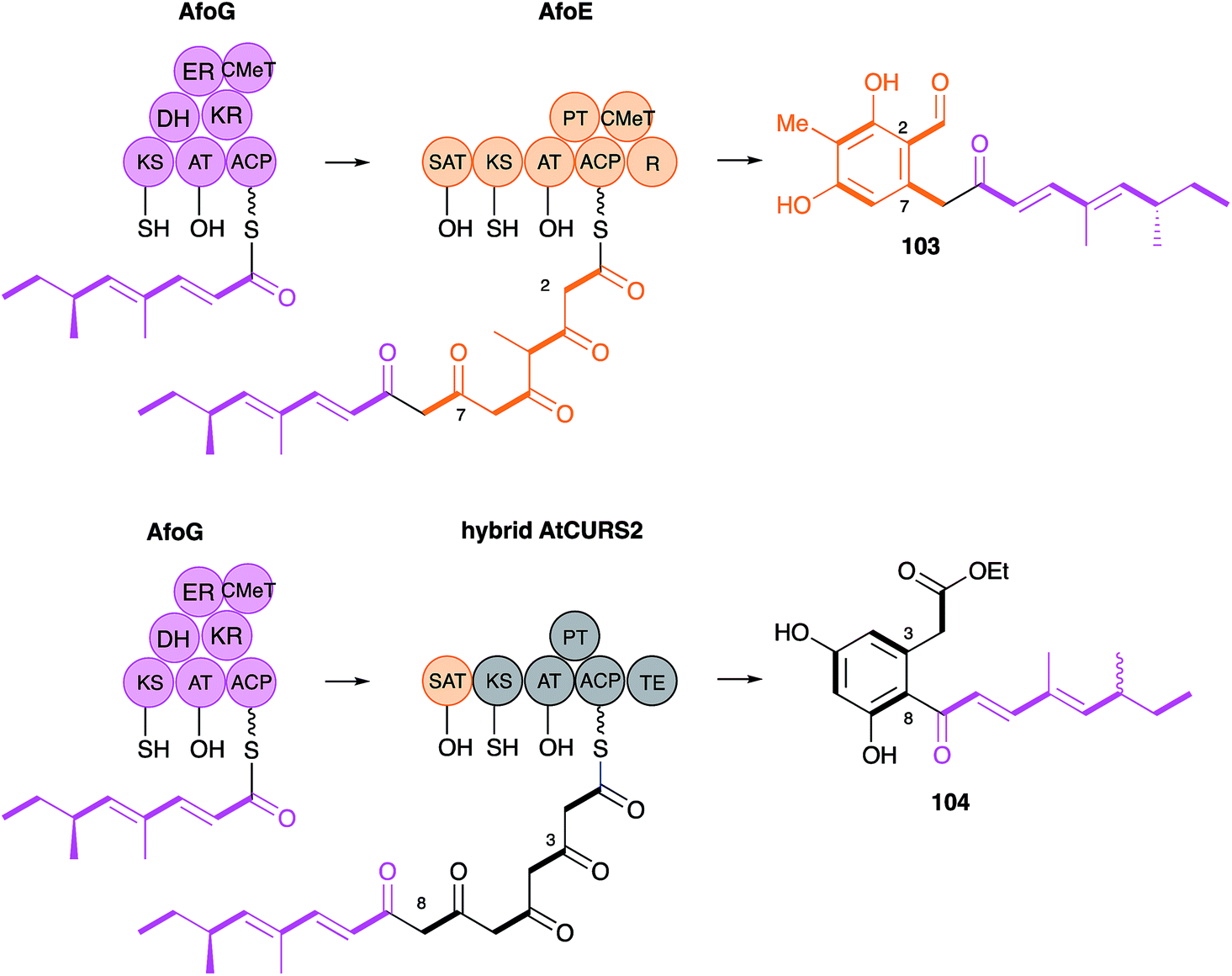 | ||
| Scheme 13 Engineered hybrid azaphilone and DAL pathways. | ||
Thus, significant progress has been made in the field of understanding and engineering RAL and DAL biosynthesis since the first discovery of 16 in the 1950s. Enough is now known to engineer the basic carbon skeletons and cyclisation patterns at-will in a process which is competitive with synthetic chemistry. Future advances will have to address the question of titre, and will no-doubt focus on expanding the range of post-PKS tailoring steps which can be rationally incorporated into the synthetic pathways. New developments in the creation of modular and controllable expression systems in fungi, such as those reported by Lazarus and coworkers,100 will underpin efforts to move synthesis of this class compounds out of the synthetic chemistry laboratory, and into the sphere of molecular and microbiology.
7. Griseofulvin
Griseofulvin 9 is a useful orally available antifungal agent used in human medicine, and is included in the World Health Organisation's list of essential drugs. Raistrick and coworkers first reported 9 as an unusual chlorinated metabolite of Penicillium griseofulvum in early 1939, after around 10 years of work.101 Chemical analysis was able to provide a preliminary structure, and capture the 6,5,6 ring system, but it was not until 1952 when Grove, MacMillan102 and coworkers at ICI determined the distinctive spirocyclic structure.103 Earlier work by Grove had already identified the antifungal properties of 9 when he showed that griseofulvin and the so called ‘curling factor’ isolated from P. janczewskii were identical. Macmillan also reported one of the very first examples of precursor-directed biosynthesis when he fed chloride-starved cultures of P. griseofulvum with KBr and isolated bromogriseofulvin.104 ICI and Glaxo developed and commercialised griseofulvin in a joint venture during the 1950s and 1960s.105Birch reported the first studies of the biosynthesis of griseofulvin 9 using [1-14C]-acetate, which were important for confirming the acetate hypothesis of polyketide biosynthesis. Feeding studies with P. griseofulvum showed that 9 is constructed from a linear chain of seven acetates.106 Rhodes and coworkers at Glaxo experimented with fermentation conditions which led to the production of putative precursors of 9. For example limiting chloride concentration in the media led to the production of various griseophenones 105-106 and 111, while inhibiting C1 metabolism gave different methylation isomers 105–108.107 Combination of this work with radio-labelling (including the use of 36Cl) then gave various labelled intermediates which were supplemented back to producing cultures. These experiments showed that griseophenone C 106 is converted to griseophenone B 107 which is in-turn a precursor of 9 itself and the shunt metabolite griseophenone A 108.108 Harris and coworkers considerably expanded the known pathway by feeding tritium labelled synthetic intermediates, and this revealed the fuller pathway shown in Scheme 14 in which the unmethylated pentahydroxybenzophenone 111 is the likely first enzyme free intermediate. Feeding of [1,2-13C2]-acetate by Simpson and Holker was used to elucidate the acetate folding pattern,109 while feeding of [5′-2H]-griseophenone B 107 led to the elucidation of the cryptic stereochemistry of the griseofulvin methylene by 2H NMR during the 1970s.110
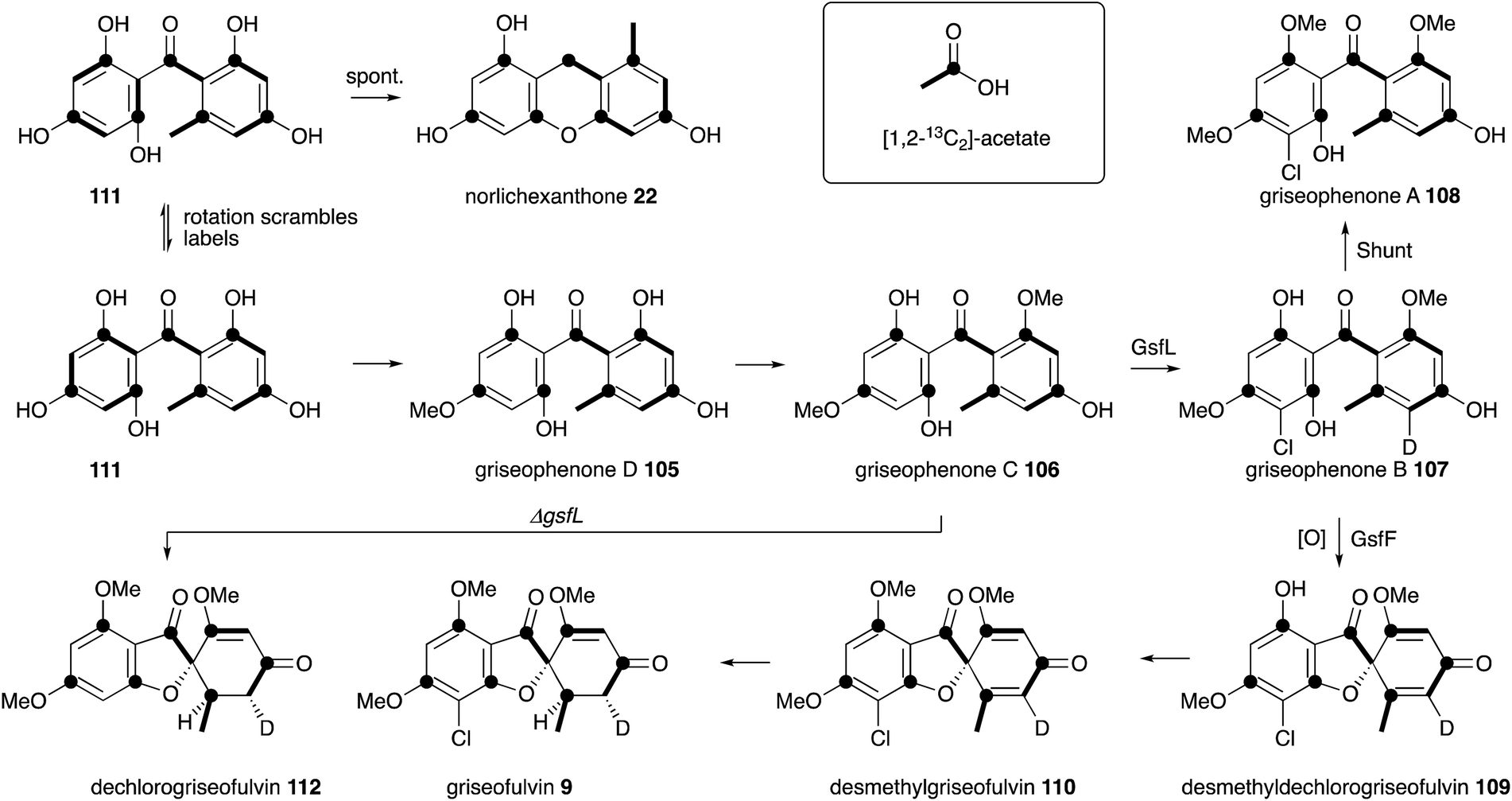 | ||
| Scheme 14 Biosynthesis of griseofulvin 9 deduced by classical labelling and feeding experiments. | ||
Once again a period of more than 30 years had to elapse before further progress could be made in understanding the molecular steps of biosynthesis. Tang and coworkers sequenced the genome of Penicillium aethiopicum, a known griseofulvin 9 producer, using 454 methodology to generate 1522 contigs with N50 of 149 Kb. Out of a total of 30 PKS genes, 7 encoded non-reducing PKS, and one of these genes was embedded in a cluster also encoding a halogenase and three methyltransferases.111 This BGC was thus designated as the most likely to encode the biosynthesis of 9. Knockout experiments targeting the core pks, gfsA, proved this hypothesis correct. The GfsA PKS does not encode a classic C-terminal thiolesterase (TE) domain (e.g. see Section 6) and so it likely releases the heptaketide product 111 by internal Claisen reaction (Scheme 15A). The three methyltransferases (GsfB, GsfC and GsfD) encoded by the cluster and the halogenase (GsfL) must then provide key intermediate griseophenone B 107. Knockout of the halogenase gene gsfL resulted in accumulation of dechlorogriseofulvin 112 in P. aethiopicum. It was expected that a copper-dependent enzyme would be involved in formation of the spirocycle, in analogy to known chemistry in the geodin pathway (Section 3). The cluster does not encode such a protein; however, a cytochrome P450 enzyme encoded by gsfF was speculated to fulfil this role.
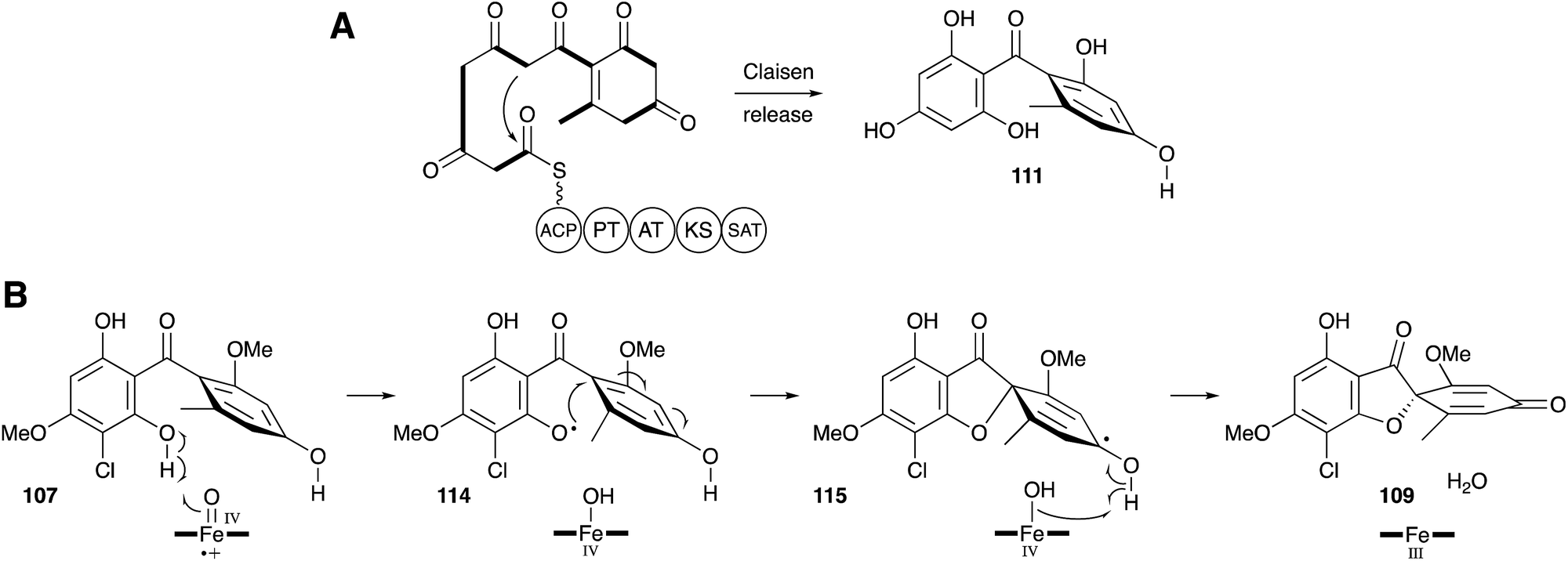 | ||
| Scheme 15 Key griseofulvin 9 biosynthetic steps revealed by Tang, Houk and coworkers. | ||
Tang and coworkers have more recently proven all of the steps of biosynthesis by reconstructing the complete pathway in vitro using purified proteins, malonyl CoA and cofactors.112 This confirms the order of steps, and the remarkable oxidative formation of the grisan system 109 by the cytochrome P450 enzyme GsfF. The mechanism of this step has been probed computationally and the results indicate that hydrogen atom abstraction from a phenolic OH of 107 to give 114, followed by direct spiran ring formation to 115 before collapse to 109 is the most likely mechanism. The alternative possibility of initial epoxidation of the toluene ring is of too high energy to be competitive (Scheme 15B).113
Once again the early work of Raistrick has reached a high level of maturity and a near complete understanding of the pathway and key metabolic steps has been achieved. Again the key advances have come after full pathway reconstruction in a heterologous host – a technique which is increasingly shown to give unambiguous answers to questions regarding the order and selectivity of the chemical steps. Comparison of the griseofulvin and geodin pathways also illustrate that nature can find more than one route for the synthesis of compounds with potent bioactivities (Scheme 16).
 | ||
| Scheme 16 Early isotopic feeding experiments to deduce the origin of mycophenolic acid. | ||
8. Mycophenolic acid
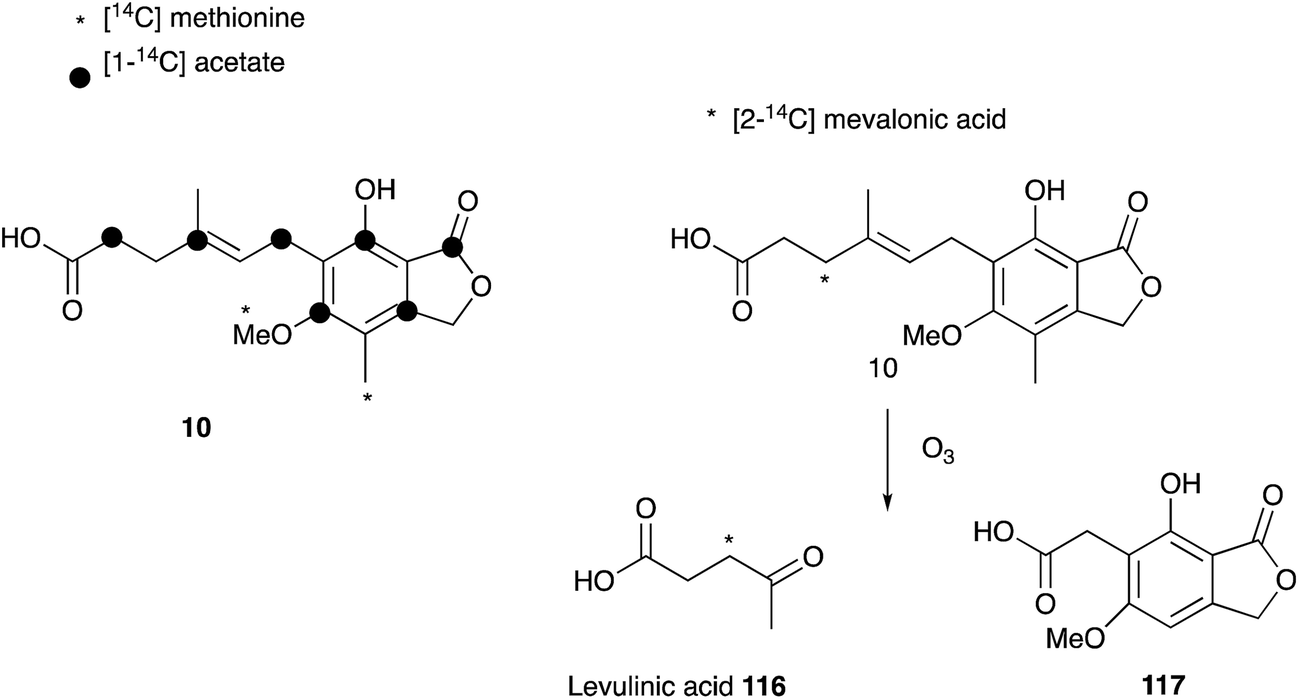
Mycophenolic acid 10 was first described by Gosio in the 1890's as a metabolite of Penicillium brevicompactum isolated from maize.114 Gosio had already discovered that an extract of P. brevicompactum showed antibiotic activity against Bacillus anthrax, and as such mycophenolic acid was the first antibacterial compound purified from any source.115 Over the following century 10 was reported to display antifungal,116 antiviral,117,118 antitumor,119,120 antipsoriasis121 and immunosuppressing122,123 activity, and it is now clinically important. However, 10 was only named mycophenolic acid by Alsberg & Black who purified it from another maize colonising fungus, Penicillium stoloniferum in 1913.124 It was Raistrick who recognized that the described compounds are identical, and, after studying the secondary metabolite production of 15 related fungal strains, 12 were found to produce 10.125,126 Although Alsberg & Black had identified the correct chemical formula of C17H20O6, the successful structure elucidation of 10 by extensive chemical reaction studies took Raistrick and co-workers until 1952.127–129Shortly after structure elucidation, the biosynthesis of 10 became of interest to Arthur Birch, Herchel Smith and coworkers. The origin of the aromatic methyl and the methoxy group was confirmed by feeding [14C]-methionine.130 Further labelling studies with [1-14C]-acetate confirmed equal integration of acetate into the aromatic nucleus and side chain, which was consistent with polyketide biosynthesis, but also a possible terpene origin. Feeding of [2-14C]-mevalonic acid resulted in labelled 10, and label was located in the side-chain by degradation to levulinic acid 116. This led to the proposal of a geranyl precursor as terpene moiety.131
However, later feeding experiments with advanced precursors containing C15 or C10 side chains showed that C15 is the preferred terpene chain length to be incorporated into 10.132 Two mechanism were proposed and proven equally important for the degradation of the C15 farnesyl chain to the observed C7 side chain in 10.133 Either stepwise oxidation of the terminal alkene134–136 of 116 to the C12 intermediate 113 (Scheme 17A), or direct oxidation of the central alkene137 of 116 (Scheme 17B), can occur.
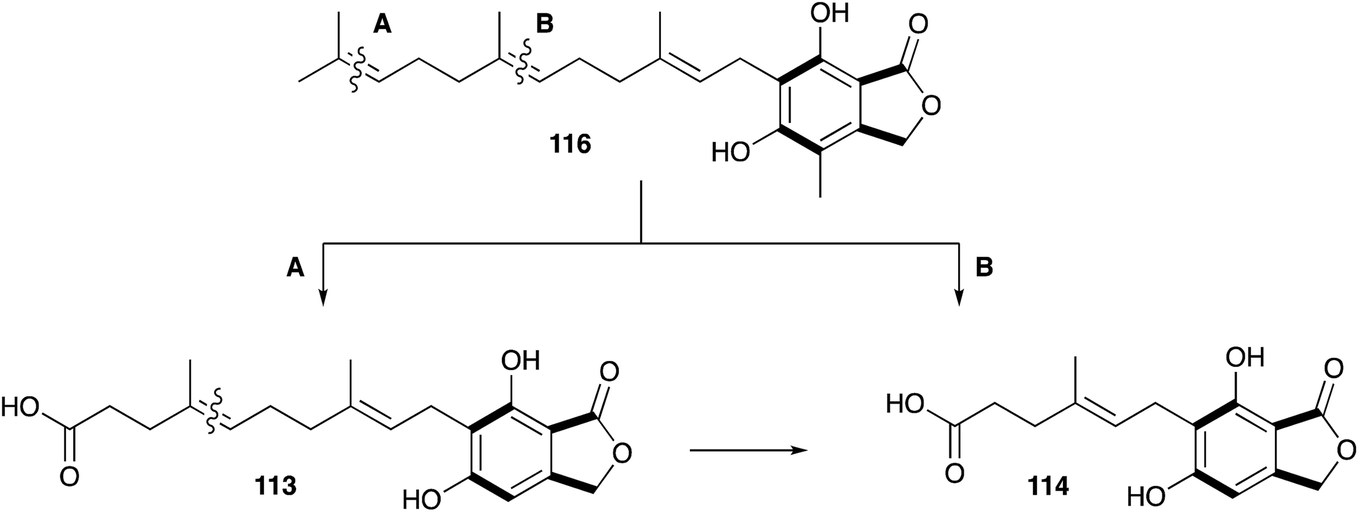 | ||
| Scheme 17 Oxidative processing of the C15 side chain towards mycophenolic acid. | ||
In 2011 the BGC encoding 10 biosynthesis was first identified in P. brevicompactum by Nielsen and coworkers. The strategy employed knowledge about its bioactivity.138 Mycophenolic acid inhibits inosine monophosphate dehydrogenase (IMPDH) and thus de novo purine biosynthesis. The genome of P. brevicompactum IBT23078 was mined for a gene encoding an IMPDH which confers resistance to 10. In this way Nielsen and coworkers identified BGC of approx. 25 Kb which contains 7 open reading frames (ORF), encoding a NR-PKS (MpaC), a chimeric P450-hydrolase (known as MpaDE), a prenyltransferase (MpaA), an oxidative cleavage enzyme (MapH), an O-methyltransferase (MapG), an IMPDH (MapF) and a protein of unknown function (MpaB). Homologous clusters were subsequently identified in one additional strain of P. brevicompactum139 and in Penicillium roqueforti,140 both known producers of 10.
The first three steps were determined by heterologous expression and knockout experiments.133,141,142 The NR-PKS synthesises the tetraketide 119, which is hydroxylated at position 6 by the P450 domain of the chimeric MpaDE to give 120. The hydrolase domain of MpaDE then acts as a lactone synthase yielding 121.137 The intermediate steps, prenylation of 121 to 116 and oxidative cleavage of the C15 to C7 side chain are proposed to be catalysed by MpaA (prenyltransferase) and MpaH (oxidative cleavage enzyme) but need further experimental confirmation. These early steps of meroterpenoid biosynthesis are similar to many others (e.g. see the comprehensive review of Abe and Matsuda)143 in that an electron-rich aromatic nucleus serves as the prenyl acceptor. However, unusually in the case of mycophenolic acid (and related compounds such as yanuthone D)144 oxidative cyclisation of the terpene does not occur (Scheme 18).
 | ||
| Scheme 18 Overall pathway to mycophenolic acid 10. | ||
A SAM-dependent O-methyltransferase was purified from P. stoloniferum and shown to catalyse the methylation of 4-OH in 122 in 1975.145 Due to technical limitations at the time no DNA sequence is available, but it is most likely to be MpaG. Further in vitro assays of recombinant MpaG′ (from P. brevicompactum NRRL 864) in 2015 confirmed these initial findings and showed a relatively broad substrate selectivity, which could be useful for generation of analogues of 10.134
Thus while impressive advances have been made, a full molecular description of mycophenolic acid biosynthesis is still lacking. It is likely, as in other cases discussed here, that full pathway expression will lead to an understanding of all the steps of biosynthesis and conclusively link these to each gene of the BGC. Once this is complete, then engineering efforts can commence.
9. Tropolones
Raistrick, Birkenshaw and Chambers first reported the isolation of stipitatic acid 4, isolated from Penicillium stipitatum (later renamed as Talaromyces stipitatus) in 1942. Extensive chemical derivatisation determined the compound to be aromatic with the formula C8H6O5, but a combination of wartime conditions and a lack of a theoretical framework underpinning concepts of aromaticity meant that the structure could not be solved. However, chemical similarities to other fungal compounds such as puberulic acid 3 (first reported in 1932),146,147 which also defied structural elucidation, led Raistrick to conclude that these compounds formed a new class of fungal metabolites. The structure was solved in 1945 when Michael Dewar realised that 4 must be a new type of aromatic system consisting of a 7-membered ring he named a tropolone. In a parallel paper Dewar also solved the long mystery of the structure of colchicine which he also showed to contain a tropolone,148 masked as its methyl ether.After 1945 the study of tropolone synthesis and biosynthesis became highly topical. For example, Alexander Todd and coworkers reported the first synthesis of 4 in 1950,149 and Robert Robinson speculated on the biosynthetic origin from “the condensation of polyhydric phenols with formaldehyde or its biological equivalent”.150 The first experimental investigations into fungal tropolone biosynthesis came from Bentley and coworkers during the early 1960s, with the development of systematic methods for purification151 and controlled degradation of 4.152 These methods allowed an investigation using 14C labelled precursors, and confirmed a pathway utilising acetate, malonate and a C1 source (Scheme 19).153
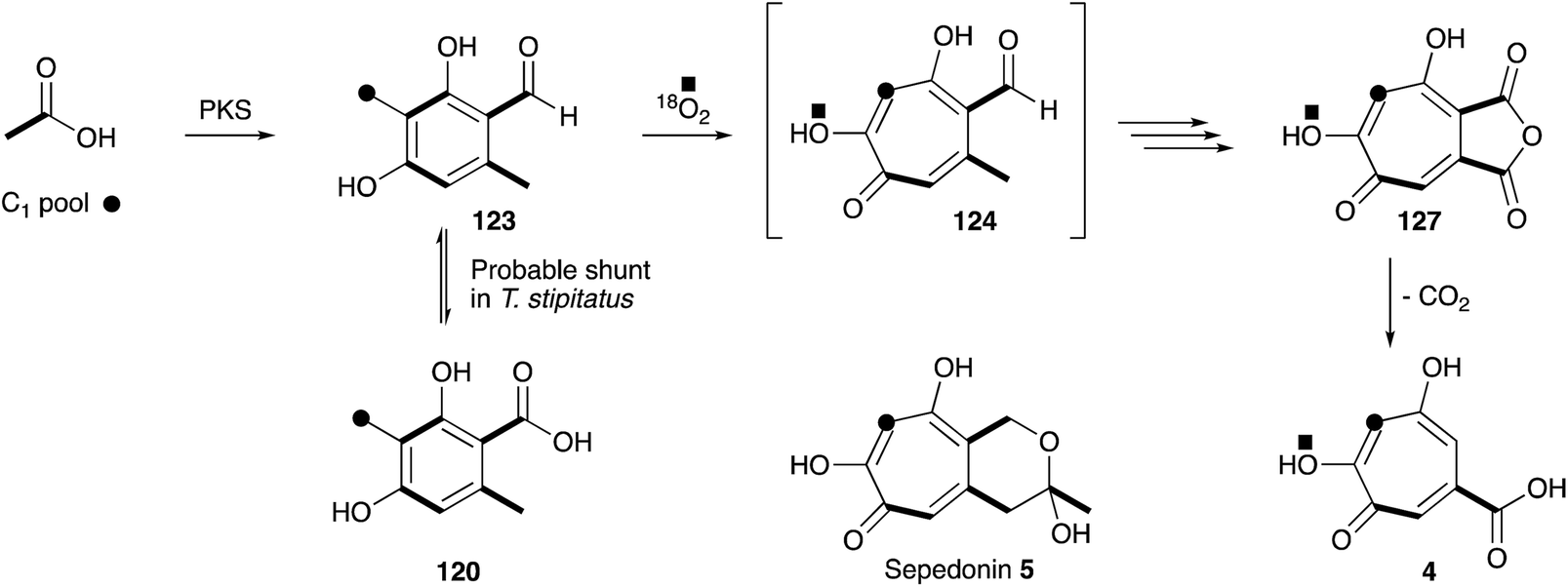 | ||
| Scheme 19 Early labelling results obtained during the study of the biosynthesis of stipitatic acid 4. | ||
The next concrete result came from the group of Ian Scott who showed that 14C-labelled 3-methylorsellinic acid 120 was incorporated into stipitatonic acid 127,154 which Bentley had already shown is decarboxylated to 4 (Scheme 19).155 Later work by Bryant and Light showed that methylorcinaldehyde 123 is incorporated much more efficiently.156 Vining and coworkers were the first to use 13C labels and NMR in the field of tropolone biosynthesis when they showed that sepedonin 5 is derived from acetate and formate in only the second reported use of 13C NMR for the direct elucidation of a biosynthetic pathway.157 The results clearly show that the C1-derived carbon is inserted between carbons derived from acetate. Scott and coworkers then used 18O2 incubations, contrasted with dual 14C–18O-labelled acetate feeding experiments, to differentiate mechanisms involving ring expansion by a migration or by a ring-opening/ring-closing sequence.158 Analysis of the produced stipitatonic acid 127 by mass spectrometry showed an enhancement of the M2+ peak, and no evidence of an M4+ peak, fully in agreement with the proposed migration mechanism.
No further original information was available for around 40 years until genomic sequence data for Talaromyces stipitatus became publically available. The fact that 3-methylorcinaldehyde 123 is likely to be the first enzyme free intermediate stimulated a search of the genome using the sequence of the PKS methylorcinaldehyde synthase (MOS) from Acremonium strictum.159 This highlighted a BGC encoding a homologous PKS (TropA), an FAD-dependent oxygenase (TropB) and a non-heme iron dependent oxygenase (TropC).160In vitro assay of purified TropB and TropC showed that the first step is oxidative dearomatisation of the polyketide to form the dienone 128 (Scheme 20). TropC then attempts to hydroxylate the adjacent methyl, but pinacol-type rearrangement gives the tropolone nucleus of 124. Competing deformylation also affords the trihydroxybenzene 129 at least in vitro. Knockout experiments of all the other genes in the BGC then illustrated how the pathway involves sequential oxidation of the C-1 and C-9 positions, followed by late anhydride hydrolysis and decarboxylation.161
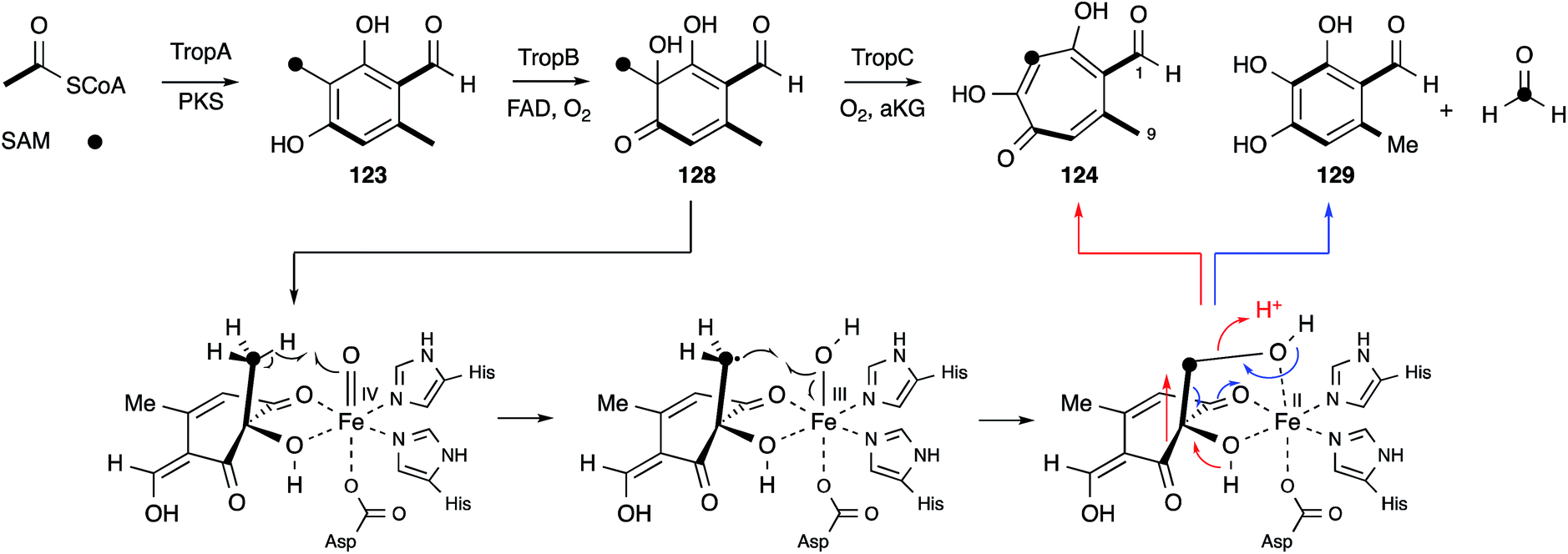 | ||
| Scheme 20 Likely molecular steps during the oxidative ring expansion to form tropolones in fungi. | ||
Elucidation of the early steps of the stipitatic acid 4 pathway also shed light on other related processes in fungal secondary metabolite biosynthesis. For example, TropB, responsible for the oxidative dearomatisation step, has homologues in the sorbicillinoid (SorbC)162 and azaphilone (MrPigN)81 pathways where it is also involved in the oxidative dearomatisation of polyketide aldehydes – a key step in the activation of the otherwise poorly reactive aromatic system. A TropC analogue, which in the stipitatic acid 4 pathway catalyses the ring expansion, is involved in hydroxylation of an aromatic methyl group in the citrinin pathway (Section 5).80 Meanwhile the nrPKS for all three pathways are closely related, producing aromatic polyketide aldehydes, differing in chain-length and methylation pattern, but all with a distinctive reductive release. These chemical observations thus point strongly to the evolution and close relationships of the biosynthetic pathways leading to these different classes of polyketides. Thus the continuing study of the classic Raistrick compounds such as 4 inevitably leads to insights and understanding of many other fungal pathways. Several other classes of fungal metabolites are known to feature tropolones in their structures, or as putative precursors or intermediates. Such compounds include Xenovulene A,163 the epolones,164 pycnidione165 and the eupenifeldins.166 However no sequence or molecular information has yet been revealed in these cases, and while it is tempting to speculate that their tropolone moieties may be derived as discussed above, other modes of tropolone biosynthesis are known in plants and bacteria.167
10. Other compounds and outstanding problems
Many of the classic Raistrick compounds have continued to yield fascinating insights into fungal secondary metabolism through the past decade, as new genomic and molecular methods have built on the foundations formed from isotopic feeding experiments. Much of the work reported in the literature has focussed on the more biologically (e.g. mycophenolic acid 10, griseofulvin 9) significant or structurally interesting (e.g. maleidrides 6, tropolones 3) metabolites where much new chemistry has been revealed. In many cases understanding the biosynthesis at the genomic and molecular levels is revealing hitherto unsuspected relationships, such as between geodin 8 and ravenelin 7 (Section 3) or between citrinin 2, the tropolones 3, the sorbicillins and the azaphilones (Sections 3 and 9). Such relationships will no doubt help to uncover evolutionary relationships between pathways, and this understanding will also aid in the future rational redesign of individual biosynthetic steps (e.g. as exemplified in the DALs, Section 6) and new pathways in their entirety. It seems likely that the Raistrick compounds will continue to be of interest in this field because of the significant depth of knowledge accrued over the past 80–90 years.Other compounds which might be considered as of more minor structural or biological significance, such as the fruit spoilage mycotoxin patulin 14 produced by Penicillium species and the mycotoxin terrein 13 from Aspergillus species remain to be fully investigated. For example, in the case of patulin 14 a biosynthetic gene cluster has been reported,168 and some P450 enzymes have been isolated,169 but no detailed pathway investigations have yet been made. The pathway should include interesting ring contracting steps. Likewise the BGC for terrein biosynthesis has also been revealed in A. terreus,170 but no detailed pathway has yet been reported, and again fascinating oxidative ring contractions are likely to be revealed. In the case of kojic acid 1 the genes have been identified,171,172 and biotechnological processes are already being engineered to increase the production of this valuable metabolite through a rather straight-forward pathway from glucose.173 Significant progress has been made in elucidating the early steps of the emodin pathway to chrysophanol, but later steps to many of the ergochromes and reduced xanthones remain cryptic. Likewise, recent progress in understanding the molecular mechanisms of maleidride biosynthesis have revealed the new proteins involved, but not yet the chemical mechanisms and selectivity factors which must be at play, or how these systems could be extended to explain the biosynthesis of the octadrides or linear family members (Scheme 21).
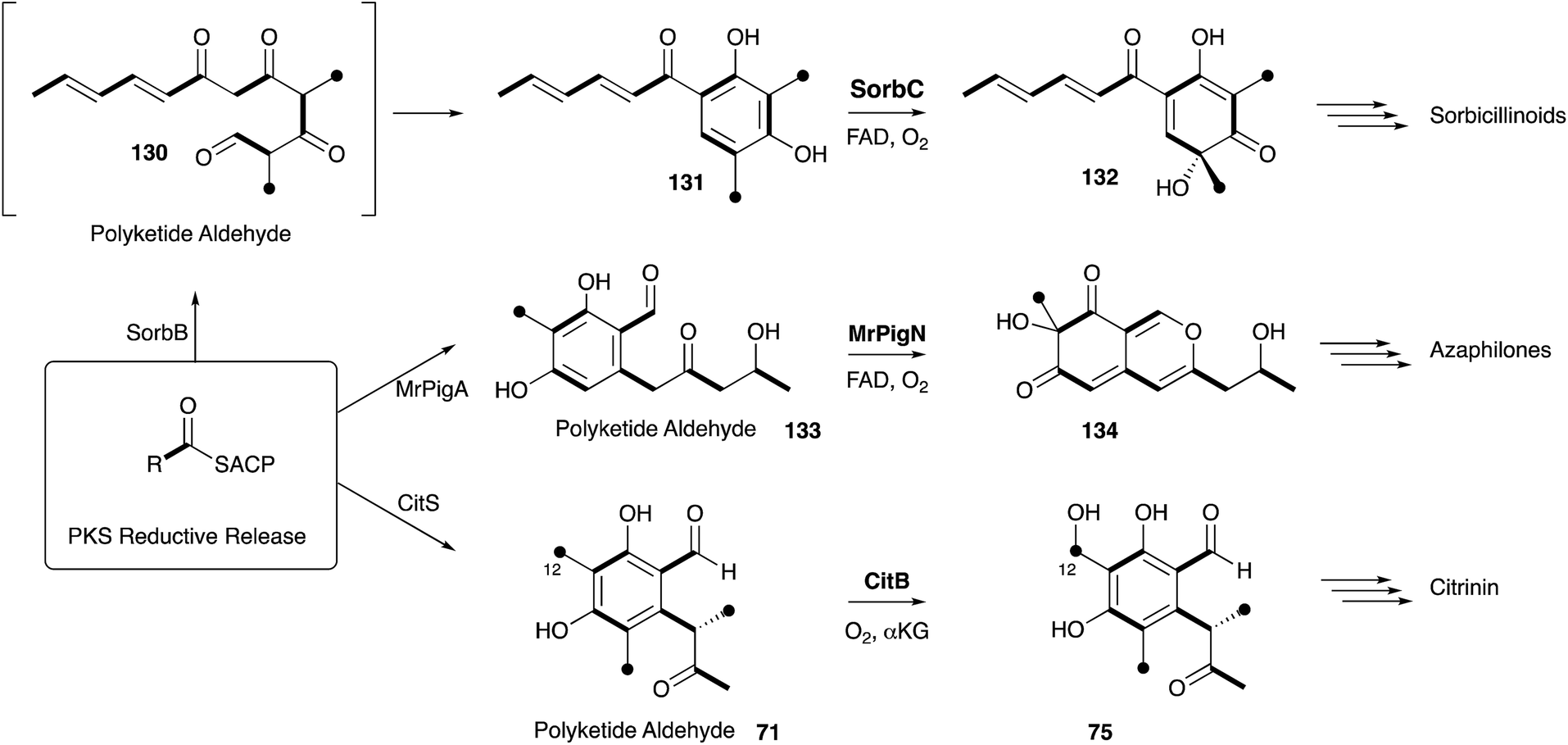 | ||
| Scheme 21 Chemical relationships between the biosynthesis of sorbicillinoids, azaphilones and citrinin. | ||
Thus it seems certain that interest in the Raistrick compounds, and their use as models for understanding and exploiting fungal biosynthetic pathways will continue into the future.
11. Conflicts of interest
There are no conflicts to declare.12. Acknowledgements
RJC thanks current and past group members who used the theme of Raistrick Compounds for a series of stimulating group meetings.13. References
- J. H. Birkinshaw, Biogr. Mem. Fellows R. Soc., 1972, 18, 488–509 CrossRef
.
- R. Bentley, Nat. Prod. Rep., 2006, 23, 1046–1062 RSC
- B. R. Clark, R. J. Capon, E. Lacey, S. Tennant and J. H. Gill, Org. Biomol. Chem., 2006, 4, 1520 CAS
- P. W. Clutterbuck, R. Lovell and H. Raistrick, Biochem. J., 1932, 26, 1907–1918 CrossRef CAS PubMed
- D. J. Cram, J. Am. Chem. Soc., 1948, 70, 440–441 CrossRef CAS PubMed
- A. J. Birch, P. Fitton, E. Pride, A. Ryan, H. Smith and W. Whalley, J. Chem. Soc., 1958, 4576–4581 RSC
- J. Barber and J. Staunton, J. Chem. Soc., Chem. Commun., 1979, 1098–1099 RSC
- U. Sankawa, Y. Ebizuka, H. Noguchi, Y. Isikawa, S. Kitaghawa, Y. Yamamoto, T. Kobayashi and Y. Iitak, Tetrahedron, 1983, 39, 3583–3591 CrossRef CAS
- T. Shimizu, H. Kinoshita and T. Nihira, Appl. Environ. Microbiol., 2007, 73, 5097–5103 CrossRef CAS PubMed
- M. Dewar, Nature, 1945, 155, 50–51 CrossRef CAS
- R. Thomas, Biochem. J., 1961, 78, 748–758 CrossRef CAS PubMed
- M. Ahuja, Y.-M. Chiang, S.-L. Chang, M. B. Praseuth, R. Entwistle, J. F. Sanchez, H.-C. Lo, H.-H. Yeh, B. R. Oakley and C. C. C. Wang, J. Am. Chem. Soc., 2012, 134, 8212–8221 CrossRef CAS PubMed
- D. Saha, R. Fetzner, B. Burkhardt, J. Podlech, M. Metzler, H. Dang, C. Lawrence and R. Fischer, PLoS One, 2012, 7, e40564 CAS
- E. E. Stinson, W. B. Wise, R. A. Moreau, A. J. Jurewicz and P. E. Pfeffer, Can. J. Chem., 1986, 64, 1590–1594 CrossRef CAS
- J. Dasenbrock and T. J. Simpson, J. Chem. Soc., Chem. Commun., 1987, 1235–1242 RSC
- M. Wenderoth, C. Pinecker, B. Voß and R. Fischer, Fungal Genet. Biol., 2017, 101, 55–60 CrossRef CAS PubMed
- R. A. Jacobson and R. Adams, J. Am. Chem. Soc., 1924, 46, 1312–1316 CrossRef CAS
- H. Raistrick, R. Robinson and D. E. White, Biochem. J., 1936, 30, 1303–1314 CrossRef CAS PubMed
- H. Raistrick and G. Smith, Biochem. J., 1936, 30, 1315–1322 CrossRef CAS PubMed
- C. T. Calam, P. W. Clutterbuck, A. E. Oxford and H. Raistrick, Biochem. J., 1947, 41, 458–463 CrossRef CAS PubMed
- D. H. R. Barton and A. I. Scott, J. Chem. Soc., 1958, 1767–1776 RSC
- A. J. Birch, T. J. Simpson and P. W. Westerman, Tetrahedron Lett., 1975, 16, 4173–4177 CrossRef
- S. Gatenbeck and L. Malmström, Acta Chem. Scand., 1969, 23, 3493–3497 CrossRef CAS
- R. F. Curtis, C. H. Hassall and D. R. Parry, J. Chem. Soc., Perkin Trans. 1, 1972, 240–245 RSC
- J. G. Hill, T. T. Nakashima and J. C. Vederas, J. Am. Chem. Soc., 1982, 104, 1745–1748 CrossRef CAS
- H. Fujimoto, H. Flasch and B. Franck, Chem. Ber., 1975, 108, 1224–1228 CrossRef CAS
- I. Fujii, Y. Ebizuka and U. Sankawa, Chem. Pharm. Bull., 1982, 30, 2283–2286 CrossRef CAS
- I. Fujii, H. Iijima, Y. Ebizuka and U. Sankawa, Chem. Pharm. Bull., 1983, 31, 337–340 CrossRef CAS
- K. X. Huang, I. Fujii, Y. Ebizuka, K. Gomi and U. Sankawa, J. Biol. Chem., 1995, 270, 21495–21502 CrossRef CAS PubMed
- I. Fujii, Y. Ebizuka and U. Sankawa, J. Biochem., 1988, 103, 878–883 CrossRef CAS PubMed
- Z. G. Chen, I. Fujii, Y. Ebizuka and U. Sankawa, Phytochemistry, 1995, 38, 299–305 CrossRef CAS
- T. P. Nicholson, B. A. Rudd, M. Dawson, C. M. Lazarus, T. J. Simpson and R. J. Cox, Chem. Biol., 2000, 8, 157–178 CrossRef
- R. D. Couch and G. M. Gaucher, J. Biotechnol., 2004, 108, 171–177 CrossRef CAS PubMed
- M. T. Nielsen, J. B. Nielsen, D. C. Anyaogu, D. K. Holm, K. F. Nielsen, T. O. Larsen and U. H. Mortensen, PLoS One, 2013, 8, e72871–10 CAS
- Y.-M. Chiang, E. Szewczyk, A. D. Davidson, R. Entwistle, N. P. Keller, C. C. C. Wang and B. R. Oakley, Appl. Environ. Microbiol., 2010, 76, 2067–2074 CrossRef CAS PubMed
- J. F. Sanchez, R. Entwistle, J.-H. Hung, J. Yaegashi, S. Jain, Y.-M. Chiang, C. C. C. Wang and B. R. Oakley, J. Am. Chem. Soc., 2011, 133, 4010–4017 CrossRef CAS PubMed
- L. Neubauer, J. Dopstadt, H.-U. Humpf and P. Tudzynski, Fungal Biol. Biotechnol., 2016, 3(2), 1–14 Search PubMed
- M. A. Schätzle, S. M. Husain, S. Ferlaino and M. Müller, J. Am. Chem. Soc., 2012, 134, 14742–14745 CrossRef PubMed
- B. H. Howard and H. Raistrick, Biochem. J., 1950, 46, 49–53 CrossRef CAS PubMed
- T. J. Simpson, ChemBioChem, 2012, 13, 1680–1688 CrossRef CAS PubMed
- S. Griffiths, C. H. Mesarich, B. Saccomanno, A. Vaisberg, P. J. G. M. de Wit, R. J. Cox and J. Collemare, Proc. Natl. Acad. Sci. U.S.A., 2016, 113, 6851–6856 CrossRef CAS PubMed
- B. H. Howard and H. Raistrick, Biochem. J., 1949, 44, 227–233 CrossRef CAS PubMed
- N. Wijkman, Justus Liebigs Ann. Chem., 1931, 485, 61–73 CrossRef CAS
- H. Raistrick and G. Smith, Biochem. J., 1933, 27, 1814–1819 CrossRef CAS PubMed
- J. E. Baldwin, D. H. R. Barton, J. L. Bloomer, L. M. Jackman, L. Rodriguez-Hahn and J. K. Sutherland, Experientia, 1962, 18, 345 CrossRef CAS PubMed
- T. Hamor, I. Paul, J. Monteath Robertson and G. Sim, Experientia, 1962, 18, 352–354 CrossRef CAS PubMed
- D. H. R. Barton and J. K. Sutherland, J. Chem. Soc., 1965, 1769–1774 RSC
- R. I. Crane, P. Hedden, J. MacMillan and W. B. Turner, J. Chem. Soc., Perkin Trans. 1, 1973, 194–197 RSC
- G. Büchi, K. M. Snader, J. D. White, J. Z. Gougoutas and S. Singh, J. Am. Chem. Soc., 1970, 92, 6638–6641 CrossRef
- G. A. Sulikowski, F. Agnelli, P. Spencer, J. M. Koomen and D. H. Russell, Org. Lett., 2002, 4, 1447–1450 CrossRef CAS PubMed
- M. Nakajima, K. Itoi, Y. Takamatsu, S. Sato, Y. Furukawa, K. Furuya, T. Honma, J. Kadotani, M. Kozasa and T. Haneishi, J. Antibiot., 1991, 44, 1065–1072 CrossRef CAS PubMed
- M. Futagawa, D. E. Wedge and F. E. Dayan, Pestic. Biochem. Physiol., 2002, 73, 87–93 CrossRef CAS
- M. Saleem, H. Hussain, I. Ahmed, S. Draeger, B. Schulz, K. Meier, M. Steinert, G. Pescitelli, T. Kurtán, U. Flörke and K. Krohn, Eur. J. Org. Chem., 2010, 2011, 808–812 CrossRef
- A. J. Szwalbe, K. Williams, D. E. O'Flynn, A. M. Bailey, N. P. Mulholland, J. L. Vincent, C. L. Willis, R. J. Cox and T. J. Simpson, Chem. Commun., 2015, 51, 17088–17091 RSC
- J. L. Bloomer, C. E. Moppett and J. K. Sutherland, J. Chem. Soc. C, 1968, 588–591 RSC
- R. K. Huff, C. E. Moppett and J. K. Sutherland, J. Chem. Soc., Perkin Trans. 1, 1972, 2584 RSC
- J. E. Baldwin, A. Beyeler, C. Keats, G. J. Pritchard, R. M. Adlington and D. J. Watkin, Tetrahedron, 1999, 55, 7363–7374 CrossRef CAS
- N. P. Mulholland, J. L. Vincent, A. M. Bailey, C. L. Willis and R. J. Cox, Angew. Chem., Int. Ed., 2016, 55, 6784–6788 CrossRef PubMed
- R. Fujii, A. Minami, S. Nagamine, I. Takeuchi and H. Oikawa, Org. Lett., 2015, 17, 5658–5661 CrossRef CAS PubMed
- K. Williams, A. J. Szwalbe, C. Dickson, T. R. Desson, N. P. Mulholland, J. L. Vincent, J. M. Clough, A. M. Bailey, C. P. Butts, C. L. Willis, T. J. Simpson and R. J. Cox, Chem. Commun., 2017, 53, 7965–7968 RSC
- J. Bai, D. Yan, T. Zhang, Y. Guo, Y. Liu, Y. Zou, M. Tang, B. Liu, Q. Wu, S. Yu, Y. Tang and Y. Hu, Angew. Chem., 2017, 129, 4860–4864 CrossRef
- B. Bonsch, V. Belt, C. Bartel, N. Duensing, M. Koziol, C. M. Lazarus, A. M. Bailey, T. J. Simpson and R. J. Cox, Chem. Commun., 2016, 52, 6777–6780 RSC
- M. Isaka, M. Tanticharoen and Y. Thebtaranonth, Tetrahedron Lett., 2000, 41, 1657–1660 CrossRef CAS
- A. C. Hetherington and H. Raistrick, Philos. Trans. R. Soc., B, 1931, 220, 269–295 CrossRef
- F. P. Coyne, H. Raistrick and R. Robinson, Philos. Trans. R. Soc., B, 1931, 220, 297–300 CrossRef
- T. S. Gore, T. B. Panse and K. Venkataraman, Nature, 1946, 157, 333–333 CrossRef CAS PubMed
- D. J. Cram, J. Am. Chem. Soc., 1948, 70, 440–441 CrossRef CAS PubMed
- J. P. Brown, N. J. Cartwright, A. Robertson and W. B. Whalley, Nature, 1948, 162, 72–73 CAS
- E. Schwenk, G. J. Alexander, A. M. Gold and D. F. StevenS, J. Biol. Chem., 1958, 233, 1211–1213 CAS
- A. J. Birch, P. Fitton, E. Pride, A. J. Ryan, H. Smith and W. B. Whalley, J. Chem. Soc., 1958, 4576–6 RSC
- R. B. Woodward, Angew. Chem., 1956, 68, 13–20 CrossRef CAS
- R. Carter, M. Garson and J. Staunton, J. Chem. Soc., Chem. Commun., 1979, 1097–1098 RSC
- J. Barber, R. H. Carter, M. J. Garson and J. Staunton, J. Chem. Soc., Perkin Trans. 1, 1981, 2577–2583 RSC
- J. Barber and J. Staunton, J. Chem. Soc., Chem. Commun., 1979, 1098–1099 RSC
- U. Sankawa, Y. Ebizuka, H. Noguchi, Y. Isikawa, S. Kitaghawa, Y. Yamamoto, T. Kobayashi and Y. Iitak, Tetrahedron, 1983, 39, 3583–3591 CrossRef CAS
- H. Hajjaj, A. Klaebe, M. Loret, G. Goma, P. Blanc and J. Francois, Appl. Environ. Microbiol., 1999, 65, 311–314 CAS
- P. C. Y. Woo, C.-W. Lam, E. W. T. Tam, K.-C. Lee, K. K. Y. Yung, C. K. F. Leung, K.-H. Sze, S. K. P. Lau and K.-Y. Yuen, Sci. Rep., 2014, 4, 6728–8 CrossRef CAS PubMed
- L. E. Bingle, T. J. Simpson and C. M. Lazarus, Fungal Genet. Biol., 1999, 26, 209–223 CrossRef CAS PubMed
- T. Shimizu, H. Kinoshita and T. Nihira, Appl. Environ. Microbiol., 2007, 73, 5097–5103 CrossRef CAS PubMed
- K. Sakai, H. Kinoshita, T. Shimizu and T. Nihira, J. Biosci. Bioeng., 2008, 106, 466–472 CrossRef CAS PubMed
- Y. He and R. J. Cox, Chem. Sci., 2016, 7, 2119–2127 RSC
- P. A. Storm and C. A. Townsend, Chem. Commun., 2017, 54, 50–53 RSC
- W. Chen, R. Chen, Q. Liu, Y. He, K. He, X. Ding, L. Kang, X. Guo, N. Xie, Y. Zhou, Y. Lu, R. J. Cox, I. Molnár, M. Li, Y. Shao and F. Chen, Chem. Sci., 2017, 8, 4917–4925 RSC
- P. V. Divekar, H. Raistrick, T. A. Dobson and L. C. Vining, Can. J. Chem., 1965, 43, 1835–1848 CrossRef CAS
- P. A. Storm, D. A. Herbst, T. Maier and C. A. Townsend, Cell Chem. Biol., 2017, 24, 1–11 CrossRef PubMed
- R. A. Cacho, J. Thuss, W. Xu, R. Sanichar, Z. Gao, A. Nguyen, J. C. Vederas and Y. Tang, J. Am. Chem. Soc., 2015, 137, 15688–15691 CrossRef CAS PubMed
- D. M. Roberts, C. Bartel, A. Scott, D. Ivison, T. J. Simpson and R. J. Cox, Chem. Sci., 2017, 8, 1116–1126 RSC
- E. Liddle, A. Scott, L.-C. Han, D. Ivison, T. J. Simpson, C. L. Willis and R. J. Cox, Chem. Commun., 2017, 53, 1727–1730 RSC
- H. Raistrick and F. A. H. Rice, J. Chem. Soc. C, 1971, 3069–3072 RSC
- O. C. Musgrave, J. Chem. Soc., 1956, 4301–4305 RSC
- A. J. Birch, O. C. Musgrave, R. W. Rickards and H. Smith, J. Chem. Soc., 1959, 3146–3147 RSC
- H. D. Munro, O. C. Musgrave and R. Templeton, J. Chem. Soc. C, 1967, 947–948 RSC
- K. Arai, B. Rawlings, Y. Yoshizawa and J. Vederas, J. Am. Chem. Soc., 1989, 111, 3391–3399 CrossRef CAS
- Y. Yoshizawa, Z. Li, P. Reese and J. Vederas, J. Am. Chem. Soc., 1990, 112, 3212–3213 CrossRef CAS
- Y. Xu, P. Espinosa-Artiles, V. Schubert, Y. M. Xu, W. Zhang, M. Lin, A. A. L. Gunatilaka, R. Sussmuth and I. Molnar, Appl. Environ. Microbiol., 2013, 79, 2038–2047 CrossRef CAS PubMed
- Y. Xu, T. Zhou, Z. Zhou, S. Su, S. A. Roberts, W. R. Montfort, J. Zeng, M. Chen, W. Zhang, M. Lin, J. Zhan and I. Molnár, Proc. Natl. Acad. Sci. U.S.A., 2013, 110, 5398–5403 CrossRef CAS PubMed
- Y. Xu, T. Zhou, S. Zhang, L.-J. Xuan, J. Zhan and I. Molnár, J. Am. Chem. Soc., 2013, 135, 10783–10791 CrossRef CAS PubMed
- Y. Xu, T. Zhou, S. Zhang, P. Espinosa-Artiles, L. Wang, W. Zhang, M. Lin, A. A. L. Gunatilaka, J. Zhan and I. Molnár, Proc. Natl. Acad. Sci. U.S.A., 2014, 111, 12354–12359 CrossRef CAS PubMed
- J. Bai, Y. Lu, Y.-M. Xu, W. Zhang, M. Chen, M. Lin, A. A. L. Gunatilaka, Y. Xu and I. Molnár, Org. Lett., 2016, 18, 1262–1265 CrossRef CAS PubMed
- K. A. K. Pahirulzaman, K. Williams and C. M. Lazarus, Meth. Enzymol., 2012, 517, 241–260 CrossRef CAS PubMed
- A. E. Oxford, H. Raistrick and P. Simonart, Biochem. J., 1939, 33, 240–248 CrossRef CAS PubMed
- J. MacMillan, Annu. Rev. Plant Physiol., 1996, 47, 1–21 CrossRef CAS PubMed
- J. F. Grove, J. MacMillan, T. P. C. Mulholland and M. A. T. Rogers, J. Chem. Soc., 1952, 3977–3987 RSC
- J. MacMillan, J. Chem. Soc., 1954, 2585–2587 RSC
-
Glaxo: A History to 1962, ed. R. P. T. Davenport-Hines and J. Slinn, Cambridge University Press, 1992 Search PubMed
- A. J. Birch, R. A. Massy-Westropp, R. W. Rickards and H. Smith, J. Chem. Soc., 1958, 360–365 RSC
- A. Rhodes, B. Boothroyd, P. Mcgonagle and G. A. Somerfield, Biochem. J., 1961, 81, 28–37 CrossRef CAS PubMed
- A. Rhodes, G. A. Somerfield and M. P. McGonagle, Biochem. J., 1963, 88, 349–357 CrossRef CAS PubMed
- T. J. Simpson and J. Holker, Phytochemistry, 1977, 16, 229–233 CrossRef CAS
- Y. Sato, T. Oda and H. Saitô, J. Chem. Soc., Chem. Commun., 1978, 135–136 RSC
- Y.-H. Chooi, R. Cacho and Y. Tang, Chem. Biol., 2010, 17, 483–494 CrossRef CAS PubMed
- R. A. Cacho, Y.-H. Chooi, H. Zhou and Y. Tang, ACS Chem. Biol., 2013, 8, 2322–2330 CrossRef CAS PubMed
- J. M. Grandner, R. A. Cacho, Y. Tang and K. N. Houk, ACS Catal., 2016, 6, 4506–4511 CrossRef CAS PubMed
- B. Gosio, G. Accad. Med. Torino, 1893, 61, 484 Search PubMed
- R. Bentley, Chem. Rev., 2000, 100, 3801–3825 CrossRef CAS PubMed
- R. Nicoletti, M. De Stefano, S. De Stefano, A. Trincone and F. Marziano, Mycopathologia, 2004, 158, 465 CrossRef CAS PubMed
- M. S. Diamond, M. Zachariah and E. Harris, Virology, 2002, 304, 211 CrossRef CAS PubMed
- K. Borroto-Esoda, F. Myrick, J. Feng, J. Jeffrey and P. Furman, Antimicrob. Agents Chemother., 2004, 48, 4387 CrossRef CAS PubMed
- F. Kavanagh, J. Bacteriol., 1947, 54, 761 CAS
- R. D. Torrenegra, J. E. Baquero and J. S. Calderon, Rev. Latinoam. Quim., 2005, 33, 76 CAS
- W. Epinette, C. Parker, E. Jones and M. Greist, J. Am. Acad. Dermatol., 1987, 17, 962 CrossRef CAS PubMed
- J. C. Cline, J. D. Nelson, K. Gerzon, R. H. Williams and D. C. Delong, Appl. Microbiol., 1969, 18, 14 CAS
- C. M. Smith, L. J. Fontenelle, H. Muzik, H. Unger, L. W. Brox and J. F. Henderson, Biochem. Pharmacol., 1974, 23, 2727 CrossRef CAS PubMed
- C. L. Alsberg and O. F. Black, U.S. Department of Agriculture, Bureau of Plant Industry, Bulletin No. 270, 1913.
- W. P. Clutterbuck, A. E. Oxford and H. Raistrick, Biochem. J., 1932, 26, 1441–1458 CrossRef PubMed
- A. E. Oxford and H. Raistrick, Biochem. J., 1933, 27, 634–653 CrossRef CAS PubMed
- H. Birkinshaw, H. Raistrick and D. J. Ross, Biochem. J., 1952, 50, 630 CrossRef PubMed
- J. Birkinshaw, A. Bracken, E. N. Morgan and H. Raistrick, Biochem. J., 1948, 43, 216 CrossRef CAS PubMed
- W. P. Clutterbuck and H. Raistrick, Biochem. J., 1933, 27, 654 CrossRef PubMed
- A. J. Birch, R. J. English, R. A. Massy-Westropp, M. Slaytor and H. Smith, Proc. Chem. Soc., London, 1957, 204–204 CAS
- A. J. Birch, R. J. English, R. A. Massy-Westropp and H. Smith, Proc. Chem. Soc., London, 1957, 233–234 CAS
- L. Canonica, W. Kroszczynski, B. M. Ranzi, B. Rindone, E. Santaniello and C. Scolastico, J. Chem. Soc., Perkin Trans. 1, 1972, 2639 RSC
- L. Colombo, C. Gennari, D. Potenza and C. Scolastico, J. Chem. Soc., Perkin Trans. 1, 1980, 365 Search PubMed
- C. P. Nulton and I. M. Campbell, Can. J. Microbiol., 1978, 24, 199–201 CrossRef CAS PubMed
- L. Colombo, C. Gennari, D. Potenza and C. Scolastico, J. Chem. Soc., Chem. Commun., 1979, 1021–1022 RSC
- D. L. Doerfler, L. A. Ernst and I. M. Campbell, J. Chem. Soc., Chem. Commun., 1980, 329–330 RSC
- L. Colombo, C. Gennari and C. Scolastico, J. Chem. Soc., Chem. Commun., 1978, 434–434 RSC
- T. B. Regueira, K. R. Kildegaard, B. G. Hansen, U. H. Mortensen, C. Hertweck and J. Nielsen, Appl. Environ. Microbiol., 2011, 77, 3035 CrossRef CAS PubMed
- W. Zhang, S. Cao, L. Qiu, F. Qi, Z. Li, Y. Yang, S. Huang, F. Bai, C. Liu, X. Wan and S. Li, ChemBioChem, 2015, 16, 565 CrossRef CAS PubMed
- A. Del-Cid, C. Gil-Durán, I. Vaca, J. F. Rojas-Aedo, R. O. García-Rico, G. Levicán and R. Chávez, PLoS One, 2016, 11, e0147047 Search PubMed
- B. G. Hansen, B. Salomonsen, M. T. Nielsen, J. B. Nielsen, N. B. Hansen, K. F. Nielsen, T. B. Regueira, J. Nielsen, K. R. Patil and U. H. Mortensen, Appl. Environ. Microbiol., 2011, 77, 3044–3051 CrossRef CAS PubMed
- B. G. Hansen, E. Mnich, K. F. Nielsen, J. B. Nielsen, M. T. Nielsen, U. H. Mortensen, T. O. Larsen and K. R. Patil, Appl. Environ. Microbiol., 2012, 78, 4908–4913 CrossRef CAS PubMed
- Y. Matsuda and I. Abe, Nat. Prod. Rep., 2015, 33, 26–53 RSC
- D. K. Holm, L. M. Petersen, A. Klitgaard, P. B. Knudsen, Z. D. Jarczynska, K. F. Nielsen, C. H. Gotfredsen and T. O. Larsen, Chem. Biol., 2014, 21, 519–529 CrossRef CAS PubMed
- W. L. Muth and C. H. Nash, Antimicrob. Agents Chemother., 1975, 8, 321 CrossRef CAS PubMed
- J. H. Birkinshaw and H. Raistrick, Biochem. J., 1932, 26, 441–453 CrossRef CAS PubMed
- M. Iwatsuki, S. Takada, M. Mori, A. Ishiyama, M. Namatame, A. Nishihara-Tsukashima, K. Nonaka, R. Masuma, K. Otoguro, K. Shiomi and S. Omura, J. Antibiot., 2011, 64, 183–188 CrossRef CAS PubMed
- M. Dewar, Nature, 1945, 155(3927), 141–142 CrossRef CAS
- R. Corbett, A. Johnson and A. Todd, J. Chem. Soc., 1950, 147–149 RSC
- R. Robinson, Chem. Ind., 1951, 12 Search PubMed
- R. Bentley and C. Thiessen, J. Biol. Chem., 1963, 238, 1880–1888 CAS
- R. Bentley, J. Biol. Chem., 1963, 238, 1889–1894 CAS
- R. Bentley, J. Biol. Chem., 1963, 238, 1895–1902 CAS
- A. I. Scott, H. Guilford and E. Lee, J. Am. Chem. Soc., 1971, 93, 3534–3536 CrossRef CAS PubMed
- R. Bentley and C. P. Thiessen, J. Biol. Chem., 1963, 238, 3811–3816 CAS
- R. Bryant and R. Light, Biochemistry, 1974, 13, 1516–1522 CrossRef CAS PubMed
- A. Mcinnes, D. Smith and L. Vining, J. Chem. Soc., Chem. Commun., 1971, 325–326 RSC
- A. I. Scott and K. J. Wiesner, J. Chem. Soc., Chem. Commun., 1972, 1075–1077 RSC
- A. M. Bailey, R. J. Cox, K. Harley, C. M. Lazarus, T. J. Simpson and E. Skellam, Chem. Commun., 2007, 4053–4055 RSC
- J. Davison, A. Al Fahad, M. Cai, Z. Song, S. Y. Yehia, C. M. Lazarus, A. M. Bailey, T. J. Simpson and R. J. Cox, Proc. Natl. Acad. Sci. U.S.A., 2012, 109, 7642–7647 CrossRef CAS PubMed
- A. Al Fahad, A. Abood, T. J. Simpson and R. J. Cox, Angew. Chem., Int. Ed., 2014, 53, 7519–7523 CrossRef CAS PubMed
- A. A. Fahad, A. Abood, K. M. Fisch, A. Osipow, J. Davison, M. Avramović, C. P. Butts, J. Piel, T. J. Simpson and R. J. Cox, Chem. Sci., 2014, 5, 523–527 RSC
- M. E. Raggatt, T. J. Simpson and M. I. ChicarelliRobinson, Chem. Commun., 1997, 2245–2246 RSC
- P. Cai, D. Smith, B. Cunningham, S. Brown-Shimer, B. Katz, C. Pearce, D. Venables and D. Houck, J. Nat. Prod., 1998, 61, 791–795 CrossRef CAS PubMed
- G. H. Harris, K. Hoogsteen, K. C. Silverman, S. L. Raghoobar, G. F. Bills, R. B. Lingham, J. L. Smith, H. W. Dougherty, C. Cascales and F. Peláez, Tetrahedron, 1993, 49, 2139–2144 CrossRef CAS
- F. Mayerl, Q. Gao, S. Huang, S. Klohr, J. Matson, D. Gustavson, D. Pirnik, R. Berry, C. Fairchild and W. Rose, J. Antibiot., 1993, 46, 1082–1088 CrossRef CAS PubMed
- R. J. Cox and A. Al Fahad, Curr. Opin. Chem. Biol., 2013, 17, 532–536 CrossRef CAS PubMed
- A.-R. Ballester, M. Marcet-Houben, E. Levin, N. Sela, C. Selma-Lázaro, L. Carmona, M. Wisniewski, S. Droby, L. González-Candelas and T. Gabaldón, Mol. Plant-Microbe Interact., 2015, 28, 232–248 CrossRef CAS PubMed
- M. P. Artigot, N. Loiseau, J. Laffitte, L. Mas-Reguieg, S. Tadrist, I. P. Oswald and O. Puel, Microbiology, 2009, 155, 1738–1747 CrossRef CAS PubMed
- C. Zaehle, M. Gressler, E. Shelest, E. Geib, C. Hertweck and M. Brock, Chem. Biol., 2014, 21, 719–731 CrossRef CAS PubMed
- Y. Terabayashi, M. Sano, N. Yamane, J. Marui, K. Tamano, J. Sagara, M. Dohmoto, K. Oda, E. Ohshima, K. Tachibana, Y. Higa, S. Ohashi, H. Koike and M. Machida, Fungal Genet. Biol., 2010, 47, 953–961 CrossRef CAS PubMed
- Y. Terabayashi, M. Sano, N. Yamane, J. Marui, K. Tamano, J. Sagara, M. Dohmoto, K. Oda, E. Ohshima, K. Tachibana, Y. Higa, S. Ohashi, H. Koike and M. Machida, Fungal Genet. Biol., 2010, 47, 953–961 CrossRef CAS PubMed
- R. Yamada, T. Yoshie, S. Wakai, N. Asai-Nakashima, F. Okazaki, C. Ogino, H. Hisada, H. Tsutsumi, Y. Hata and A. Kondo, Microb. Cell Fact., 2014, 13, 71–78 CrossRef PubMed
Footnote |
| † Dedicated to professor T. J. Simpson to mark his retirement. |
| This journal is © The Royal Society of Chemistry 2018 |