
An Fe(II) metal–organic framework as a visible responsive photo-Fenton catalyst for the degradation of organophosphates†
Wen-Juan
Li‡
a,
Yue
Li‡
ab,
Di
Ning
a,
Qiao
Liu
a,
Lan
Chang
a and
Wen-Juan
Ruan
 *ab
*ab
aCollege of Chemistry, Nankai University, Tianjin 300071, China. E-mail: wjruan@nankai.edu.cn
bKey Laboratory of Advanced Energy Materials Chemistry (Ministry of Education), Nankai University, Tianjin 300071, China
First published on 24th November 2017
Abstract
An Fe(II) metal–organic framework with micro-sized particles was synthesized. This material could serve as a visible responsive photo-Fenton catalyst to induce the mineralization of a series of organophosphates, and exhibited high activity, high stability and wide pH tolerance. The degradation mechanism of organophosphates in this photocatalytic system was illustrated by different methods.
Organophosphates are a large kind of pesticides used in modern agriculture for pest control.1 Containing ∼150 different chemicals, this common class of compounds accounts for more than one third of the total pesticides used in the world.2 Because of their broad use and persistence under natural conditions, the residues of organophosphates have been commonly detected in surface and ground water and have become a serious environmental problem in many countries.3 Most organophosphates are highly toxic, and could attack the nervous system to induce a series of toxic symptoms.4 The carcinogenicity and teratogenicity of long periods of exposure to organophosphates were also illustrated by some studies.5 Due to the wide distribution and adverse effects of organophosphates, the development of efficient treatment technologies for this kind of contaminant is in urgent need.
Advanced oxidation processes (AOPs), which are characterized by the in situ generation of ˙OH under mild conditions, have been the subject of interest for the decomposition of recalcitrant pollutants.6 Among various AOPs, photo-Fenton technology is extremely suitable for the treatment of organophosphates, because it usually shows the highest efficiency in the degradation of this kind of pollutant.7 Compared with the traditional photo-Fenton system which has the drawbacks of iron sludge production and narrow pH range, the heterogeneous version of this technology is more favorable for industrial application.8 However, most of the developed heterogeneous photo-Fenton catalysts are limited to inorganic minerals, including clay-based Fe nanocomposites,9 goethite,10 and Fe-exchanged zeolites.11 These photocatalysts can only work under the irradiation of UV light that comprises a small fraction of the solar spectrum. The modulation of the coordination sphere of metal ions in the heterogeneous photo-Fenton catalyst is the most direct way to extend its response wavelength, and the development of metal–organic frameworks (MOFs) has made this strategy possible. Constructed by metal ions and bridging ligands, MOFs present incomparable flexibility of chemical composition.12,13 Because their absorption could be conveniently changed by the reasonable selection of organic ligands, MOFs show great potential as a new kind of photocatalytic material.14,15
In this work, an Fe(II) MOF with micro-sized particles (Fe-pydc) was synthesized as a heterogeneous photo-Fenton catalyst. Under visible light irradiation, a series of organophosphates could be efficiently degraded, and even mineralized by this MOF catalyst. The degradation mechanism was also preliminarily discussed.
Fe-pydc was obtained as bronzing powder from the solvothermal reaction between FeSO4 and 3,5-pyridinedicarboxylic acid (H2pydc) in the mixed solvent of H2O–DMF (v/v = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). The SEM observation showed that this sample presents a micro-sized prism morphology (Fig. 1a). The sharp edges and smooth surfaces of the particles exhibit their good crystallinity. The crystallinity of this sample was further confirmed by the well-defined diffraction peaks recorded in the powder X-ray diffraction (PXRD) pattern (Fig. 1b). All the peaks have close positions with those of the simulated pattern from the previously reported bulk phase of [Mn(pydc)·2H2O] (Fig. S1, ESI†),16 indicating that our sample presents the same framework structure.
:1). The SEM observation showed that this sample presents a micro-sized prism morphology (Fig. 1a). The sharp edges and smooth surfaces of the particles exhibit their good crystallinity. The crystallinity of this sample was further confirmed by the well-defined diffraction peaks recorded in the powder X-ray diffraction (PXRD) pattern (Fig. 1b). All the peaks have close positions with those of the simulated pattern from the previously reported bulk phase of [Mn(pydc)·2H2O] (Fig. S1, ESI†),16 indicating that our sample presents the same framework structure.
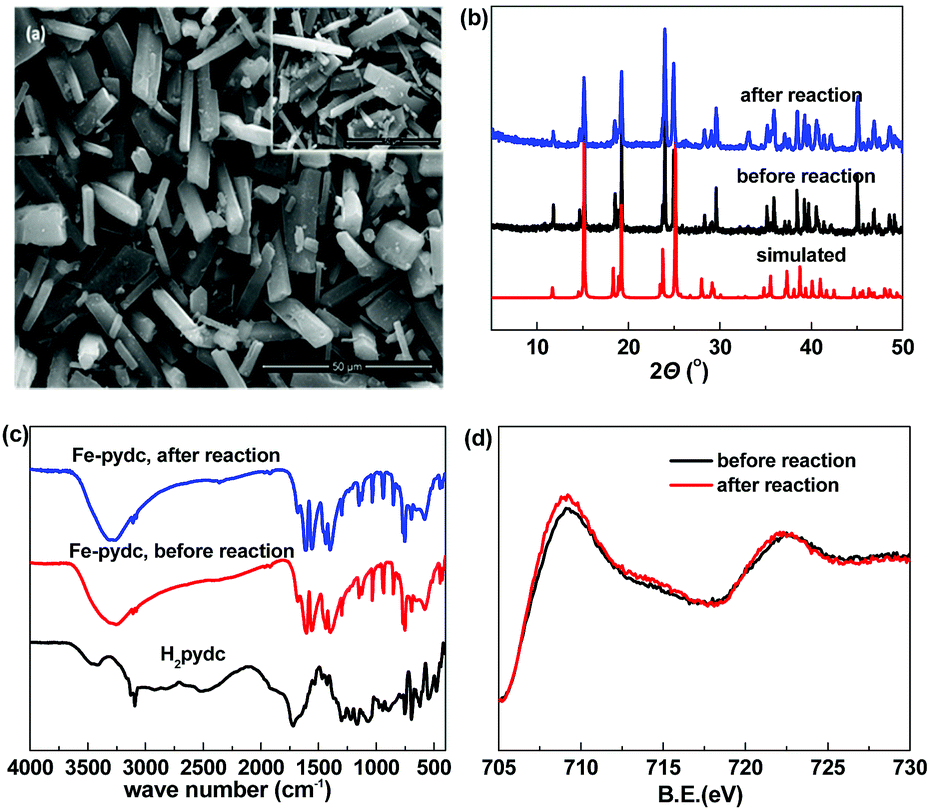 | ||
| Fig. 1 (a) SEM images, (b) PXRD patterns, (c) FT-IR spectra and (d) XPS spectra of the Fe-pydc sample. Inset of (a): SEM image of the used Fe-pydc catalyst. | ||
The structure of Fe-pydc was further characterized by elemental analysis, thermogravimetry, FT-IR and XPS. According to the elemental analysis, the contents of C, H and N of our sample are 31.40%, 2.49% and 5.28%, respectively. Combined with the framework structure obtained by PXRD comparison, the empirical formula of Fe-pydc was speculated to be [Fe(pydc)(H2O)2]·0.5H2O (MW: 265.97; calcd: 31.61% C, 3.03% H, 5.27% N). This formula was validated by the thermogravimetric curve (Fig. S2, ESI†), in which two stages of weight loss were recorded and their measured and calculated values have good consistency. The first weight loss in the temperature range of 100–173 °C could be attributed to the loss of 2.5 H2O molecules with the insertion of 0.5 oxygen atoms to oxidize Fe(II) to Fe(III) (obsd 13.9%, calcd 13.9%). When the sample was heated above 364 °C, the second stage of weight loss occurred (obsd 52.8%, calcd 56.0%), which corresponds to the removal of organic components with the residue of Fe2O3. Fig. 1c gives the FT-IR spectrum of Fe-pydc. The broad band around 3316 cm−1 is assigned to the O–H stretching vibration of coordinated H2O molecules.17 The stretching vibration of the C–H bonds on the pyridine ring of the pydc2− ligand gives absorption peaks at 3080 and 3109 cm−1.18 The carboxyl groups give two groups of doublet peaks around 1580 and 1419 cm−1, while no characteristic band is observed in the range of 1690–1730 cm−1, which implies the complete deprotonation of H2pydc upon the reaction with metal ions.19 The bands of Fe–O at 852, 768 and 694 cm−1 confirm the coordination of the pydc2− ligand to the Fe ion.20 The chemical valence of Fe in Fe-pydc was examined by XPS analysis. As shown by the Fe 2p binding spectrum (Fig. 1d), the Fe 2p1/2 and Fe 2p3/2 peaks of Fe-pydc are centered respectively at 722.5 and 709.1 eV, which are consistent with Fe(II) species.21,22
The UV-Vis diffuse reflectance spectrum (Fig. S3, ESI†) shows that Fe-pydc displays a very broad adsorption band covering the range from 200 to 600 nm. Neither the Fe2+ ion nor pydc2− ligand has absorption above 400 nm, so the visible absorption of Fe-pydc can only be attributed to the charge transfer transition between the ligand and metal ion. Such an extended coverage in the visible spectrum makes it possible for Fe-pydc to utilize most of the energy from sunlight.
Bis(p-nitrophenyl) phosphate (BNPP) was selected as the primary substrate to investigate the photocatalytic property of Fe-pydc. As shown in Fig. 2a, this substrate could be rapidly degraded under visible irradiation in the suspension containing Fe-pydc and H2O2. Under the photocatalysis of only 0.01 g L−1 Fe-pydc, nearly complete elimination (conv. > 90%) of 0.05 mM of BNPP was achieved in 90 min, showing the high activity of our MOF sample. In control experiments, little degradation occurred in the presence of H2O2 or Fe-pydc alone under visible irradiation. When the reaction was carried out in the dark (with the coexistence of H2O2 and Fe-pydc), the BNPP consumption rate was much slower than that under irradiation. These results demonstrate that both Fe-pydc and H2O2 are indispensable for the degradation of BNPP, and visible irradiation indeed remarkably accelerates this reaction. We also tested the degradation of other organophosphates, such as paraoxon-ethyl (DENP) and tris(4-nitrophenyl) phosphate (TNPP), in this photocatalytic system (Fig. S4, ESI†). These two compounds (0.05 mM) could be completely decomposed within 180 and 300 min, respectively. The high removal rates observed in these experiments show the wide applicability and high efficiency of our MOF catalyst in the treatment of organophosphates.
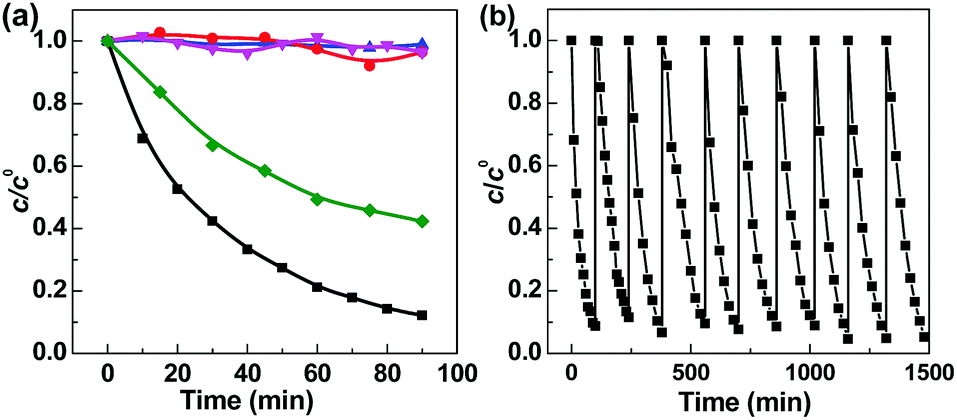 | ||
Fig. 2 (a) Degradation of BNPP under different conditions: (black) visible irradiation in the presence of Fe-pydc and H2O2, (red) visible irradiation in the presence of Fe-pydc without H2O2, (blue) visible irradiation in H2O2 solution without Fe-pydc, (green) dark reaction in the presence of Fe-pydc and H2O2, and (magenta) after the degradation of BNPP, the catalyst was separated while BNPP and H2O2 were added to the initial concentrations. 0.01 g L−1 Fe-pydc, c0BNPP = 0.05 mM, 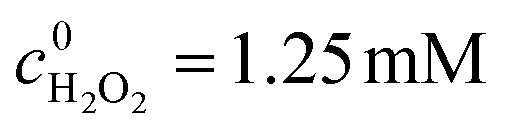 , 200 mL H2O, irradiated with a 300 W Xe lamp cutoff below 420 nm. (b) Recycling experiments of Fe-pydc in BNPP degradation. At the end of each run, BNPP and H2O2 were supplemented to the initial concentrations. , 200 mL H2O, irradiated with a 300 W Xe lamp cutoff below 420 nm. (b) Recycling experiments of Fe-pydc in BNPP degradation. At the end of each run, BNPP and H2O2 were supplemented to the initial concentrations. | ||
After the completion of photocatalysis, Fe-pydc was separated from the reaction medium by filtration, while H2O2 and BNPP were replenished to the original concentrations. The irradiation of the filtrate caused no remarkable reaction (Fig. 2a), which not only proves that Fe-pydc assuredly works as a heterogeneous photocatalyst in the reaction but also implies its satisfactory stability. Further evidence supporting the stability of Fe-pydc came from the characterization of the used Fe-pydc photocatalyst and recycling experiments. In general, the PXRD diffraction peaks, FT-IR spectrum, Fe 2p binding energies and morphology of the used Fe-pydc were almost the same as those of the as-synthesized sample (Fig. 1), which manifests that the structure of Fe-pydc was hardly changed during the reaction. In recycling experiments, the catalytic activity of Fe-pydc was maintained after 10 consecutive runs of photodegradation without remarkable decrease (Fig. 2b). This reusability is superior to most of the recently reported MOF photocatalysts.23–26
To investigate the pH tolerance of Fe-pydc, a group of degradation experiments were conducted with different initial acidities (Fig. 3). Before irradiation and H2O2 addition, a solution of HClO4 or NaOH was added to a suspension containing Fe-pydc and BNPP to modulate its pH to the target value. As exhibited in Fig. 3, the degradation of BNPP could smoothly proceed in the pH range of 2–6. Although the reaction rate slightly reduced at pH = 7, the concentration of BNPP could still be more than halved in 1 h. The reaction could only be stopped when the pH value was enhanced to above 8, probably due to the decomposition of the Fe-pydc photocatalyst. These results indicate that Fe-pydc can work in environments from acidic to neutral. This wide pH tolerance is much extended relative to the traditional photo-Fenton system (2.5–3.5)27 and more suitable for the treatment of real effluents.
 | ||
Fig. 3 Degradation kinetics of BNPP at different pH values in the photocatalytic system of Fe-pydc. 0.01 g L−1 Fe-pydc, c0BNPP = 0.05 mM,  , 200 mL H2O, irradiated with a 300 W Xe lamp cutoff below 420 nm. , 200 mL H2O, irradiated with a 300 W Xe lamp cutoff below 420 nm. | ||
In our photocatalytic system, H2O2 acts as an exogenous oxidant, so its utilization efficiency directly affects the running cost of this technology in water purification. The concentration measurement of H2O2 showed that the decompositions of H2O2 and BNPP occurred synchronously with the consumption ratio of 5.96:1 (Fig. 4a). We also investigated the photocatalytic reaction of H2O2 (Fig. 4b). In the absence of BNPP, H2O2 was basically stable under the photocatalysis of Fe-pydc. No remarkable concentration change of H2O2 was observed during 50 min of irradiation. The consumption of H2O2 occurred only after the artificial addition of BNPP. Since the catalytic decomposition of H2O2 by Fe-pydc under visible irradiation is negligible, H2O2 is expected to be consumed stoichiometrically by the reaction with BNPP in the photocatalytic system and thus give high utilization efficiency.
 | ||
Fig. 4 (a) Consumption of H2O2 with the degradation of BNPP. 0.01 g L−1 Fe-pydc, c0BNPP = 0.05 mM,  , 200 mL H2O, irradiated with a 300 W Xe lamp cutoff below 420 nm. (b) Comparison of the photocatalytic decompositions of H2O2 in the presence and absence of BNPP. , 200 mL H2O, irradiated with a 300 W Xe lamp cutoff below 420 nm. (b) Comparison of the photocatalytic decompositions of H2O2 in the presence and absence of BNPP. | ||
As in traditional photo-Fenton processes, ˙OH was speculated to be the main active species in the photocatalytic system of Fe-pydc. Electron spin resonance (ESR) with 5,5-dimethyl-1-pyrroline N-oxide (DMPO) as a spin-trapper was employed to examine the generation of ˙OH (Fig. S5, ESI†). When H2O2 was injected into the suspension containing Fe-pydc and BNPP, the characteristic signal of the DMPO–˙OH adduct (fourfold peaks with intensity ratio of 1:2:2:1) appeared.28 The irradiation of the suspension with visible light could largely enhance the intensity of this signal, showing that the generation of ˙OH was facilitated. This result is in accordance with the much higher degradation rate of photocatalysis than that of the dark reaction. It is reasonable that the visible light induced ligand to metal charge transfer smoothens the regeneration of Fe(II) species in Fe-pydc and thus enhances the catalytic efficiency.
To illustrate the degradation mechanism of organo-phosphates in this photocatalytic system, the oxidation intermediates of BNPP were identified by ESI-MS. Other than the peak of the original substrate (m/z = 339.0 [BNPP − H]−), additional peaks at m/z 354.9, 234.3, 218.3 and 138.1 were shown in the negative mode mass spectrum (Fig. 5). Since ˙OH works as the main active species in the reaction system and the benzene ring is prone to the attack of this radical, it is rational that the hydroxylated product of BNPP (1 in Scheme 1, m/z = 354.9) was observed to be the main degradation intermediate. The peaks at m/z 218.3 and 138.1 could be assigned to mono(p-nitrophenyl) phosphate (2) and 4-nitrophenol (3), the hydrolysis products of BNPP. Since the contact of BNPP with Fe-pydc (without H2O2) caused no remarkable reaction, this hydrolysis process is likely to be induced by the ipso-addition of ˙OH to the benzene ring.29 When the substrate got through one hydroxylation and one hydrolysis reaction, it produced intermediate 4, which gave MS peak at m/z 234.3. Based on the above results, we conclude that both the hydroxylation of the benzene ring and the hydrolysis of the P–O bond are the primary degradation pathways of BNPP in the photocatalytic system of Fe-pydc.
 | ||
| Fig. 5 ESI-MS of the degraded solution of BNPP. | ||
 | ||
| Scheme 1 Speculated degradation pathway of BNPP. | ||
The deep oxidation of BNPP was investigated by total organic carbon (TOC) measurement and ion chromatograph (Fig. 6). The TOC of the degraded solution decreased almost linearly with time from 7.20 to 1.95 ppm in 110 min. This ∼73% TOC removal shows that BNPP could be completely mineralized in this photocatalytic system. The mineralization of BNPP was also validated by the quantification of PO43− and NO3−, the final oxidation products of P and N. In the same period of reaction, 75% of P and 95% of N were converted to the corresponding inorganic ions.
 | ||
Fig. 6 Variation of TOC and concentration changes of PO43− and NO3− during the photocatalytic degradation of BNPP. 0.01 g L−1 Fe-pydc, c0BNPP = 0.05 mM,  , 200 mL H2O, irradiated with a 300 W Xe lamp cutoff below 420 nm. , 200 mL H2O, irradiated with a 300 W Xe lamp cutoff below 420 nm. | ||
In summary, a MOF-based heterogeneous photo-Fenton catalyst was developed and successfully applied to the degradation of organophosphates. Generating ˙OH as the main active species, Fe-pydc could induce the hydroxylation and hydrolysis of this kind of pollutant. Further oxidation would lead to the complete mineralization of organo-phosphates and convert C, P and N elements respectively to CO2, PO43− and NO3−. Fe-pydc could work in acidic to neutral environments, and exhibited high activity, stability, reusability and H2O2 utilization efficiency in our experiments. The results of this work illustrate the wide potential of MOFs in the photocatalytic degradation of recalcitrant pollutants.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21673121 and 21307062) and the Tianjin Research Program of Application Foundation and Advanced Technology (14JCQNJC08000).Notes and references
- K. V. Ragnarsdottir, J. Geol. Soc., 2000, 157, 859–876 CrossRef CAS.
- K. D. Racke, in Organophosphates Chemistry, Fate, and Effects, ed. P. E. L. Janice and E. Chambers, Academic Press, Boston, 1992, pp. 47–78 Search PubMed.
- H. Tse, M. Comba and M. Alaee, Chemosphere, 2004, 54, 41–47 CrossRef CAS PubMed.
- J. E. Casida and G. B. Quistad, Chem. Res. Toxicol., 2004, 17, 983–998 CrossRef CAS PubMed.
- B. Eskenazi, A. R. Marks, A. Bradman, K. Harley, D. B. Barr, C. Johnson, N. Morga and N. P. Jewell, Environ. Health Perspect., 2007, 115, 792–798 CrossRef CAS PubMed.
- M. Tokumura, A. Sugawara, M. Raknuzzaman, M. Habibullah-Al-Mamun and S. Masunaga, Chemosphere, 2016, 159, 317–325 CrossRef CAS PubMed.
- K. Ikehata and M. G. El-Din, J. Environ. Eng. Sci., 2006, 5, 81–135 CrossRef CAS.
- J. J. Pignatello, E. Oliveros and A. MacKay, Crit. Rev. Environ. Sci. Technol., 2006, 36, 1–84 CrossRef CAS.
- J. Feng, X. Hu and P. L. Yue, Environ. Sci. Technol., 2004, 38, 269–275 CrossRef CAS PubMed.
- M.-J. Liou and M.-C. Lu, J. Hazard. Mater., 2008, 151, 540–546 CrossRef CAS PubMed.
- M. B. Kasiri, H. Aleboyeh and A. Aleboyeh, Appl. Catal., B, 2008, 84, 9–15 CrossRef CAS.
- T. Zhang and W. Lin, Chem. Soc. Rev., 2014, 43, 5982–5993 RSC.
- H. Furukawa, K. E. Cordova, M. O’Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- Y. Horiuchi, T. Toyao, M. Saito, K. Mochizuki, M. Iwata, H. Higashimura, M. Anpo and M. Matsuoka, J. Phys. Chem. C, 2012, 116, 20848–20853 CAS.
- D. Sun, Y. Fu, W. Liu, L. Ye, D. Wang, L. Yang, X. Fu and Z. Li, Chem. – Eur. J., 2013, 19, 14279–14285 CrossRef CAS PubMed.
- X. Haitao, Z. Nengwu, X. Hanhuia, W. Yonggang, Y. Ruyi, Y. Enyi and J. Xianglin, J. Mol. Struct., 2002, 610, 47–52 CrossRef.
- Q. Wu, N. Xing, X. Liu, L. Xu, X. Ma, Z. Yan and Y. Xing, Polyhedron, 2015, 87, 390–397 CrossRef CAS.
- A. Nataraj, V. Balachandran, T. Karthick, M. Karabacak and A. Atac, J. Mol. Struct., 2012, 1027, 1–14 CrossRef CAS.
- H.-Y. Wang, S. Gao, L.-H. Huo, S. W. Ng and J.-G. Zhao, Cryst. Growth Des., 2008, 8, 665–670 CAS.
- Q.-B. Bo, G.-X. Sun and D.-L. Geng, Inorg. Chem., 2010, 49, 561–571 CrossRef CAS PubMed.
- A. P. Grosvenor, B. A. Kobe, M. C. Biesinger and N. S. McIntyre, Surf. Interface Anal., 2004, 36, 1564–1574 CrossRef CAS.
- T. Yamashita and P. Hayes, Appl. Surf. Sci., 2008, 254, 2441–2449 CrossRef CAS.
- T. Araya, M. Jia, J. Yang, P. Zhao, K. Cai, W. Ma and Y. Huang, Appl. Catal., B, 2017, 203, 768–777 CrossRef CAS.
- S. E.-d. H. Etaiw and M. M. El-bendary, Appl. Catal., B, 2012, 126, 326–333 CrossRef CAS.
- L. Ai, C. Zhang, L. Li and J. Jiang, Appl. Catal., B, 2014, 148, 191–200 CrossRef.
- Z.-Q. Li, M. Zhang, B. Liu, C.-Y. Guo and M. Zhou, Inorg. Chem. Commun., 2013, 36, 241–244 CrossRef CAS.
- P. Bautista, A. F. Mohedano, J. A. Casas, J. A. Zazo and J. J. Rodriguez, J. Chem. Technol. Biotechnol., 2008, 83, 1323–1338 CrossRef CAS.
- X.-j. Yang, X.-m. Xu, J. Xu and Y.-f. Han, J. Am. Chem. Soc., 2013, 135, 16058–16061 CrossRef CAS PubMed.
- X. Li and W. S. Jenks, J. Am. Chem. Soc., 2000, 122, 11864–11870 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, framework structure, TGA curve and absorption spectrum of Fe-pydc, ESR spectrum of the reaction system, and degradation kinetics of DENP and TNPP. See DOI: 10.1039/c7nj03815a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2018 |