
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Microwave-assisted synthesis of organically functionalized hexa-molybdovanadates†
H.
Karoui
and
C.
Ritchie
 *
*
School of Chemistry, The University of Melbourne, Parkville, Victoria 3010, Australia. E-mail: critchie@unimelb.edu.au
First published on 8th December 2017
Abstract
The microwave-assisted reaction of (TBA)4[β-Mo8O24] and (TBA)3[H3V10O28] with pentaerythritol or tris(hydroxymethyl)aminomethane yields polyanions with the general formula (TBA)2[V3Mo3O16(O3–R)] (R: C5H8OH – 1; R: C4H6NH2 – 3). Post-synthetic esterification of 1 yields the acylated derivative 2, with all compounds being characterized in the solid and solution state.
Isopolyoxometalates are oligomeric molecular metal oxides with notable structural diversity, while incorporation of heteroatoms into the metal oxide framework in the form of organic,1 organometallic,2 main group,3 transition metal,4 lanthanoid5,6 and actinoid7 components yield heteropolyoxometalates. Consequently, the properties of polyoxometalates (POMs) are equally extensive, resulting in their investigation in research areas encompassing molecular magnetism,8 catalysis,9–11 nano-materials,12,13 energy storage,14,15 and sensing applications.16,17
Modification of POM surfaces by covalently grafting organic ligands has been a long-standing strategy to generate hybrid molecular materials via one-pot or post-synthetic methodologies.18,19 The selection of synthetic approach is driven by the most efficient method to yield analytically pure products due to difficulties associated with the separation of POM mixtures.20 Post-synthetic modification is therefore most commonly utilized when targeting the introduction of sophisticated organic components that are challenging to cleanly install in one step.18,19 Despite the impressive range of inorganic–organic hybrids reported to date, the number of polyoxometalate (POM) platforms available for functionalisation is somewhat limited. The most intensively studied alkoxo functionalised POMs include the hexavanadate Lindqvist [V6O19{R}2]2− and the one and two-sided Anderson-Evans [M(Mo6O24){(OCH2)3C–R}1], [M(Mo6O24){(OCH2)3C–R}2] (R = CH3, NO2, CH2OH, NH2…) species.21,22 Additionally, the Wells-Dawson phosphovanadotungstates of general formula [RC(CH2O)3V3P2W15O59]6− (R = CH3, NO2, CH2OH) firstly reported by Hill et al. have been utilized as building blocks for structural and compositional diversification.23 Overall these compounds can generally be prepared in good yield and relatively high purity.
Recently, we reported the initial findings of our investigation targeting the microwave-assisted synthesis of molybdovanadates.24 As part of that work we discussed the facile preparation of the alkoxo-functionalized molybdovanadate (TBA)2[V3Mo3O16{C5H9O3}] from (TBA)4[β-Mo8O24], (TBA)3[H3V10O28] and tris(hydroxymethyl)ethane.24 As a continuation of that study, we established that the methodology could be extended to yield more synthetically useful derivatives.
Herein, we report the microwave-assisted synthesis and characterisation of alcohol, acetyl and amine terminated molybdovanadates of general formula (TBA)2[V3Mo3O16(O3–R)]; where R = (C5H9O1) 1, (C7H11O2) 2 and (C4H8N) 3. The three compounds are characterised in the solid state by single crystal X-ray diffraction, FT-IR and elemental analysis, meanwhile the solution stability and structural isomer distributions of 1–3 in acetonitrile using ESI-MS, UV-Vis, and NMR spectroscopy are reported.
Compounds 1 and 3 were prepared by reacting (TBA)4[β-Mo8O26] and (TBA)3[H3V10O28] in acetonitrile with either pentaerythritol (Tris-OH) or tris(hydroxymethyl)aminomethane (Tris-NH2) respectively, in the stoichiometric ratios found in the products. Microwave irradiation of the reaction mixture for 5 minutes at 110 °C and 4 bar, results in the formation of a dark brown solution that is subsequently purified via ether precipitation multiple times to yield crystalline products of 1 and 3 in 56% and 43% yield based on vanadium (see the ESI†). An acylation reaction of compound 1 with acetic anhydride, N,N-dimethylaminopyridine (DMAP) and triethylamine (TEA) in acetonitrile afforded compound 2 in 11% yield. The compositions of 1–3 were confirmed by ESI-MS (Fig. S11–S13 and Tables S1–S3 in ESI†), with the spectra of each compound containing two distinctive isotopic envelopes that are attributed to the mono- and di-anions of the parent clusters. The mono-anion envelopes for 1–3 are centred at m/z 1072.52, 1114.64 and 1072.37 respectively.
Crystallographic analysis of 1–3 reveals a mixture of structural isomers as observed for (TBA)2[V3Mo3O16{C5H9O3}], with the tripodal ligand being condensed to a vanadium rich triad, ({V3} or {V2Mo1}).22 This non-random distribution of V and Mo results in the polyanion metal–oxo core having C3v – 1A–3A, Cs – 1B–3B or C1 – 1C–3C point symmetry (Fig. 1). Despite the inability to distinguish isomers due to the averaging nature of X-ray diffraction, assignment of three distinct crystallographic metal sites with the following occupancies (site A – V, 0.75; Mo, 0.25, site B – V, 0.5; Mo, 0.5, site C – V, 0.25; Mo, 0.75) is possible (Scheme 1). Correspondingly, the metal–oxo bond lengths within the hexametallates can be clearly differentiated for the three crystallographically defined sites A–C (Fig. 1, Table 1). As expected the vanadium rich site A has the shortest average terminal M–Ot bonds (M–Ot; 1.615 Å), with the corresponding molybdenum rich site C having the longest average terminal M–Ot bonds (Mo–Ot; 1.666 Å). The molybdenum rich and ligand free triad ({Mo3} or {Mo2V1}) is somewhat compressed in comparison to the ligand grafted triad. Additional crystallographic details are provided in the ESI† (Table S7).
 | ||
| Fig. 1 Simplified mixed polyhedral, ball and stick representation of the structural isomers A–C and their distributions for compounds 1–3. Vanadium, orange spheres; molybdenum, blue spheres; carbon, black spheres; hydrogen, white spheres; oxygen, red spheres. | ||
 | ||
| Scheme 1 Synthetic scheme for the preparation of 1 and 3. Carbon, black spheres; hydrogen, white spheres; nitrogen, blue spheres; molybdenum, light blue spheres; oxygen, red spheres; vanadium, orange spheres. Metal site occupancies: site A: V 0.75, Mo 0.25 purple polyhedra; site B: V 0.5, Mo 0.5 teal polyhedra; site C: V 0.25, Mo 0.75 green polyhedra. | ||
| Compound | Site A | Site B | Site C | ||||||
|---|---|---|---|---|---|---|---|---|---|
| M–Ot | M–Ob | M–Oc | M–Ot | M–Ob | M–Oc | M–Ot | M–Ob | M–Oc | |
| 1 | 1.615 | 1.820–2.054 | 2.231 | 1.650 | 1.848–2.012 | 2.281 | 1.665 | 1.921 | 2.356 |
| 2 | 1.614 | 1.712–2.087 | 2.247 | 1.640 | 1.812–2.088 | 2.284 | 1.664 | 1.871–1.957 | 2.299 |
| 3 | 1.615 | 1.825–2.048 | 2.228 | 1.639 | 1.880–2.090 | 2.277 | 1.667 | 1.921 | 2.365 |
In addition to the crystallographic comparisons, NMR has proven very informative, with excellent correlation between the experimentally observed and theoretically predicted distribution of structural isomers for [V3Mo3O16(O3C5H9)]2−, with no structural re-arrangement in MeCN at room temperature. We anticipated that the same distribution and stability would be observed for 1–3, however, our findings reveal a more complicated situation (Fig. 1). Assignment of the 1H NMR spectrum of freshly prepared samples of 1 and 2 in D3-acetonitrile revealed the same preferential substitution pattern (A > B > C) as for [V3Mo3O16{C5H9O3}]2− with slightly higher populations of isomers B and C (Fig. 1 and Fig. S1, S2, ESI†). Unexpectedly this trend is reversed for the amine terminated polyanion (3C > 3B > 3A) with the relative populations determined by integration of 1H peak areas (Fig. 1 and Fig. S3, ESI†). The 1H NMR spectra of 1–3 resemble that of the previously reported [V3Mo3O16{C5H9O3}]2−, with deshielded methylene proton resonances between 4.50 and 5.25 ppm being due to their proximity to the electron deficient VV or MoVI (Fig. 1 and Tables S4–S6, ESI†). The C3v symmetry of isomers 1A–3A results in a single resonance for the methylene −(CH2)3–R protons of the grafted ligand, meanwhile a broad singlet and two sets of broad doublets- are observed for the Cs symmetrical 1B–3B isomers –(CHA2)(CHBC)2–R (Fig. S1–S3 and Tables S4–S6, ESI†). The singlet is observed for the two equivalent protons bisected by the mirror plane with the pair of doublets (J = 11.5 Hz) due to coupling between the two diastereotopic geminal protons HB and HC (Fig. 2). The meridional arrangement of the V atoms in isomers 1–3C/C′ results in chiral polyanions, where each of the methylene protons are inequivalent. Significant peak overlap complicates the unambiguous assignment of the methylene protons, nonetheless two doublets of doublets with coupling constants of (J = 11.5 Hz and 1.5 Hz) attributed to geminal and 4-bond “W” coupling are clearly identifiable in 2C/C′. Integration of peak areas associated with the methylene (3.9–4.02 ppm) and methyl (2.83–3.01 ppm) resonances of acetyl functionalised 2 assists with the assignment and relative population of isomers A–C.
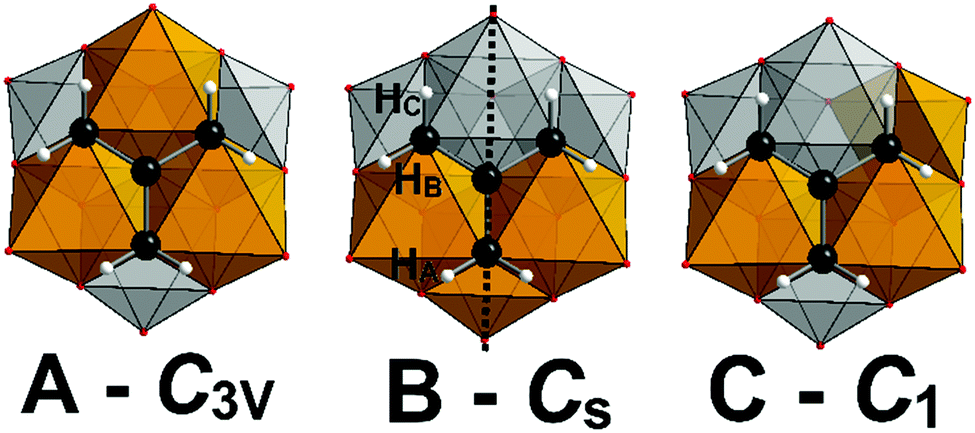 | ||
| Fig. 2 Mixed polyhedral, ball and stick representation of isomers A–C emphasising the methylene proton environments. Vanadium, orange polyhedra; molybdenum, grey polyhedra; carbon, black spheres; hydrogen, white spheres; oxygen, red spheres. | ||
Cyclic voltammetric analysis of compounds 1–3 in MeCN were conducted to gain some understanding of their electrochemical properties. All three compounds possess a one-electron quasi-reversible reduction wave (with E1/2 = −0.584 V vs. Fc/Fc+ for 1, E1/2 = −0.615 V vs. Fc/Fc+ for 2 and E1/2 = −0.572 V vs. Fc/Fc+ for 3); that we have assigned as reduction of one VV to VVI, based on previous data reported by Zubieta et al. on hexavanadates.25 Comparison of this redox couple with hexavanadate derivatives indicates a significant electron withdrawing influence of the three electron-deficient MoVI centres, with the one-electron reduced derivatives of [V2VV1IVMo3O16(OCH2)3–R]3− being stabilized with respect to the nitroso derivative of the structurally related hexavanadate [V5VV1IVO13{(OCH2)3CNO2}2]3− in line with the more positive first reduction wave.25 Furthermore, the insensitivity of the UV-Vis spectra to modifications of the organic ligand is in agreement with this observation, with the lowest lying electronic transition (for 2, 365 nm, ε = 0.7670 × 104 M−1 cm−1) being assigned as an LMCT band (Fig. 3).
 | ||
| Fig. 3 Cyclic voltammogram of 0.1 mM 2 in a 0.1 M TBA-PF6 electrolyte solution showing a quasi-reversible one-electron redox process. | ||
In conclusion, we report the extension of our previously established microwave-assisted synthesis of the hexamolybdovanadate [V3Mo3O16{C5H9O3}]2− to include alcohol 1 and amine 3 terminated derivatives. Post-synthetic derivatization of 1 to yield acetylated 2 verifies that this POM platform holds significant potential as a new building block that can be utilized in a wide range of functional materials. Attempts to separate the isomeric mixtures of these compounds is ongoing.
We thank the Australian Research Council (DE130100615) and the University of Melbourne for financial support.
Conflicts of interest
There are no conflicts to declare.Notes and references
- Z. Peng, Angew. Chem., Int. Ed., 2004, 43, 930–935 CrossRef CAS PubMed.
- B. Artetxe, S. Reinoso, L. San Felices, P. Vitoria, A. Pache, J. Martín-Caballero and J. M. Gutiérrez-Zorrilla, Inorg. Chem., 2015, 54, 241–252 CrossRef CAS PubMed.
- A. Tézé, M. Michelon and G. Hervé, Inorg. Chem., 1997, 36, 505–509 CrossRef.
- H. Lv, J. Song, Y. V. Geletii, J. W. Vickers, J. M. Sumliner, D. G. Musaev, P. Kögerler, P. F. Zhuk, J. Bacsa, G. Zhu and C. L. Hill, J. Am. Chem. Soc., 2014, 136, 9268–9271 CrossRef CAS PubMed.
- C. Ritchie, V. Baslon, E. G. Moore, C. Reber and C. Boskovic, Inorg. Chem., 2012, 51, 1142–1151 CrossRef CAS PubMed.
- C. Ritchie, E. G. Moore, M. Speldrich, P. Kögerler and C. Boskovic, Angew. Chem., Int. Ed., 2010, 49, 7702–7705 CrossRef CAS PubMed.
- S. S. Mal, M. H. Dickman and U. Kortz, Chem. – A Eur. J., 2008, 14, 9851–9855 CrossRef CAS PubMed.
- S. Cardona-Serra, J. M. Clemente-Juan, E. Coronado, A. Gaita-Ariño, N. Suaud, O. Svoboda, R. Bastardis, N. Guihéry and J. J. Palacios, Chem. – A Eur. J., 2015, 21, 763–769 CrossRef CAS PubMed.
- S.-S. Wang and G.-Y. Yang, Chem. Rev., 2015, 115, 4893–4962 CrossRef CAS PubMed.
- H. Lv, Y. V. Geletii, C. Zhao, J. W. Vickers, G. Zhu, Z. Luo, J. Song, T. Lian, D. G. Musaev and C. L. Hill, Chem. Soc. Rev., 2012, 41, 7572–7589 RSC.
- K. Kastner, A. J. Kibler, E. Karjalainen, J. A. Fernandes, V. Sans and G. N. Newton, J. Mater. Chem. A, 2017, 5, 11577–11581 CAS.
- S. Landsmann, M. Luka and S. Polarz, Nat. Commun., 2012, 3, 1299 CrossRef PubMed.
- J. Li, X. Li, J. Xu, Y. Wang, L. Wu, Y. Wang, L. Wang, M. Lee and W. Li, Chem. – A Eur. J., 2016, 22, 15751–15759 CrossRef CAS PubMed.
- S. Herrmann, C. Ritchie and C. Streb, Dalton Trans., 2015, 44, 7092–7104 RSC.
- S. Herrmann, N. Aydemir, F. Nägele, D. Fantauzzi, T. Jacob, J. Travas-Sejdic and C. Streb, Adv. Funct. Mater., 2017, 27, 1700881 CrossRef.
- M. Ortiz, A. M. Debela, M. Svobodova, S. Thorimbert, D. Lesage, R. B. Cole, B. Hasenknopf and C. K. O’Sullivan, Chem. – Eur. J., 2017, 23, 10597 CrossRef CAS PubMed.
- A. M. Kaczmarek, J. Liu, B. Laforce, L. Vincze, K. Van Hecke and R. Van Deun, Dalton Trans., 2017, 46, 5781–5785 RSC.
- A. Proust, B. Matt, R. Villanneau, G. Guillemot, P. Gouzerh and G. Izzet, Chem. Soc. Rev., 2012, 41, 7605–7622 RSC.
- A. Dolbecq, E. Dumas, C. R. Mayer and P. Mialane, Chem. Rev., 2010, 110, 6009–6048 CrossRef CAS PubMed.
- C. Yvon, A. Macdonell, S. Buchwald, A. J. Surman, N. Follet, J. Alex, D.-L. Long and L. Cronin, Chem. Sci., 2013, 4, 3810–3817 RSC.
- P. R. Marcoux, B. Hasenknopf, J. Vaissermann and P. Gouzerh, Eur. J. Inorg. Chem., 2003, 2406–2412 CrossRef CAS.
- D. Hou, G.-S. Kim, K. S. Hagen and C. L. Hill, Inorg. Chim. Acta, 1993, 211, 127–130 CrossRef CAS.
- Y. Hou and C. L. Hill, J. Am. Chem. Soc., 1993, 115, 11823–11830 CrossRef CAS.
- S. Spillane, R. Sharma, A. Zavras, R. Mulder, C. A. Ohlin, L. Goerigk, R. A. J. O’Hair and C. Ritchie, Angew. Chem., Int. Ed., 2017, 56, 8568–8572 CrossRef CAS PubMed.
- Q. Chen, D. P. Goshorn, C. P. Scholes, X. L. Tan and J. Zubieta, J. Am. Chem. Soc., 1992, 114, 4667–4681 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1553741–1553743. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7nj02820b |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2018 |